

Abstract
We recently demonstrated that interleukin-1β (IL-1β) increases system xc− (cystine/glutamate antiporter) activity in mixed cortical cell cultures, resulting in an increase in hypoxic neuronal injury when glutamate clearance is impaired. Herein, we demonstrate that neurons, astrocytes and microglia all express system xc− subunits (xCT, 4F2hc, RBAT) and are capable of cystine import. However, IL-1β stimulation increases mRNA for xCT— the light chain that confers substrate specificity— in astrocytes only; an effect blocked by the transcriptional inhibitor actinomycin D. Additionally, only astrocytes show an increase in cystine uptake following IL-1β exposure; an effect associated with a change in xCT protein. The increase in cystine uptake that follows IL-1β is lacking in astrocytes derived from mice harboring a mutation in Slc7a11 (sut gene), which encodes for xCT, and in wild-type astrocytes treated with the protein synthesis inhibitor cycloheximide. IL-1β does not regulate the light chain of the amino acid transporter, LAT2, or the expression and function of astrocytic excitatory amino acid transporters (EAATs), demonstrating some target selectivity. Finally, the enhanced neuronal vulnerability to hypoxia that followed IL-1β treatment in our mixed culture system was not observed in chimeric cultures consisting of wild-type neurons plated on top of sut astrocytes. Nor was it observed in wild-type cultures treated with a system xc− inhibitor or an NMDA receptor antagonist. Overall, our data demonstrate that IL-1β selectively regulates system xc− activity in astrocytes and that this change is specifically responsible for the deleterious, excitotoxic effects of IL-1β found under hypoxic conditions.
Keywords: primary cell culture, xCT, cystine/glutamate antiporter, hypoxia
Introduction
System xc− is a heteromeric amino acid transporter consisting of two subunits: xCT — the light chain that confers substrate specificity — and a heavy chain (4F2hc or RBAT) thought to target the transporter to the plasma membrane (Bassi et al. 2001; Sato et al. 1999). The import of cystine via system xc− is directly coupled to glutamate export, occurring in a Na+-independent, Cl−-dependent manner with 1:1 stoichiometry (Bannai 1986; Reichelt et al. 1997). Although, system xc− is best known for its role in the synthesis of the antioxidant molecule glutathione (GSH) (Bannai et al. 1989; Bridges et al. 2001; Dun et al. 2006; Lewerenz et al. 2009; Miura et al. 1992; Sato et al. 1995; Watanabe and Bannai 1987), enhanced transporter activity has been reported to contribute to neuronal and oligodendrocyte injury both in vitro and in vivo (Barger and Basile 2001; Chung et al. 2005; Domercq et al. 2007; Fogal et al. 2007; Qin et al. 2006; Savaskan et al. 2008; Sontheimer 2008).
System xc− subunits and activity have been demonstrated to be dynamically regulated. For instance, xCT expression and/or the activity of system xc− is enhanced following deprivation of certain cellular amino acids or exposure to lipopolysaccharide (LPS), nitric oxide (NO), dibutyryl cAMP (dBcAMP), or to electrophilic reagents such as diethyl maleate (DEM) (Bridges et al. 2001; Gochenauer and Robinson 2001; Miura et al. 1992; Sato et al. 2004). Recently, we demonstrated that the cytokine, IL-1β, enhances system xc− activity (i.e. increases Vmax) in a mixed cortical cell culture system (Fogal et al. 2007). While increased activity is not toxic alone, presumably because system XAG− (glutamate transport) is sufficient to prevent the toxic accumulation of extracellular glutamate – under conditions where glutamate uptake is compromised (i.e., hypoxia), this IL-1β-mediated enhancement of system xc− activity contributed to an enhancement of extracellular glutamate levels, which resulted in excitotoxic neuronal cell death (Fogal et al. 2005; Fogal et al. 2007). Although we previously determined that this enhancement in hypoxic neuronal injury in mixed cortical cell culture was dependent on astrocyte IL-1RI signaling, the cell type that demonstrated an increase in transporter activity was not ascertained (Fogal et al. 2007). As our cultures contain predominantly neurons and astrocytes with some contaminating microglia, the cellular and molecular target of system xc− enhancement by IL-1β was examined herein using purified populations of primary astrocyte, neuron, and microglial cultures. Results indicate that IL-1β regulates the expression and activity of system xc− in astrocytes exclusively and that glutamate released via astrocytic system xc− directly underlies the neurotoxic propensity of IL-1β under hypoxic conditions.
Part of the work has been published in abstract form (Jackman et al. 2009; Jackman and Hewett 2009).
Materials and Methods
Cell culture
Cell culture media and experimental buffer compositions were as follows: Media stock (MS): L-glutamine-free modified Eagle’s medium (Earl’s salt; MediaTech) supplemented with L-glutamine, glucose, and sodium bicarbonate to a final concentration of 2.0, 25.7, and 28.2 mM, respectively; Glial plating media: MS containing 10% fetal bovine serum (FBS; Hyclone) and 10% calf serum (CS; Hyclone), 10 ng/ml epidermal growth factor (Invitrogen), 50 IU penicillin, and 50 μg/ml streptomycin (Gibco/BRL); Neuronal plating media: Neurobasal media containing 1x B27 (Invitrogen), 2 mM L-glutamine, 50 IU penicillin and 50 μg/ml streptomycin; Mixed culture plating media: MS containing 5% CS, 5% bovine growth serum (BGS, Hyclone), 50 IU penicillin, and 50 μg/ml streptomycin. Microglia growth media: DMEM (high glucose; Gibco) containing 5% FBS, 2mM L-glutamine, 50 IU penicillin, 50 μg/ml streptomycin and 50% LADMAC (ATCC)-conditioned media to supply colony stimulating factor-1 (Sklar et al. 1985). To produce LADMAC conditioned media, the LADMAC cell line (CRL-2420, ATCC,) was grown to confluence in DMEM containing 5% FBS, 2 mM L-glutamine, 50 IU penicillin and 50 μg/ml streptomycin in 75 cm2 flasks for ~14 days followed by harvesting, centrifugation (720 × g; 3 min) and filtering of the culture supernatant, which is then stored at −80°C. Maintenance media: MS containing 10% CS and 50 IU penicillin/50 μg/ml streptomycin; HBSS (mM): 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 15 glucose, 20 HEPES, 10 NaOH, and 0.01 glycine (pH 7.4). Balanced Salt Solution [BSS (mM)]: 116 NaCl, 5.4 KCl, 0.8 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, 1.8 CaCl2, 0.01 glycine, 2 L-glutamine, 1x MEM amino acids (Invitrogen), and 5 or 20 mM glucose (BSS5 or BSS20, respectively).
