

SUMMARY
Host factors required for viral replication are ideal drug targets because they are less likely than viral proteins to mutate under drug-mediated selective pressure. Although genome-wide screens have identified host proteins involved in influenza virus replication, limited mechanistic understanding of how these factors affect influenza has hindered potential drug development. We conducted a systematic analysis to identify and validate host factors that associate with influenza virus proteins and affect viral replication. After identifying over one thousand host factors that co-immunoprecipitate with specific viral proteins, we generated a network of virus-host protein interactions based on the stage of the viral lifecycle affected upon host factor down-regulation. Using compounds that inhibit these host factors, we validated several proteins, notably Golgi-specific brefeldin A resistant guanine nucleotide exchange factor (GBF1) and JAK1, as potential antiviral drug targets. Thus, virus-host interactome screens are powerful strategies to identify targetable host factors and guide antiviral drug development.
INTRODUCTION
Viruses, which rely on host cellular functions to replicate, hijack the host cell machinery and re-wire it for their own needs. A comprehensive understanding of host-virus interactions would greatly improve our understanding of the viral life cycle and be invaluable in identifying strategies to prevent or treat potentially deadly virus infections.
Influenza viruses cause annual epidemics and recurring pandemics, which have claimed millions of lives and had a considerable impact on public health and the global economy. Recent sporadic human infections with avian viruses of the H5N1 and H7N9 subtypes have raised concerns about the pandemic potential of these viruses (Gao et al., 2013; Li et al., 2014; Webster and Govorkova, 2006; Yen and Webster, 2009). Two antiviral drugs (that inhibit the ion channel (M2) or neuraminidase (NA) proteins) are available (Davies et al., 1964; Hayden, 2001), but the emergence of drug-resistant viruses has become a serious problem (Bright et al., 2005; Bright et al., 2006; Dawood et al., 2009; Nicoll et al., 2008). Therefore, there is an urgent need to identify targets for antiviral drugs.
In recent years, six genome-wide screens have identified a total of 1,449 human genes (including 110 human orthologs of Drosophila genes) with potential roles in the life cycle of influenza virus (Brass et al., 2009; Hao et al., 2008; Karlas et al., 2010; Konig et al., 2010; Shapira et al., 2009; Sui et al., 2009). Meta-analyses revealed limited overlap among these studies (de Chassey et al., 2012; Mehle and Doudna, 2010; Watanabe et al., 2010). This limited overlap may be caused by differences in the experimental conditions of the screens. Also, the experimental methods used in the screens might be suboptimal to investigate the whole life cycle of influenza viruses [e.g., using non-permissive cells for influenza virus infection and/or non-authentic influenza virus (i.e., recombinant viruses possessing reporter genes)]. Moreover, the criteria used to determine the candidate host factors likely differed among the screens and each screen might include a number of false positives. More importantly, most of these studies validated only subsets of potential host interaction factors, and only a few of the validated candidates were assessed for their function(s) in the viral life cycle. We, therefore, used authentic influenza virus and a human cell line permissive for influenza virus replication to conduct a systematic analysis of influenza viral host interaction partners, which was followed by extensive validation studies and a systematic assessment of the functional roles of these host proteins in influenza virus replication,. This information was then used to identify targets for antiviral drugs.
RESULTS AND DISCUSSION
Identification of host proteins that co-precipitate with 11 viral proteins of influenza A virus and are involved in viral replication
We first attempted to establish a comprehensive map of viral-host protein interactions in human embryonic kidney (HEK) 293 cells, which support influenza virus replication (Hatta et al., 2007; Le Ru et al., 2010). Eleven FLAG-tagged viral proteins (i.e., PB2, PB1, PA, HA, NP, NA, M1, M2, NS1, NS2, and PB1-F2, which represent all of the viral proteins with the exception of the recently identified potential accessory factors) of an influenza A virus (A/WSN/33, H1N1 subtype; WSN) were individually expressed in HEK 293 cells, and then immunoprecipitated with an anti-FLAG antibody. Mass spectrometry analyses of the co-precipitated proteins identified 1,292 host proteins in total: 388, 322, 304, 351, 574, 675, 659, 531, 113, 42, and 81 host proteins co-precipitated with the viral PB2, PB1, PA, HA, NP, NA, M1, M2, NS1, NS2, and PB1-F2 proteins, respectively (Figure 1 and Table S1; note that the data for NS2 were reported previously (Gorai et al., 2012)).
Figure 1. Overview of a systematic study to elucidate the physical and functional host-viral interactions in influenza virus replication, and to identify antiviral drugs.

(A, B) Schematic diagram of the identification of host proteins that co-precipitated with 11 influenza A viral proteins and affected viral replication. (A) Mass spectrometry analysis identified 1,292 host proteins that co-immunoprecipitated with one or more of the 11 FLAG-tagged influenza viral proteins. (B) To identify host factors that affect virus replication, cells were transfected with siRNAs targeted to each of the 1,292 candidate host genes and were then infected with influenza virus. Virus titers and cell viability were then determined. We identified 323 host genes whose mRNA levels were down-regulated, while virus titers were reduced by more than two log10 units compared with a control (299 host factors) or increased by more than one log10 unit (24 host factors). (C) To better understand the role of the identified host factors, we performed mechanistic studies assessing different steps in the viral life cycle for our ‘top hits’, that is, 91 host factors whose siRNA-mediated down-regulation reduced viral replication in cultured cells by at least three log10 units while retaining >80% cell viability. (D) To identify antiviral drugs for influenza virus, we searched for drugs targeting the 299 host factors identified here and selected 11 drugs for in vitro testing. See also Figures S1–4 and Tables S1–3.
