

Abstract
Background
Short-term administration of Galactosamine to experimental animals causes liver damage and acute liver failure (ALF), as well as acute renal failure in some cases. The aim of our study was to describe kidney disorders that developed in the course of galactosamine-induced liver failure.
Material/Methods
Sprague-Dawley rats were randomly divided into 2 groups: a study group administered galactosamine intraperitoneally and a control group administered saline.
Results
All the animals in the study group developed liver damage and failure within 48 h, with significant increase of alanine (p<0.001), aspartate aminotransferases (p<0.0001), bilirubin (p<0.004), and ammonia (p<0.005) and decrease of albumin (p<0.001) concentrations. Acute renal failure was observed in all test animals, with a significant increase in creatinine (p<0.001) and urea (p<0.001) concentrations and a decrease in creatinine clearance (p<0.0012). Moreover, osmotic clearance (p<0.001), daily natriuresis (p<0.003), and fractional sodium excretion (p<0.016) decreased significantly in this group of animals. The ratio of urine osmolality to serum osmolality did not change. Histopathology of the liver revealed massive necrosis of hepatocytes, whereas renal histopathology showed no changes.
Conclusions
Acute renal failure that developed in the course of galactosamine-induced ALF was of a functional nature, with the kidneys retaining the ability to concentrate urine and retain sodium, and there were no renal changes in the histopathological examination. It seems that the experimental model of ALF induced by galactosamine can be viewed as a model of hepatorenal syndrome that occurs in the course of acute damage and liver failure.
Keywords: Acute Kidney Injury; Disease Models, Animal; Galactosamine; Hepatorenal Syndrome; Liver Failure, Acute
Background
Galactosamine (GAL) is a 6-carbon amino sugar derivative of galactose. Under physiological conditions, it is a component of specific glycoprotein hormones, such as follicle-stimulating hormone or luteinizing hormone [1]. GAL is a potent hepatotoxic substance, which can cause hepatocyte death both by necrosis and apoptosis. It inhibits the synthesis of liver RNA via the production of uridine diphosphate hexosamines, which block the transcription of genetic material [2]. According to 1 study, GAL seems to sensitize liver cells to other hepatotoxic agents (e.g., intestinal endotoxins), and TNF-α may be a major mediator of this damage [3]. Due to the damage that GAL causes to liver cells, it is used in experimental models of acute liver failure (ALF).
Short-term administration of GAL to experimental animals causes liver damage and ALF [4]. Kepler et al. [5] showed that GAL administered to rats caused diffuse inflammation and liver damage. Successive studies confirmed the hepatotoxic potential of GAL, with a significant difference noted in the susceptibility of different animal species to the toxic effects of the compound [3]. Rats and rabbits proved to be sensitive to GAL, but mice were resistant, and the sensitivity of dogs depended on the type of substance used for general anaesthesia [6–9]. An ideal model of ALF is still missing. Although the GAL-based model is not perfect and does not fully reflect the picture of human ALF, the general consensus is that it is an appropriate model for studying the pathogenesis and treatment of ALF, particularly in small experimental animals [4].
Few studies have examined renal complications in the course of experimental GAL-induced ALF. Anand et al. [2] showed that acute renal failure occurred in the absence of changes in renal histopathology, in addition to acute liver damage, in rats following administration of GAL. Javlé et al. [10] and Makin et al. [11] observed a significant reduction in renal blood flow, in addition to hemodynamic changes in the splanchnic vascular bed following the administration of GAL to rats. We also found that animals rapidly developed functional acute renal failure, the development and the degree of which depended on the strain of animal tested, in addition to acute damage and liver failure, following intoxication with GAL [12]. However, the previous studies investigated the impact of other factors on the development of ALF and associated complications rather than the ALF model itself. Thus, the evaluation of renal disorders in those studies was only a background, not a primary objective.
The mechanisms underlying the transition from liver damage to renal failure have not been fully explained, and the lack of knowledge on these mechanisms makes it difficult to study the pathogenesis and treatment of ALF and its complications in humans. Therefore, the aim of this study was to describe all detectable kidney disorders that developed in the course of GAL-induced experimental liver failure.
Material and Methods
Animals
The research project was approved by the Local Ethical Committee for Experiments on Animals. The study included 24 randomly selected male Sprague-Dawley rats. All weighed 200–250 g and were obtained from the Department of Laboratory Animals of the Polish Mother’s Memorial Hospital Research Institute, Lódź, Poland. All animals were kept in standard cages, fed a standard diet, and had free access to water and food. They were all maintained under a natural 12-h diurnal day/night cycle, at a constant temperature of 22±2°C and constant humidity of 45–50%. The experiments were performed between 10.00 and 18.00 h on naturally moving animals performing normal activities. During the experiments, the animals were placed individually in metabolic cages with free access to food and water. All experiments were performed in accordance with the principles of The Animals Scientific Procedures Act 1986.
Chemicals
D-α-galactosamine hydrochloride (Sigma-Aldrich, Germany) was administered intraperitoneally to the rats in the test group at a concentration of 200 mg/ml in physiological saline. The control group received an equivalent volume of saline.
Experimental protocols
The animals were randomly divided into 2 groups (n=12 in each): a control group (Group 1) and a test group (Group 2) in which the acute liver damage was induced. Group 1 received 1 ml of 0.9% saline intraperitoneally, and Group 2 received 1.1 g of GAL intraperitoneally per kg body weight in a solution of 0.9% saline at a concentration of 200 mg/ml.