Primary astrocyte cultures were derived from pooled cortices of day 1–3 postnatal CD1 mouse pups (Charles River Laboratories) or from mouse pups derived from sut heterozygous breeding pairs (JAX; Stock # 001310) or sut control mice (C3H/HeSnJ; JAX; Stock # 000661) essentially as described (Trackey et al. 2001) save for the addition β-mercaptoethanol (β-ME; 55 μM) to the glial plating medium of the sut cultures to maintain viability and growth (Shih et al. 2006). Il1r1 wild-type and null mutant astrocytes were cultured from cerebral cortices of single pups derived from Il1r1 heterozygous breeding pairs (JAX; stock # 003245). Following dissection of the cerebral cortices, the rest of the brain was used for genotyping as described [http://jaxmice.jax.org/strain/003245.html]. Cells from cerebral cortices were dissociated, plated (Falcon Primaria; BD Biosciences) and once confluent, then treated with 8 μM β-D-arabinofuranoside (AraC; Sigma) once for 4–7 days to reduce the number of microglia. Purified astrocyte cultures were generated by removing residual microglia by treatment with 75 mM L-leucine methyl ester for 60–90 min, one day prior to experimentation (Hamby et al. 2006a). Cultures were ≤ 35 days in vitro at the time of experimentation. Microglia cultures were prepared by plating dissociated cortical cells from CD1 mouse pups (1–3 days) in T25 tissue culture flasks (2 hemisphere/5 ml/flask) in glial plating medium. Fourteen to 21 days later, the culture medium was supplemented with HEPES buffer to a final concentration of 25 mM and flasks shaken overnight at 150 rpm (37°C). The supernatant containing dislodged microglia was collected, spun (3 min; 720 × g) and the resulting pellet resuspended in microglial growth media and plated in 15 mm 24-well plates (Corning) (Hamby et al. 2006b). Primary neuronal cultures were derived from dissociated cortical cells of embryonic day 15 CD1 mouse fetuses using a modification of the protocol of Brewer and colleagues (Brewer et al. 1993). Two days after plating in neuronal plating medium, cultures were treated once with 1 μM AraC for two days, then media was partially replenished (½ volume exchange) twice weekly. Experiments were performed on pure neuronal cultures after 7–10 days in vitro. Mixed cortical cell cultures containing predominantly neurons and astrocytes with a small amount of contaminating microglia were prepared by isolating cortices obtained from embryonic day 15 mouse fetuses and plating them on a confluent layer of astrocytes in mixed culture plating media (Trackey et al. 2001). After 7 days in vitro, mixed cultures were treated with 8 μM AraC for 2 days then switched into maintenance media. The media was changed after 5 and 9 days in vitro, and one day prior to experimentation, cells were placed into MS. Experiments were performed on mixed cortical cultures after 13–14 days in vitro. All cultures were maintained at 37°C in a humidified 6% CO2-containing atmosphere.
IL-1β Treatment
Cells were treated with 3 ng/ml recombinant murine IL-1β (R&D Systems) for various times in an incubation buffer of MS (neurons and astrocytes) or microglial growth media (microglia) both supplemented with 0.1% fatty-acid free BSA (Sigma). Cells were then returned to a humidified 37°C normoxic (21% O2) incubator containing 6% CO2.
Radiolabeled L-cystine and D-aspartate uptake
System xc− specific 14C-L-cystine (PerkinElmer) and system XAG−-mediated 3H-D-aspartate (PerkinElmer) uptake was performed as previously described (Fogal et al. 2007). Cultures were washed into a HEPES buffered salt solution (HBSS; 3 × 750 μL) and allowed to equilibrate for 10 min (25°C). For cystine uptake, cells were incubated in HBSS containing 3 μM 14C-L-cystine (1 μCi/ml), 27 μM unlabeled cystine, 1mM D-aspartate and 0.5 mM acivicin (Biomol). D-aspartate and acivicin were included in the uptake buffer to block system XAG− and γ-glutamyltranspeptidase, respectively, thereby ensuring system xc− specific cystine uptake. Uptake was terminated by washing in ice-cold PBS (3 × 750 μL). For D-aspartate uptake, cells were incubated in HBSS containing 0.1 μCi/ml 3H-D-aspartate and varying concentrations of unlabeled D-aspartate (25°C) for 5 min and uptake terminated by washing cells with an ice-cold Na+-free choline stop buffer containing 116 mM choline chloride, 0.8 mM MgSO4, 1 mM KH2PO4, 10 mM HEPES, 5 mM KOH, 10 mM glucose, 0.9 mM CaCl2, and 5 mM non-radioactive D-aspartate. Varying concentrations of unlabeled D-aspartate were used to span the Km of the different astrocytic glutamate transporters (Danbolt 2001), thus ensuring that the concentration of non-radioactive D-aspartate used was not rate-limiting. To measure amino acid uptake, cells were lysed with warm 0.5% SDS and accumulated radioactivity estimated using a liquid scintillation counter. Uptake was normalized to total protein as determined by the BCA Assay (Pierce).
Reverse transcriptase-PCR analysis
Three or four wells of cells grown in 24 well tissue culture plates were combined, the RNA was extracted (TRIzol, Invitrogen) and then suspended in 20 μL RNase-free water. RNA was quantified spectrophotometrically at 260 nm and first-strand cDNA synthesized from 0.5–1μg RNA using M-MLV reverse transcriptase (400 U, Invitrogen) and oligo (d)T primers (Promega) as previously described (Hewett et al. 1999). Reactions were performed in 20 μL volumes at 40–42°C for 1 hr. Each RNA sample was incubated similarly in the absence of reverse transcriptase to test for genomic DNA contamination (none detected). PCR amplimer pairs for analysis of specific cDNAs are as follows:
| Amplimers | Annealing Temp. | ||
|---|---|---|---|
| xCT | 5′-AAACTTGCTAAGCTCTGTGTTGG-3′ | (sense) | 63 °C |
| 5′-CACATCACATGTTTGTACACTCG-3′ | (antisense) | ||
| 4F2hc | 5′-GCCCAATTCACAAGAACCAG-3′ | (sense) | 60 °C |
| 5′-CCTAGTGACTAGGGATTCAG-3′ | (antisense) | ||
| RBAT | 5′-TTGCCATTTATCCAGGAAGC-3′ | (sense) | 60 °C |
| 5′-AGTTTGTGGGTAGCCAGGTG-3′ | (antisense) | ||
| β-actin | 5′-GTGGGCCGCTCTAGGCACCAA-3′ | (sense) | 63 °C |
| 5′-CTCTTTGATGTCACGCACGATTTC-3′ | (antisense) |
PCR was performed on 1 μL of cDNA using Taq DNA polymerase (1 U, Invitrogen) in a total volume of 25 μL in a Bio-Rad iCycler thermal cycler. Each cycle consisted of a denaturation step (94 °C; 30 sec), an annealing step (45 sec), and a primer extension step (72 °C, 1 min). PCR products were separated by electrophoresis in 2% agarose with 1 kb size markers (Invitrogen) and visualized by ethidium bromide (BioRad) using a UV transilluminator.
Quantitative Real-time PCR (qPCR)
RNA was isolated and first-strand cDNA synthesized as described above. qPCR was performed using mouse-specific primer pairs [Taqman Gene Expression Assays, Applied Biosystems: xCT (Mm00442530_m1); 4F2hc (Mm00500521_m1); RBAT (Mm00486218_m1); EAAT-1 (Mm00600697_m1); EAAT-2 (Mm00441457_m1); LAT2 (Mm00444250_m1)] per manufacturer’s instructions. Reactions were run in the Applied Biosystems Fast Real-Time PCR System and relative quantification performed using the comparative cycle threshold method (ΔΔCT), where CT values of the transcript of interest were normalized to β-actin CT values from the same sample, then compared to a calibrator CT value (untreated cells) to determine the relative fold increase in mRNA. β-actin CT values were unaffected by IL-1β treatment. Results were collected and analyzed using Applied Biosystems software. Statistics were performed on the logarithmic retransformation (i.e. geometric means) of 2−ΔΔCT values. Preliminary experiments were performed to establish that the amplification efficiency for each of the primer pairs was >94%.
