

Abstract
Liver eosinophilia has been associated with incidences of drug-induced liver injury (DILI) for more than 50 years, although its role in this disease has remained largely unknown. In this regard, we recently showed for the first time that eosinophils played a pathogenic role in a mouse model of halothane-induced liver injury (HILI). However, the signaling events that drove hepatic expression of eosinophil associated chemokines, eotaxins, eosinophil infiltration, and subsequent HILI were unclear. We now provide evidence implicating hepatic epithelial derived cytokine thymic stromal lymphopoietin (TSLP) and type 2 immunity, in particular interleukin-4 (IL-4) production, in mediating hepatic eosinophilia and injury during HILI. TSLP was constitutively expressed by mouse hepatocytes and increased during HILI. Moreover, the severity of HILI was reduced in mice deficient in either the TSLP receptor (TSLPR) or IL-4 and was accompanied by decreases in serum levels of eotaxins and hepatic eosinophilia. Similarly, concanavalin A-induced liver injury, where type 2 cytokines and eosinophils play a significant role in its pathogenesis, was also reduced in TSLPR-deficient mice. Studies in vitro revealed that mouse and human hepatocytes produce TSLP and eotaxins in response to treatment with combinations of IL-4 and pro-inflammatory cytokines IL-1β and tumor necrosis factor-α.
Conclusion
This report provides the first evidence implicating roles for hepatic TSLP signaling, type 2 immunity, and eosinophilia in mediating liver injury caused by a drug.
Keywords: drug-induced liver injury, concanavalin A, type 2 immunity, eotaxins, eosinophils
Drug-induced liver injury (DILI) is a major health concern as it can lead to significant patient morbidity and mortality (1) and it is difficult to predict which new drugs will cause injury and who will be susceptible to this disease. Consequently, there is a need to better understand the mechanisms that lead to or protect against DILI and identify clinically relevant biomarkers of DILI. Eosinophilia, whether peripheral or hepatic, has been widely associated with instances of DILI (2, 3), although its role in DILI remains largely unknown. In this regard, we recently reported that eosinophils can play a pathogenic role in a mouse model of halothane-induced liver injury (HILI) (4). We found that eosinophils infiltrated the liver during early stages of injury, accumulated exclusively around areas of necrosis, and increased in number proportionally to the degree of injury (4). The severity of HILI was significantly reduced when eosinophils, but not neutrophils, were selectively removed with antibodies or when eosinophils were deleted genetically (4). Moreover, eotaxins appeared to attract eosinophils to the liver following halothane treatment (4). However, the signaling events that drove eotaxin expression, eosinophil infiltration, and subsequent HILI were not elucidated in that study.
Type 2 immune responses, traditionally referred to as T helper cell type 2 responses, are characterized by their dependence upon effector cytokines including interleukin-4 (IL-4), IL-5, and IL-13 and are known to drive eosinophil infiltration during allergic diseases of the skin and lungs (5). A main regulator of type 2 immunity is thymic stromal lymphopoietin (TSLP), a cytokine primarily derived from epithelial cells that exerts its effect through a heterodimeric receptor complex comprised of the TSLP receptor (TSLPR) and IL-7 receptor alpha (IL-7Rα) (6). Since it was known that TSLP signaling plays a pathogenic role in mouse models of allergic inflammation in the lung (7, 8) and skin (9) that was mediated in part by type 2 cytokines, including IL-4, and eosinophils, we decided to determine whether similar signaling could play a role in the mouse model of HILI. In this report, we provide evidence for this hypothesis by showing that mice deficient in TSLPR or IL-4 were resistant to HILI and that IL-4 can play a role in activating hepatocytes to secrete both TSLP and eotaxins. Similarly, it appears TSLP signaling may play a pathogenic role in other diseases of the liver as TSLPR-deficient mice were also resistant to concanavalin A-induced liver injury, which is an established model of natural killer T (NKT) cell-mediated hepatitis, where type 2 responses, in particular IL-4, and eosinophils, play a prominent role in mediating toxicity (10, 11).
Materials and Methods
Animals and Treatments
Wild-type (WT) Balb/cJ (000651) and IL4-/- (002496) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Tslpr-/- mice were developed as described previously (12) and backcrossed >10 generations to the Balb/c background. All animals employed in this study were 7 to 10 week old females with weights ranging between 18-22 g. Animals were acclimated for at least 6-7 days to a 12-hour light/dark cycle in a humidity and temperature-controlled, specific-pathogen-free environment in microisolator autoclaved cages. Mice were allowed autoclaved food and water ad libitum. All maintenance of animals conformed to the guidelines for humane treatment set by the Association for Assessment and Accreditation for Laboratory Animal Care International's Guide for the Care and Use of Laboratory Animals and by the National Institutes of Health. Animals were injected intraperitoneally with distilled halothane (30 mmol/kg) (Sigma, St. Louis, MO) dissolved in olive oil (Mild Olive Flavor Originale, Star Fine Foods, Fresno, CA) at a final concentration of 0.30 mmol/mL or vehicle only. In separate studies, mice were injected intravenously through the lateral tail vein with 10 mg/kg of concanavalin A (Type V) (Sigma) (1.25 mg/ml dissolved in sterile PBS). In other studies, 2 ng of recombinant mouse IL-4 (rIL-4) (BD Biosciences, San Jose, CA) was administered intravenously (0.1 mL) through the lateral tail vein of WT and IL4−/− mice 12 hours following halothane treatment. Recombinant IL-4 was dissolved in sterile PBS, pH 7.4, containing 1% bovine serum albumin (BSA) Cohn Fraction V (Sigma) for protein stabilization.
Sera and Tissue Collection, Assessment of Liver Injury, and Detection of Trifluoroacetylated (TFA)-Protein Adducts in Liver Homogenates of Mice
Mouse sera and liver tissues were collected and used for assessing liver injury by measurement of alanine aminotransferase (ALT) activities and histopathological analysis of hematoxylin and eosin (H&E) stained liver sections as reported previously (4). In addition, liver homogenates from mice were prepared and SDS-PAGE immunoblot analysis of TFA-protein adducts and β-tubulin as reported (4).
Isolation of Hepatic Leukocytes and Flow Cytometric Analysis of Hepatic Eosinophils
Mouse hepatic leukocytes were isolated and stained to quantify eosinophils by flow cytometry as reported previously (4). Briefly, cells gated by flow cytometry as CD11c- CD11b+ Gr-1low Siglec-Fhigh were quantified as eosinophils. The absolute number of eosinophils was calculated by multiplying their percentage by the total number of viable hepatic leukocytes per liver.
Isolation and Treatment of Primary Mouse Hepatocytes and Hepa 1-6 cells with Cytokines
Cultures of primary female Balb/cJ mouse hepatocytes and Hepa 1-6 cells (CRL-2026, ATCC, Manassas, VA) were used in this study (see Supporting Material for detailed methods).
Treatment of Cultured Human Primary Hepatocytes with Cytokines
Plateable cryopreserved human hepatocytes from three different donors (donors: HH1020, HH1026, HH1031; In Vitro ADMET Laboratories, Columbia, MD) were used in this study (see Supporting Material for detailed methods).
