

Abstract
In the present study, poly(ethylene glycol)-b-poly(ε-caprolactone) micelles loaded with etoposide (ETO) were formulated and further conjugated with pentapeptide Glu-Ile-Leu-Asp-Val (EILDV) to target α4β1 integrin receptor overexpressed on metastatic tumor cell. Using a distinct ratio of carboxyl-terminated poly(ethylene glycol)-block-poly(ε-caprolactone) (HOOC–PEG-b-PCL) to methoxy-poly(ethylene glycol)-block-poly(ε-caprolactone (CH3O–PEG-b-PCL) polymers, we formulated a series of micellar formulations having different surface densities of EILDV and observed optimum cellular uptake of micelles with 10% EILDV surface density by B16F10 cells. The cytotoxicity of EILDV-conjugated micelles was observed close to 1.5-fold higher than plain ETO after 72 h of drug incubation, demonstrating controlled release of drug inside the cell after enhanced intracellular uptake with the ability to selectively target cancer cells. In addition, EILDV-conjugated micelles inhibited the migration of B16F10 cells effectively compared with plain ETO and non-conjugated micellar formulations when cells were treated with equivalent cytotoxic concentration of the drug, i.e., IC25. B16F10 cells treated with EILDV-conjugated micelles showed a significant reduction in the attachment of cells to the substrate-coated plate compared with non-conjugated micellar formulations, implying retention of the biological activity of EILDV after coupling to micelles. Furthermore, the in vivo experimental metastasis assay conducted on C57BL/6 mice demonstrated significant activity of EIDLV-conjugated micelles in the reduction of pulmonary metastatic nodule formation in both pretreatment and post-treatment methods. In conclusion, EIDLV-conjugated micelles showed higher efficacy in the treatment of metastasis and would be a promising approach in the treatment of metastasis.
Keywords: Metastasis, EILDV, B16F10 cells, Cell adhesion, Etoposide, Integrin
Introduction
Metastasis, the ability of cells in a primary tumor to invade other tissues and to spread throughout the body, is the underlying cause of most cancer deaths (Leber and Efferth 2009). Detection of cancer at an early stage before it has spread is possible to treat successfully by surgery/local irradiation or by chemotherapy. However, in most cases when “primary” tumor is detected, cancer cells have already moved from their primary site and settled in other organs to continue their secondary growth, gaining a “head start” in the race against malignancy that patients rarely win (Bagi 2002). The cascade of metastasis consists of five basic steps: (1) invasion and migration; (2) intravasation; (3) circulation; (4) extravasation; and (5) colonization, proliferation, and angiogenesis (Chamber et al. 2002; Leber and Efferth 2009). During this complex process, the adhesive interaction of metastasizing tumor cells to the extracellular matrix (ECM) components (fibronectin, collagen, laminin, and vitronectin) appears a key part in metastasis (Bidard et al. 2008; Brooks et al. 2010).
Integrins, the most important cell surface receptors that mainly bind to ECM proteins, consist of two non-covalently associated transmembrane glycoproteins subunits, α and β (Janes and Watt 2006; Kuphal et al. 2005; Villard et al. 2006). There are 18 α- and 8 β-subunits of integrins, which combine to form at least 24 different heterodimers, which are involved in key biological processes such as adhesion, signaling, migration, proliferation, survival, angiogenesis, oncogenesis, and metastasis (Mizejewski 1999; Wan et al. 2009). The binding of these integrins to a particular ECM protein during cell adhesion can be complex phenomena, but usually involves the recognition of a specific target sequence of the ligand (ECM components; Singleton and Menino 2005). Among various integrin receptors, the α4β1 integrin receptor binds to two different ligands, the endothelial cell surface protein vascular cell adhesion molecule-1 (VCAM-1) and the extracellular matrix component fibronectin (Mould et al. 1994). Wu et al. (1995) reported that the expression of α4β1 integrin in the CHO cell line enables the cells to adhere, spread, and migrate in response to VCAM-1. Furthermore, Mould et al. (1994) also suggested an α4β1-mediated melanoma cell adhesion and migration using human metastatic melanoma cell lines A375-SM. The adhesion and migration of lymphatic endothelial cells were also promoted by α4β1 integrin during growth factor and tumor-induced lymphangiogenesis, which facilitates tumor metastasis (Garmy-Susini et al. 2010). It has been reported that α4β1 integrin binds to three different site of fibronectin (Mould et al. 1994). Two of these (represented by peptides CS1 and CS5) are present separately and independently in the alternatively spliced IIICS, while the third site resides in the adjacent constitutively expressed HepII domain.