Immunoblotting
Protein expression was determined by Western Blot analysis. Astrocytes in 24-well plates were washed twice with ice-cold PBS then incubated in 50 μL lysis buffer [50 mM Tris, pH 8.0, 1.0% Nonidet-P40 (NP40), 150 mM NaCl, and protease inhibitor cocktail (Roche)] for 30 minutes on ice. Cells were harvested by scraping, lysates from eight wells pooled, cellular debris removed by centrifugation (10,000 × g; 15 min; 4°C), and the resulting supernatants stored at −20°C. After thawing, proteins were concentrated via ethanol precipitation. Two volumes of ice-cold ethanol were added to the cell lysates. This mixture was stored at −20°C overnight, then spun (14,000 × g; 30 min; 4° C) and the pelleted protein resuspended in 1x urea buffer (50 mM Tris, pH 6.8, 2.5% glycerol, 5% SDS, 4 M Urea, 10 mM DTT, 0.02% bromophenol blue). One hundred μg protein (BCA assay; Pierce) was separated by 10% SDS-PAGE under reducing conditions and electrophoretically transferred to a nitrocellulose membrane (0.2 μm; Bio-Rad). Proteins of interest were detected sequentially using species-specific Western Breeze Immunodetection kits (Invitrogen) per manufacturer’s instructions. Primary antibodies and incubation times were as follows: xCT (2 μg/ml; rabbit polyclonal; Novus Biologicals; 2 h at 37° C), β-actin (0.3 μg/ml; mouse monoclonal; Sigma; 2 h at 37° C) or CD98 (4F2hc; 0.4 μg/ml; goat polyclonal; Santa Cruz; overnight at 4° C). Results were recorded on X-ray film (FujiFilm). Digitized images were analyzed by computer-assisted densitometry (Gel-Pro Analyzer) and xCT and 4F2hc protein normalized to their respective β-actin levels.
Hypoxia
Mixed cortical cell cultures were placed into an anaerobic chamber (Thermolabs) containing a gas mixture of 5% CO2, 10% H2, and 85% N2 (<0.2% O2). Culture medium was replaced by thorough exchange with a deoxygenated balanced salt solution (BSS5). Cells were placed in a 37°C incubator within the chamber for 4–5 hr and then returned to a 37°C, 6% CO2-containing normoxic (21% O2) incubator. Parallel cultures within the same plate were placed into deoxygenated BSS20 to assess for neuronal injury unrelated to the experimental paradigm. Importantly, in the presence of 20 mM glucose, cells can resist neuronal injury for nearly 12 hr of oxygen deprivation (Fogal et al., 2005). Neuronal cell death was assessed 20–24 hr later.
Measurement of neuronal cell death
Neuronal cell death was quantitatively determined by measurement of lactate dehydrogenase (LDH) released into the culture supernatant as described previously (Uliasz and Hewett 2000). Data are expressed as a percentage of total neuronal LDH activity (defined as 100%), determined by assaying the supernatant of parallel cultures exposed to 200 μM NMDA for 20–24 hr. Since primary cortical astrocytes do not contain NMDA receptors (Backus et al. 1989; Chan et al. 1990; Janssens and Lesage 2001) nor have been shown by us to be injured by oxygen deprivation times up to 12 hr (Fogal et al. 2005), changes in LDH activity can be used as a specific marker of neuronal injury in this system.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism (Version 4.03, GraphPad Software, Inc.) as described in each figure legend. Significance was assessed at p < 0.05.
Results
We previously demonstrated in a mixed cortical cell culture system that IL-1β potentiated neuronal injury induced by hypoxia (Fogal et al. 2005) via a process dependent on increased system xc− activity and impaired glutamate clearance (Fogal et al. 2007). Although, it was determined that this increase required astrocyte IL-1 receptor I (IL-1RI) signaling, the specific cell types responding were not ascertained (Fogal et al. 2007). Hence, initial experiments utilized purified cell culture preparations of neurons, astrocytes, and microglia to identify which cell types express xCT – the system xc− light chain –and 4F2hc and RBAT –the system xc− heavy chains – demonstrate functional cystine uptake, and respond to IL-1β by increasing system xc− activity.
Under basal conditions, xCT, 4F2hc, and RBAT transcripts are present in mixed cortical cell cultures and in each of the purified populations of cells: astrocytes, neurons, and microglia (Figure 1). Further, all culture preparations functionally express system xc− as determined by their ability to import radiolabeled cystine (Figure 2A–C; white bars). However, only astrocytes respond to IL-1β treatment (3 ng/ml; 20–24 hr) with an enhancement in cystine uptake (Figure 2A; black bars). No change in uptake was observed in pure neuronal or microglial cultures following IL-1β exposure at any time point assessed (Figure 2B,C).
Figure 1. Cellular system xc− expression.
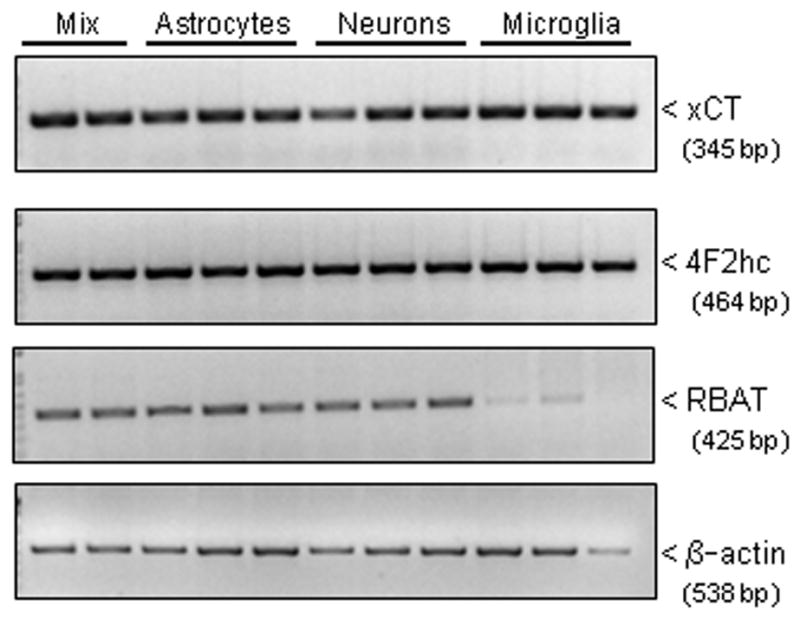
(A) Total RNA was isolated from unstimulated mixed cortical cell cultures (mix; lanes 1–2), pure astrocytes (lanes 3–5), pure neurons (lanes 6–8), and pure microglia (lanes 9–11), reverse transcribed, and PCR performed using specific primers for xCT (33 cycles), 4F2hc (33 cycles), RBAT (33 cycles) and β-actin (23 cycles) in separate reactions.
Figure 2. Astrocytes increase cystine uptake following IL-1β treatment.

Astrocytes (A; n=4), neurons (B; n=4–8) and microglia (C; n=6–10) were treated with vehicle (white bars) or IL-1β (3 ng/ml; black bars) for 20–24 hr following which cells were washed and incubated with a buffer containing 14C-L-cystine (3 μM) and uptake was determined over time as indicated. Data are expressed as mean ± SEM 14C-L-cystine uptake in pmol/mg protein. An asterisk (*) indicates a significant between-group difference as determined by a two-way ANOVA followed by Bonferroni’s post hoc test. Significance was set at p < 0.05.