The co-precipitated host proteins may be specific binding partners of influenza viral proteins with essential or non-essential functions in the viral life cycle. Alternatively, they may be non-specific, that is, false-positive, binding partners resulting from experimental artifacts such as the overexpression of viral proteins in our assay and/or the absence of other viral components. Therefore, to identify host factors that are specifically involved in virus replication, we transfected HEK 293 cells with siRNAs targeted to each of the 1,292 candidate host genes (two siRNAs for each host gene were used, as shown in Table S2; AllStars Negative Control siRNA (QIAGEN) was used as a negative control), infected cells with WSN virus at 24 h post-transfection, and then harvested the culture supernatants for virus titration at 48 h post-infection. Virus titers were determined by using plaque assays, which are a well-established and reliable method of virus titration in influenza virus research. In parallel, we examined the cell viability of siRNA-transfected HEK 293 cells by using the CellTiter-Glo assay, which determines the number of viable cells in culture by quantifying the ATP levels and thereby signaling the presence of metabolically active cells (see also Experimental Procedures). We identified 323 host genes whose mRNA levels were down-regulated, as confirmed by qRT-PCR, that reduced virus titers by more than two log10 units (299 host factors) or increased virus titers by more than one-log10 unit (24 host factors) compared with the control siRNA (see Experimental Procedures; Figure S1 and Table S2). Moreover, down-regulation of these host factors did not reduce cell viability by more than 40% (Table S2). For the set of 323 host factors that co-immunoprecipitated with viral proteins and affected influenza virus replication, we generated a network of virus-host protein interactions that may have critical roles in influenza virus replication (Figure 2). Sixty-three of these host factors had been identified previously (Brass et al., 2009; Hao et al., 2008; Karlas et al., 2010; Konig et al., 2010; Shapira et al., 2009; Sui et al., 2009) (Table S3 and Figure S2). Gene Ontology and pathway analyses revealed that the host factors identified here are involved in various cellular functions, including many ‘house-keeping’ processes such as transcription, translation, cell cycle, and mRNA splicing mechanisms (Supplemental text, Figure S3 and Table S4).
Figure 2. Network of host-influenza viral protein interactions.
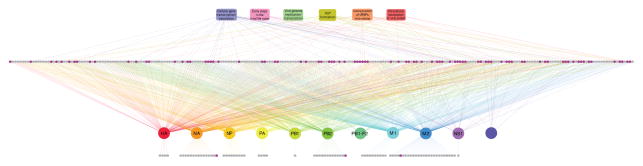
Interactions among the viral proteins and the 323 host factors identified here (gray and magenta circles) were visualized by using Cytoscape (http://cytoscape.org/). ‘Top hits’ (for a definition, see text and legend to Figure 1) are shown in magenta. Also shown are the steps in the viral life cycle affected by down-regulation of the respective host factor. The network image is fully-zoomable on the monitor. See also Tables S2 and S5.
The roles of the identified host factors in the influenza virus life cycle
Most of the previous genome-wide screens identified the affected viral life cycle step(s) for only a limited number of the host factors identified in their experiments (Brass et al., 2009; Hao et al., 2008; Karlas et al., 2010; Konig et al., 2010; Shapira et al., 2009; Sui et al., 2009). Because such information is essential for a better mechanistic understanding of the viral life cycle and to identify drug targets, we performed mechanistic studies for our ‘top hits’, namely the 91 host factors whose siRNA-mediated down-regulation reduced viral replication in cultured cells by at least three log10 units while retaining >80% cell viability (Figures 1–3, and Table S5). As described above, we confirmed that the siRNAs targeting the 91 ‘top hits’ decreased mRNA levels by qRT-PCR.
Figure 3. Effects of siRNA-mediated down-regulation of the 91 ‘top hits’ on the influenza virus life cycle.

A summary of the effects of siRNA-mediated down-regulation of the 91 ‘top hits’ on influenza virus replication steps. The percentages indicate the relative efficiency compared with the negative control and correspond to the values presented in Table S5. For factors with a significant effect on the early steps in the viral life cycle, polymerase activity, or VLP formation, we did not test the efficiency of vRNA and NP virion incorporation; these factors are shown in gray in the respective columns.
First, we assessed whether siRNA-mediated down-regulation of the ‘top hits’ affected cellular gene transcription and/or translation by measuring the expression levels of the Renilla luciferase reporter protein expressed under the control of a cellular RNA polymerase II promoter. We found that siRNAs targeting 28 host factors significantly reduced the activity of Renilla luciferase (expressed under the control of a cellular RNA polymerase II promoter) by more than 80% (p value < 0.05; Figure 3, and Tables S5A and B), suggesting that depletion of these host factors inhibited cellular transcription and/or translation, which indirectly inhibited influenza virus replication. Nonetheless, some of these factors may also have specific roles in the viral life cycle. For example, down-regulation of several host factors in this category also affected the intracellular localization of influenza virus proteins as determined by immunofluorescence studies that detected the viral HA, NA, NP, and M1 proteins in siRNA-treated and virus-infected cells (Figures 4, Figure S4, and Table S5A).
Figure 4. Effects of selected siRNAs targeting the 91 ‘top hits’ on the intracellular localization of viral proteins in infected cells.

To examine whether the down-regulation of the 91 ‘top hits’ affects the intracellular localization of the viral proteins in virus-infected cells, siRNA-transfected HEK 293 cells were infected with 200 pfu of WSN virus per well of a 24-well tissue culture plate, fixed at 12 h post-infection, and then stained with an anti-HA, anti-NA, anti-NP, or anti-M1 antibody. The intracellular localization of HA (A), NA (B), and NP (C) is shown. None of the siRNAs affected M1 localization. See also Figure S4 and Table S5.
To test the role of the ‘top hits’ in viral genome replication and transcription, we measured the activity of the viral replication complex (which comprises the PB2, PB1, PA, and NP proteins) based on its ability to replicate a virus-like RNA encoding the firefly luciferase reporter protein in a mini-replicon assay as described previously (Octaviani et al., 2010). We identified nine host factors (BUB3, CCDC56, CLTC, CYC1, NIBP, ZC3H15, C14orf173, CTNNB1, and ANP32B) that are critical for viral genome replication and transcription because their down-regulation significantly decreased the relative viral RNA polymerase activity by more than 50% compared with a control (p value < 0.05; Tables S5A and B; see also Figure 3 for an overview). None of these factors affected host protein synthesis, suggesting a specific effect on viral replication. BUB3, a mitotic checkpoint protein, and CTNNB1, catenin (cadherin-associated protein) beta 1, were also detected in the genome-wide screens conducted by Brass et al. (Brass et al., 2009), Shapira et al. (Shapira et al., 2009), and Karlas et al. (Karlas et al., 2010) (Table S3A). CTNNB1 is part of a complex that forms adherens junctions and plays a role in cytoskeleton formation. Several of the factors identified here, including CTNNB1, share cellular interaction partners (Figure S5), suggesting that they may function in similar cellular processes. With the exception of BUB3 and CLTC, the host factors identified in the polymerase activity assay did not co-immunoprecipitate with components of the viral replication complex (Table S1A), suggesting an indirect effect on viral replication. This finding may have important implications for drug development given that the inhibitory effects of drugs directed at these host factors may not be overcome easily by ‘escape’ mutations in viral proteins.