The pentapeptide sequence Glu-Ile-Leu-Asp-Val (EILDV), present in an alternatively spliced segment of fibronectin, is recognized by both the α4β1 and α4β7 integrins and exhibited its antimetastatic activity (Kaneda et al. 1997; Singleton and Menino 2005; Yamamoto et al. 1994). Various other peptide analogs like cRGD and YIGSR also showed their antimetastatic activity, but to a lesser extent compared with EILDV (Yamamoto et al. 1994). To exert an antimetastatic effect, large amounts of such peptides are needed due to their enzymatic degradation and rapid renal clearance in vivo (Mu et al. 1999). To circumvent these, the bioconjugation of these peptides with PEG and poly(styrene-co-maleic acid) has been reported to enhance their antimetastatic activity with longer blood circulation (Maeda et al. 1997; Mu et al. 1999; Yamamoto et al. 1994). The utilization of EILDV as a targeting moiety for recognition of tumor cells overexpressing the α4β1 integrin receptor could be a better alternative in tumor targeting with possible use in conditions of tumor metastasis. Various other peptides like YIGSR and cRGD have been used as targeting moieties for nanosized carriers to target laminin and αvβ5 receptors, respectively, and have showed their potential greater than plain drug (Lopez-Barcons et al. 2004; Nasongkla et al. 2004; Oba et al. 2007). To the best of our knowledge, the use of EILDV as a targeting moiety for nanocarriers loaded with an anticancer drug the treatment of metastasis has not been explored, and this is the first report of an EILDV-conjugated micellar system for active tumor targeting.
The present study was focused on the design and development of polymeric micelles loaded with the anticancer drug, etoposide (ETO), where EILDV was conjugated to the surface of the micelles. Various in vitro evaluations were carried out using highly metastatic B16F10 cell lines to assess the effectiveness of EILDV-conjugated micellar formulations. Moreover, experimental metastasis studies were conducted using B16F10 melanoma mouse models to examine the effect of micellar formulations in the prevention of metastasis.
Materials and methods
Materials
ETO was a kind gift from Cadila Pharmaceutical Ltd. (Ahmedabad, India). Monomethoxy poly(ethylene glycol) of molecular weights 2 and 5 kDa, ε-caprolactone (CL), HCl in diethyl ether (1.0 M), 2,4,6 trinitro benzenesulfonic acid (TNBS), calcium hydride, and propidium iodide were purchased from Sigma-Aldrich (Mumbai, India). α-Carboxy ω-hydroxyl polyethylene glycol (HOOC-PEG-OH) of molecular weights 2 and 5 kDa were purchased from Jenkem Technology Inc. (TX, USA). EILDV was a kind gift from the National Institute of Research in Reproductive Health, Mumbai, India. Toluene, dichloromethane (DCM), diethyl ether, and triethylamine of analytical grade were purchased from S.D. Fine-Chem Ltd., (Mumbai, India). DCM was dried and distilled using calcium hydride before use. CL was dried over calcium hydride for 48 h and distilled under reduced pressure before use. Plastic wares were purchased from Tarson Products Pvt. Ltd. (Mumbai, India). Fetal bovine serum (FBS) was purchased from Biowest (Genetix, India). Iscove’s minimum Dulbecco’s medium (IMDM), MTT A.R. (3-(4,5-dimethyl-2-yl)-2,5-diphenyl tetrazolium bromide), and RNAse were procured from Himedia Lab. Pvt. Ltd. (Mumbai, India). All other chemicals were of analytical grade and used without purification.