Since IL-1β is known to regulate the expression of various genes, we next assessed whether its treatment (3 ng/ml) alters xCT steady-state mRNA expression in purified astrocyte, neuronal, and microglial cultures using quantitative PCR. Consistent with the uptake data, IL-1β elicits a time-dependent increase in xCT mRNA in astrocyte (Figure 3A), but not neuron or microglial cultures (Figure 3B), such that a 4–8 hr incubation produces a ≈12-fold increase in astrocytic xCT mRNA (Figure 3A). Interestingly, the transcripts for the system xc− heavy chains do not increase following IL-1β stimulation (Figure 3C). The increase in astrocytic steady-state xCT mRNA that follows IL-1β treatment (3 ng/ml; 6 hr) is not observed in astrocytes derived from IL-1RI −/− mice (Figure 4A). Further, simultaneous exposure of pure astrocytes with IL-1β (3 ng/ml; 6 hr) and the transcriptional inhibitor actinomycin D (10 μg/ml; 6 hr) blocks the IL-1β-mediated increase in xCT mRNA expression, whereas basal levels remain unchanged (Figure 4B). Consistent with the qPCR data, IL-1β treatment enhances proteins levels of astrocyte xCT (Figure 5A,B), whereas 4F2hc levels are unaffected (Figure 5A,C). Finally, concomitant exposure of astrocytes to IL-1β (3 ng/ml) and actinomycin D (12.5 μg/ml; 24 hr), prevents the IL-1β-mediated enhancement in cystine uptake, as does incubation with the protein synthesis inhibitor cycloheximide (1 μg/ml; 24 hr) (Figure 6).
Figure 3. IL-1β selectively increases astrocytic xCT mRNA.

(A) Pure astrocytes (n=4) were treated with IL-1β (3ng/ml) or its vehicle for the indicated durations and xCT mRNA assessed via qPCR. Data are expressed as mean ± SEM fold change in xCT mRNA compared to untreated cells (0 h). (B) Neurons (n=4) and microglia (inset; n=4) were treated with IL-1β (3ng/ml) or its vehicle for the indicated durations and xCT mRNA expression was assessed via qPCR. Data are expressed as mean ± SEM fold change in xCT mRNA compared to untreated cells (0 h). (C) Astrocytes (n=3) were treated with IL-1β (3ng/ml) or its vehicle for the indicated durations and 4F2hc and RBAT (inset) mRNA expression was assessed via qPCR. Data are expressed as mean ± SEM fold change in 4F2hc and RBAT mRNA compared to untreated cells (0 h). An asterisk (*) denotes values different from 0 hr as assessed by one-way ANOVA. Significance was set at p < 0.05.
Figure 4. IL-1R1 signaling and transcription are required for the enhancement in astrocyte xCT mRNA expression.

(A) Astrocytes (n=4–5 from single pup dissections) were treated with IL-1β (3 ng/ml) or its vehicle for 6 h. Thereafter, triplicate or quadruplicate culture wells were pooled, total RNA isolated, reverse transcribed and xCT and β-actin expression assessed using quantitative PCR. Data are expressed as mean ± SEM fold change in xCT mRNA compared to Il1r1 +/+ untreated cells (-IL-1β). (B) Astrocytes (n= 5) were treated with IL-1β (3 ng/ml) or its vehicle in the presence and absence of actinomycin D (Act D; 10 μg/ml) and xCT mRNA expression assessed via qPCR 6 hr later. Data are expressed as mean ± SEM fold change in xCT mRNA compared to untreated cells (-IL-1β, -Act D). An asterisk (*) denotes values different from control and a pound sign (#) indicates values that significantly differ from IL-1β-treated conditions as assessed by two-way ANOVA followed by Bonferroni’s post hoc test. Significance was set at p <0.05.
Figure 5. IL-1β increases xCT protein expression.

(A) Pure astrocyte cultures were incubated with vehicle or 3 ng/ml IL-1β in the absence or presence of cycloheximide (CHX 1 μg/ml) for 20–24 hr. Cells were harvested, whole cell lysates prepared, and 100 μg protein was separated by SDS-PAGE (10% gel). Western blot analysis was performed using antibodies directed against xCT, 4F2hc, and β-actin (loading control). Protein from unstimulated C6 glioma cells was used as a positive control for xCT protein. Lane 1, Basal; Lane 2, IL-1β; Lane 3, IL-1β+CHX, Lane 4, C6 positive control. Representative of two blots. (B,C) Films were scanned and densitometry performed using Gelpro Analyzer software. (B) xCT and (C) 4F2hc protein levels were normalized to their corresponding β-actin protein levels and expressed as a fold increase (mean ± SEM; n = 2) over control (basal; set to 1).
Figure 6. Protein synthesis is required for the enhancement of astrocyte system xc− activity that follows IL-1β treatment.
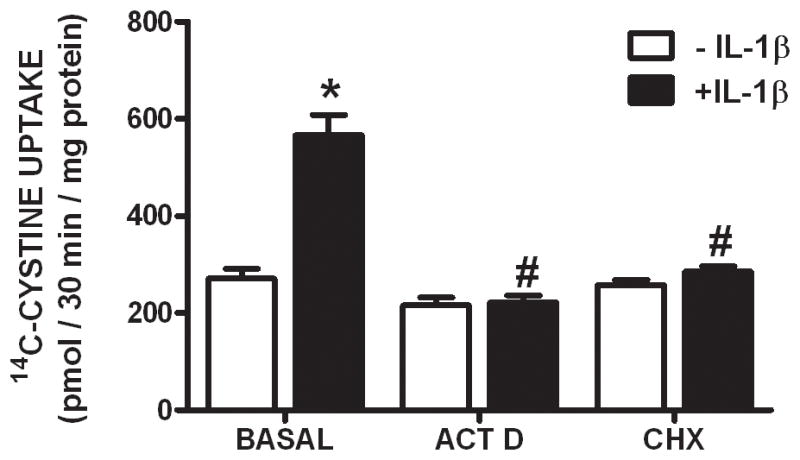
Pure astrocyte cultures (n =11–12) were treated with actinomycin D (ACT D; 12.5 μg/ml) or cycloheximide (CHX; 1 μg/ml) in the absence (white bars) or presence (black bars) of IL-1β (3 ng/ml) for 20–24 hr following which 14C-L-cystine uptake (3 μM labeled + 27μM unlabeled; 25°C) was determined. Data are expressed as mean ± SEM 14C-L-cystine uptake in pmol/30 min/mg protein. An asterisk (*) denotes values different from control (-IL-1β) and a pound sign (#) indicates values different from IL-1β-treated conditions as assessed by two-way ANOVA followed by Bonferroni’s post hoc test. Significance was set at p < 0.05.
We next set out to ascertain whether the astrocytic excitatory amino acid transporters (EAATs/system XAG−) are coordinately regulated by IL-1β, since glutamate efflux via system xc− is balanced by system XAG−-mediated glutamate uptake (Danbolt 2001; McBean 2002). Treatment of astrocytes with IL-1β (3 ng/ml) does not enhance EAAT-1 (aka GLAST) or EAAT-2 (aka GLT-1) mRNA expression (Figure 7A) in the same time frame as it does xCT (Figure 3A). Additionally, there is no difference in astrocytic 3H-D-aspartate uptake–used here as a measure of EAAT activity– following a 24 hr exposure to IL-1β as compared to control (Figure 7B). Finally, the mRNA expression of LAT-2, a light chain of the System L amino acid transporter, is also unchanged by IL-1β stimulation (Figure 7A), further demonstrating target specificity of the IL-1β response.
Figure 7. IL-1β does not regulate mRNA expression or activity of system XAG− amino acid transporters.