Urine was collected daily for 24 h starting from the 24th to 48th hour after the administration of saline or GAL and analyzed immediately after collection. Blood samples of 6-ml volume were collected 48 h after the administration of GAL or saline from the beating heart of the animals under deep general anaesthesia. Biochemistry was determined in serum or urine using an Integra 700 autoanalyzer (Roche, USA), and reagents for the determination of bilirubin, aspartate aminotransferase, alanine aminotransferase, albumin, sodium, potassium, creatinine and urea were obtained from Roche (Germany). The ammonia level in the plasma was determined with EDTA-K3 anticoagulant using a Vitros 5600 autoanalyzer, (Johnson & Johnson, USA), and reagents for ammonia determination were obtained from Johnson & Johnson. An automatic osmometer (Knauer, Germany) was used to measure the osmolality of the urine and serum. Urine specific gravity was measured with a Junior II Miditron analyser (Roche Diagnostics, France), and reagents were purchased from Roche Diagnostics (France). Proteinuria was measured daily using an AU 680 analyser (Beckman Coulter, USA), and reagents for the determination of proteinuria were purchased from Beckman Coulter (USA).
Creatinine clearance was calculated according to the following formula:
where creatu is the creatinine concentration in urine, creats is the creatinine concentration in blood serum, and VU 24 h is the daily urine volume.
Osmolality clearance was calculated according to the following formula:
where U osm is the osmolality of the urine, V U 24 h is the daily urine volume, and P osm is the plasma osmolality.
The plasma osmolality was calculated according to the following formula:
where Nas is the sodium concentration in the blood serum, Ks is the potassium concentration in blood serum, and ureas is the urea concentration in blood serum.
Free water clearance was calculated according to the following formula:
where V U 24 h is the daily urine volume, and osmol cl is the osmolality clearance.
The excreted fraction of sodium was calculated according to the following formula:
where Nau is the sodium concentration in urine, Nas is the sodium concentration in blood serum, creatu is the creatinine concentration in urine, and creats is the creatinine concentration in blood serum.
Free water reabsorption was calculated according to the following formula:
where osmol cl is the osmolality clearance, and V U 24 h is the daily urine volume.
Preparation of liver and kidney sections
After blood collection, the liver and kidney of each of the test animals were prepared for histopathological examination. The organs were placed in formalin, embedded in paraffin, cut into ultra-thin sections, and finally stained with hematoxylin and eosin. Next, 2 independent pathologists evaluated the preparations under a light microscope.
Statistical analysis
Statistical analyses were performed with the Student’s t-test and an analysis of variance when multiple comparisons were needed. Where necessary, the Mann-Whitney U test was applied for the analysis of nonparametric data. A p value <0.05 was assumed as the level of significance. All data are expressed as means ± standard deviation.
Results
Profile of liver parameters after GAL-induced intoxication
Table 1 shows the liver parameters of the rats, indicating the development of acute liver damage and failure. Compared to the control group, the administration of GAL at a dose of 1.1 g/kg body weight caused severe liver damage and the development of ALF after 48 h, with a statistically significant increase in plasma levels of alanine aminotransferase (p<0.001), aspartate aminotransferase (p<0.0001), bilirubin (p<0.004), and ammonia (p<0.005) and a statistically significant decrease in albumin (p<0.001) (Table 1, Figure 1). GAL administration led to the development of hepatic encephalopathy in all animals tested, while 10 animals developed hepatic coma (83%) in clinical assessment.
Table 1.
Liver parameters in rats after galactosamine injection.
| Gr. (n) | ALTs IU/L | ASTs IU/L | Bils mg/dl | Ammons μmol/l | Albumins g/dl |
|---|---|---|---|---|---|
| 1 (12 sham | 56.2±9.9 | 252.5±149.8 | 0.4±0.26 | 52.8±38.1 | 2.9±0.1 |
| 2 (12) gal | 2098.6±886.1 | 1624.12±692.92 | 3.43±1.35 | 275.7±73.7 | 2.6±0.1 |
| gr 2 v gr 1 − p | p<0.001* | p<0.0001* | p<0.004* | p<0.005* | p<0.001* |
Bils – serum bilirubin; ASTs – serum aspartate aminotransferase; ALTs – serum alanine aminotransferase; albumins – serum albumin; ammons – serum ammonium; p – value of p. Biochemical parameters of the liver were evaluated 48 hours after saline or gal injection. Values are means ±SE, significance – p<0.05.
p<0.05.
Gr. 1 – sham, Gr. 2 – given 1.1 g/kg gal.
Figure 1.

Profile of liver parameters after galactosamine intoxication. Within 48 h, administration of galactosamine caused severe liver damage and the development of acute liver failure with an increase of the serum levels of ALT, bilirubin, and ammonia. ALT – alanine aminotransferase. * p<0.05, values are means ±SD.
Histopathological examination of the liver
Histopathological evaluation of the liver of the test animals showed generalized and massive hepatocyte necrosis (Figure 2A). In addition, diffused degenerative changes were visible, including small and giant cells, infiltrations of eosinophilic granulocytes, portal tract fibrosis and hyperemia. No changes were observed in the control group, which received only saline (Figures 2B).
