

Abstract
Purpose of Review
Hirschsprung’s Disease (HSCR) is characterized by an absence of ganglion cells in the distal hindgut, extending from the rectum to a variable distance proximally, and results from a failure of cranial-caudal neural crest cell migration. Hirschsprung’s-Associated Enterocolitis (HAEC) is a condition with classic manifestations that include abdominal distention, fever and foul-smelling stools, and is a significant and life-threatening complication of HSCR. The purpose of this review is to critically evaluate recent findings regarding the pathophysiology of HAEC.
Recent Findings
Several recent studies have investigated the etiology of HAEC in humans and mouse models. These studies suggest that alterations in the intestinal barrier, including goblet cell number and function and Paneth cell function, impaired gastrointestinal mucosal immunity, including B-lymphocyte trafficking or function and secretory IgA production, and dysbiosis of the intestinal microbiota may contribute to the development of HAEC.
Summary
Recent studies add to the body of literature suggesting that the intestinal defects observed in HSCR are not restricted to the aganglionic segment but extend to the mucosal immune system within and beyond the gastrointestinal tract. Future studies further dissecting mechanisms of HAEC and validating these findings in human patients will allow for development of directed therapeutic interventions.
Keywords: Hirschsprung’s disease, Hirschsprung disease, aganglionosis, enterocolitis, microbiome
Introduction
Since the Danish pediatrician Harald Hirschsprung described two cases of “Constipation in newborns due to dilatation and hypertrophy of the colon” to the International Congress for Children’s Diseases in Berlin in 1886[1], a great deal has been discovered regarding the etiology, pathophysiology, optimal medical and surgical management and follow-up care of children suffering from the disease bearing his name. Hirschsprung’s disease (HSCR, Online Mendelian Inheritance in Man #142623) is characterized by an absence of enteric nervous system (ENS) ganglion cells in the myenteric and submucosal plexues of the distal hindgut, resulting from failure of proximal to distal migration of neural crest cells in the gastrointestinal (GI) tract[2]. Most children present during the neonatal period with delayed passage of meconium, abdominal distention, feeding difficulty, and other signs of distal bowel obstruction. HSCR is managed surgically with removal of the aganglionic segment of bowel and re-establishing intestinal continuity by a surgical “pull-through” procedure to bring innervated bowel down to the anus while still preserving sphincter function. Unrecognized, HSCR leads to progressive bowel distention, the development of Hirschsprung’s-Associated Enterocolitis (HAEC), and death[3]. HAEC is a condition with classic manifestations that include abdominal distention, fever and foul-smelling stools, symptoms similar to those described by Professor Hirschsprung in his original two patients. HAEC accounts for the majority of morbidity and mortality in neonates with HSCR[3,4]. Furthermore, the incidence of HAEC appears to be unchanged from the pre-operative to post-operative period in both animals and humans and can present at any time from the newborn period into adulthood[5-7]. Current therapy for HAEC is relatively non-specific, and consists of intravenous antibiotics, rectal irrigations and bowel rest[8,9]. However, recent research has shed light on the pathogenesis of HAEC and may hold promise for the future development of targeted therapies. The purpose of this review is to critically review recent studies investigating the pathophysiology of HAEC.
Historical investigations into HAEC pathophysiology
The gut-associated lymphoid tissue (GALT) is the largest lymphoid organ in the body and is charged with providing protection against a variety of antigens that may gain access to the host, including food particles, commensal and pathogenic bacteria and their toxins. Historically, investigations have focused primarily on histopathologic changes present in HAEC-affected bowel in postulating an etiology for the disease[10,11]. The majority of these studies conclude that distal obstruction is causative in the development of HAEC. However, the clinical observation of continued susceptibility to HAEC in the post-pull-through period argues against obstruction as the sole etiology[5-7].