(A) Astrocytes (n= 3–4) were treated with IL-1β (3ng/ml) or its vehicle for the indicated durations and EAAT-1, EAAT-2, and LAT2 mRNA expression assessed via qPCR. Data are expressed as mean ± SEM fold change in mRNA compared to untreated cells (0 h). (B) Pure astrocyte cultures (n =10) were treated with vehicle (white bars) or IL-1β (3 ng/ml; black bars) for 20–24 hr following which 3H-D-aspartate (0.1 μCi/ml labeled + 1–100 μM unlabeled; 25°C) uptake was determined. Data are expressed as mean ± SEM 3H-D-aspartate uptake in cpm × 103/5 min/mg protein. No significant between-group differences were found via two-way ANOVA.
To elucidate the functional importance of these findings and definitively test whether astrocyte-mediated alterations in system xc− activity contribute to the development and progression of inflammatory (IL-1β-enhanced) hypoxic neuronal injury, astrocytes derived from sut mice – which carry a functional mutation in xCT (Chintala et al. 2005) – were utilized. Since no PCR genotyping protocol currently exists to detect the truncated slc7a11 (sut) gene, astrocytes were derived from the pooled cortices of the progeny obtained from heterozygous breeders (sut/+). If Mendelian inheritance is followed (¼ +/+, ½ sut/+, ¼ sut/sut), cultures should possess an estimated 50% reduction in functional sut gene expression. Indeed, astrocytes derived from sut animals demonstrate 61 ± 6% less cystine uptake compared to WT astrocytes when cultured under basal conditions (Figure 8A). Moreover, sut astrocytes do not respond to IL-1β with an increase in cystine uptake as do cultures derived from wild-type controls (Figure 8A). Finally, the enhanced neuronal vulnerability to hypoxia that follows IL-1β treatment in our mixed culture system (Fogal et al. 2007) –which is recapitulated herein (Figure 8B,9)– is not observed in chimeric cultures consisting of wild-type neurons plated on top of sut astrocytes (Figure 8B). Nor is it observed in wild-type cultures treated with a system xc− inhibitor (50 μM LY367385) or an NMDA receptor antagonist (10 μM MK-801) (Figure 9), as was similarly demonstrated by us previously (Fogal et al., 2007). Together, these data demonstrate that IL-1β selectively regulates system xc− activity in astrocytes and that this change is specifically responsible for the deleterious, excitotoxic effects of IL-1β found under hypoxic conditions.
Figure 8. Cystine uptake and hypoxic neuronal cell death are reduced in cultures containing sut astrocytes.

(A) Pure astrocyte cultures (n = 5–6) derived from either wild-type (white bars) or sut mice (black bars) [all cultured w/55 μM β-ME] were treated with vehicle or IL-1β (3 ng/ml) for 20–24 hr after which 14C-L-cystine uptake was determined. Data are expressed as mean ± SEM 14C-L -cystine uptake in pmol/30 min/mg protein. (B) Chimeric mixed cortical cell cultures were obtained by plating wild-type neurons on astrocytes derived from sut mice (black bars). These and control cultures (WT neurons on WT astrocytes; white bars) were treated with 1 ng/ml IL-1β or vehicle for 20–24 hr, washed, and then deprived of oxygen for 5 hr. The percentage of total neuronal cell death was determined 20–24 hr later (n = 4 cultures pooled from two independent experiments). An asterisk (*) indicates a significant within-group difference, while a pound (#) sign indicates a significant between-group difference as determined by a two-way ANOVA followed by Bonferroni’s post hoc test. Significance was set at p < 0.05.
Figure 9. Ionotropic glutamate receptor and system xc− antagonism prevent IL-1β-mediated hypoxic neuronal injury.
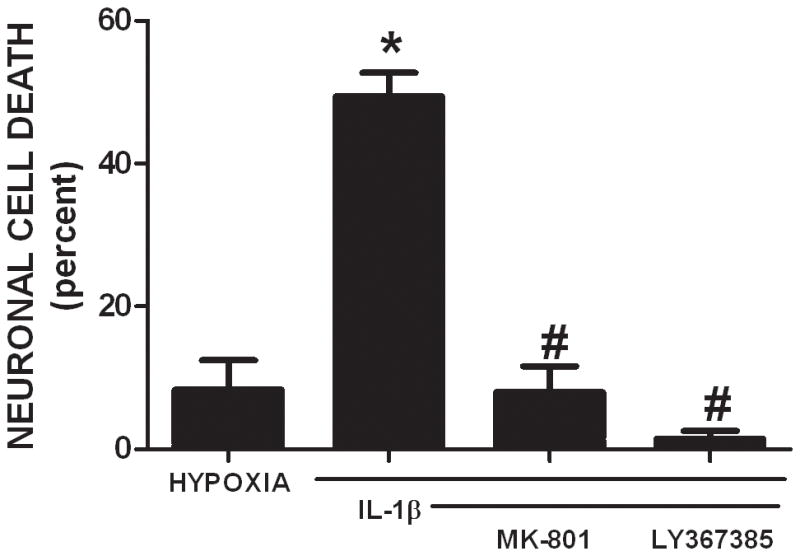
Mixed cortical cell cultures were treated with 3 ng/ml IL-1β for 20–24 hr, washed, and then deprived of oxygen for 4 hr. The ionotropic glutamate receptor antagonist MK-801 (10 μM) and the system xc− antagonist LY367385 (50 μM) were added at the initiation of hypoxia. The percentage of total neuronal cell death was determined 20–24 hr later (n = 5–6 cultures pooled from 2 independent experiments). An asterisk (*) denotes values different from control untreated cultures (hypoxia) and a pound sign (#) indicates values different from IL-1β-treated conditions as assessed by one-way ANOVA followed by a Student-Newman-Keul’s post hoc test. Significance was set at p < 0.05.
Discussion
Several studies have demonstrated that system xc− is an important contributor to the ambient extracellular glutamate levels that bathe the central nervous system (Augustin et al. 2007; Baker et al. 2002a; Baker et al. 2002b; Featherstone and Shippy 2008; Jabaudon et al. 1999; Melendez et al. 2005; Warr et al. 1999). Additionally, system xc− activity has been demonstrated to control synapse strength and courtship behavior in drosophila (Grosjean et al. 2008), as well as, drug seeking and sensitization behavior in rodents (Baker et al. 2008; Baker et al. 2002a; Moran et al. 2005). Finally, its activity can also contribute to neuropathology. For instance, export of glutamate via system xc− produces an excitotoxic necrosis that aids in glioma tumor growth, migration, and invasion (Lyons et al. 2007; Savaskan et al. 2008; Sontheimer 2008; Ye et al. 1999; Ye and Sontheimer 1999). Further, the deleterious effect of Aβ-, sAPP- or LPS-treated microglia or IL-1β-treated mixed cultures on neuronal and oligodendrocyte survival in vitro has been shown to be caused by system xc− -mediated excitotoxicity (Barger and Basile 2001; Domercq et al. 2007; Fogal et al. 2007; Piani and Fontana 1994; Qin et al. 2006). Thus, understanding the regulation of system xc− at the cellular and molecular level is of great import. Toward this end, the central observation of this study is that IL-1β enhances the functional expression of system xc− in astrocytes specifically and selectively via a process dependent on IL-1R1 signaling and de novo protein synthesis. Of pathological relevance, this IL-1β-mediated increase in astrocytic system xc− activity enhances neuronal injury initiated by hypoxia.