Figure 2.
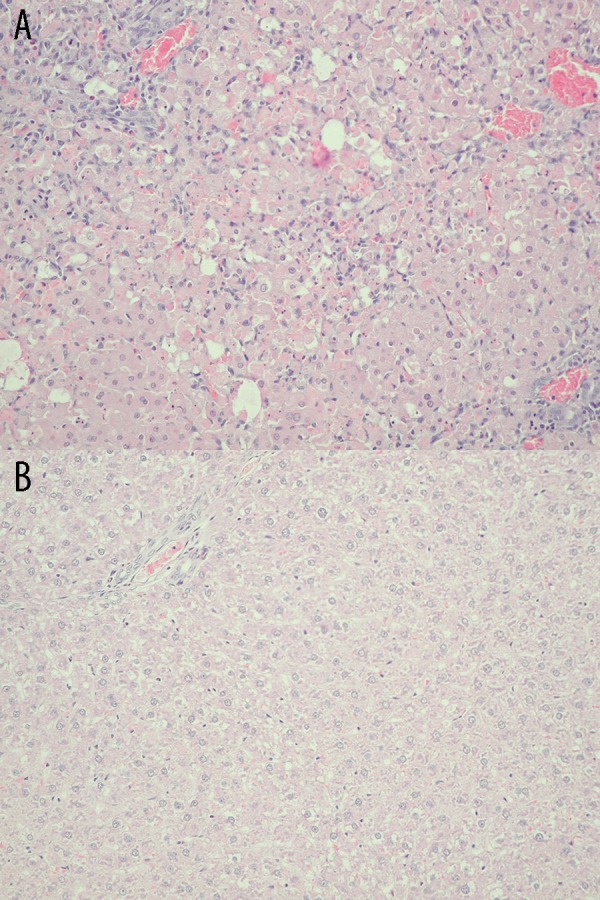
(A) Histopathological examination of the liver in sham rats. Lack of necrosis of hepatocytes in the liver from sham rats (Group 1) at 48 h after saline injection. Hematoxylin and eosin staining, light microscope, magnification ×20. (B) Histopathological examination of the liver in test rats. Diffuse necrosis of hepatocytes in the liver from test rats (Group 2) at 48 h after galactosamine injection. Diffused degenerative changes, including small and giant cells, infiltrations of eosinophilic granulocytes, portal tract fibrosis, and hyperemia. Hematoxylin and eosin staining, light microscope, magnification ×20.
Profile of kidney parameters after GAL-induced intoxication
Table 2 shows the renal parameters of the rats, indicating the development of acute renal failure. Compared to the control group, the administration of GAL at a dose of 1.1 g/kg bodyweight resulted in elevated indices of liver damage and failure, as well as the development of acute renal failure. The test group also showed a significant increase in serum concentrations of creatinine (p<0.001) and urea (p<0.001), a reduction in creatinine clearance (p<0.0012), and a reduction in daily urine volume (p<0.003) (Table 2, Figure 3). The water and electrolyte balance of the test group were typical of functional renal failure, with a significant decrease in osmotic clearance (p<0.001), daily natriuresis (p<0.003), fractional sodium excretion (p<0.016), and daily diuresis (p<0.003), and an increase in the urine specific gravity and osmolality, although the last 2 parameters were statistically insignificant. In addition, the ratio of urine osmolality to plasma osmolality did not change significantly; the ratio of the concentration of creatinine in urine to serum creatinine significantly decreased (p<0.0052); free water clearance increased (p <0.001), and free water reabsorption decreased (p<0.001). Moreover, the daily loss of proteins significantly decreased (p<0.007) (Table 2, Figure 3). Other parameters of water and electrolyte balance dysfunction and acute renal failure are presented in Table 2.
Table 2.
Renal parameters in rats after galactosamine injection.
| Gr. (n) | Creats mg/dl | Ureas mg/dl | Creat cl ml/min | Nas mmol/l | Ks mmol/l | Osmolp mosmol/kg | Creatu mg/dl | Ureau mg/dl | Nau mmol/l | Ku mmol/l | Nau 24 h mmol/24 h | FENa% | Osmolu mosmol/kg | Osmol cl ml/24 h | FW cl ml/24 h | Osmu/osmp | Creatu/creats | FW reab ml/24 h | SGu | Proteinu (mg/dl) | Proteinu 24 h mg/24 h | 24 h urine ml/24 h |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (12) sham | 0.47 ±0.04 | 33.7 ±3.8 | 0.89 ±0.4 | 142.6 ±1.4 | 5.82 ±0.46 | 308.1 ±3.5 | 50.4 ±14.3 | 3244 ±973.9 | 52 ±15.79 | 191. ±39.2 | 0.61 ±0.23 | 0.4 ±0.018 | 1480.7 ±69.3 | 57.3 ±13.9 | -45.3 ±10.9 | 4.8 ±0.21 | 108.2 ±38 | 45.3 ±10.9 | 1.001 ±0.003 | 193.25 ±72.63 | 22.86 ±10.86 | 11.9 ±2.9 |
| 2 (12) GAL | 0.76 ±0.09 | 80.1 ±10.1 | 0.18 ±0.12 | 142.3 ±1.1 | 5.90 ±0.47 | 323.2 ±3.9 | 37.9 ±22.4 | 2484.6 ±1787.1 | 46.25 ±12.73 | 70.8 ±27.2 | 0.24 ±0.15 | 0.17 ±0.12 | 1528.6 ±95.5 | 24.09 ±10.7 | -19.04 ±8.6 | 4.7 ±0.31 | 49.5 ±27.7 | 19.04 ±8.6 | 1.005 ±0.006 | 143.75 ±72.61 | 8.12 ±5.91 | 5.05 ±2.1 |
| gr 2 v gr 1 − p | p<0.001* | p<0.001* | p<0.0012* | p<0.71 | p<0.76 | p<0.003* | p<0.23 | p<0.34 | p<0.46 | p<0.001* | p<0.003 | p<0.016* | p<0.30 | p<0.001* | p<0.001* | p<0.27 | p<0.0052* | p<0.001* | p<0.17 | p<0.22 | p<0.007 | p<0.003* |
p<0.05.