Cell culture and animals
The B16F10 melanoma cell line was purchased from the National Center for Cell Sciences (Pune, India) and maintained in IMDM supplemented with 10% FBS containing penicillin (100 U/ml) and streptomycin (100 μg/ml) in a humidified atmosphere containing 5% CO2 at 37°C.
Pathogen-free female C57BL/6 mice 6–8 weeks of age were procured from Animal House, Advanced Centre for Treatment, Research and Education in Cancer (Navi Mumbai, India). All animal studies were carried out under the guidelines compiled by the Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Culture, Government of India, and all the study protocols were approved by the Institutional Animal Ethics Committee of the Advanced Centre for Treatment, Research and Education in Cancer, Navi Mumbai. The animals were maintained in a room (23 ± 2°C and 60 ± 10% humidity) under a 12-h light/dark cycle. Food and water were given ad libitum.
Synthesis and characterization
The synthesis of HOOC-PEG-PCL was carried out as reported earlier with slight modifications (Kim et al. 2004; Lee et al. 2007). The typical synthesis procedure of HOOC-PEGx-PCLy (where x = 2 kDa and y = 3.5 kDa) of molecular weight 5.5 kDa was followed as described. Briefly, 1 g of HOOC-PEG-OH (0.5 mmol, Mn = 2 kDa) was azeodistilled twice with dry toluene (50 ml) to remove water. Ten milliliters of dried DCM was added to HOOC-PEG-OH followed by the addition of CL (1.74 g, 15.33 mmol) using a syringe. The polymerization was initiated by the addition of 1.0 M solution of HCl in diethyl ether (1.5 ml, 1.5 mmol); the reaction was maintained at 25°C for 24 h with vigorous stirring under nitrogen atmosphere. The reaction was terminated by the addition of 0.1 ml of triethylamine; the copolymer was collected by precipitation of the filtrate in cold diethyl ether and washed thrice with cold methanol to remove residual monomer. HOOC-PEG5-PCL7 of molecular weight 12 kDa was synthesized by a similar method using HOOC-PEG-OH of molecular weight 5 kDa. MPEG-PCL, i.e., MPEG2-PCL3.5 and MPEG5-PCL7, of molecular weights 5.5 and 12 kDa were also synthesized by a similar method using monomethoxy poly(ethylene glycol) of molecular weights 2 and 5 kDa, respectively. Gel permeation chromatography was performed to determine the molecular weight and polydispersity index as reported elsewhere (Kim et al. 2004; Shen et al. 2008).
Preparation and characterization of EILDV-conjugated micelles
ETO-loaded polymeric micelles were prepared by co-solvent evaporation method (Aliabadi et al. 2005). Briefly, 40 mg of MPEG2–PCL3.5 and 4.45 mg of HOOC–PEG2–PCL3.5 or 40 mg of MPEG5–PCL7 and 4.45 mg of HOOC–PEG5–PCL7 were dissolved in 4 ml of acetone along with 2 mg of ETO and injected dropwise to 5 ml of distilled water under stirring at room temperature. Stirring was continued until complete evaporation of acetone, and the residual amount of acetone was removed by a rotary vacuum evaporator.
Peptide EILDV was conjugated to the prepared micelles as described earlier with slight modifications by the formation of an amide bond between the terminal amine group of EILDV and the surface-free carboxyl group of micelles (Lopez-Barcons et al. 2004). Briefly, 1-(3-dimethylaminopropyl)-3-ethylcarbodimide hydrochloride and N-hydroxysuccinimide dissolved in phosphate buffer, pH 4.5, were added to the micellar solution at a four-fold higher molar concentration to the HOOC–PEG–PCL copolymer used in micellar formulation and stirred at room temperature for 15 min, followed by pH adjustment to 7.5 with 0.1 N NaOH. The required amount of peptide EILDV dissolved in PBS, pH 7.4, was added (molar ratio of peptide/HOOC–PEG–PCL, 1:1) to the micellar solution and incubated for 3 h under mild stirring at room temperature. The micellar solution was dialyzed using a dialysis tube (MWCO 12,000) against distilled water to remove unconjugated peptide and finally filtered through a 0.45-μm membrane filter to remove non-entrapped drug and polymer aggregates. The EILDV-conjugated micelles prepared with polymers of molecular weights 5.5 and 12 kDa were denoted as EPCL235 and EPCL570.