Demonstration of the expression of xCT, 4F2hc and RBAT mRNA in purified populations of neurons, astrocytes and microglia indicate that each cell type has the molecular machinery necessary for the formation of a functional system xc− antiporter (Figure 1). That these transcripts are translated to functional protein is demonstrated by the ability of each cell type to take up cystine in a system xc− -dependent manner (Figure 2). While others have demonstrated xCT mRNA expression in neurons (Dun et al. 2006; Ogawa et al. 2008) and retinal Muller cells (Mysona et al. 2009; Tomi et al. 2003), this is the first report of xCT mRNA expression in astrocytes and microglia, although xCT protein expression has been demonstrated in all three cell types (Burdo et al. 2006; Domercq et al. 2007; Dun et al. 2006; La Bella et al. 2007). Previous studies have also demonstrated functional system xc− activity in neurons (Dun et al. 2006; Murphy et al. 1990), astrocytes (Bender et al. 2000; Cho and Bannai 1990; Lewerenz et al. 2009; Pow 2001; Tang and Kalivas 2003), and microglia (Barger and Basile 2001; Barger et al. 2007; Domercq et al. 2007; Nakamura et al. 2003; Piani and Fontana 1994).
Interestingly, of the three cell types studied, only astrocytes respond to IL-1β by increasing the mRNA of the system xc− light chain xCT – though not the heavy chains (Figure 3A,C) – and by increasing cystine uptake (Figure 2A). Additionally, IL-1β induces expression of xCT protein in astrocytes, which can be completely blocked by concomitant incubation with cycloheximide (Figure 5). As increases in mRNA do not always translate to similar changes in protein expression, it is not surprising that a ≈12-fold increase in xCT mRNA expression (Figures 3,4) only resulted in a approximate four-fold change in xCT protein expression (Figure 5). Additionally, the discrepancy between changes in protein levels (4 fold) and the increase in cystine uptake (2 fold) (Figures 2,6) may occur as a function of the experimental system utilized to specifically isolate cystine transport via system xc−. The presence of 1mM D-aspartate –used to inhibit XAG−-mediated cystine uptake – likely alters the driving force required for optimal system xc− activity, as has been previously described (Reichelt et al. 1997).
Nevertheless, conclusive demonstration that the IL-1β-mediated enhancement in cystine uptake is mediated by astrocytic system xc− comes from our observation that astrocytes derived from sut animals harboring a functional mutation in the xCT gene (Chintala et al. 2005; Swank et al. 1996) fail to demonstrate this effect (Figure 8A). Both of these results (xCT upregulation and increased activity) are consistent with several studies, in neural and non-neural systems, which demonstrate an association between xCT mRNA expression and system xc− activity (Bridges et al. 2001; Dun et al. 2006; Mysona et al. 2009; Sato et al. 2001; Sato et al. 2004; Tomi et al. 2003). The lack of coordinate regulation of the subunits might not be too surprising as the heavy chains are utilized by other transport systems and as such exist in cells in excess (Stevens and Vo 1998; Verrey et al. 2004). Thus, they need not be dynamically regulated with their partners. It should be noted, however, that there is at least one study that describes a parallel increase in xCT and 4F2hc mRNA occurring in response to LPS (Sato et al. 2001).
The fact that microglia system xc− components (Figure 3B) and activity (Figure 2C) are unaffected by treatment with IL-1β is consistent with other studies demonstrating the inability of IL-1β to alter system xc− activity in cells of the macrophage/monocyte lineage (Piani and Fontana 1994; Sato et al. 1995). This finding may be due to the fact that microglia have a low ratio of signaling (i.e. IL-1RI) to decoy (i.e. IL-1RII) receptors making them unresponsive to IL-1β in either their resting or activation states (Pinteaux et al. 2002). Whether this same mechanism accounts for the inability of IL-1β to alter system xc− components and activity in neurons remains to be determined. Additionally, it is possible that the differential signaling that follows IL-1RI activation in neurons and astrocytes (Srinivasan et al. 2004) fosters xCT regulation in one cell type and not the other.
The IL-1β-mediated increase in steady-state xCT mRNA is due, at least in part, to the initiation of transcription following IL-1RI activation as this response was ablated in astrocytes derived from IL1r1 null mice or via concomitant treatment with actinomycin D (Figure 4). Whether the latter occurs via activation of transcription factors known to facilitate transcription of the xCT promoter in response to amino acid deprivation, LPS, and oxidative stress (e.g., Nrf2 and ATF4) (Lewerenz et al. 2009; Sasaki et al. 2002; Sato et al. 2004) remains to be determined. The failure of IL-1β to increase xCT protein expression (Figure 5) and cystine uptake (Figure 6) in the presence of cycloheximide suggests that de novo synthesis and subsequent insertion of a functional transporter into the membrane are required for the regulation of system xc− by IL-1β. Cycloheximide also prevented the enhancement of transporter activity mediated by LPS in mouse microglia (Piani and Fontana 1994), by glucose/glucose oxidase (i.e. oxidative stress) in human endothelial cells (Miura et al. 1992), and by an NO donor in retinal pigment epithelial cells (Bridges et al. 2001). Nevertheless, the requirement of transcription and translation may be cell, species, and/or stimulus-specific, as the work of Barger and colleagues showed that transcriptional and translational inhibitors were largely ineffective in blocking the LPS-mediated enhancement of glutamate release mediated by system xc− in rat microglial cultures (Barger et al. 2007). Additionally, post-transcriptional regulation of system xc− components and activity in astrocytes in response to the antibiotic ceftriaxone has recently been described (Lewerenz et al. 2009).
Not only does IL-1β demonstrate cellular specificity, but it appears to show target specificity as well. IL-1β had no effect on the expression of the system L transporter light chain, LAT2, or the expression and function of astrocytic excitatory amino acid transporters, EAAT-1 and EAAT-2 (Figure 7). System L, and LAT-2 in particular, was chosen as a potential target as it is expressed in brain (Segawa et al. 1999), it mediates the uptake of the neutral amino acids cysteine and methionine (Oxender et al. 1977; Sato et al. 1987; Segawa et al. 1999), and because methionine can be converted in the brain to cysteine via transulfuration, a process linked to GSH biosynthesis/homeostasis (Vitvitsky et al. 2006). EAATs were assessed as it is possible that glutamate import machinery could be cooperatively regulated to maintain low extracellular glutamate levels. The lack of effect on the EAATs may not be surprising considering the abundance at which they are expressed in astrocytes (Bergles and Jahr 1997; Lehre and Danbolt 1998). Additionally, this agrees with previous studies that note no alterations in system XAG− function in the face of increased system xc− activity (Lewerenz et al. 2009; Mysona et al. 2009).
The data described herein coupled with our previous studies (Fogal et al., 2005, 2007) suggest the following scenario (Supplemental Figure 1). As System xc− is an obligate exchanger, the import of cystine is coupled to glutamate export. Under physiological conditions, the accumulation of glutamate is prevented by its rapid clearance from the extracellular space via system XAG−. Consequently, no neuronal toxicity is observed — demonstrating that increased activity of system xc− is not inherently injurious (Fogal et al., 2005). In contrast, when glutamate uptake is impaired, increased system xc− activity can result in the accumulation of extracellular glutamate and subsequent excitotoxic neuronal cell death (Fogal et al. 2007). This work advances our previous study by demonstrating unequivocally that IL-1β enhances system xc− activity in astrocytes exclusively (Figure 2) and that this increase is responsible for the potentiation of neuronal injury found under hypoxic conditions. To wit, sut astrocytes – with impaired functional system xc− – neither increase cystine uptake in response to IL-1β (Figure 8A) nor support the ability of IL-1β to mediate hypoxic neuronal injury when co-cultured with neurons (Figure 8B).