To determine whether the ‘top hits’ are involved in the early steps of the viral life cycle, we next infected siRNA-treated cells with a replication-incompetent PB2-knockout virus whose polymerase PB2-coding region was replaced with that of the Renilla luciferase reporter protein, as described previously (Ozawa et al., 2011) (see Experimental Procedures). Due to the lack of a functional PB2 gene, reporter expression is indicative of virus binding, internalization, and/or limited replication driven by the polymerase complex associated with the viral RNA segments of the infecting virions. Twenty-three host factors including several proteasome components affected these early steps in the viral life cycle. siRNAs targeting fourteen of these 23 host factors reduced cellular transcription/replication, whereas those targeting the remaining nine human genes (BAG3, BRD8, CCDC135, DDX55, DPM3, EEF2, IGF2BP2, KRT14, and S100A4) appeared to have influenza virus-specific effects (Figure 3 and Tables S5A and B). None of the siRNAs targeting these nine factors reduced viral replication as assessed in mini-replicon assays (see above; Tables S5A and B), suggesting that they have important roles in an earlier step in the viral life cycle such as virus binding, internalization, and/or transport of viral ribonucleoprotein (vRNP) complexes to the nucleus (see below).
Next, we attempted to determine whether the 91 ‘top hits’ are critical for processes late in the viral life cycle, such as virion formation. To this end, we treated cells with siRNAs to the ‘top hits’ and examined the effect on the formation of influenza virus-like particles (VLPs) derived from the M1, HA, and NA proteins (see Experimental Procedures). The efficiency of VLP production was calculated as the amount of M1 and HA in VLPs in the supernatant compared with the amount of M1 and HA in cell lysates. After removing the host genes that have general effects on cellular replication/transcription, we identified 15 host genes (ASCC3L1, BRD8, C19orf43, DDX55, DKFZp564K142, DPM3, EEF2, FAM73B, FLJ20303, GBF1, NCLN, C14orf173, XPO1, LRPPRC and RCN1) whose siRNA-mediated down-regulation decreased the average of VLP production by more than 50% compared with the control, although there were no statistically significant differences (Figure 3 and Tables S5A and C). None of these factors was identified in previous genome-wide screens (Brass et al., 2009; Hao et al., 2008; Karlas et al., 2010; Konig et al., 2010; Shapira et al., 2009; Sui et al., 2009). Four of these host factors (BRD8, DDX55, DPM3, and EEF2) also affected the early steps in the viral life cycle and may thus play a role in the intracellular transport of viral proteins and/or vRNP complexes, functions that are essential during the early and late stages of infection. Hence, these host factors may be interesting targets for drug development.
Among the 91 ‘top hits’, there were 35 host factors whose siRNA-mediated down-regulation did not affect the virus life cycle steps assessed to this point. Therefore, we determined whether depletion of these factors affected the incorporation efficiency of vRNPs into progeny virions (see Experimental Procedures). The vRNP incorporation efficiency was determined by dividing the amount of viral RNA or NP protein in viruses harvested from the culture supernatants by the amount of viral RNA and NP in the cell lysates. The down-regulation of 28 host factors decreased the incorporation efficiency of vRNA and/or NP by more than 50% compared with a control (Figure 3 and Table S5D); these differences were statistically significant (p value < 0.05), with the exception of MYH10, whose p value was 0.06 (Table S5D). In addition, several of these 28 host factors share common cellular interaction partners; for example, CIRBP and HNRNPK interact with RBMX, an RNA-binding protein with roles in RNA transcription and splicing (indicated with a green arrow in Figure S5E, and Table S5J). Based on this information, future studies could be directed at assessing the exact roles of CIRBP, HNRNPK, and potentially RBMX, in the packaging of influenza vRNPs.
Further evaluation of the host factors described above and their cellular interaction partners revealed a group of cellular factors likely involved in the intracellular transport of influenza vRNP. For example, SUMO2, a cellular protein involved in protein sumoylation and localization (indicated with a blue arrow in Figures S5B, D, and E), interacts with seven different host factors (BRD8, PSMD11, SF3B2 (Figures S5B and D), NSUN2, SNRNP200 (Figure S5D), THOC2, and TRIM28 (Figure S5E) whose down-regulation affected early steps in the viral life cycle, VLP formation, and/or vRNP packaging (Figures S5B, D, and E, and Tables S5E–J). This is consistent with a previous study that suggested a role for sumoylation in the influenza viral life cycle (Wu et al., 2011). Host factors like SUMO2 that affect several steps in the viral life cycle may therefore be attractive targets for antiviral compounds.
Insights into host factors involved in the influenza virus life cycle
On the basis of the analyses in this study, influenza virus-host protein interactions can be mapped to individual steps of the influenza virus life cycle (Figure 5), as outlined below.
Figure 5. Putative roles of identified host factors in the influenza virus life cycle.

Influenza virus is internalized by receptor-mediated endocytosis. The viral ribonucleoprotein (vRNP) complexes containing the 8 viral genome RNAs (depicted by orange bars) are transported into the nucleus where replication and transcription of the viral genome take place. vRNPs formed with newly synthesized viral RNA, NP, and viral polymerase proteins are transported from the nucleus to the cytoplasm. HA and NA are processed posttranslationally during their transport from the ER to the Golgi apparatus. In the late stage of the viral life cycle, virion components are transported to the plasma membrane and progeny viruses then bud from the cells. The light orange boxes indicate individual steps of the influenza virus life cycle; the gray boxes indicate host cellular processes that are likely involved. Host factors identified in this study are grouped according to the viral life cycle steps they affected; light green circles indicate host factors identified in previous studies. Among the ‘Known host factors’, only XPO1 (also known as CRM1) was identified in this study. See also Table S5.