24 h urine – twenty-four-hour urine collection; creats – serum creatinine; ureas – serum urea; creat cl – creatinine clearance; Nas – serum sodium; Ks – serum potassium; osmolp – plasma osmolality; creatu – urine creatinine; ureau – urine urea; Nau – urine sodium; Ku – urine potassium; Nau 24 h – 24 h urine sodium excretion; osmolu – urine osmolality; osmol cl – osmolal clearance; FW cl – free water clearance; FENa% – ejected fraction of sodium; osmu/osmp – urine osmolality/plasma osmolality ratio; creatu/creats – urine creatinine/serum creatinine ratio; FW reab – free water reabsorption; SGu – specific gravity of urine; proteinu – concentration of protein in urine; proteinu 24 h – 24h proteinuria, p – value of p. Biochemical parameters were evaluated 48 hours after saline or gal injection. 24h urine samples were collected during 24 hours from 24th to 48th hour after saline or gal injection and evaluated 48 hours after saline or gal injection. Values are means ±SE, significance – p<0.05. Group 1 – sham, group 2 – given 1.1 g/kg of GAL.
Figure 3.

Profile of renal parameters after galactosamine intoxication. Galactosamine administration resulted within 48 h in the development of acute functional renal failure with a reduction in creatinine clearance, fractional sodium excretion and osmotic clearance. * p<0.05, values are means ±SD.
Histopathological examination of the kidney
Renal histopathology showed no changes in the test group (Figure 4A) or the control group (Figure 4B).
Figure 4.
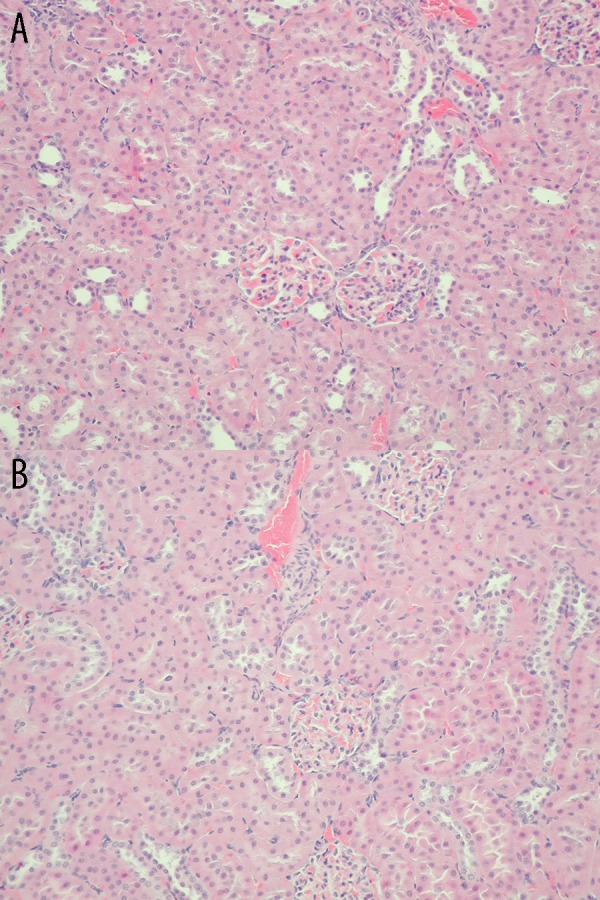
(A) Histopathological examination of the kidney in sham rats. Lack of morphological changes in the kidney from sham SDR rats (Group 1) at 48 h after saline injection. Hematoxylin and eosin staining, light microscope, magnification ×20. (B) Histopathological examination of the kidney in test rats. Lack of morphological changes in the kidney from test rats (Group 2) at 48 h after galactosamine injection. Hematoxylin and eosin staining, light microscope, magnification ×20.