RNA Isolation and RT-PCR Analysis
Total RNA was isolated from liver sections, freshly isolated hepatocytes, infiltrating hepatic leukocytes, and cultured cells as reported previously (4). Relative gene expression was determined by quantitative RT-PCR analysis using validated gene-specific assays (Applied Biosystems, Carlsbad, CA) for mouse IL1β, IL4, IL7Rα, TNFα, CRLF2 (referred to throughout the manuscript as TSLPR), and mouse and human CCL11, CCL24, CCL26 (human only isoform), TSLP, and β-Actin.
Measurement of Mouse and Human Cytokines and Chemokines in Cell Culture Supernatants and Sera from Mice
Mouse and human protein levels in cell culture supernatants or sera from mice of CCL11, CCL24, CCL26, and TSLP were quantified using their respective DuoSet® ELISA kits (R&D Systems) following manufacturers protocols. Serum levels of mouse IL-4, IL-1β, and TNF-α were quantified using their respective ELISA Ready-Set-Go® kits (eBioscience) following manufacturers protocols.
Statistical Analysis
All data presented are reported as means ± standard error of the mean (SEM). Statistical significance between two groups was determined by the two-tailed Student t test, while statistical differences between multiple groups were determined by one-way Analysis of Variance with Newman-Keuls post-test analysis. Differences were considered significant when P values <0.05.
Results
TSLP, TSLP Receptor, and Interleukin-4 are Induced in Mouse Liver During HILI
TSLP and TSLPR expression was assessed in a mouse model of HILI (13), where eosinophils play a pathogenic role (4). As reported, treatment of female Balb/cJ mice with halothane increased serum ALT activities in a time-dependent manner beginning at 12 hours post treatment (Figure 1A). In accord with the serum ALT activities, histological evaluation of liver sections from halothane-treated animals revealed centrilobular necrotic lesions that peak at 24 hours post treatment (Figure 1B). TSLP mRNA increased in liver homogenates from mice treated with halothane relative to liver homogenates from vehicle-treated control mice at 12, 18, and 24 hours post-treatment (Figure 1C). This was similar to the observed gene expression changes of eotaxin-1 (CCL11) in liver homogenates in response to halothane-treatment (4). Hepatocytes appeared to be the source of TSLP, as TSLP mRNA was enriched in hepatocytes relative to liver homogenates and hepatic leukocytes isolated from naïve Balb/cJ mice (see Supporting Material Figure S1A). Serum levels of TSLP were also detected 18 and 24 hours post-treatment only from mice treated with halothane (Figure 1D). In addition, TSLPR and IL7Rα mRNA increased in liver homogenates from mice treated with halothane (Figure 1C). We also found that IL4 mRNA increased in liver homogenates in response to HILI at 12 hours post-treatment and then declined (Figure 1E), whereas IL-4 protein was detected in serum only from mice treated with halothane at 12, 18, and 24 hours post-treatment (Figure 1F). This finding may be due in part to the fact that expression levels of mRNA and protein do not always correlate and that a significant amount of serum IL-4 may have been derived from one or more sites other than the liver.
Figure 1.
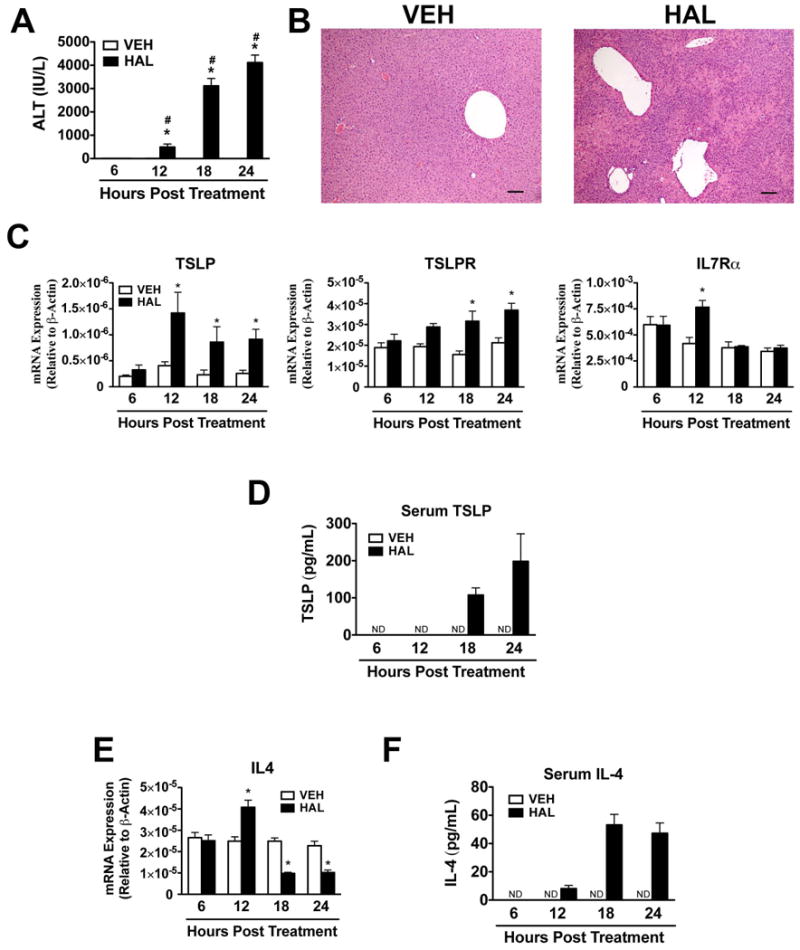
Hepatic TSLP and IL-4 are induced during HILI in mice. (A) Female Balb/cJ mice were injected intraperitoneally with halothane (30 mmol/kg) or vehicle (olive oil), and serum ALT activities were determined 6, 12, 18, and 24 hours post-treatment in mice treated with vehicle or halothane. Four independent experiments were performed with N=19-20 per group. *P<0.05 vs. vehicle treated control mice at the same timepoint. #P<0.05 vs. the previous timepoint within the same treatment. (B) Representative photomicrographs of H&E-stained liver sections from mice showing necrotic lesions in mice treated with halothane for 24 hours. Scale bars = 100 μm. (C) Levels of TSLP, TSLPR, and IL7Rα mRNA from liver homogenates at various timepoints following vehicle or halothane treatment with N=10-15 mice per group from three independent experiments. (D) Serum protein levels of TSLP in mice treated with vehicle or halothane with N=6 per group. Shown are means ± SEM, *P<0.05 vs. vehicle treated control mice at the same timepoint. ND=not detected. (E) Levels of IL4 mRNA from liver homogenates at various timepoints following vehicle or halothane treatment with N=10-15 mice per group from three independent experiments. (F) Serum protein levels of IL-4 in mice treated with vehicle or halothane with N=6 per group. All data shown as means ± SEM, *P<0.05 vs. vehicle treated control mice at the same timepoint. ND=not detected.