Influenza virus infection begins with attachment of the viral hemagglutinin (HA) protein to a cell surface receptor, followed by internalization of the virus into cells, facilitated by endocytosis (Marsh and Helenius, 2006; Matlin et al., 1981; Rust et al., 2004; Sieczkarski and Whittaker, 2002). HA mediates the fusion between the viral and endosomal membranes, leading to the release of vRNPs into the cytoplasm (Stegmann et al., 1990). vRNP complexes are then transported through the cytoplasm to the nuclear core complex. We identified several host factors involved in these early steps in the viral life cycle (Figure 3, and Table S5), perhaps by facilitating intracellular transport of influenza vRNPs, as discussed earlier. For example, BRD8 (also known as SMAP or SMAP2) plays a role in intracellular vesicle trafficking (Tanabe et al., 2006) and binds to M1, the viral matrix protein, which is a major structural component of influenza virions. The interaction of M1 with BRD8 may affect intracellular vesicle trafficking and hence the transport of incoming and newly synthesized virus components to and from the plasma membrane.
The nuclear import of influenza vRNP is mediated by importins, which are part of the active import machinery of the host cell nuclear pore complex (NPC) (Deng et al., 2006; Gabriel et al., 2008; O’Neill et al., 1995; Resa-Infante et al., 2008; Tarendeau et al., 2007; Tarendeau et al., 2008; Wang et al., 1997). Once the vRNPs are transported into the nucleus, the replication and transcription of influenza virus genomic RNA is facilitated by the viral polymerase subunits (PB1, PB2 and PA) and the nucleoprotein, NP (reviewed in (Engelhardt and Fodor, 2006; Palese, 2007)). Here, we identified several host factors that appear to be important for influenza viral RNA replication (summarized in Figure 3). Influenza virus also uses cellular machinery for nuclear export of vRNP complexes. Consistent with previous studies (Boulo et al., 2007; Elton et al., 2001; Neumann et al., 2000), we found that down-regulation of XPOI (also known as CRM1) suppresses vRNP nuclear export (Figure 4C). Moreover, our 91 ‘top hits’ include 5 other host factors whose suppression caused NP accumulation in the nucleus (Figure 4C), suggesting that these factors are likely involved in the nuclear export of influenza vRNPs.
In the late stage of the virus replication cycle, virion components, such as vRNPs, M1, and the viral envelope proteins (HA, NA and M2) are transported to the plasma membrane where progeny virions bud (Nayak et al., 2004). Although relatively few host factors are currently known to be involved in these steps, here we identified several host factors with potential roles in the late stages of infection (see also earlier discussion of a potential role for BRD8; Table S5). Brass et al. (Brass et al., 2009) showed that depletion of subunits of the COPI complex reduced HA expression levels on the cell surface, suggesting a role for the COPI complex in the transport of influenza viral envelope proteins (i.e., HA and NA) to the cell surface. In agreement with their findings, we found that depletion of GBF1 (Golgi-specific brefeldin A resistant guanine nucleotide exchange factor 1), which associates with the COPI complex, reduced the efficiency of VLP formation (Figure 3), supporting the importance of this pathway in influenza virus replication.
Taken together, the mapping of virus-host protein interactions to individual steps of the influenza virus life cycle provides a systematic understanding of the mechanism of virus replication that can be further utilized to explore targets for antiviral therapy.
Exploration of targets for antiviral therapy based on 299 host factors whose suppression reduced influenza virus replication
To identify targets for antiviral therapy, we queried the DrugBank database (Wishart et al., 2006), IPA (Ingenuity), and the database of pharmaceutical companies (Millipore, Sigma Aldrich, and Selleck Chemicals) for chemicals that suppress the functions of the 299 host factors whose suppression reduced virus titers by more than two log10 units. We identified 61 drugs (including bortezomib and colchicine, which are known to interfere with influenza virus replication (Patterson et al., 1979; Widjaja et al., 2010) and therefore could serve as positive controls) that inhibited 44 of the 299 host factors (Table S6A).
Of these 61 drugs, we chose 11 for further testing based on their availability. To test the efficacy of these drugs on influenza virus replication, we infected HEK 293 and A549 (human lung epithelial) cells with WSN virus, treated them with the 11 selected drugs (Table S6B), and measured virus titers 48 h post-infection. Histone acetyltransferase inhibitor II (100 μM), genistein (100 μM), 2,3-Butanedione 2-Monoxime (30 mM), and WP1130 (50 μM) reduced virus titers by more than 5 log10 units in at least one of the cell lines tested, but also markedly decreased cell viability (Figure S6). In accordance with previous studies (Patterson et al., 1979; Widjaja et al., 2010), bortezomib (0.2 μM) and colchicine (2.5 μM) reduced virus titers by 4-log10 units and 2-log10 units, respectively, in A549 cells without causing major cell toxicity (Figure S6B). Interestingly, golgicide A (10 μM) and ruxolitinib (30 μM), which were not previously known to interfere with influenza virus replication, markedly decreased virus titers in at least one of the cell lines tested. Ruxolitinib (30 μM) did not cause notable cell toxicity, whereas golgicide A (10 μM) decreased the viability of HEK293, but not that of A549 cells (Figure 6, Table S6B, and Figure S6). Golgicide A inhibits the cis-Golgi localized guanine nucleotide exchange factor GBF1, which co-immunoprecipitated with M2. siRNAs directed to GBF1 decreased virus titers by more than 5 log10 units (Table S2), altered the intracellular localization of HA and NA (Figures 4A and B), and decreased the efficiency of VLP production (Figure 3), indicating the potential involvement of GBF1 in the transport of viral surface proteins. More importantly, ruxolitinib reduced virus titers by 3 log10 units at a concentration of 30 μM, which had no effect on cell viability (Figure 6). This compound inhibits JAK1, a tyrosine kinase that co-immunoprecipitated with several viral proteins (Table S2). siRNA-mediated down-regulation of JAK1 reduced virus titers by more than 5-log10 units (Table S2), inhibited virion formation (Figure 7A), and decreased the efficiency of vRNP incorporation into virions in virus-infected cells (Figure 7B, and Tables S5A and D). In addition, siRNAs to JAK1 reduced the efficiency of VLP formation to 57.7% (see Table S5C), which was slightly above our cutoff of 50%, and explains why JAK1 was not identified as a ‘hit’ in our VLP assays. These results demonstrate that JAK1 may play an important role in the late steps of influenza virus replication cycle.