Infectious etiologies for HAEC, both viral and bacterial, have been proposed, but conflicting data abounds and no causative organism has been identified[11-15]. Barrier function against pathogens and foreign particles, provided by a mucus gel layer composed of several glycoproteins and secretory IgA (sIgA), has been studied in HSCR. HSCR patients with HAEC demonstrate increased IgA-containing plasma cells but decreased in luminal sIgA along the length of resected aganglionic bowel as compared to patients without HAEC[16]. Multiple studies have demonstrated decreased turnover and alterations in subsets of mucins, a major component of the luminal barrier, in HSCR patients[17]. Both of these observations extend to the ganglionated as well as aganglionic regions of the bowel, underscoring the concept that abnormalities in HSCR may extend beyond the aganglionic segment.
The lymphocyte populations present within the bowel wall have been studied in HSCR. No differences in lymphocyte numbers have been demonstrated in the distal bowel of HSCR or HAEC patients vs. normal neonates[18]. However, there are increased lymphocytes in the ganglionated bowel of HSCR patients that develop diversion colitis (HAEC equivalent)33. Taken as a whole, it is likely that the development of HAEC is multifactorial, with contribution from an altered microbiome, impaired innate immune and barrier defense, and impaired adaptive immunity (Figure 1).
Figure 1.
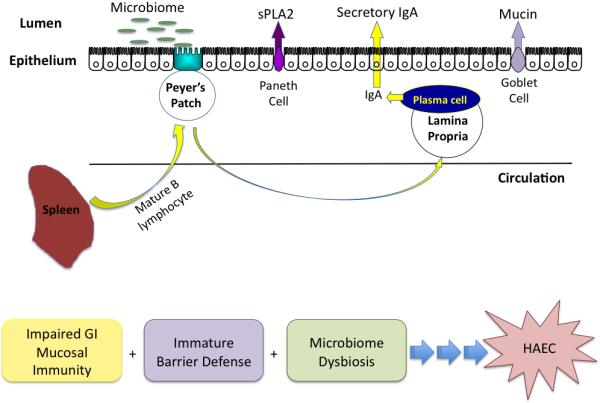
Pathogenesis of HAEC.
Based on recent studies, the etiology of HAEC likely includes impaired mucosal immunity, immature barrier defense and an altered microbiome. These studies suggest that alterations in the intestinal barrier, including goblet cell number and function and Paneth cell function, impaired gastrointestinal mucosal immunity, including B-lymphocyte trafficking or function and secretory IgA production, and dysbiosis of the intestinal microbiota may contribute to the development of HAEC.
Genetics of HSCR and mouse models of HSCR/HAEC
Multiple genetic defects have been associated with HSCR, most commonly mutations of rearranged during transfection (Ret) and of Endothelin receptor B (EdnrB), both of which are involved in ENS formation[19]. Recently, Ret has been shown to be necessary for the development of Peyer’s patches (PP), the primary inductive site for GI host defense, which suggests a potential developmental link between the ENS and GI mucosal immunity[20]. Unfortunately, Ret−/− animals exhibit a severe phenotype with an ENS absent distal to the stomach and renal agenesis, resulting in death shortly after birth and limiting their utility in studying HAEC. The second most common gene defect identified in patients with HSCR are mutations of Endothelin receptor B (EdnrB). EdnrB and its ligand, endothelin 3 (ET-3), regulate enteric neural crest cell proliferation, migration and differentiation. Naturally occurring mutations of both EdnrB and ET-3 have been found in mice, rabbits and horses. In mice, the piebald lethal strain lacks EdnrB and the spotted lethal strain lacks ET-3. Importantly, murine models of EdnrB mutation display aganglionosis of the distal hindgut, mimicking the most common clinical finding in HSCR patients[21,22].
Impaired Mucosal Immunity
Multiple lines of investigation point to interactions between the ENS and GI mucosal immune system. Anatomically, enteric neurons innervate the intestinal mucosa, including the gut-associated lymphoid tissue[23]. Enteric ganglia are positioned along the base, corona and interfollicular regions of PP[24]. Other investigators have demonstrated functional alterations in immune cell function by the ENS, likely related to the presence of neuropeptide receptors on the surface of immune cells[25]. It is also proposed that invasion of enteric nerves via the PP serves as a route of infection of the central nervous system by ingested prions[26].