Halothane-Induced Liver Injury is Attenuated in Mice Deficient in TSLP Signaling
To determine whether TSLP/TSLPR signaling plays a role in HILI, we assessed HILI in TSLPR-deficient (Tslpr-/-) mice. This mouse model has been used extensively to study the role of TSLP and TSLPR signaling in leukocyte development and differentiation (12) as well as in the pathogenesis and progression of allergic lung (7, 8) and skin inflammation (9). The severity of HILI was significantly reduced at 12, 18, and 24 hours post-treatment in Tslpr-/- mice relative to WT control animals (Figure 2A). The decrease in serum ALT activity 24 hours post-treatment with halothane corresponded with a significant reduction in the size of necrotic lesions in the livers of Tslpr-/- mice relative to WT controls (Figure 2B). Similar results were obtained when littermate Tslpr+/+ controls were used in the place of WT mice (data not shown). TFA-protein adducts, which appear to initiate HILI (14), are formed when liver proteins are covalently labeled by the trifluoroacetyl chloride metabolite of halothane. Consequently, we verified that the decrease in the severity of HILI was not due to a decrease in TFA-protein adduct formation, as there was no substantial difference in TFA-protein adducts detected in liver homogenates from WT and Tslpr-/- mice 8 hours after halothane-treatment (Figure 2C). 8 hours is a timepoint at which there was no noticeable liver damage (Figure 2A), where TFA-protein adducts could be leaked from necrotic hepatocytes.
Figure 2.
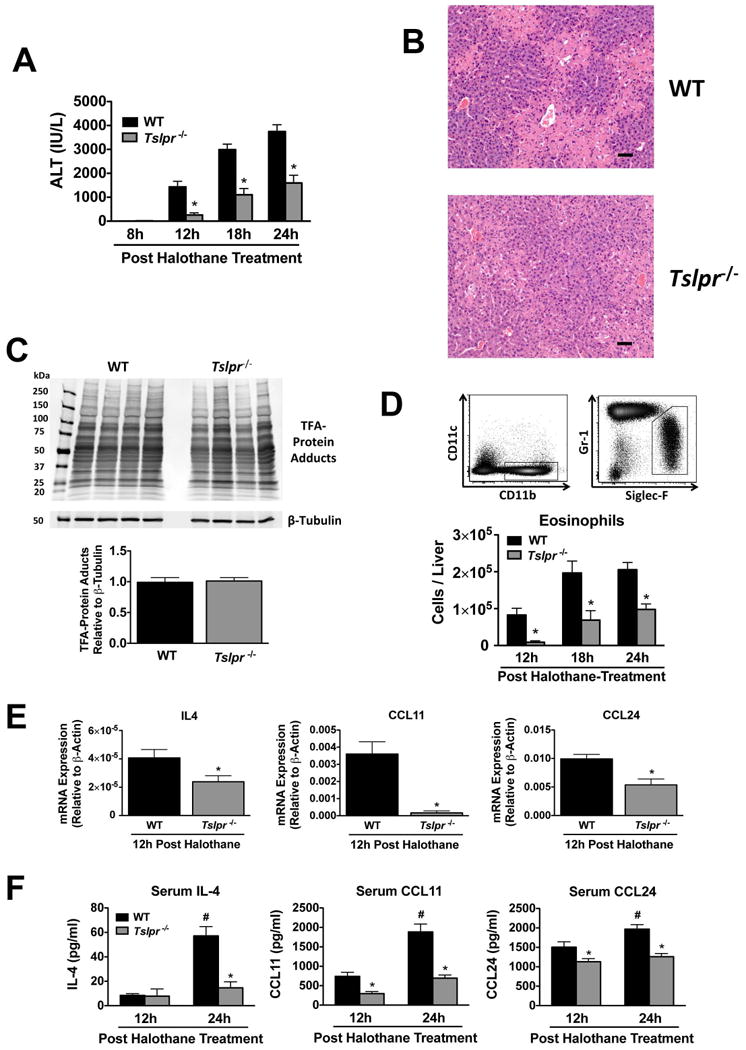
The severity of HILI was significantly attenuated in mice deficient in TSLPR. (A) Serum ALT activities at 8, 12, 18, and 24 hours post-halothane treatment in WT and Tslpr/- mice with N=5 for 8 hours and N=14-15 for 12, 18, and 24 hours. (B) Representative photomicrographs of H&E stained liver sections 24 hours after halothane treatment in WT and Tslpr-/- mice (scale bar = 50 μm). (C) Immunochemical detection of TFA-protein adducts and β-tubulin in homogenates from individual livers excised from WT and Tslpr-/- mice 8 hours post halothane-treatment (N=4 per group). Molecular markers (kDa) are indicated on the left and the relative abundance of TFA-protein adducts per group as measured by densitometry below (D) Representative flow cytometric density dot plots for hepatic eosinophils (CD11c- CD11b+ Gr-1low Siglec-Fhigh) from WT mice at 24 hours post treatment with halothane with a summary of the total number of hepatic eosinophils per liver from WT and Tslpr-/- mice 12, 18, and 24 hours post-halothane treatment with N=9-10 per group from two independent experiments. (E) Levels of IL4, CCL11, and CCL24 mRNA from liver homogenates from WT and Tslpr-/- mice sacrificed 12 hours post-halothane treatment with N=9-10 per group from two independent experiments. *P<0.05 vs. WT mice. (F) Serum protein levels of IL-4, CCL11, and CCL24 in WT and Tslpr-/- mice 12 and 24 hours post-halothane treatment with an N=9-10 per group from two independent experiments. *P<0.05 vs. WT mice and #P<0.05 vs. the previous timepoint within the group. All data was reported as means ± SEM.
Next we examined the levels of hepatic eosinophils in Tslpr-/- and WT mice during HILI. Hepatic eosinophils were quantified by flow cytometry as viable CD11c-, CD11b+, Gr-1low, Siglec-Fhigh cells (4) (representative gating scheme is depicted in Figure 2D). Similar to serum ALT activities, the number of hepatic eosinophils was significantly lower at 12, 18, and 24 hours after halothane treatment in Tslpr-/- mice relative to WT mice (Figure 2D), consistent with eosinophils contributing to HILI (4) and being regulated by TSLP (8, 9). There was no difference in the numbers of hepatic eosinophils in vehicle-treated Tslpr-/- and WT mice at any time-point (data not shown).
In order to determine whether the decrease in the severity of HILI and numbers of hepatic eosinophils observed in Tslpr-/- relative to WT mice may be due in part to decreases in the type 2 effector cytokine IL-4 as well as eotaxins CCL11 and CCL24, we examined hepatic expression of these factors in Tslpr-/- mice in relation to WT controls 12 hours post treatment with halothane. This timepoint was selected because it is first time at which liver injury is observed (Figure 1A) and also the point at which the greatest hepatic induction of eotaxins (4) and IL-4 (Figure 1E) occurs. Liver mRNA levels of IL4, CCL24, and particularly CCL11 were reduced in Tslpr-/- mice 12 hours post halothane treatment relative to WT controls (Figure 2E), as were serum protein levels of CCL11 and CCL24 at 12 and 24 hours and IL-4 at 24 hours post-treatment with halothane of Tslpr-/- mice relative to WT animals (Figure 2F).