Figure 6. Effects of selected drugs on virus titers and cell viability in virus-infected cells.

HEK 293 (A) or A549 (B) cells were infected with WSN virus at an MOI of 0.001. One hour later, cells were washed and incubated with medium containing the indicated concentration of golgicide A (A) and ruxolitinib (B). DMSO (final concentration, 1%) was used as a control. Forty-eight hours later, culture supernatants were harvested for virus titration and cell viability was determined by using CellTiter-Glo. Average and standard deviation of 3 replicates are shown. The p value was calculated with Welch’s t-test compared with a non-targeting siRNA control. To control for the multiplicity effect, p values were adjusted using Benjamini-Hochberg’s procedure keeping the false discovery ratio < 0.05. Asterisk indicates that the adjusted p value is < 0.05. See also Figure S6 and Table S6.
Figure 7. Role of JAK1 in influenza virus replication.

(A) siRNA targeting JAK1 reduced the formation of virus particles from virus-infected cells. HEK 293 cells transfected with the indicated siRNAs were infected with WSN virus at an MOI of 5. At 12 h post-infection, cells were subjected to transmission electron microscopy. (B) Depletion of JAK1 inhibited vRNP incorporation into virions. siRNA directed at JAK1 reduced relative levels of NP and vRNA in virions released from HEK 293 cells infected with WSN virus. See also Table S5C.
Given the known function of JAK1 in immune responses, its inhibition would have been expected to increase, rather than decrease, virus titers. Several JAK1 inhibitors are approved or are in advanced clinical testing for the treatment of myelofibrosis, rheumatoid arthritis, and psoriasis in humans (Cutolo and Meroni, 2013; Pardanani et al., 2011; Quintas-Cardama et al., 2011), demonstrating their therapeutic potential. Overall, the identification of JAK1 as a previously unrecognized target to control influenza virus infection underscores the power of our approach, providing a proof-of-concept for virus-host interactome screens to identify host factors that could be targets for antiviral drugs.
In summary, we present a comprehensive study to better understand interactions between influenza viral and host proteins. We used this information to identify drug targets and found that inhibition of GBF1 and JAK1 (which had not been considered targets for anti-influenza viral drugs) drastically reduced influenza virus titers in cultured cells with no appreciable effect on cell viability. These findings demonstrate that this comprehensive experimental approach has considerable potential for identifying drug targets.
EXPERIMENTAL PROCEDURES
Cells and viruses
HEK 293 and A549 (human lung epithelial) cells were cultured in DMEM (Sigma-Aldrich, St. Louis, USA) supplemented with 10% FCS (10% FCS/DMEM) and antibiotics at 37 °C in 5% CO2. Madin–Darby canine kidney (MDCK) cells were cultured in Eagle’s MEM (GIBCO) with 5% NCS at 37 °C in 5% CO2. Influenza A/WSN/33 virus (WSN; H1N1) was generated by use of reverse genetics, as described previously (Neumann et al., 1999) and propagated in MDCK cells. Virus was titrated by plaque assay in MDCK cells (Tobita et al., 1975). The PB2-knockout virus possessing the Renilla luciferase gene (PB2-KO/Rluc virus) was generated with pPolIPB2(120)Rluc(120) (encoding Renilla luciferase flanked by 120 N- and C-terminal nucleotides derived from the PB2 segment) and plasmids encoding the remaining seven viral RNA segments, and propagated and titrated in MDCK cells stably expressing the PB2 protein as described previously (Ozawa et al., 2011).
Plasmids
Plasmids expressing viral RNAs or proteins were generated as previously described (Li et al., 2009). For co-immunoprecipitation analysis, the coding regions of the viral PB2, PB1, PA, HA, NP, NA, M1, M2, NS1, NS2, and PB1-F2 proteins were cloned into the expression vector pCAGGS/MCS (Niwa et al., 1991) in-frame with a FLAG-tag sequence at either the 5′ or 3′ end. Consequently, expression plasmids encoding N- or C-terminally FLAG-tagged viral proteins were generated.
Antibodies
Mouse antibodies recognizing the FLAG epitope (F1804) or β-actin (clone AC-74) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Mouse anti-M1 antibody (WS-27/52), mouse anti-HA antibody (WS3-54), and mouse anti-NA antibody (WS5-29) were kindly provided by Dr. Emi Takashita (National Institute of Infectious Diseases, Tokyo, Japan). Mouse anti-Aichi NP antibody (2S-347/3) and rabbit anti-WSN virus antibody (R309) were available in our laboratory.
Pull-down assay and mass spectrometry
Pull-down assays with HEK293 cells individually expressing the eleven FLAG-tagged viral proteins, followed by mass spectrometry, were performed as described previously (Gorai et al., 2012) and in the Supplemental Experimental Procedures.
siRNA treatment
For each candidate host factor identified by mass spectrometric analysis, two siRNAs were selected from a predesigned genome-wide human siRNA library (FlexiTube siRNA; QIAGEN) (Table S2). AllStars Negative Control siRNA (QIAGEN) was used as a negative control. The siRNA against the NP gene of WSN virus (GGA UCU UAU UUC UUC GGA GUU) was purchased from Sigma-Aldrich. HEK 293 cells were transfected twice with 25 nM (final concentration, 50 nM) of siRNA duplexes using RNAiMAX Reagent (Invitrogen). At 24 h after the second transfection, cell viability was measured by using the CellTiter-Glo assay system (Promega) according to the manufacturer’s instructions. To assess influenza viral replication, two parallel sets of siRNA-transfected cells were infected with 50 plaque-forming units (pfu) of WSN virus per well of a 24-well tissue culture plate at 24 h after the second siRNA transfection. Forty-eight hours post-infection, supernatants were harvested and virus titers were determined by means of plaque assays in MDCK cells.