Recently, Ret has been shown to be necessary for the formation of PP. In an elegant series of experiments[20,27], it has been shown that PP generation involves the aggregation of CD4+CD3−IL-7Ra+c-Kit+CD11c− lymphoid tissue inducer (LTi) cells, CD4−CD3−c-Kit+IL-7Ra−CD11c+Ret+ lymphoid tissue initiator (LTin) cells, and mesenchymal lymphoid tissue organizer (LTo) cells, leading to formation of PP primordia in the midgut of mice by embryonic day 16.5. Specifically, while NCC Ret signaling during ENS development is in cis, LTin cells respond to unconventional, in trans, Ret signaling. These Ret−/− animals fail to form PP and animals with a hypomorphic allele of Ret (Ret51/51) that exhibit aganglionosis limited to the distal colon demonstrate a reduced number of PP.
Reduced splenic size has been noted in EdnrB−/− animals[28]. These animals have abnormal splenic architecture and reduced total lymphocytes in the spleen. In a separate series of experiments, using mice with a neural crest-specific deletion of EdnrB that develop HAEC by 4 weeks of age, another group has demonstrated lymphopenia (mature B-lymphocytes) of the small bowel PP with decreased production of sIgA (Gosain et.al., unpublished data). The decreased sIgA production was only identified in the small bowel, not in respiratory mucosa (nasal and bronchial airways), indicating a gut specific defect in IgA production. To further dissect this mechanism, the authors evaluated the spleen histology and composition as previous reports have indicated the spleen as the primary site for IgA producing mature B-lymphocytes. Interestingly, the spleen of the knockout mice did demonstrate decreased size and altered composition with decreased B-lymphocytes in the germinal centers and marginal zone suggesting that the etiology for the specific gut defect in IgA production may be based in the spleen.
To differentiate between the immune system findings being a primary defect related to the endothelin receptor mutation, versus secondary to bowel obstruction from aganglionosis, another group has recently characterized mice with an induced bowel obstruction and found that they have similar changes in stress hormone levels and alterations in B lymphocyte development to those seen in EdnrB−/− mice[29*]. Furthermore, bone marrow transplant from EdnrB−/− mice to wild type animals failed to recapitulate the immune phenotype found in the EdnrB−/− mice. They interpret these findings to mean that bowel obstruction is the cause of the immune system changes seen in mice with aganglionosis. However, these results do not explain the continued susceptibility of these mice to HAEC following surgical pull-through[7], and fail to evaluate the possibility that immune system dysfunction must be coupled with alterations in the microbiome and mucosal barrier in order to lead to HAEC (final common pathway).
Impaired Mucosal Barrier Defense
Changes in colonic epithelium have been postulated as a causative factor in the development of HAEC through alterations in intestinal stem cell differentiation leading to perturbations in structure and function of the epithelial lining. Goblet cells are responsible for mucus secretion in the lumen of the GI tract, which facilitates trapping of harmful bacterial and viral pathogens. The mucus layer plays a key role in maintaining the integrity of the colonic epithelium and is composed of an extracellular matrix of water and glycosylated molecules with antimicrobial properties. Alterations in goblet cell structure and function have been found inflammatory bowel disease (IBD), with a decreased number of goblet cells observed in patients with active versus quiescent disease. A recent study investigated goblet cell structure and function in a murine model of HAEC and demonstrated that knockout mice that developed HAEC by post-natal day 21 had increased goblet cell number, size and proliferation in the aganglionic region, as compared to wild type animals[30*]. Similar results were also noted in humans, where the authors found that overall goblet cell size was similar however, the intracellular acidic and neutral mucin concentrations was greatly decreased compared to control groups. The authors extended this finding by performing electrophysiological measurements on colon segments and stool studies and found that knock-out mice exhibit fecal dehydration compared to wild type mice and that the transepithelial electrical resistance, a marker for epithelial barrier function, is greatly enhanced in knock-out animals with HAEC. These results were partially explained through increased resident time of the stool in the colon leading to enhanced dehydration and alterations in tight cellular junctions during development in aganglionic segments of colon. Finally in addition to alterations in goblet cell number, the mucin produced and subsequent mucus layer established demonstrated significantly hindered movement of nanoparticles on both the macro-scale (increased viscosity) and nano-scale (nanoparticle and mucin interactions) that resulted in decreased effective mesh pore size. Ongoing work will dissect the mechanism by which altered goblet cell structure and function in addition to mucus cell layer composition interact with the innate immune system and altered luminal microbiome.