Halothane-Induced Liver Injury is Attenuated in Mice Deficient in IL-4
Since IL-4 is often associated with TSLP signaling (9, 15) and can stimulate eotaxin expression and resultant eosinophil infiltration in mouse liver (11), we investigated whether IL-4 may be playing a pathogenic role during HILI. Similar to Tslpr-/- mice, the severity of HILI was reduced in IL4-/- mice relative to WT animals (Figure 3A and B) and was not due to diminished TFA-protein adduct formation (Figure 3C). Moreover, hepatic eosinophils in IL4-/- mice were reduced at 18 and 24 hours post halothane treatment relative to WT control mice (Figure 3D), while levels of hepatic eosinophils remained unchanged between the groups when animals were treated with vehicle only (data not shown). As expected, hepatic IL4 mRNA was undetected in IL4-/- mice (Figure 3E), whereas liver mRNA expression (Figure 3E) and serum levels (Figure 3F) of CCL11 and CCL24 12 hours post halothane treatment did not differ between IL4-/- and WT mice. However, there were decreases in serum levels of CCL11 and CCL24 between IL4-/- mice and WT controls 24 hours post treatment with halothane (Figure 3F).
Figure 3.
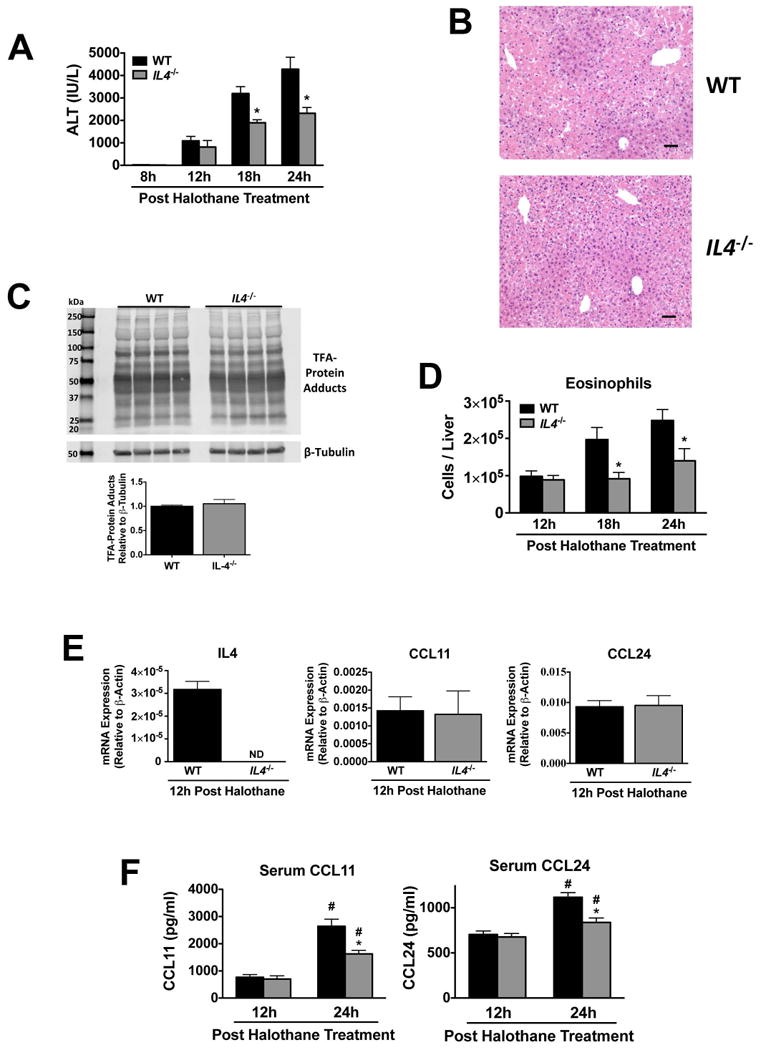
The severity of liver injury, numbers of hepatic eosinophils, and eotaxins were significantly attenuated in IL4-/- mice during HILI. (A) Serum ALT activities at 8, 12, 18, and 24 hours post-halothane treatment in WT and IL4-/- mice with N=5 for 8 hours and N=12-13 for 12, 18, and 24 hours. (B) Representative photomicrographs of H&E stained liver sections 24 hours after halothane treatment in WT and IL4-/- mice (scale bar = 50 μm). (C) Immunochemical detection of TFA-protein adducts and β-tubulin in homogenates from individual livers excised from WT and IL4-/- mice 8 hours post halothane-treatment (N=4 per group). Molecular markers (kDa) are indicated on the left and relative abundance of TFA-protein adducts per group as measured by densitometry below. (D) Summary of total number of hepatic eosinophils (CD11c- CD11b+ Gr-1low Siglec-Fhigh) from WT and IL4-/- mice at 12, 18 and 24 hours post-halothane treatment with N=9-10 per group from two independent experiments. (E) Levels of IL4, CCL11, and CCL24 mRNA from liver homogenates from mice sacrificed 12 hours post-halothane treatment with N=9-10 per group from two independent experiments. ND=not detected. (F) Serum protein levels of CCL11 and CCL24 in WT and IL4-/- mice 12 and 24 hours post-halothane treatment with an N=9-10 per group from two independent experiments. *P<0.05 vs. WT mice at same timepoint, #P<0.05 vs. previous timepoint within the same group. All data reported as means ± SEM.
To confirm that IL-4 has a pathologic role in HILI, we attempted to enhance the susceptibility of the IL4-/- mice to HILI by injecting them with rIL-4 12 hours post halothane treatment, a timepoint at which hepatic expression and serum levels of IL-4 rise in response to HILI (Figure 1E and F, respectively). rIL-4 increased the severity of HILI (Figure 4A and B) and numbers of hepatic eosinophils (Figure 4C) to levels greater than those observed in IL4-/- mice treated with PBS. Injection of rIL-4 failed to cause liver injury or increase the number of hepatic eosinophils in the absence of halothane treatment (data not shown).
Figure 4.
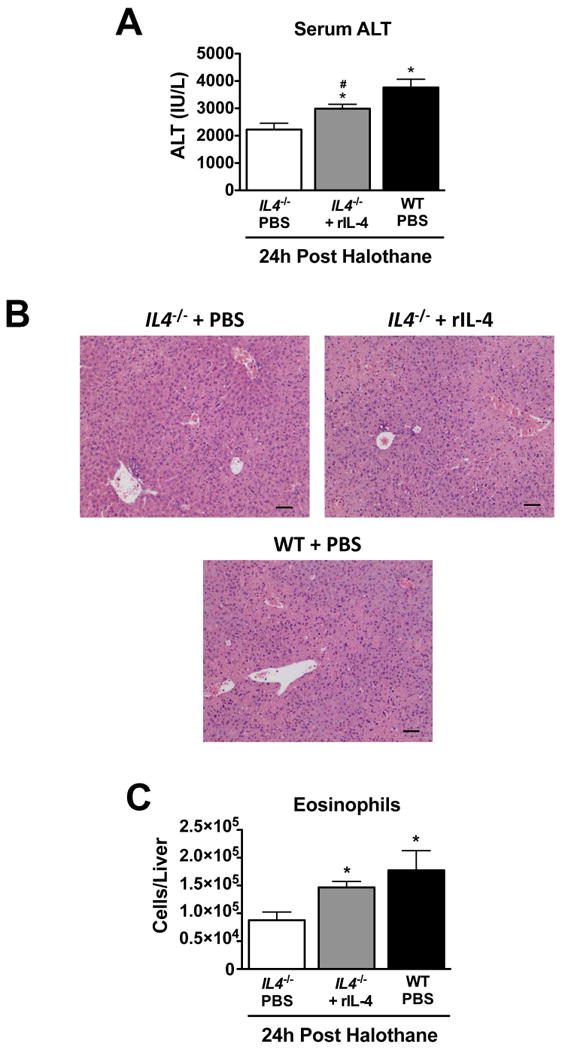
Recombinant IL-4 increased the susceptibility of IL4-/- mice to HILI. IL4-/- and WT mice were injected with 2.0 ng of rIL-4 12 hours post halothane-treatment or vehicle only (PBS) and sacrificed 12 hours later. (A) Serum ALT activities obtained 24 hours after halothane-treatment from IL4-/- and WT mice treated with rIL-4 or PBS. (B) Representative photomicrographs of H&E stained liver sections 24 hours after halothane treatment in WT and IL4-/- mice treated in the with rIL-4 or PBS (scale bar = 50 μm). (C) Summary of total number of hepatic eosinophils (CD11c- CD11b+ Gr-1low Siglec-Fhigh) from PBS or rIL-4 treated WT and IL4-/- mice at 24 hours post-halothane treatment. All data reported as means ± SEM with N=13-15 from three independent experiments. *P<0.05 vs. IL4-/- PBS-treated mice, #P<0.05 vs. WT PBS-treated mice.