Quantitative reverse transcription-PCR
To confirm the down-regulation of host genes by their respective target siRNAs, quantitative reverse transcription-PCR (qRT-PCR) experiments were performed. HEK 293 cells, transfected twice with 25 nM of siRNA (final concentration, 50 nM), were lysed at 48 h post-transfection and total RNA was extracted by using the Maxwell 16 LEV simplyRNA Tissue Kit (Promega). Reverse transcription was performed by using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan) or SuperScript III Reverse Transcriptase (Invitrogen). The synthesized cDNA was subjected to quantitative PCR with primers specific for each gene by using the THUNDERBIRD SYBR qPCR Mix (TOYOBO). The relative mRNA expression levels of each gene were calculated by the ΔΔCt method using beta-actin as internal control. Primer sequences are available upon request.
PB2-KO/Rluc virus assay
HEK 293 cells were transfected twice with 25 nM of siRNA. At 24 h after the second transfection, cells were infected with a replication-incompetent PB2-knockout WSN virus (PB2-KO/Rluc virus) that possesses a reporter gene encoding Renilla luciferase in place of the coding region of the polymerase PB2 protein. At 8 h post-infection, cells were subjected to the Renilla Luciferase Assay System (Promega). Luminescence was detected by use of the GloMax-96 Microplate Luminometer (Promega). This virus lacks the capacity to synthesize PB2; therefore, only limited replication can occur, which is dependent on the PB2 protein associated with infecting vRNP complexes.
Immunofluorescence microscopy
HEK 293 cells transfected twice with 25 nM of the respective siRNA or AllStars Negative Control siRNA were infected with 200 pfu of WSN virus per well of a 24-well tissue culture plate at 24 h after the second siRNA transfection, and were then used to the immunofluorescence assay as described in the Supplemental Experimental Procedures.
Western blotting
Cells or viruses were lysed with Tris-Glycine SDS sample buffer (Invitrogen), heated for 10 min at 95 °C, and then immediately chilled on ice prior to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). SDS-PAGE was performed on 4%–20% Mini-PROTEAN TGX gradient gels (Bio-Rad) for 30 min at 15–20 mA, followed by Western blotting as described in the Supplemental Experimental Procedures.
VLP formation assay
HEK 293 cells transfected twice with 25 nM siRNA were transfected with pCAGGS plasmids expressing the viral proteins HA, NA, and M1 by using TransIT293 transfection reagent (Mirus, Madison, USA). At 48 h after plasmid transfection, cells were lysed with SDS Sample Buffer Solution (Wako) with 100 mM dithiothreitol. The culture supernatant containing the released virus-like particles (VLPs) was also harvested and centrifuged at 3,000 × g at 4 °C for 5 min to remove cell debris. Supernatant was layered onto PBS-containing 30% (w/v) sucrose in an ultracentrifuge tube. Ultracentrifugation was performed at 50,000 rpm at 4 °C for 1 h in a SW55Ti Rotor. The pellet was lysed in SDS Sample Buffer Solution (Wako) with 100 mM dithiothreitol and subjected to Western blotting as described above.
Mini-replicon assay
Mini-replicon assays to determine the activity of the influenza viral RNA polymerase were performed as described previously (Octaviani et al., 2010). HEK 293 cells were transfected twice with 25 nM siRNA. At 24 h after the second transfection, cells were transfected with pCAGGS plasmids expressing the viral proteins PA, PB1, PB2, and NP, and with a plasmid expressing luciferase from a virus-like RNA encoding firefly luciferase (pPolINP(0)luc2(0)) (Octaviani et al., 2010). A plasmid expressing Renilla luciferase under the control of a cellular RNA polymerase II promoter (i.e., the chicken β-actin promoter) was used as a transfection control. At 48 h after plasmid transfection, cells were subjected to a luciferase assay using the Dual-Glo Luciferase assay system (Promega). Luminescence was detected with the GloMax-96 Microplate Luminometer (Promega). The activity of the viral RNA polymerase was calculated as the activity of the firefly luciferase normalized to that of the Renilla luciferase. The efficiency of cellular gene transcription and/or translation was determined based on the expression levels of the Renilla luciferase reporter protein.
vRNP incorporation assay
HEK 293 cells transfected twice with 25 nM of the respective siRNA or AllStars Negative Control siRNA were infected with WSN virus at a multiplicity of infection (MOI) of 5. At 12 h after infection, the medium containing the released virions was centrifuged at 3,000 × g at 4 °C for 5 min to remove cell debris. Supernatant was layered onto PBS containing 30% (w/v) sucrose in an ultracentrifuge tube. Ultracentrifugation was performed at 50,000 rpm and 4 °C for 1 h in a SW55Ti Rotor. Virus-containing pellets were homogenized in PBS and viral RNAs were extracted by using the Maxwell 16 LEV simplyRNA Tissue Kit according to the manufacturer’s instructions. To quantify viral RNA in cells and supernatants, strand-specific real-time RT-PCR was performed as previously described (Kawakami et al., 2011) with slight modification. Total RNA was reverse transcribed with SuperScript III Reverse Transcriptase and influenza gene-specific primers to which a 19 nucleotide ‘tag’ sequence had been added at the 5′ end (for example: vRNAtag_NP_1F; GGCCGTCATGGTGGCGAATAGCAAAAGCAGGGTAGATAATCACTC). Quantitative PCR was carried out using the THUNDERBIRD Probe qPCR Mix with primers specific for the ‘tag’ (vRNAtag; GGCCGTCATGGTGGCGAAT) and the viral gene sequence (WSN-NP_100R; GTTCTCCATCAGTCTCCATC), and with a probe labeled with 6-FAM/ZEN/IBFQ (IDT, WSN-NP_46–70; ATGGCGACCAAAGGCACCAAACGA T).
Effects of antiviral drugs on influenza virus titers in cultured cells
To test the effects of the selected drugs on viral replication in vitro, HEK 293 or A549 cells were infected with WSN virus at an MOI of 0.001. One hour later, cells were washed and medium containing the indicated drug concentrations was added. DMSO (final concentration, 1%) was used as a control. Forty-eight hours later, culture supernatants were harvested for virus titration; cell viability was determined in the mock-infected cells in the presence of drugs by using CellTiter-Glo.