Dysbiosis of the Intestinal Microbiome
Several microbes (Clostridium difficile, Escherichia coli, rotavirus) have been postulated as the causative agent(s) in HAEC, however no studies have documented a specific pathogen as the etiology. Intestinal microbiome composition is largely regulated by mucosal secretions, innate immune cells (Paneth cells) and secretion of antimicrobial molecules including secretory phospholipase A2 (sPLA2), lysozyme and defensins/cryptidins from the intestinal crypts[3,9,31*]. One group hypothesized that disturbances in the microbiota and mucosal innate immune defense mechanisms precede the development of HAEC and that increased susceptibility to bacterial translocation may partially be explained through alterations in innate immunity with decreased concentrations and activity of sPLA2[31*]. EdnrB knockout mice that closely resemble the phenotypic traits observed in humans with the development of HAEC by post-natal day of life 24-26 and nearly 100% mortality by day 28 were used for the study. Using cutting-edge culture-independent techniques, they found a significant difference in overall survival and alterations in the luminal microbiome (dysbiosis) with increased concentrations of Lactobacillus species in the non-HSCR animals and decreased concentrations in the knockout animals. The group further noted increased susceptibility to enteroinvasive E.coli in quantitative tissue culture of intestinal segments from knock-out animals, explaining this finding by demonstrating decreased levels of sPLA2 thus leading to decreased cleavage of bacterial fatty acids from phospholipids in the membrane and less bacterial membrane permeability and lysis.
Another study, in a similar mouse model of HSCR/HAEC, demonstrated alterations in both the fecal and colonic mucosa-associated microbiome of EdnrB knock-out animals starting at an early age, prior to the onset of bowel obstruction or HAEC[32]. They noted similar changes in the Lactobacillus population to those noted in the study by Pierre et.al. This group also performed fecal metabolite profiling and noted lower concentrations of formate in the HSCR animals. This alteration in formate, a short chain fatty acid associated with mucosal health, suggests alterations in microbial fermentation pathways in the hindgut of HSCR animals.
The development of a “high risk luminal biome” has also been described in a single human patient by analysis of bacterial 16S rRNA genes on stool samples both before and after 3 episodes of HAEC[15]. In keeping with previous reports, no single bacterial species was identified as the causative factor but rather the authors observed changes in the luminal biome prior to the development of HAEC. Recently a prospective, randomized, double blinded, placebo-controlled, multicenter trial was performed that evaluated the probiotic prophylaxis after pull-through for HSCR to decrease the development of HAEC[33*]. Unfortunately, the study failed to demonstrate a reduced incidence of HAEC between probiotic and placebo group. However, this may be reflective of the study size or of the specific probiotic formulation chosen, rather than a flawed hypothesis.
Summary
Taken together, these recent studies support a hypothesis for the development of HAEC, which includes luminal microbial alterations in conjunction with impaired mucosal barrier function and innate immune responses, allowing for the bacterial translocation and the development of HAEC (Figure 1). These studies add to the body of literature suggesting that the intestinal defects observed in HSCR are not only restricted to the aganglionic segment but extend to the mucosal immune system within and beyond the GI tract.