Eotaxins and TSLP are Secreted by IL-4 and IL-1β or TNF-α Treatment Respectively from Mouse Liver Cells and Human Hepatocytes
One way IL-4 may promote HILI is by inducing hepatic secretion of eotaxins, as suggested by the results in a previous study where IL-4 was found to induce the expression of eotaxin genes in primary hepatocyte cultures from Balb/c mice (11). We have now extended this finding by showing that IL-4 can induce protein secretion of eotaxins in primary mouse hepatocyte cultures. The mRNA expression (Figure 5A) and protein secretion (Figure 5B) of CCL11 and CCL24 increased in mouse hepatocytes following treatment with 10 ng/ml of IL-4 for 24 hours, which was the minimum level of IL-4 at which maximal induction of eotaxins was observed (data not shown).
Figure 5.

Eotaxins and TSLP are secreted by IL-4 and IL-1β or TNF-α treatment respectively from mouse liver cells in vitro. Mouse hepatocytes were isolated from naïve WT mice and cultured for 48 hours. Gene expression (A) and protein levels in the culture supernatant (B) of CCL11 and CCL24 were determined at 24 hours post treatment with recombinant mouse IL-4 (10 ng/ml) or vehicle (N=4). *P<0.05 vs. untreated control. (C) Levels of IL-1β and TNF-α mRNA from liver homogenates of mice treated with vehicle or halothane (N=10-15 mice per group from three independent experiments). *P<0.05 vs. vehicle animals at the same timepoint. (D) Serum protein levels of IL-1β and TNF-α in mice treated with vehicle or halothane with N=6 per group. ND not detected. (E) Expression of TSLP mRNA (N=3 per group) and protein levels (N=6 per group) in the cell culture supernatant were assessed from mouse Hepa 1-6 cells treated individually or in combination for 24 hours with IL-1β (1ng/ml), TNF-α (10 ng/ml), and IL-4 (10 ng/ml). *P<0.05 vs. same IL-4 treatment in the absence of IL-1β and TNF-α, #P<0.05 vs. same IL-1β and TNF-α treatment in the absence of IL-4. (F) Expression of TSLP protein levels in the cell culture supernatant were assessed from primary mouse hepatocytes treated individually or in combination for 24 hours with IL-1β (1ng/ml), TNF-α (10 ng/ml), and IL-4 (10 ng/ml). *P<0.05 vs. same IL-4 treatment in the absence of IL-1β and TNF-α, #P<0.05 vs. same IL-1β and TNF-α treatment in the absence of IL-4. All data reported as means ± SEM with an N=6 comprised of N=3 wells from hepatocytes isolated from two individual mice.
It is known that early inflammatory cytokines IL-1β and TNF-α can induce TSLP secretion from cultured epithelial cells (16-18) and that IL-4 can augment the activities of IL-1β or TNF-α in inducing TSLP secretion in vitro (16, 18). Based on these findings and our discovery that gene expression of IL-1β and TNF-α were elevated in liver homogenates as early as 12 hours following halothane treatment (Figure 5C) as were serum levels relative to vehicle treated mice (Figure 5D), it seemed plausible that IL-1β, TNF-α, and IL-4 could have similar effects on inducing hepatic TSLP during HILI. We tested this idea initially in a mouse Hepa 1-6 cell line. The concentrations of recombinant IL-1β (1 ng/ml) and TNF-α (10 ng/ml) were selected based on previous reports (17, 18). IL-1β and TNF-α had little effect on TSLP mRNA and protein secretion when treated individually, but when combined together induced TSLP in Hepa 1-6 cells (Figure 5E). IL-4 also had no effect alone on TSLP mRNA and protein secretion from Hepa 1-6 cells, but synergistically induced TSLP levels over 25-fold in relation to untreated wells in the presence of IL-1β or TNF-α and 70-fold in the presence of both IL-1β and TNF-α (Figure 5E). In contrast to Hepa 1-6 cells, treatment of primary mouse hepatocytes with either IL-1β or TNF-α alone or in combination did not induce TSLP protein secretion (Figure 5F). However, similar to the results observed in Hepa 1-6 cells IL-4 treatment alone did not significantly increase TSLP protein secretion but when combined with either IL-1β or TNF-α and in the presence of both IL-1β and TNF-α increased TSLP levels relative to the same cytokine treatments in the absence of IL-4.
We also examined the effects of recombinant human IL-1β, TNF-α, and IL-4 treatment on the production of TSLP and eotaxins from cultured human hepatocytes from 3 separate donors. Unlike mice, the human TSLP gene encodes for two splice variant transcripts. The long transcript represents the inducible variant that encodes the fully functional TSLP protein (17, 19), while the function of the truncated protein remains unknown. Treatment of human hepatocytes with IL-1β or TNF-α induced TSLP mRNA 3-fold, while a 10-fold increase was observed when cells were treated with both IL-1β and TNF-α relative to untreated controls (Figure 6A). Protein levels of TSLP in cell culture supernatants were also induced by IL-1β and TNF-α (Figure 6B) analogous to the influence these cytokines had on TSLP mRNA. In contrast, IL-4 did not appear to augment the ability of IL-1β or TNF-α to induce TSLP mRNA production and protein secretion from human hepatocytes (Figure 6A and B). Closer examination revealed that similar to mouse liver cells (Figure 5E and F) IL-4 treatment augmented the activities of IL-1β and TNF-α in inducing TSLP mRNA and secretion in hepatocytes from donor HH1026 (see Supporting Material Figure S2).
Figure 6.