Electron microscopy
HEK 293 cells transfected twice with 25 nM of siRNA targeting JAK1 or AllStars Negative Control siRNA were infected with WSN virus at an MOI of 5. At 12 h after infection, the cells were processed for ultrathin section electron microscopy as described previously (Noda et al., 2006). The samples were examined with a Tecnai F20 electron microscope (FEI).
Identification of host proteins involved in influenza virus replication
For each set of siRNAs, the virus titers obtained from cells treated with gene-specific siRNAs were compared with those obtained from cells treated with AllStars Negative Control siRNA. siRNAs that decreased cell viability by more than 40% relative to that of AllStars Negative Control siRNA-treated cells were removed from further analysis. LOESS regression was applied to adjust for the effect of cell viability on virus titers (Figure S1). The normalized virus titers were fitted to a linear model and a two-sided, unpaired student’s t-test was used to compare the mean virus titers in cells treated with gene-specific siRNAs with those in cells treated with AllStars Negative Control siRNA. All p-values were Holm’s adjusted for multiple comparisons. Initially, we found 325 host genes whose siRNA-mediated down-regulation reduced virus titers by more than 2-log10 units (300 host factors) or increased virus titers by more than 1-log10 unit (25 host factors) compared with the control siRNA (Figure S1 and Table S2). However, the siRNAs targeting ARHGEF19 had opposite effects on virus production, that is, one of the siRNAs targeting ARHGEF19 decreased virus titers, whereas the other siRNA increased virus titers (Table S2); thus, ARHGEF19 was originally counted twice as having virus titer-enhancing and -decreasing properties, respectively. After eliminating ARHGEF19 from both lists, we arrived at a total of 323 genes involved in influenza virus replication: 299 host genes whose suppression decreased virus production and 24 host genes whose suppression increased virus production.
Gene Ontology and pathway analyses
We performed Gene Ontology and pathway analysis using two web-resources, DAVID (Huang et al., 2008) and ConsensusPathBD (Kamburov et al., 2011). The results were filtered to leave only entries with Hochberg corrected p-value < 0.01 in DAVID. We examined the ‘Biological Process’ and ‘Cellular Component’ information and manually removed GO categories that were largely overlapping as described in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Naomi Fujimoto, Izumi Ishikawa, Kazue Goto, Manami Katoh, Ai Kakumoto, Kei Takahashi and Fujimi Arai for technical support. We thank Susan Watson and Krisna Wells for editing the manuscript. We thank Dr. Emi Takashita for mouse anti-M1 antibody (WS-27/52), mouse anti-HA antibody (WS3-54), and mouse anti-NA antibody (WS5-29). This work was supported by National Institute of Allergy and Infectious Diseases Public Health Service research grants (RO1 AI080598 and R56 AI099275), by ERATO, and by the Strategic Basic Research Programs of Japan Science and Technology Agency (Japan Science and Technology Agency).
Footnotes
AUTHOR CONTRIBUTIONS
T.W., E.K., J.E.S., T.J.S.L., Y.M., Y.T., H.K.H., K.F., H.G., S.F., S.W., M.O., H.K., and Y.K. designed the study; T.W., E.K., Y.T., H.K.H., T.G., T. K., E.T., A.N., R.T., M. K., M. Y., Y. S-T., H. K., N.N., H.F., Y.S., T.N., and S.W. performed the experiments; J.E.S., T.J.S.L., and Y.M. conducted computational analyses; T.W., E.K., J.E.S., T.J.S.L., Y.M., Y.T., H.K.H., K.F., S.W., G.N., M.O., H.K., and Y.K. analyzed the data; T.W., E.K., J.E.S., T.J.S.L., Y.M., Y.T., T.N., G.N., M.O., and Y.K. wrote the manuscript; and Y.K. oversaw the project. T.W. and E.K. contributed equally to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boulo S, Akarsu H, Ruigrok RW, Baudin F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007;124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, Davis XM, Povinelli L, Cox NJ, Klimov AI. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–1181. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA. 2006;295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- Cutolo M, Meroni M. Clinical utility of the oral JAK inhibitor tofacitinib in the treatment of rheumatoid arthritis. J Inflamm Res. 2013;6:129–137. doi: 10.2147/JIR.S35901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies WL, Grunert RR, Haff RF, McGahen JW, Neumayer EM, Paulshock M, Watts JC, Wood TR, Hermann EC, Hoffmann CE. Antiviral Activity of 1-Adamantanamine (Amantadine) Science. 1964;144:862–863. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Andre P, Lotteau V. Genetic screens for the control of influenza virus replication: from meta-analysis to drug discovery. Mol Biosyst. 2012;8:1297–1303. doi: 10.1039/c2mb05416g. [DOI] [PubMed] [Google Scholar]
- Deng T, Engelhardt OG, Thomas B, Akoulitchev AV, Brownlee GG, Fodor E. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J Virol. 2006;80:11911–11919. doi: 10.1128/JVI.01565-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elton D, Simpson-Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J Virol. 2001;75:408–419. doi: 10.1128/JVI.75.1.408-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt OG, Fodor E. Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol. 2006;16:329–345. doi: 10.1002/rmv.512. [DOI] [PubMed] [Google Scholar]
- Gabriel G, Herwig A, Klenk HD. Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog. 2008;4:e11. doi: 10.1371/journal.ppat.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- Gorai T, Goto H, Noda T, Watanabe T, Kozuka-Hata H, Oyama M, Takano R, Neumann G, Watanabe S, Kawaoka Y. F1Fo-ATPase, F-type proton-translocating ATPase, at the plasma membrane is critical for efficient influenza virus budding. Proc Natl Acad Sci U S A. 2012;109:4615–4620. doi: 10.1073/pnas.1114728109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, Ahlquist P, Kawaoka Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta M, Hatta Y, Kim JH, Watanabe S, Shinya K, Nguyen T, Lien PS, Le QM, Kawaoka Y. Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog. 2007;3:1374–1379. doi: 10.1371/journal.ppat.0030133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden FG. Perspectives on antiviral use during pandemic influenza. Philos Trans R Soc Lond B Biol Sci. 2001;356:1877–1884. doi: 10.1098/rstb.2001.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Research. 2011;39:D712–D717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J Virol Methods. 2011;173:1–6. doi: 10.1016/j.jviromet.