Stealth micellar formulations, i.e., MPCL235 and MPCL570, were prepared by a similar method using the synthesized copolymers MPEG2–PCL3.5 and MPEG5–PCL7, respectively. Percent drug entrapment of micelles was determined by dissolving an aliquot of micelles in acetonitrile and the absorbance of the drug quantified at 285 nm using a UV–visible spectrophotometer (UV 1700, PharmaSpec, Shimadzu, Japan). Particle size and the zeta potential of micelles were determined using Zetasizer, Nano-ZS (Malvern Inst., UK). The amount of peptide EILDV bound to micelles was determined by the developed TNBS assay method, measuring unconjugated peptide found in the dialysis medium (Habeeb 1966; Snyder and Sobocinski 1975).
Cellular uptake
The effect of the different surface densities of EILDV (i.e., 5%, 10%, and 20%) on the cellular uptake of micelles was investigated by formulating a series of micelles using a blend of copolymers HOOC–PEG–PCL and MPEG–PCL at various ratios. The cellular uptakes of EILDV-conjugated micelles, non-conjugated micelles, and plain ETO were quantified by an HPLC method as reported earlier (Kang et al. 2005). Briefly, 4 × 104 B16F10 cells per milliliter were added to 90-mm tissue culture plates and allowed to grow up to 90% confluency. Cells were incubated with plain ETO or micellar formulations (drug concentration, 20 μg/ml) at 37°C. After 2 h, the medium was aspirated and the cells were washed three times with ice-cold PBS to stop further uptake. Cells were harvested using saline EDTA and centrifuged at 1,000 rpm for 15 min. After resuspending in PBS, cells were lysed by a probe sonicator (Branson Sonifier S-450, USA) for 3 min at 60% duty cycle followed by the addition of 1 ml of 6 M hydrochloric acid for the precipitation of proteins. The mixture was extracted with 3.0 ml of dichloromethane, vortexed for 2 min, and centrifuged for 15 min at 2,500 rpm. The upper organic layer was collected and evaporated to dryness with N2. The residue was reconstituted in 80 μl mobile phase and analyzed using HPLC.
The HPLC system was composed of a UV–visible detector (SPD-20A, Prominence), sampling injector (model 231 XL, Gilson), and a syringe pump (model 402, Gilson). The separation was performed on a reverse-phased C18 HPLC column (Phenomenex Luan 5μ C18 (2) 250 × 4.6-mm i.d., 5 μm). The mobile phase, acetonitrile/water/glacial acetic acid (35:61:1, v/v/v), was run at a flow rate of 1 ml/min and the column effluent monitored by a UV detector set at 254 nm. Diazepam was used as an internal standard and the samples extracted using a liquid–liquid extraction process. The protein content in the cell lysate was measured by the Bradford’s method using bovine serum albumin (BSA) as a standard. The percentage uptake of drug was calculated using the following formula as reported earlier (Yuan et al. 2008):
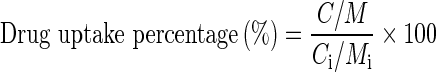 |
where C is the intracellular concentration of ETO measured by HPLC, M is the unit weight (in milligrams) of cellular protein after incubation, Ci is the initial concentration of ETO, and Mi is the initial unit weight (in milligrams) of cellular protein.
Cytotoxicity assay
The cytotoxicity of ETO-loaded, EILDV-conjugated and non-conjugated micelles against B16F10 cells was assessed by the MTT assay (Dua and Gude 2006). Briefly, B16F10 cells were seeded in 96-well plates at a density of 4,000 cells/100 μl per well. The cells were allowed to grow and stabilize for 24 h at 37°C with 5% CO2. Subsequently, the cells were exposed to a series of doses of plain ETO and micellar formulations and incubated for 24 h. Post-treatment cells in each well were washed with PBS, and to this, 20 μl of MTT in PBS (5 mg/ml) and 80 μl of complete media were added and incubated for 4 h at 37°C. Plates were then centrifuged at 1,500 rpm for 20 min. The medium was aspirated from the wells, and 100 μl of DMSO was added to each well and kept on a shaker for 10 min to dissolve formazan crystals. The optical density was measured using an ELISA plate reader (Molecular Devices, Spectra Max 190 with Softmax Pro) at 540 nm with a reference wavelength of 690 nm. Cytotoxicity studies at time points of 48 and 72 h were carried out by seeding B16F10 cells at a density of 2,000 cells/100 μl per well and 1,500 cells/100 μl per well, respectively. The IC50 values of various formulations were calculated by plotting a graph of percent cell viability vs. concentrations.