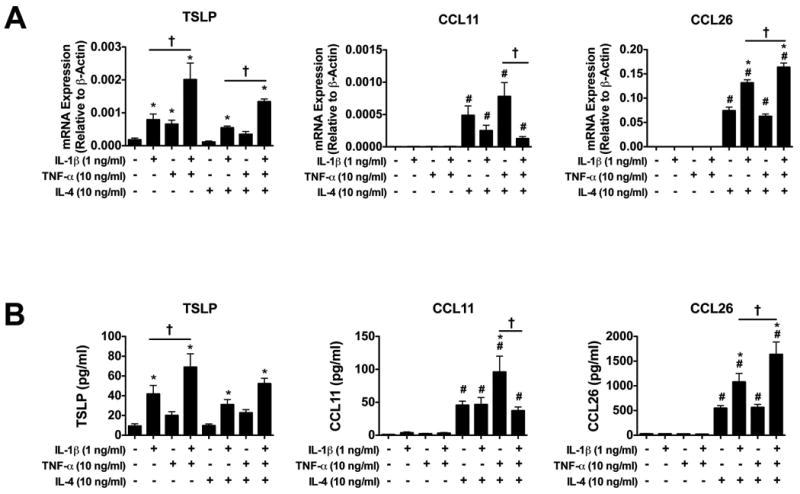
IL-4, IL-1β, and TNF-α stimulated eotaxin and TSLP expression and secretion from primary cultured human hepatocytes. Plateable cryopreserved hepatocytes from 3 donors (HH1020, HH1026, HH1031) were cultured for 2 days. On day 3, the cells were treated for 24 hours individually or in combination with recombinant human IL-1β (1 ng/ml), TNF-α (10 ng/ml), and IL-4 (10 ng/ml). Expression of TSLP, CCL11, and CCL26 mRNA in the cells (A) and protein levels of TSLP, CCL11, and CCL26 from cell culture supernatant (B) were assessed 24 hours post treatment. All data reported as means ± SEM with an N=9 comprised of N=3 wells from hepatocytes isolated from three individual donors. All data reported as means ± SEM. *P<0.05 vs. same IL-4 treatment in the absence of IL-1β and TNF-α, #P<0.05 vs. same IL-1β and TNF-α treatment in the absence of IL-4. †P<0.05 vs. the two bracketed treatment groups.
IL-4 treatment of the human hepatocytes also induced mRNA of CCL11 by 50-fold over the untreated control, with a corresponding 10-fold increase in protein levels in cell culture supernatant (Figure 6A and B). The effect of IL-4 on CCL11 protein secretion was enhanced by the presence of TNF-α, but was ablated when IL-1β was added to TNF-α and IL-4. IL-4 treatment significantly induced CCL26 mRNA expression (upwards of 10,000-fold) and protein secretion (upwards of 100-fold) in human hepatocytes, which was enhanced by IL-1β alone and TNF-α when in combination with IL-1β (Figure 6A and B). Unlike CCL11 and CCL26, CCL24 mRNA expression increased only 2-fold in response to IL-4 treatment (data not shown).
The Severity of Concanavalin A-Mediated Hepatitis is Significantly Attenuated in Mice Deficient in TSLPR
In order to determine whether TSLP signaling plays a pathogenic role in other diseases of the liver, we evaluated the susceptibility of Tslpr-/- mice to concanavalin A-mediated hepatitis, an established mouse model in which IL-4 and eosinophils play a critical role in its hepatotoxicity (10, 11). The severity of concanavalin A-mediated hepatitis was decreased even more in Tslpr-/- mice relative to WT mice (Figure 7A and B) than that found in studies of HILI (Figure 2A and B).
Figure 7.
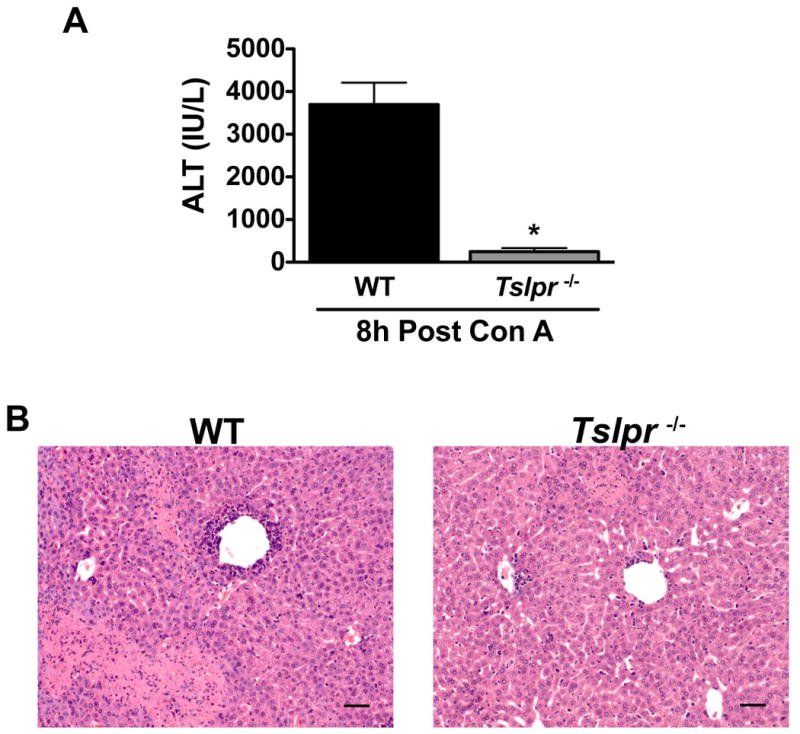
Mice deficient in TSLPR are protected against concanavalin A-mediated liver injury. Female WT or Tslpr-/- mice were administered concanavalin A (10 mg/kg) intravenously and sacrificed 8 hours post-treatment. Serum ALT activities (A) and representative photomicrographs of H&E stained liver sections (B) from WT and Tslpr-/- mice treated for 8 hours with concanavalin A. (Scale bar = 50 μm). *P<0.05 vs. WT mice. Data reported as means ± SEM with N=19-20 from four independent experiments.
Discussion
TSLP functions as an initiator and regulator of allergic inflammatory diseases of the skin and lung (6), which is attributed in mice to its prominent role in driving and sustaining type 2 responses, including production of IL-4, and infiltration of eosinophils (8, 9). Although the signaling mechanisms that lead to the secretion of TSLP in vivo are not known, early inflammatory signals including IL-1β and TNF-α can induce TSLP expression and secretion from epithelial cells (16-18) and may play a role in this process. TSLP in turn can induce expression and secretion of type 2 cytokines including IL-4 from leukocytes (9, 15).