In summary, we have analyzed expression of xCT mRNA and the activity of system xc− in purified astrocyte, neuron, and microglial cultures in the presence and absence of IL-1β, a cytokine known to be upregulated in and to contribute to various neurological disorders [for review see (Fogal and Hewett 2008)]. The results unequivocally demonstrate that astrocytes increase system xc− expression and activity in response to IL-1β, whereas neurons and microglia do not. The enhancement in astrocytic transporter activity requires transcription and translation, demonstrates specificity for the xCT subunit, and is responsible for the increase in hypoxic inflammatory (IL-1β-mediated) neuronal cell death.
Supplementary Material
The import of cystine via system xc− is directly coupled to glutamate export, occurring in a Na+-independent, Cl−-dependent manner with 1:1 stoichiometry. XAG− transports glutamate into the cell to maintain the driving force for system xc− (Reichelt et al. 1997) (left panel). Under hypoxic conditions, enhanced export of glutamate via system xc− in combination with reduced glutamate clearance by system XAG− produces an accumulation of extracellular glutamate which contributes to excitotoxic neuronal cell death (right panel). [Based on results described herein as well as those found in Fogal et al., 2005, 2007)].
Acknowledgments
Supported by grant NS051445 to SJH and JAH. While working on this project NAJ was supported by T32 NS041224. She is currently supported by NS066745 and AG035036.
References
- Augustin H, Grosjean Y, Chen K, Featherstone DE. Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J Neurosci. 2007;27(1):111–123. doi: 10.1523/JNEUROSCI.4770-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backus KH, Kettenmann H, Schachner M. Pharmacological characterization of the glutamate receptor in cultured astrocytes. J Neurosci Res. 1989;22(3):274–82. doi: 10.1002/jnr.490220307. [DOI] [PubMed] [Google Scholar]
- Baker DA, Madayag A, Kristiansen LV, Meador-Woodruff JH, Haroutunian V, Raju I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology. 2008;33(7):1760–72. doi: 10.1038/sj.npp.1301532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Shen H, Kalivas PW. Cystine/glutamate exchange serves as the source for extracellular glutamate: modifications by repeated cocaine administration. Amino Acids. 2002a;23(1–3):161–2. doi: 10.1007/s00726-001-0122-6. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi Z, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002b;22(20):9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261(5):2256–63. [PubMed] [Google Scholar]
- Bannai S, Sato H, Ishii T, Sugita Y. Induction of cystine transport activity in human fibroblasts by oxygen. J Biol Chem. 1989;264(31):18480–18484. [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursos protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101(5):1205–13. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi MT, Gasol E, Manzoni M, Pineda M, Riboni M, Martín R, Zorzano A, Borsani G, Palacín M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc−. Pflugers Arch. 2001;442(2):286–96. doi: 10.1007/s004240100537. [DOI] [PubMed] [Google Scholar]
- Bender AS, Reichelt W, Norenberg MD. Characterization of cystine uptake in cultured astrocytes. Neurochem Int. 2000;37:269–276. doi: 10.1016/s0197-0186(00)00035-8. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19(6):1297–308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum free medium combination. J Neurosci Red. 1993;35(5):567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Bridges C, Kekuda R, Wang H, Prasad P, Mehta P, Huang W, Smith S, Ganapathy V. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42(1):47–54. [PubMed] [Google Scholar]
- Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc− in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54(5):549–57. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- Chan PH, Chu L, Chen S. Effects of MK-801 on glutamate-induced swelling of astrocytes in primary cell culture. J Neurosci Res. 1990;25(1):87–93. doi: 10.1002/jnr.490250111. [DOI] [PubMed] [Google Scholar]
- Chintala S, Li W, Lamoreux ML, Ito S, Wakamatsu K, Sviderskaya EV, Bennett DC, Park YM, Gahl WA, Huizing M, et al. Slc7a11 gene controls production of pheomelanin pigment and proliferation of cultured cells. PNAS USA. 2005;102(31):10964–10969. doi: 10.1073/pnas.0502856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Bannai S. Uptake of glutamate and cysteine in C-6 glioma cells and in cultured astrocytes. J Neurochem. 1990;55(6):2091–7. doi: 10.1111/j.1471-4159.1990.tb05800.x. [DOI] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25(31):7101–10. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Domercq M, Sánchez-Gómez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc− and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178(10):6549–56. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- Dun Y, Mysona B, Van Ells T, Amarnath L, Ola MS, Ganapathy V, Smith SB. Expression of the cystine-glutamate exchanger (xc−) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324(2):189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Shippy SA. Regulation of synaptic transmission by ambient extracellular glutamate. Neuroscientist. 2008;14(2):171–81. doi: 10.1177/1073858407308518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogal B, Hewett JA, Hewett SJ. Interleukin-1beta potentiates neuronal injury in a variety of injury models involving energy deprivation. J Neuroimmunol. 2005;161(1–2):93–100. doi: 10.1016/j.jneuroim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Fogal B, Hewett SJ. Interleukin-1β: a bridge between inflammation and excitotoxicity? J Neurochem. 2008;106:1–23. doi: 10.1111/j.1471-4159.2008.05315.x. [DOI] [PubMed] [Google Scholar]
- Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System x(c)− activity and astrocytes are necessary for interleukin-1β-mediated hypoxic neuronal injury. J Neurosci. 2007;27(38):10094–105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gochenauer GE, Robinson MB. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc(−) subunits. J Neurochem. 2001;78(2):276–86. doi: 10.1046/j.1471-4159.2001.00385.x. [DOI] [PubMed] [Google Scholar]
- Grosjean Y, Grillet M, Augustin H, Ferveur JF, Featherstone DE. A glial amino-acid transporter controls synapse strength and courtship in Drosophila. Nat Neurosci. 2008;11(1):54–61. doi: 10.1038/nn2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby ME, Uliasz TF, Hewett SJ, Hewett JA. Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J Neurosci Methods. 2006a;150(1):128–137. doi: 10.1016/j.jneumeth.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Uliasz TF, Hewett SJ, Hewett JA. Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J Neurosci Methods. 2006b;150:128–137. doi: 10.1016/j.jneumeth.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Hewett J, Hewett S, Winkler S, Pfeiffer S. Inducible nitric oxide synthase expression in cultures enriched for mature oligodendrocytes is due to microglia. J Neurosci Res. 1999;57(3):411. doi: 10.1002/(sici)1097-4547(19990801)57:3<411::aid-jnr14>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci U S A. 1999;96(15):8733–8. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman N, Hewett JA, Hewett SJ. Society for Neuroscience Online. Chicago, IL: Neuroscience Meeting Planner; 2009. Enhanced astrocytic system xc− expression and activity mediate the neurotoxic effect of interleukin-1beta. Program No. 545.2. [Google Scholar]
- Jackman N, Hewett SJ. Interleukin-1beta regulates astrocytic system xc− activity and expression. J Neurochem. 2009;108(S1):117. [Google Scholar]
- Janssens N, Lesage AS. Glutamate receptor subunit expression in primary neuronal and secondary glial cultures. J Neurochem. 2001;77(6):1457–74. doi: 10.1046/j.1471-4159.2001.00369.x. [DOI] [PubMed] [Google Scholar]