Cell migration assay
Cell migration assay was performed to estimate the migration of B16F10 cells after drug treatment (Dua and Gude 2008). B16F10 cells at a density of 2 × 104 cells/ml in complete medium were added to 40-mm tissue culture plates and incubated for 48 h. The cell monolayer was treated with ETO and micellar formulations at half IC50 (48 h) concentration and incubated at 37°C with 5% CO2 for 48 h. Wounds were created with a sterile plastic tip and the peeled off cells removed by washing with PBS twice. A 0-h time point wound created plate was kept as the reference plate and fixed immediately. Remaining plates were incubated for 48 h in the presence of IMDM containing 0.1% FBS (to allow cell migration only and not proliferation). The plates were fixed with cold methanol and stained with 0.5% crystal violet. Migration of cell from the edge of the injured monolayer was measured using Laser Capture Microdissection Microscope (LCMM, Zeiss, Germany) along with Palm Robo software at 25 different positions. The experiment was performed six times, and percent cell migration was calculated using following equation:
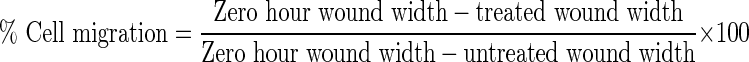 |
Cell adhesion study
B16F10 cell adhesion after treatment with plain ETO and micellar formulation to substrate-coated plates was carried out as reported earlier, with slight modifications (Dua et al. 2007; Lopez-Barcons et al. 2004). Briefly, flat-bottom 96-well plates were coated with 50 μl of EILDV (0.2 mg/ml in PBS) and the plates incubated overnight at 37°C. Plates were washed with PBS and the uncoated space blocked with 1% BSA in PBS and incubated for 2 h at 37°C. Plates were again washed with PBS, and to these, substrate-coated plates, 3 × 105 cells/ml in IMDM containing 0.1% BSA treated with plain ETO and micellar formulations for 48 h at a dose of half IC50 values (48 h) were added. Cells were further incubated at 37°C for 1 h, and non-adherent cells were removed giving two washes with PBS. The adherent cells were quantified using the MTT assay and expressed as the relative percent adhesion to the respective total unwashed cells.
Confocal microscopy
B16F10 cells at a density of 2 × 104 cells/ml were seeded in 40-mm tissue culture plates containing heat sterilized coverslips. When cells reached 70% confluency, these were treated with 6-coumarin-loaded micellar formulations (300 μg/ml) in complete media for 1 h (Panyam et al. 2003). Cells were washed with PBS twice and fixed with 4% paraformaldehyde in PBS for 15 min at 37°C. Coverslips were mounted using 4% DABCO (fluorescent mounting medium), and confocal images were taken using FITC filter (Ex (λ) 495 nm, Em (λ) 520 nm) by a confocal laser scanning microscope (Zeiss LSM S10 Meta).
In vitro treatment of B16F10 melanoma cells and its effect on inhibition of lung metastasis (pretreatment)
The in vitro-treated B16F10 cell-induced metastasis was performed as reported previously (Ratheesh et al. 2007). Briefly, sub-confluent B16F10 cells were treated with plain ETO and micellar formulations at a subtoxic dose (IC25 value, 48 h) and incubated for 48 h at 37°C. After two washes with PBS, the cells were harvested and 0.1 ml of PBS containing 0.1 million B16F10 cells (control and treated) were inoculated to C57BL/6 mice via the tail vein. The mice were randomly assigned to six groups (five mice/group); one group was assigned as a control which received untreated B16F10 cells. All mice were killed on the 21st day of cell inoculation; their lungs were excised and the numbers of pulmonary metastatic nodules (lung colonies) on the surface of the lung were counted under a dissecting microscope. The percent inhibitions in pulmonary metastatic nodule formation were calculated with respect to the number of nodules present in the untreated control group.