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, et al. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ru A, Jacob D, Transfiguracion J, Ansorge S, Henry O, Kamen AA. Scalable production of influenza virus in HEK-293 cells for efficient vaccine manufacturing. Vaccine. 2010;28:3661–3671. doi: 10.1016/j.vaccine.2010.03.029. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhou L, Zhou M, Chen Z, Li F, Wu H, Xiang N, Chen E, Tang F, Wang D, et al. Epidemiology of human infections with avian influenza A(H7N9) virus in China. N Engl J Med. 2014;370:520–532. doi: 10.1056/NEJMoa1304617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Watanabe T, Hatta M, Watanabe S, Nanbo A, Ozawa M, Kakugawa S, Shimojima M, Yamada S, Neumann G, et al. Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J Virol. 2009;83:4153–4162. doi: 10.1128/JVI.02642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol. 1981;91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Doudna JA. A host of factors regulating influenza virus replication. Viruses. 2010;2:566–573. doi: 10.3390/v2020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak DP, Hui EK, Barman S. Assembly and budding of influenza virus. Virus Res. 2004;106:147–165. doi: 10.1016/j.virusres.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, Hughes MT, Kawaoka Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000;19:6751–6758. doi: 10.1093/emboj/19.24.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hughes M, Perez DR, Donis R, Hoffmann E, et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A. 1999;96:9345–9350. doi: 10.1073/pnas.96.16.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll A, Ciancio B, Kramarz P. Observed oseltamivir resistance in seasonal influenza viruses in Europe interpretation and potential implications. Euro Surveill. 2008;13 doi: 10.2807/ese.13.05.08025-en. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, Kawaoka Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439:490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- O’Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem. 1995;270:22701–22704. doi: 10.1074/jbc.270.39.22701. [DOI] [PubMed] [Google Scholar]
- Octaviani CP, Ozawa M, Yamada S, Goto H, Kawaoka Y. High level of genetic compatibility between swine-origin H1N1 and highly pathogenic avian H5N1 influenza viruses. J Virol. 2010;84:10918–10922. doi: 10.1128/JVI.01140-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa M, Victor ST, Taft AS, Yamada S, Li C, Hatta M, Das SC, Takashita E, Kakugawa S, Maher EA, et al. Replication-incompetent influenza A viruses that stably express a foreign gene. J Gen Virol. 2011;92:2879–2888. doi: 10.1099/vir.0.037648-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P, editor. Orthomixoviridae. 5. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- Pardanani A, Vannucchi AM, Passamonti F, Cervantes F, Barbui T, Tefferi A. JAK inhibitor therapy for myelofibrosis: critical assessment of value and limitations. Leukemia. 2011;25:218–225. doi: 10.1038/leu.2010.269. [DOI] [PubMed] [Google Scholar]
- Patterson S, Oxford JS, Dourmashkin RR. Studies on the mechanism of influenza virus entry into cells. J Gen Virol. 1979;43:223–229. doi: 10.1099/0022-1317-43-1-223. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10:127–140. doi: 10.1038/nrd3264. [DOI] [PubMed] [Google Scholar]
- Resa-Infante P, Jorba N, Zamarreno N, Fernandez Y, Juarez S, Ortin J. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS One. 2008;3:e3904. doi: 10.1371/journal.pone.0003904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Lakadamyali M, Zhang F, Zhuang X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol. 2004;11:567–573. doi: 10.1038/nsmb769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. 2002;76:10455–10464. doi: 10.1128/JVI.76.20.10455-10464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmann T, White JM, Helenius A. Intermediates in influenza induced membrane fusion. EMBO J. 1990;9:4231–4241. doi: 10.1002/j.1460-2075.1990.tb07871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui B, Bamba D, Weng K, Ung H, Chang S, Van Dyke J, Goldblatt M, Duan R, Kinch MS, Li WB. The use of Random Homozygous Gene Perturbation to identify novel host-oriented targets for influenza. Virology. 2009;387:473–481. doi: 10.1016/j.virol.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Kon S, Natsume W, Torii T, Watanabe T, Satake M. Involvement of a novel ADP-ribosylation factor GTPase-activating protein, SMAP, in membrane trafficking: implications in cancer cell biology. Cancer Sci. 2006;97:801–806. doi: 10.1111/j.1349-7006.2006.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarendeau F, Boudet J, Guilligay D, Mas PJ, Bougault CM, Boulo S, Baudin F, Ruigrok RW, Daigle N, Ellenberg J, et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat Struct Mol Biol. 2007;14:229–233. doi: 10.1038/nsmb1212. [DOI] [PubMed] [Google Scholar]
- Tarendeau F, Crepin T, Guilligay D, Ruigrok RW, Cusack S, Hart DJ. Host determinant residue lysine 627 lies on the surface of a discrete, folded domain of influenza virus polymerase PB2 subunit. PLoS Pathog. 2008;4:e1000136. doi: 10.1371/journal.ppat.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobita K, Sugiura A, Enomoto C, Furuyama M. Plaque assay and primary isolation of influenza A viruses in an established line of canine kidney cells (MDCK) in the presence of trypsin. Medical microbiology and immunology. 1975;162:9–14. doi: 10.1007/BF02123572. [DOI] [PubMed] [Google Scholar]
- Wang P, Palese P, O’Neill RE. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J Virol. 1997;71:1850–1856. doi: 10.1128/jvi.71.3.1850-1856.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. 2010;7:427–439. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, Govorkova EA. H5N1 influenza--continuing evolution and spread. N Engl J Med. 2006;355:2174–2177. doi: 10.1056/NEJMp068205. [DOI] [PubMed] [Google Scholar]
- Widjaja I, de Vries E, Tscherne DM, Garcia-Sastre A, Rottier PJ, de Haan CA. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J Virol. 2010;84:9625–9631. doi: 10.1128/JVI.01048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Jeng KS, Lai MM. The SUMOylation of matrix protein M1 modulates the assembly and morphogenesis of influenza A virus. J Virol. 2011;85:6618–6628. doi: 10.1128/JVI.02401-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HL, Webster RG. Pandemic influenza as a current threat. Curr Top Microbiol Immunol. 2009;333:3–24. doi: 10.1007/978-3-540-92165-3_1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.