- La Bella V, Valentino F, Piccoli T, Piccoli F. Expression and developmental regulation of the cystine/glutamate exchanger (xc−) in the rat. Neurochem Res. 2007;32(6):1081–90. doi: 10.1007/s11064-006-9277-6. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18(21):8751–7. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, Wiedau-Pazos M, Schubert D, Maher P, Methner A. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67(19):9463–71. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23(7):299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Vuthiganon J, Kalivas PW. Regulation of extracellular glutamate in the prefrontal cortex: focus on the cystine glutamate exchanger and group I metabotropic glutamate receptors. J Pharmacol Exp Ther. 2005;314(1):139–47. doi: 10.1124/jpet.104.081521. [DOI] [PubMed] [Google Scholar]
- Miura K, Ishii T, Sugita Y, Bannai S. Cystine uptake and glutathione level in endothelial cells exposed to oxidative stress. Am J Physiol. 1992;262:C50–C58. doi: 10.1152/ajpcell.1992.262.1.C50. [DOI] [PubMed] [Google Scholar]
- Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25(27):6389–93. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4(6):1624–1633. [PubMed] [Google Scholar]
- Mysona B, Dun Y, Duplantier J, Ganapathy V, Smith SB. Effects of hyperglycemia and oxidative stress on the glutamate transporters GLAST and system xc− in mouse retinal Muller glial cells. Cell Tissue Res. 2009;335(3):477–88. doi: 10.1007/s00441-008-0742-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Ohmaki M, Murakami K, Yoneda Y. Involvement of protein kinase C in glutamate release from cultured microglia. Brain Res. 2003;962(1–2):122–8. doi: 10.1016/s0006-8993(02)03979-3. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Saito Y, Nishio K, Yoshida Y, Ashida H, Niki E. Gamma-tocopheryl quinone, not alpha-tocopheryl quinone, induces adaptive response through up-regulation of cellular glutathione and cysteine availability via activation of ATF4. Free Radic Res. 2008;42(7):674–87. doi: 10.1080/10715760802277396. [DOI] [PubMed] [Google Scholar]
- Oxender DL, Lee M, Moore PA, Cecchini G. Neutral amino acid transport systems of tissue culture cells. J Biol Chem. 1977;252(8):2675–9. [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system xc− in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152(7):3578–85. [PubMed] [Google Scholar]
- Pinteaux E, Parker LC, Rothwell NJ, Luheshi GN. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J Neurochem. 2002;83(4):754–63. doi: 10.1046/j.1471-4159.2002.01184.x. [DOI] [PubMed] [Google Scholar]
- Pow DV. Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. doi: 10.1002/glia.1037. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc− and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1–40. J Neurosci. 2006;26(12):3345–56. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt W, Stabel-Burow J, Pannicke T, Weichert H, Heinemann U. The glutathione level of retinal Muller glial cells is dependent on the high-affinity sodium-dependent uptake of glutamate. Neuroscience. 1997;77(4):1213–24. doi: 10.1016/s0306-4522(96)00509-x. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277(47):44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- Sato H, Fujiwara K, Sagara J, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem J. 1995;310 (Pt 2):547–51. doi: 10.1042/bj3100547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang H, Ishii T, Mann GE, Bannai S. Effect of oxygen on induction of the cystine transporters by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biol Chem. 2001;276(13):10407–10412. doi: 10.1074/jbc.M007216200. [DOI] [PubMed] [Google Scholar]
- Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutatmate transporter gene by amino acid deprivation. Biochem Biophys Res Commun. 2004;325(1):109–116. doi: 10.1016/j.bbrc.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274(17):11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Sato H, Watanabe H, Ishii T, Bannai S. Neutral amino acid transport in mouse peritoneal macrophages. J Biol Chem. 1987;262(27):13015–9. [PubMed] [Google Scholar]
- Savaskan NE, Heckel A, Hahnen E, Engelhorn T, Doerfler A, Ganslandt O, Nimsky C, Buchfelder M, Eyupoglu IY. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med. 2008;14(6):629–632. doi: 10.1038/nm1772. [DOI] [PubMed] [Google Scholar]
- Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. Identification and functional characterization of a Na+-independent neutral amino acid transporter with broad substrate selectivity. J Biol Chem. 1999;274(28):19745–51. doi: 10.1074/jbc.274.28.19745. [DOI] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26(41):10514–23. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar MD, Tereba A, Chen BD, Walker WS. Transformation of mouse bone marrow cells by transfection with a human oncogene related to c-myc is associated with the endogenous production of macrophage colony stimulating factor 1. J Cell Physiol. 1985;125(3):403–12. doi: 10.1002/jcp.1041250307. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105(2):287–95. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Jospeh DJ, Friedman W. Cell type-specific interleukin-1 beta signaling in the CNS. J Neurosci. 2004;24(29):6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens BR, Vo CB. Membrane transport of neuronal nitric oxide synthase substrate L-arginine is constitutively expressed with CAT1 and 4F2hc, but not CAT2 or rBAT. J Neurochem. 1998;71(2):564–70. doi: 10.1046/j.1471-4159.1998.71020564.x. [DOI] [PubMed] [Google Scholar]
- Swank RT, Reddington M, Novak EK. Inherited prolonged bleeding time and platelet storage pool deficiency in the subtle gray (sut) mouse. Lab Anim Sci. 1996;46(1):56–60. [PubMed] [Google Scholar]
- Tang X, Kalivas PW. Bidirectional modulation of cystine/glutamate exchanger activity in cultured cortical astrocytes. Ann NY Acad Sci. 2003;1003:472–475. doi: 10.1196/annals.1300.056. [DOI] [PubMed] [Google Scholar]
- Tomi M, Funaki T, Abukawa H, Katayama K, Kondo T, Ohtsuki S, Ueda M, Obinata M, Terasaki T, Hosoya K. Expression and regulation of L-cystine transporter, system xc-, in the newly developed rat retinal Müller cell line (TR-MUL) Glia. 2003;43(3):208–17. doi: 10.1002/glia.10253. [DOI] [PubMed] [Google Scholar]
- Trackey JL, Uliasz TF, Hewett SJ. SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem. 2001;79:445–455. doi: 10.1046/j.1471-4159.2001.00584.x. [DOI] [PubMed] [Google Scholar]
- Uliasz TF, Hewett SJ. A microtiter trypan blue absorbance assay for the quantitative determination of excitotoxic neuronal injury in cell culture. J Neurosci Methods. 2000;100(1–2):157–163. doi: 10.1016/s0165-0270(00)00248-x. [DOI] [PubMed] [Google Scholar]
- Verrey F, Closs EI, Wagner CA, Palacin M, Endou H, Kanai Y. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 2004;447(5):532–42. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem. 2006;281(47):35785–93. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- Warr O, Takahashi M, Attwell D. Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol. 1999;514 (Pt 3):783–93. doi: 10.1111/j.1469-7793.1999.783ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages. J Exp Med. 1987;165:628–640. doi: 10.1084/jem.165.3.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci. 1999;19(24):10767–77. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Research. 1999;59:4383–4391. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The import of cystine via system xc− is directly coupled to glutamate export, occurring in a Na+-independent, Cl−-dependent manner with 1:1 stoichiometry. XAG− transports glutamate into the cell to maintain the driving force for system xc− (Reichelt et al. 1997) (left panel). Under hypoxic conditions, enhanced export of glutamate via system xc− in combination with reduced glutamate clearance by system XAG− produces an accumulation of extracellular glutamate which contributes to excitotoxic neuronal cell death (right panel). [Based on results described herein as well as those found in Fogal et al., 2005, 2007)].