Discussion
Renal complications in the course of experimental ALF have not been sufficiently studied. This work is the first report in the current literature to present a detailed description of water and electrolyte balance disorders and the development of acute renal failure as a consequence of acute damage and liver failure. In the present study, renal failure developed rapidly in a typical manner, with a parallel increase in nitrogen retention parameters and a decrease in creatinine clearance and daily diuresis. The osmotic clearance, daily natriuresis, and fractional sodium excretion values decreased, whereas those of urine osmolality and its specific gravity increased. Hence, the kidneys retained the ability to concentrate urine and retained osmotically effective osmolytes. The aforementioned, together with the lack of changes in the histopathological analyses of the kidneys, suggests that the kidneys remained functioning. The unchanged urine to plasma osmolality and urine to serum creatinine ratio after GAL intoxication also indicate that GAL was not directly toxic to the kidneys. The preserved renal tubular function and intact structure of the whole nephron also point to the functional nature of the kidney disorders. Other studies found similar findings, reporting no adverse effect of GAL in cell lines of renal tubules, even at much higher concentrations than those applied in our study [2,13]. These observations are also consistent with previous studies of the effect of GAL in experimental animals that reported only hepatotoxic effects [10,11].
Various methods can be used to induce acute renal failure in surgical, pharmacological, and infectious ALF models [3,14]. Surgical models include partial or total hepatectomy, temporary or permanent hepatic artery ligation, or ligation of the common bile duct [15–17]. Pharmacological models are based on the hepatotoxic properties of compounds, such as GAL, carbon tetrachloride, acetaminophen, thioacetamide, concanavaline A, or lipopolysaccharides [3,9,18–20]. Infectious models attempt to infect experimental animals with different hepatotropic viruses [21]. None of the aforementioned models provide an ideal ALF model because they do not accurately reflect clinical, biochemical, and histopathological ACF observed in humans. However, their diversity and the use of different animal species provide sufficient tools to study the pathogenesis and therapeutic possibilities of ALF. Terblanche and Hickman [22] described 6 features of an ideal model of ALF: 1) it should be reversible (i.e., react to the potential treatment; 2) it should be reproducible, with recurrent, characteristic endpoints; 3) the damage and liver failure should be a direct cause of death in experimental animals; 4) it should have a ‘therapeutic window’ (ie, sufficient time to implement the proposed treatment); 5) it should be possible to use the model in large experimental animals, where the treatment could include artificial liver support systems; and 6) the techniques and toxins or viruses applied should be as safe as possible for researchers and other individuals undertaking the experiments.
The presented model of GAL-induced intoxication of animals meets most of the above criteria. It is reproducibly established that liver damage and failure are the direct cause of death of the animals, it has an appropriate therapeutic window, it can be used in large experimental animals, and it is safe for scientists carrying out the experiments. Although the partial reversibility may be a limitation, the results of some studies suggested that certain substances can slow down the development of GAL-induced ALF [23,24]. As noted previously, the model of ALF used in the present study is employed less frequently than other models, and its value is probably underappreciated [5]. However, it has recently become more widely accepted due to standardization of methodology and standardization of clinical, morphological, and biochemical parameters of ALF [2,25]. In the model employed in the present study, the animals were intoxicated with a GAL dose of approximately 1 g/kg bodyweight at a concentration of 200 mg/ml in saline solution, which was administered intraperitoneally [26]. Experiments can be performed after 48 h when acute damage of hepatic parenchyma develops, leading to liver failure. The damage results in a very significant increase in the level of cellular enzymes alanine and aspartate aminotransferases, as well as bilirubin, in serum [27]. Simultaneously, the synthetic activity (decreased albumin) and metabolic and detoxifying functions of the liver decline (increased concentration of ammonia), coagulopathy develops, and the histopathological picture is dominated by massive necrosis of hepatocytes [28]. Severe liver damage leads to hemodynamic changes in the splanchnic vascular bed, with portal pressure, venous inflow, and intrahepatic portosystemic shunt all increased. Mean arterial pressure is reduced, mainly due to decreased peripheral vascular resistance, and the myocardial ejection fraction increases, which leads to the development of hyperdynamic circulation [29]. The vascular bed in the kidneys becomes contracted, particularly within the cortical vessels [11]. Renal function tests demonstrate a marked decline by more than 50% in renal blood flow and the glomerular filtration rate, with a simultaneous increase in urea and creatinine in serum [10]. Daily excretion of sodium is reduced, but the excretion of fractional sodium varies only slightly. The following parameters do not change: osmolality of the urine, ratio of urine osmolality to plasma osmolality, and free water clearance [2].
It seems that the GAL-induced experimental ALF model in the present study meets the rigorous criteria of an appropriate ALF model proposed by Terblanche and Hickman [22]. However, taking into account previous descriptions of the model and, above all, our results, the question arises as to whether it is solely a model of acute liver failure. Acute renal failure develops in this model in a relatively short time (about 48 h) after intoxication of animals with GAL. As it appears, it is only of functional nature. The deterioration of the glomerular filtration rate is accompanied by a reduction in osmotic clearance and daily natriuresis and an increase in urine osmolality. The ratio of urine osmolality to plasma osmolality does not change. Therefore, the kidneys retain the ability to concentrate urine, and their histopathological examination shows no changes. The clinical situation in which acute functional renal failure develops in the course of acute damage and liver failure is known as hepatorenal syndrome (HRS) [30,31]. In this syndrome, liver disease is the only etiological factor of kidney failure. In contrast, renal failure with oliguria, hyperazotemia, and hyponatremia develops in the absence of clinical, laboratory, and histological characteristics of any known renal disease, leading to renal failure [32,33]. This is the case in the ALF model described above and presented in our study.