We now provide evidence for the first time that TSLP signaling plays a pathologic role in liver inflammation initiated by the hepatotoxic drug halothane. First, halothane-treatment of mice resulted in liver injury that was accompanied by increased liver expression of TSLP, its receptor components, and IL-4 as well as elevated serum levels of the cytokines (Figure 1). We also showed the hepatic expression and serum levels of IL-1β and TNF-α increased during the early phase of HILI in mice (Figure 5C and D) and that mouse and human hepatocytes secreted TSLP in response to these factors (Figure 5E and F and Figure 6B, respectively). It is possible that TSLP in the liver exerts its effect on various leukocyte populations to confer a type 2 immune response which may play a role in mediating HILI. In this regard, TSLPR and IL-7Rα were detected on the surface of CD4+ T cells, CD8+ T cells, NKT cells, and eosinophils isolated from naïve mice and those treated with vehicle and halothane (refer to Supporting Material Figure S1C and D, respectively). Second, the severity of HILI in mice deficient in TSLPR was lower than in WT controls, as were hepatic expression and serum levels of IL-4 and eotaxins, and hepatic eosinophils (Figure 2A and B, E and F, and D, respectively). These findings are consistent with previous reports that Tslpr-/- mice exhibited attenuated disease severity, levels of type 2 cytokines including IL-4, and numbers of eosinophils at the site of inflammation in mouse models of asthma (8) and atopic dermatitis (9). Third, we provided evidence indicating that IL-4 also had a pathogenic role in HILI by showing that IL-4 deficient mice were less susceptible than WT type mice to HILI and had depressed levels of hepatic eosinophils (Figure 3A and B, and D, respectively) that were reversed significantly when mice were treated with recombinant IL-4 (Figure 4A and B, and C, respectively). IL-4 was similarly reported to be involved in antigen-induced lung inflammation when IL-4 deficient mice experienced decreased disease severity and numbers of eosinophils in the lungs compared to WT controls (20). Fourth, our finding that concanavalin A-induced liver injury was nearly abolished in Tslpr-/- mice (Figure 7) provided additional support for a role of TSLP signaling in injury to the liver because elevated levels of IL-4, eotaxins, and eosinophils play prominent roles in its pathogenesis (10, 11, 21). Although these findings provide significant evidence for TSLP having a pathologic role in HILI, additional mechanistic studies will be needed to establish this role for TSLP.
The mechanism by which IL-4 caused liver injury in the mouse model of HILI was investigated in hepatocyte cultures. We found that IL-4 directly stimulated expression and secretion of eosinophil eotaxins in cultured mouse (Figure 5A and B, respectively) and human hepatocytes (Figure 6). IL-4 also augmented the activities of IL-1β and TNF-α to induce TSLP expression and secretion in mouse liver cells (Figure 5E and F) and to a lesser extent in human hepatocytes from 1 of the 3 donors (Figure S3), supporting previous reports of synergistic effects of IL-4, IL-1β, and TNF-α on production of TSLP from epithelial cells (16, 18). These findings suggest TSLP and IL-4 may act in a feed-forward inflammatory cascade in the liver to drive eosinophil infiltration during HILI. At this point in remains unknown what leukocytes are signaled by TSLP and what the source of IL-4 is during HILI. Although NKT cells appear to be the main source of IL-4 in concanavalin A-mediating liver injury (21), the role these cells play in HILI in mice remains controversial (22, 23).
TSLP signaling may be involved in instances of DILI caused by other drugs because IL-4 is associated in the pathogenesis of diclofenac-, flutamide-, and methimazole-induced liver injury in mice (24-26). In contrast to these reports, IL-4 plays a protective role in a mouse model of acetaminophen-induced liver injury (27). In addition, genetic variants that resulted in high IL-4 transcription were associated with cases of diclofenac-induced hepatotoxicity (28), suggesting increased type 2 responses may lead to enhanced susceptibility to DILI. Similarly, in diseases with type 2 induced pathology, genetic variants and polymorphisms in the TSLP gene are associated with disease susceptibility, such as polymorphisms in the promoter region of the TSLP gene that enhance mRNA expression in asthma (29), and variants in the genes encoding TSLP, TSLPR, and CCL26 in eosinophil esophagitis (30). It is possible that aberrant levels of TSLP, other type 2 cytokines, and/or eotaxins mediated by genetic and other modulators of gene expression may serve as potential risk factors for DILI. In addition, TSLP and type 2 cytokines may play similar roles in other diseases of the liver where hepatic eosinophilia are associated, in particular hepatic allograft rejection (31), primary sclerosing cholangitis (32), primary biliary cirrhosis (33), and chronic viral hepatitis C (34). For example, a recent report showed TSLP expression was increased in patients with chronic hepatitis C viral infection and that hepatocytes infected with hepatitis C virus secrete TSLP in vitro (35). In conclusion, this report provides the first evidence implicating TSLP in liver inflammation and injury caused by a drug, and suggests that other liver diseases where eosinophilia and type 2 immunity have been associated, may also be mediated by TSLP signaling.
Supplementary Material
Acknowledgments
We would like to acknowledge John George for maintaining the IL4-/- mouse colony and the NHLBI Flow Cytometry core facility for their help with this work. We thank Dr. Kimberly Dyer (NIAID) for her helpful discussions pertaining to eosinophils. We also thank Tami McCoy Graf for her careful review of the manuscript.
Financial Support: This work was supported by the Intramural Research Program of the National Institutes of Health and the National Heart, Lung and Blood Institute.
List of Abbreviations
- DILI
Drug-Induced Liver Injury
- HILI
Halothane-Induced Liver Injury
- IL
Interleukin
- TSLP
Thymic Stromal Lymphopoietin
- TSLPR
Thymic Stromal Lymphopoietin Receptor
- IL7Rα
Interleukin-7 Receptor Alpha
- NKT
Natural Killer T Cell
- WT
Wild-Type
- rIL-4
Recombinant Interleukin-4
- BSA
Bovine Serum Albumin
- ALT
Alanine Aminotransferase
- H&E
Hematoxylin and Eosin
- TNF-α
Tumor Necrosis Factor-α
- TFA
Trifluoroacetylated
- SEM
Standard Error of the Mean
References
- 1.Chalasani N, Fontana RJ, Bonkovsky HL, Watkins PB, Davern T, Serrano J, Yang H, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135:1924–1934. 1934 e1921–1924. doi: 10.1053/j.gastro.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pham BN, Bemuau J, Durand F, Sauvanet A, Degott C, Prin L, Janin A. Eotaxin expression and eosinophil infiltrate in the liver of patients with drug-induced liver disease. Journal of hepatology. 2001;34:537–547. doi: 10.1016/s0168-8278(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 3.Bjornsson E, Kalaitzakis E, Olsson R. The impact of eosinophilia and hepatic necrosis on prognosis in patients with drug-induced liver injury. Alimentary pharmacology & therapeutics. 2007;25:1411–1421. doi: 10.1111/j.1365-2036.2007.03330.x. [DOI] [PubMed] [Google Scholar]