Conclusions
Since Professor Hirschsprung’s description of HAEC in 1886, great strides have been made in the diagnosis and management of HSCR. However, the pathogenesis of HAEC has remained unclear. Recent studies, using genetic models of HSCR and novel laboratory techniques, suggest that alterations in the intestinal barrier, impaired GI mucosal immunity and dysbiosis of the intestinal microbiota may contribute to the development of HAEC. Worldwide collaborative efforts, such as those being undertaken by the Hirschsprung’s Disease Research Collaborative, will allow validation and testing of these findings in human patients and will potentially provide for directed therapeutic interventions.
Key Points.
HAEC is a condition with classic manifestations that include abdominal distention, fever and foul-smelling stools, symptoms similar to those described by Professor Hirschsprung in his original two patients.
Murine models of EdnrB mutation display aganglionosis of the distal hindgut, mimicking the most common clinical finding in HSCR patients.
EdnrB knockout animals display reduced splenic size, abnormal splenic architecture, reduced total lymphocytes in the spleen and decreased production of sIgA in the gut.
Altered goblet cell structure and function is observed in aganglionic segments of bowel and may be responsible for the altered surface mucus properties observed in HSCR.
Intestinal dysbiosis and impaired innate immune function can lead to increased enteric microbe invasion and may be responsible for the systemic illness observed with HAEC.
Acknowledgements
None
Financial support and sponsorship
This work was supported by the National Institutes of Health K08DK098271 (AG).
Footnotes
Conflicts of interest
None
References
- 1.Hirschsprung H. Stuhlträgheit Neugeborener in Folge von Dilation und Hypertrophie des Kolons. Jahrbuch für Kinderheilkunde. 1888;27 [Google Scholar]
- 2.McKusick-Nathans Institute of Genetic Medicine, editor. Online Mendelian Inheritance in Man, OMIM® [Internet] Johns Hopkins University; Baltimore, MD: [no date] [Google Scholar]
- 3.Demehri FR, Halaweish IF, Coran AG, Teitelbaum DH. Hirschsprung-associated enterocolitis: pathogenesis, treatment and prevention. Pediatr Surg Int. 2013;29:873–881. doi: 10.1007/s00383-013-3353-1. [DOI] [PubMed] [Google Scholar]
- 4.Pini-Prato A, Rossi V, Avanzini S, Mattioli G, Disma N, Jasonni V. Hirschsprung's disease: what about mortality? Pediatr Surg Int. 2011;27:473–478. doi: 10.1007/s00383-010-2848-2. [DOI] [PubMed] [Google Scholar]
- 5.Marty TL, Seo T, Matlak ME, Sullivan JJ, Black RE, Johnson DG. Gastrointestinal function after surgical correction of Hirschsprung's disease: long-term follow-up in 135 patients. J Pediatr Surg. 1995;30:655–658. doi: 10.1016/0022-3468(95)90682-7. [DOI] [PubMed] [Google Scholar]
- 6.Polley TZ, Coran AG, Wesley JR. A ten-year experience with ninety-two cases of Hirschsprung's disease. Including sixty-seven consecutive endorectal pull-through procedures. Ann Surg. 1985;202:349–355. doi: 10.1097/00000658-198509000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Dhall D, Cheng Z, Wang HL, Doherty TM, Bresee C, Frykman PK. Murine model of Hirschsprung-associated enterocolitis II: Surgical correction of aganglionosis does not eliminate enterocolitis. J Pediatr Surg. 2010;45:206–11. doi: 10.1016/j.jpedsurg.2009.10.035. discussion 211–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frykman PK, Short SS. Hirschsprung-associated enterocolitis: prevention and therapy. Semin Pediatr Surg. 2012;21:328–335. doi: 10.1053/j.sempedsurg.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pontarelli EM, Ford HR, Gayer CP. Recent developments in Hirschsprung's-associated enterocolitis. Curr Gastroenterol Rep. 2013;15:340. doi: 10.1007/s11894-013-0340-6. [DOI] [PubMed] [Google Scholar]