Firstly, damage to the liver in the present study caused by GAL is the only cause of acute renal failure. GAL had no direct toxic effect on the structure of kidneys in common with that reported in previous studies [2,13]. Secondly, the pathogenesis of the development of renal failure in this model corresponds with the mechanisms observed in typical HRS. It progresses from damage to the liver parenchyma to the development of portal hypertension, enlargement of the splanchnic vascular bed, reduction of the effective volume of fluid in the systemic circulation, and subsequent vascular baroreceptor stimulation, followed by activation of numerous vasoconstriction factors, including the renin-angiotensin system, sympathetic nervous system, or arginine vasopressin system. These mechanisms lead to renal cortical vasoconstriction, renal hypoperfusion, and renal failure. Javle et al. [10] observed that the administration of GAL to rats increased acute liver damage, portal pressure, venous inflow, and intrahepatic portosystemic shunting and that renal blood flow was subsequently reduced significantly, particularly in the renal cortical vessels. They also reported that the glomerular filtration rate was lower. Thirdly, the acute renal failure in the present study is of a purely functional nature. Other studies reported a similar finding. For example, Anand et al. [2] showed that following intraperitoneal administration of GAL to rats, in addition to acute liver damage, acute renal failure occurred in the absence of changes in renal histopathology. Furthermore, the kidneys in the current study retained the ability to concentrate urine, indicating the functional nature of the developing renal disorder. Therefore, it seems that the presented model is not only a model of ALF but also of HRS. However, opinions on this subject are divided, with some authors not sharing this view [2].
The model discussed is not the HRS model that develops in patients with cirrhosis and ascites. The etiology, pathogenesis, time of the emergence of symptoms and prognosis differ in these cases. The model that can mimic the mechanism of cirrhotic HRS is (e.g., chronic intoxication with carbon tetrachloride [CCl4]) administered by inhalation or orally [34,35]. CCl4 is administered for several weeks. After this time, ascites, elevated liver enzymes, hyponatremia, and hypoalbuminemia develop [36]. Peripheral vascular resistance is decreased, mean arterial pressure is reduced, and cardiac output is increased. The histopathological examination of the liver shows a typical picture of cirrhosis [37]. Renal disorders are dominated by an increase in sodium and water retention, with no apparent decrease in the glomerular filtration rate and renal blood flow [38]. However, some researchers emphasize the possibility of a direct toxic effect of CCl4 on the kidneys [39]. It is not known whether renal failure is derived solely from liver damage or additional toxic damage to the kidneys.
In contrast, intoxication of animals with GAL models the situation of patients with ALF and the development of acute renal failure during the onset of ALF. According to some authors, the model in the present study should be considered simply as a model of ALF and its complications and not as a model of typical HRS [40,41]. We believe that the difference between these opinions stems solely from the various classifications and definitions of HRS (i.e., whether acute renal failure per se falls within the definition of HRS). According to the stance and modified definition in 2007 of the International Ascites Club, HRS induced by acute liver damage is included in this classification as a type IV separate syndrome [42]. In our opinion, this view is completely legitimate. As indicated above, despite the different etiology of the mechanisms leading to renal failure, they coincide, at least partially, in both chronic and acute liver damage. These include disorders of the splanchnic vascular bed and hemodynamic changes in the systemic circulation and renal cortical vessels. Therefore, in our opinion, the experimental model of ALF caused by GAL intoxication can be simultaneously treated as a model of HRS in the course of acute damage and liver failure. In Table 3 we present short specification of the experimental model of ALF/HRS induced by galactosamine intoxication.
Table 3.
Specification of the experimental model of ALF/HRS induced by galactosamine intoxication.
| Apecies | Number/weight | Dosage of GAL | Route of intoxication | Sham | Anesthesia (for authopsy) | Confirmation of ALF | Confirmation of ARF | Confirmation of FRF | Confirmation of coma | Animals in coma (%) | Survivors – 48 h (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GAL | Rats | 24/200–250 g | 1.1 g/kg b.w. | i.p. | 0.9% saline | Chloral hydrate | ALT, AST, bilirubin, NH3, albumin, liver histology | Creatinine, urea, creatinine clearance, 24 hu collection | Osmolal clearance, FENa%, Nau24 h, 24 hu collection, kidney histology | Clinical assessment, NH3 | 83% | 100% |
GAL administration led to the development of hepatic encephalopathy in all animals tested, while 10 animals developed hepatic coma (83%). GAL – galactosamine; ALF – acute liver failure; ARF – acute renal failure; FRF – functional renal failure; b.w. – body weight; i.p. – intraperitoneally; ALT – alanine aminotransferase; AST – aspartate aminotransferase; NH3 – ammonium; 24 hu collection – 24 h collection of urine; Nau 24 h – 24 h urine excretion of sodium; FENa% – fraction of ejected sodium.
Conclusions
In the present study, acute GAL-induced intoxication of experimental animals caused acute damage and hepatic failure and secondary development of acute renal failure. This acute renal failure was of a functional nature, with the kidneys retaining the ability to concentrate urine and retain sodium, and no changes were observed in the histopathological examination. Therefore, it seems that the GAL-induced experimental model of ALF can be simultaneously treated as a model of HRS that occurs in the course of acute damage and liver failure.
Footnotes
Source of support: The study was supported by grant No. 216/KBL/12 from Ministry of Science and Higher Education
Conflicts of interest
The authors declare no conflict of interest.