- 4.Proctor WR, Chakraborty M, Chea LS, Morrison JC, Berkson JD, Semple K, Bourdi M, et al. Eosinophils mediate the pathogenesis of halothane-induced liver injury in mice. Hepatology. 2013;57:2026–2036. doi: 10.1002/hep.26196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pulendran B, Artis D. New paradigms in type 2 immunity. Science. 2012;337:431–435. doi: 10.1126/science.1221064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziegler SF. Thymic stromal lymphopoietin and allergic disease. The Journal of allergy and clinical immunology. 2012;130:845–852. doi: 10.1016/j.jaci.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. The Journal of experimental medicine. 2005;202:829–839. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, Gyarmati D, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nature immunology. 2005;6:1047–1053. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 9.He R, Oyoshi MK, Garibyan L, Kumar L, Ziegler SF, Geha RS. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11875–11880. doi: 10.1073/pnas.0801532105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis H, Le Moine A, Flamand V, Nagy N, Quertinmont E, Paulart F, Abramowicz D, et al. Critical role of interleukin 5 and eosinophils in concanavalin A-induced hepatitis in mice. Gastroenterology. 2002;122:2001–2010. doi: 10.1053/gast.2002.33620. [DOI] [PubMed] [Google Scholar]
- 11.Jaruga B, Hong F, Sun R, Radaeva S, Gao B. Crucial role of IL-4/STAT6 in T cell-mediated hepatitis: up-regulating eotaxins and IL-5 and recruiting leukocytes. Journal of immunology. 2003;171:3233–3244. doi: 10.4049/jimmunol.171.6.3233. [DOI] [PubMed] [Google Scholar]
- 12.Al-Shami A, Spolski R, Kelly J, Fry T, Schwartzberg PL, Pandey A, Mackall CL, et al. A role for thymic stromal lymphopoietin in CD4(+) T cell development. The Journal of experimental medicine. 2004;200:159–168. doi: 10.1084/jem.20031975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.You Q, Cheng L, Reilly TP, Wegmann D, Ju C. Role of neutrophils in a mouse model of halothane-induced liver injury. Hepatology. 2006;44:1421–1431. doi: 10.1002/hep.21425. [DOI] [PubMed] [Google Scholar]
- 14.Bourdi M, Amouzadeh HR, Rushmore TH, Martin JL, Pohl LR. Halothane-induced liver injury in outbred guinea pigs: role of trifluoroacetylated protein adducts in animal susceptibility. Chem Res Toxicol. 2001;14:362–370. doi: 10.1021/tx000244x. [DOI] [PubMed] [Google Scholar]
- 15.Omori M, Ziegler S. Induction of IL-4 expression in CD4(+) T cells by thymic stromal lymphopoietin. Journal of immunology. 2007;178:1396–1404. doi: 10.4049/jimmunol.178.3.1396. [DOI] [PubMed] [Google Scholar]
- 16.Bogiatzi SI, Fernandez I, Bichet JC, Marloie-Provost MA, Volpe E, Sastre X, Soumelis V. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. Journal of immunology. 2007;178:3373–3377. doi: 10.4049/jimmunol.178.6.3373. [DOI] [PubMed] [Google Scholar]
- 17.Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:914–919. doi: 10.1073/pnas.0607305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nonaka M, Fukumoto A, Ogihara N, Sakanushi A, Pawankar R, Yagi T. Synergistic induction of thymic stromal lymphopoietin by tumor necrosis factor alpha and Th2 cytokine in nasal polyp fibroblasts. American journal of rhinology & allergy. 2010;24:e14–18. doi: 10.2500/ajra.2010.24.3436. [DOI] [PubMed] [Google Scholar]
- 19.Harada M, Hirota T, Jodo AI, Doi S, Kameda M, Fujita K, Miyatake A, et al. Functional analysis of the thymic stromal lymphopoietin variants in human bronchial epithelial cells. American journal of respiratory cell and molecular biology. 2009;40:368–374. doi: 10.1165/rcmb.2008-0041OC. [DOI] [PubMed] [Google Scholar]
- 20.Webb DC, Cai Y, Matthaei KI, Foster PS. Comparative roles of IL-4, IL-13, and IL-4Ralpha in dendritic cell maturation and CD4+ Th2 cell function. Journal of immunology. 2007;178:219–227. doi: 10.4049/jimmunol.178.1.219. [DOI] [PubMed] [Google Scholar]
- 21.Toyabe S, Seki S, Iiai T, Takeda K, Shirai K, Watanabe H, Hiraide H, et al. Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. Journal of immunology. 1997;159:1537–1542. [PubMed] [Google Scholar]
- 22.Cheng L, You Q, Yin H, Holt MP, Ju C. Involvement of natural killer T cells in halothane-induced liver injury in mice. Biochemical pharmacology. 2010;80:255–261. doi: 10.1016/j.bcp.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dugan CM, Fullerton AM, Roth RA, Ganey PE. Natural killer cells mediate severe liver injury in a murine model of halothane hepatitis. Toxicol Sci. 2011;120:507–518. doi: 10.1093/toxsci/kfr005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi S, Kobayashi M, Yoshikawa Y, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. IL-4 mediates dicloxacillin-induced liver injury in mice. Toxicology letters. 2011;200:139–145. doi: 10.1016/j.toxlet.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Higuchi S, Kobayashi M, Yano A, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Involvement of Th2 cytokines in the mouse model of flutamide-induced acute liver injury. Journal of applied toxicology : JAT. 2012;32:815–822. doi: 10.1002/jat.1706. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi M, Higuchi S, Ide M, Nishikawa S, Fukami T, Nakajima M, Yokoi T. Th2 cytokine-mediated methimazole-induced acute liver injury in mice. Journal of applied toxicology : JAT. 2012;32:823–833. doi: 10.1002/jat.2731. [DOI] [PubMed] [Google Scholar]
- 27.Ryan PM, Bourdi M, Korrapati MC, Proctor WR, Vasquez RA, Yee SB, Quinn TD, et al. Endogenous interleukin-4 regulates glutathione synthesis following acetaminophen-induced liver injury in mice. Chemical research in toxicology. 2012;25:83–93. doi: 10.1021/tx2003992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aithal GP, Ramsay L, Daly AK, Sonchit N, Leathart JB, Alexander G, Kenna JG, et al. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology. 2004;39:1430–1440. doi: 10.1002/hep.20205. [DOI] [PubMed] [Google Scholar]
- 29.Harada M, Hirota T, Jodo AI, Hitomi Y, Sakashita M, Tsunoda T, Miyagawa T, et al. Thymic stromal lymphopoietin gene promoter polymorphisms are associated with susceptibility to bronchial asthma. American journal of respiratory cell and molecular biology. 2011;44:787–793. doi: 10.1165/rcmb.2009-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherrill JD, Gao PS, Stucke EM, Blanchard C, Collins MH, Putnam PE, Franciosi JP, et al. Variants of thymic stromal lymphopoietin and its receptor associate with eosinophilic esophagitis. The Journal of allergy and clinical immunology. 2010;126:160–165 e163. doi: 10.1016/j.jaci.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Groen PC, Kephart GM, Gleich GJ, Ludwig J. The eosinophil as an effector cell of the immune response during hepatic allograft rejection. Hepatology. 1994;20:654–662. doi: 10.1016/0270-9139(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe H, Ohira H, Kuroda M, Takagi T, Ishikawa H, Nishimaki T, Kasukawa R, et al. Primary sclerosing cholangitis with marked eosinophilic infiltration in the liver. Journal of gastroenterology. 1995;30:524–528. doi: 10.1007/BF02347572. [DOI] [PubMed] [Google Scholar]
- 33.Terasaki S, Nakanuma Y, Yamazaki M, Unoura M. Eosinophilic infiltration of the liver in primary biliary cirrhosis: a morphological study. Hepatology. 1993;17:206–212. [PubMed] [Google Scholar]
- 34.Tarantino G, Cabibi D, Camma C, Alessi N, Donatelli M, Petta S, Craxi A, et al. Liver eosinophilic infiltrate is a significant finding in patients with chronic hepatitis C. Journal of viral hepatitis. 2008;15:523–530. doi: 10.1111/j.1365-2893.2008.00976.x. [DOI] [PubMed] [Google Scholar]
- 35.Lee HC, Sung SS, Krueger PD, Jo YA, Rosen HR, Ziegler SF, Hahn YS. Hepatitis C virus promotes T-helper (Th)17 responses through thymic stromal lymphopoietin production by infected hepatocytes. Hepatology. 2013;57:1314–1324. doi: 10.1002/hep.26128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.