- 10.Bill AH, Chapman ND. The enterocolitis of Hirschsprung's disease: its natural history and treatment. Am J Surg. 1962;43:70–73. [Google Scholar]
- 11.Teitelbaum DH, Caniano DA, Qualman SJ. The pathophysiology of Hirschsprung's-associated enterocolitis: importance of histologic correlates. J Pediatr Surg. 1989;24:1271–1277. doi: 10.1016/s0022-3468(89)80566-4. [DOI] [PubMed] [Google Scholar]
- 12.Wilson-Storey D, Scobie WG, McGenity KG. Microbiological studies of the enterocolitis of Hirschsprung's disease. Arch Dis Child. 1990;65:1338–1339. doi: 10.1136/adc.65.12.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas DF, Fernie DS, Malone M, Bayston R, Spitz L. Association between Clostridium difficile and enterocolitis in Hirschsprung's disease. Lancet. 1982;1:78–79. doi: 10.1016/s0140-6736(82)90216-1. [DOI] [PubMed] [Google Scholar]
- 14.Hardy SP, Bayston R, Spitz L. Prolonged carriage of Clostridium difficile in Hirschsprung's disease. Arch Dis Child. 1993;69:221–224. doi: 10.1136/adc.69.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Filippo C, Pini-Prato A, Mattioli G, Avanzini S, Rapuzzi G, Cavalieri D, Di Paola M, Stefanini I, Ceccherini I, Mavilio D, et al. Genomics approach to the analysis of bacterial communities dynamics in Hirschsprung's disease-associated enterocolitis: a pilot study. Pediatr Surg Int. 2010;26:465–471. doi: 10.1007/s00383-010-2586-5. [DOI] [PubMed] [Google Scholar]
- 16.Imamura A, Puri P, O'Briain DS, Reen DJ. Mucosal immune defence mechanisms in enterocolitis complicating Hirschsprung's disease. Gut. 1992;33:801–806. doi: 10.1136/gut.33.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aslam A, Spicer RD, Corfield AP. Children with Hirschsprung's disease have an abnormal colonic mucus defensive barrier independent of the bowel innervation status. J Pediatr Surg. 1997;32:1206–1210. doi: 10.1016/s0022-3468(97)90683-7. [DOI] [PubMed] [Google Scholar]
- 18.Turnock RR, Spitz L, Strobel S. A study of mucosal gut immunity in infants who develop Hirschsprung's-associated enterocolitis. J Pediatr Surg. 1992;27:828–829. doi: 10.1016/0022-3468(92)90375-h. [DOI] [PubMed] [Google Scholar]
- 19.Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, et al. Hirschsprung disease, associated syndromes and genetics: a review. J. Med. Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- 20.Veiga-Fernandes H, Coles MC, Foster KE, Patel A, Williams A, Natarajan D, Barlow A, Pachnis V, Kioussis D. Tyrosine kinase receptor RET is a key regulator of Peyer’s Patch organogenesis. Nature. 2007;446:547–551. doi: 10.1038/nature05597. [DOI] [PubMed] [Google Scholar]
- 21.Gariepy CE, Cass DT, Yanagisawa M. Null mutation of endothelin receptor type B gene in spotting lethal rats causes aganglionic megacolon and white coat color. Proc Natl Acad Sci USA. 1996;93:867–872. doi: 10.1073/pnas.93.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Druckenbrod NR, Powers PA, Bartley CR, Walker JW, Epstein ML. Targeting of endothelin receptor-B to the neural crest. Genesis. 2008;46:396–400. doi: 10.1002/dvg.20415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vulchanova L, Casey MA, Crabb GW, Kennedy WR, Brown DR. Anatomical evidence for enteric neuroimmune interactions in Peyer's patches. J Neuroimmunol. 2007;185:64–74. doi: 10.1016/j.jneuroim.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krammer HJ, Kühnel W. Topography of the enteric nervous system in Peyer's patches of the porcine small intestine. Cell Tissue Res. 1993;272:267–272. doi: 10.1007/BF00302732. [DOI] [PubMed] [Google Scholar]