References
- 1.Wu YH, Hu SQ, Liu J, et al. Nature and mechanisms of hepatocyte apoptosis induced by D-galactosamine/lipopolysaccharide challenge in mice. Int J Mol Med. 2014;33:1498–506. doi: 10.3892/ijmm.2014.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anand R, Harry D, Holt S, et al. Endothelin is an important determinant of renal function in a rat model of acute liver and renal failure. Gut. 2002;50:111–17. doi: 10.1136/gut.50.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rahman TM, Hodgson HJF. Animal models of acute hepatic failure. Int J Mol Path. 2000;81:145–57. doi: 10.1046/j.1365-2613.2000.00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuñón MJ, Alvarez M, Culebras JM, González-Gallego J. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol. 2009;15:3086–98. doi: 10.3748/wjg.15.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keppler D, Lesch R, Reutter W, Decker K. Experimental hepatitis induced by D-galactosamine. Exp Mol Pathol. 1968;9:279–90. doi: 10.1016/0014-4800(68)90042-7. [DOI] [PubMed] [Google Scholar]
- 6.Leist M, Gantner F, Künstle G, et al. The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol Med. 1996;2:109–24. [PMC free article] [PubMed] [Google Scholar]
- 7.Blitzer BL, Waggoner JG, Jones EA, et al. A model of fulminant hepatic failure in the rabbit. Gastroenterology. 1978;74:664–71. [PubMed] [Google Scholar]
- 8.Sielaff TD, Hu MY, Rollins MD, et al. An anesthetized model of lethal canine galactosamine fulminant hepatic failure. Hepatology. 1995;21:796–804. [PubMed] [Google Scholar]
- 9.Newsome PN, Plevris JN, Nelson LJ, Hayes PC. Animal models of fulminant hepatic failure: a critical evaluation. Liver Transpl. 2000;6:21–31. doi: 10.1002/lt.500060110. [DOI] [PubMed] [Google Scholar]
- 10.Javlé P, Yates J, Kynaston HG, et al. Hepatosplanchnic haemodynamics and renal blood flow and function in rats with liver failure. Gut. 1998;43:272–79. doi: 10.1136/gut.43.2.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makin AJ, Hughes RD, Williams R. Systemic and hepatic hemodynamic changes in acute liver injury. Am J Physiol. 1997;272:G617–25. doi: 10.1152/ajpgi.1997.272.3.G617. [DOI] [PubMed] [Google Scholar]
- 12.Saracyn M, Patera J, Kocik J, et al. Strain of experimental animals and modulation of nitric oxide pathway: their influence on development of renal failure in an experimental model of hepatorenal syndrome. Arch Med Sci. 2012;8:555–62. doi: 10.5114/aoms.2012.29281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, Söderbergh H, Karlsson K, Danielsson A. Protective effect of S-adenosyl-L-methionine on bromobenzene- and D-galactosamine-induced toxicity to isolated rat hepatocytes. Hepatology. 1996;23:359–65. doi: 10.1053/jhep.1996.v23.pm0008591864. [DOI] [PubMed] [Google Scholar]
- 14.Pereira RM, dos Santos RA, Oliveira EA, et al. Development of hepatorenal syndrome in bile duct ligated rats. World J Gastroenterol. 2008;14:4505–11. doi: 10.3748/wjg.14.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y, Zhou J, Dou KF, Chen Y. A rat model for acute hepatic failure. Hepatobiliary Pancreat Dis Int. 2003;2:423–25. [PubMed] [Google Scholar]
- 16.Ytrebø LM, Sen S, Rose C, et al. Systemic and regional hemodynamics in pigs with acute liver failure and the effect of albumin dialysis. Scand J Gastroenterol. 2006;41:1350–60. doi: 10.1080/00365520600714527. [DOI] [PubMed] [Google Scholar]
- 17.Rivera-Huizar S, Rincón-Sánchez AR, Covarrubias-Pinedo A, et al. Renal dysfunction as a consequence of acute liver damage by bile duct ligation in cirrhotic rats. Exp Toxicol Pathol. 2006;58:185–95. doi: 10.1016/j.etp.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 18.McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 2012;264:387–94. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pallottini V, Martini C, Bassi AM, Romano P, et al. Rat HMGCoA reductase activation in thioacetamide-induced liver injury is related to an increased reactive oxygen species content. J Hepatol. 2006;44:368–74. doi: 10.1016/j.jhep.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Torisu T, Nakaya M, Watanabe S, et al. Suppressor of cytokine signaling 1 protects mice against concanavalin A-induced hepatitis by inhibiting apoptosis. Hepatology. 2008;47:1644–54. doi: 10.1002/hep.22214. [DOI] [PubMed] [Google Scholar]
- 21.Tunon MJ, Sanchez-Campos S, Garcia-Ferreras J, et al. Rabbit hemorrhagic viral disease: characterization of a new animal model of fulminant liver failure. J Lab Clin Med. 2003;141:272–78. doi: 10.1067/mlc.2003.30. [DOI] [PubMed] [Google Scholar]
- 22.Terblanche J, Hickman R. Animal models of fulminant hepatic failure. Dig Dis Sci. 1991;36:770–74. doi: 10.1007/BF01311235. [DOI] [PubMed] [Google Scholar]
- 23.Wu Z, Kong X, Zhang T, et al. Pseudoephedrine/ephedrine shows potent anti-inflammatory activity against TNF-α-mediated acute liver failure induced by lipopolysaccharide/D-galactosamine. Eur J Pharmacol. 2014;724:112–21. doi: 10.1016/j.ejphar.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 24.Wang LK, Wang LW, Li X, et al. Ethyl pyruvate prevents inflammatory factors release and decreases intestinal permeability in rats with D-galactosamine-induced acute liver failure. Hepatobiliary Pancreat Dis Int. 2013;12:180–88. doi: 10.1016/s1499-3872(13)60029-6. [DOI] [PubMed] [Google Scholar]
- 25.Kalpana K, Ong HS, Soo KC, et al. An improved model of galactosamine-induced fulminant hepatic failure in the pig. J Surg Res. 1999;82:121–30. doi: 10.1006/jsre.1998.5420. [DOI] [PubMed] [Google Scholar]
- 26.Arai K, Lee K, Berthiaume F, et al. Intrahepatic amino acid and glucose metabolism in a D-galactosamine-induced rat liver failure model. Hepatology. 2001;34:360–71. doi: 10.1053/jhep.2001.26515. [DOI] [PubMed] [Google Scholar]
- 27.Cuesta E, Boada J, Calafell R, et al. Fructose 1,6-bisphosphate prevented endotoxemia, macrophage activation, and liver injury induced by D-galactosamine in rats. Crit Care Med. 2006;34:807–14. doi: 10.1097/01.ccm.0000202016.60856.03. [DOI] [PubMed] [Google Scholar]
- 28.Galun E, Zeira E, Pappo O, et al. Liver regeneration induced by a designer human IL-6/sIL-6R fusion protein reverses severe hepatocellular injury. FASEB J. 2000;14:1979–87. doi: 10.1096/fj.99-0913com. [DOI] [PubMed] [Google Scholar]
- 29.Horowitz ME, Schafer DF, Molnar P, et al. Increased blood-brain transfer in a rabbit model of acute liver failure. Gastroenterology. 1983;84:1003–11. [PubMed] [Google Scholar]
- 30.Mindikoglu AL, Weir MR. Current concepts in the diagnosis and classification of renal dysfunction in cirrhosis. Am J Nephrol. 2013;38:345–54. doi: 10.1159/000355540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore CM, Van Thiel DH. Cirrhotic ascites review: pathophysiology, diagnosis and management. World J Hepatol. 2013;5:251–63. doi: 10.4254/wjh.v5.i5.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fagundes C, Barreto R, Guevara M, et al. A modified acute kidney injury classification for diagnosis and risk stratification of impairment of kidney function in cirrhosis. J Hepatol. 2013;59:474–81. doi: 10.1016/j.jhep.2013.04.036. [DOI] [PubMed] [Google Scholar]
- 33.Wonq F, Nadim NK, Kellum JA, et al. Working party proposal for a revised classification system of renal dysfunction in patients with cirrhosis. Gut. 2011;60:702–9. doi: 10.1136/gut.2010.236133. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Novoa JM, Rengel MA. A micropuncture study of salt and water retention in chronic experimental cirrhosis. Am J Physiol. 1977;232:F315–18. doi: 10.1152/ajprenal.1977.232.4.F315. [DOI] [PubMed] [Google Scholar]
- 35.López-Novoa JM, Rengel MA, Hernando L. Dynamics of ascites formation in rats with experimental cirrhosis. Am J Physiol. 1980;238:F353–57. doi: 10.1152/ajprenal.1980.238.5.F353. [DOI] [PubMed] [Google Scholar]
- 36.Rincón AR, Covarrubias A, Pedraza-Chaverrí J, et al. Differential effect of CCl4 on renal function in cirrhotic and non-cirrhotic rats. Exp Toxicol Pathol. 1999;51:199–205. doi: 10.1016/S0940-2993(99)80094-3. [DOI] [PubMed] [Google Scholar]
- 37.Clària J, Jiménez W, Ros J, et al. Pathogenesis of arterial hypotension in cirrhotic rats with ascites: role of endogenous nitric oxide. Hepatology. 1992;15:343–49. doi: 10.1002/hep.1840150227. [DOI] [PubMed] [Google Scholar]
- 38.Martin PY, Ohara M, Gines P, et al. Nitric oxide synthase (NOS) inhibition for one week improves renal sodium and water excretion in cirrhotic rats with ascites. J Clin Invest. 1998;101:235–42. doi: 10.1172/JCI626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrow JD, Awad JA, Kato T, et al. Formation of novel non-cyclooxygenase-derived prostanoids (F2-isoprostanes) in carbon tetrachloride hepatotoxicity. An animal model of lipid peroxidation. J Clin Invest. 1992;90:2502–7. doi: 10.1172/JCI116143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venkat D, Venkat KK. Hepatorenal syndrome. South Med J. 2010;103:654–61. doi: 10.1097/SMJ.0b013e3181e07751. [DOI] [PubMed] [Google Scholar]
- 41.Lata J. Hepatorenal syndrome. World J Gastroenterol. 2012;18:4978–84. doi: 10.3748/wjg.v18.i36.4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munoz SJ. The hepatorenal syndrome. Med Clin North Am. 2008;92:813–37. viii–ix. doi: 10.1016/j.mcna.2008.03.007. [DOI] [PubMed] [Google Scholar]