- 25.Pascual DW, Kiyono H, McGhee JR. The enteric nervous and immune systems: interactions for mucosal immunity and inflammation. Immunomethods. 1994;5:56–72. doi: 10.1006/immu.1994.1038. [DOI] [PubMed] [Google Scholar]
- 26.Chiocchetti R, Mazzuoli G, Albanese V, Mazzoni M, Clavenzani P, Lalatta-Costerbosa G, Lucchi ML, Di Guardo G, Marruchella G, Furness JB. Anatomical evidence for ileal Peyer's patches innervation by enteric nervous system: a potential route for prion neuroinvasion? Cell Tissue Res. 2008;332:185–194. doi: 10.1007/s00441-008-0583-y. [DOI] [PubMed] [Google Scholar]
- 27.Patel A, Harker N, Moreira-Santos L, Ferreira M, Alden K, Timmis J, Foster K, Garefalaki A, Pachnis P, Andrews P, et al. Differential RET Signaling Pathways Drive Development of the Enteric Lymphoid and Nervous Systems. Science Signaling. 2012;5:ra55–ra55. doi: 10.1126/scisignal.2002734. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Z, Wang X, Dhall D, Zhao L, Bresee C, Doherty TM, Frykman PK. Splenic lymphopenia in the endothelin receptor B-null mouse: implications for Hirschsprung associated enterocolitis. Pediatr Surg Int. 2011;27:145–150. doi: 10.1007/s00383-010-2787-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *29.Frykman PK, Cheng Z, Wang X, Dhall D. Enterocolitis causes profound lymphoid depletion in endothelin receptor B- and endothelin 3-null mouse models of Hirschsprung-associated enterocolitis. Eur J Immunol. 2014 doi: 10.1002/eji.201444737. doi:10.1002/eji.201444737. Uses bone marrow transplantation from EdnrB−/− mice to wild type mice, along with induced intestinal obstruction, to demonstrate that B lymphocyte alterations prior to HAEC may be related to the stress or bowel obstruction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *30.Thiagarajah JR, Yildiz H, Carlson T, Thomas AR, Steiger C, Pieretti A, Zukerberg LR, Carrier RL, Goldstein AM. Altered goblet cell differentiation and surface mucus properties in Hirschsprung disease. PLoS ONE. 2014;9:e99944. doi: 10.1371/journal.pone.0099944. Demonstrates altered goblet cell number and size in HSCR patients and EdnrB−/− mice, as well as changes in trans-epithelial resistance and barrier function to macro- and nano-particles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Pierre JF, Barlow-Anacker AJ, Erickson CS, Heneghan AF, Leverson GE, Dowd SE, Epstein ML, Kudsk KA, Gosain A. Intestinal dysbiosis and bacterial enteroinvasion in a murine model of Hirschsprung's disease. J Pediatr Surg. 2014;49:1242–1251. doi: 10.1016/j.jpedsurg.2014.01.060. Demonstrates that EdnrB mutant animals exhibit an altered intestinal microbiome, reduced intestinal secretory phospholipase A2 and increased susceptibility of the intestine to invasion by pathogenic bacteria prior to the development of HAEC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward NL, Pieretti A, Dowd SE, Cox SB, Goldstein AM. Intestinal aganglionosis is associated with early and sustained disruption of the colonic microbiome. Neurogastroenterology & Motility. 2012;24:874–e400. doi: 10.1111/j.1365-2982.2012.01937.x. [DOI] [PubMed] [Google Scholar]
- *33.El-Sawaf M, Siddiqui S, Mahmoud M, Drongowski R, Teitelbaum DH. Probiotic prophylaxis after pullthrough for Hirschsprung disease to reduce incidence of enterocolitis: a prospective, randomized, double-blind, placebo-controlled, multicenter trial. J Pediatr Surg. 2013;48:111–117. doi: 10.1016/j.jpedsurg.2012.10.028. Prospective, randomized trial evaluating probiotics for prophylaxis against HAEC after pull-through for HSCR. [DOI] [PubMed] [Google Scholar]