In vivo treatment of B16F10 melanoma and its effect on inhibition of lung metastasis (post-treatment)
C57BL/6 mice were injected with 0.1 million B16F10 cells in 0.1 ml of PBS through intravenous route by the tail vein (Sant et al. 2000). One group received only B16F10 cells and served as a control. On the next day of cell inoculation, plain ETO and micellar formulations were injected at a drug dose of 2 mg/kg intravenously via the tail vein. The animals were killed on the 21st day of cell inoculation and the lungs removed. The numbers of pulmonary metastatic nodule formation on the surface of the lungs were calculated and compared with the untreated control for percent inhibition in pulmonary metastatic nodule formations.
Histopathology study
After counting the pulmonary metastatic nodules, the lungs were fixed in 10% buffered formaldehyde. The tissue was processed through a histological routine with a Shandon Pathcentre Tissue Processor. Paraffin sections were cut at 5-μm thickness and stained with hematoxilin and eosin. The tissue morphology was studied under Zeiss AxioImager Z-1 microscope and images taken.
Statistical analysis
The statistical significance of the results was analyzed using SPSS (version 16). Differences between experimental groups were compared using ANOVA followed by a Bonferroni post hoc test.
Results
Characterization of synthesized copolymer and EILDV-conjugated micelles
PEG–PCL block copolymers of different molecular weights were prepared by ring opening polymerization of CL with PEG using HCl in diethyl ether as a catalyst. The molecular weights of HOOC–PEG2–PCL3.5 and HOOC–PEG5–PCL7 obtained by GPC were 5.8 and 12.8 kDa, respectively, while for MPEG2–PCL3.5 and MPEG5–PCL7, the molecular weights found were 5.9 and 12.6 kDa, respectively. The polydispersity index of the synthesized polymers was observed between 1.2 and 1.4. In addition, the GPC chromatogram exhibited only a single peak, which indicated that all the impurities were removed after purification (data not shown). Co-solvent evaporation (nanoprecipitation) method was employed for the fabrication of micellar formulations using a blend of MPEG–PCL and HOOC–PEG–PCL of similar molecular weight for the formulation of EILDV-conjugated micelles. To control the density of peptide on the surface of micelles, the HOOC–PEG–PCL copolymer was used at 10% to the total weight of polymer in the formation of micelles. Table 1 represents the characterization of micellar formulations. A major difference in particle size between the two groups of micelles (MPCL235 and EPCL235 to MPCL570 and EPCL570) was found due to differences in the molecular weights of the polymer used. The zeta potential of EPCL235 and EPCL570 were found at a lower value comparable to MPCL235 and MPCL570 due to the presence of two carboxyl groups in the EILDV peptide despite the utilization of a free carboxyl group of polymer for the conjugation of peptide (Table 1). Entrapment efficiency and the percent drug loading content of micelles were dependent on the molecular weights of the polymers utilized. The conjugation efficiency of EILDV to polymeric micelles was found >85% at a 1:1 molar ratio of peptide to functional polymer, i.e., HOOC–PEG–PCL.
Table 1.
Characterization of EILDV-conjugated and non-conjugated micelles
| Formulations code | Particle size (nm) | Polydispersity Index | Zeta potential (mV) | % drug entrapment | % drug loading | % conjugation of EILDV |
|---|---|---|---|---|---|---|
| EPCL235 | 46.2 ± 4.5 | 0.176 | −8.7 ± 1.9 | 75.3 ± 3.6 | 3.39 ± 0.16 | 85.3 ± 7.4 |
| EPCL570 | 80.3 ± 4.8 | 0.198 | −9.3 ± 2.2 | 83.9 ± 4.9 | 3.77 ± 0.22 | 89.2 ± 6.3 |
| MPCL235 | 38.5 ± 2.5 | 0.152 | −4.0 ± 0.4 | 81.1 ± 2.0 | 3.90 ± 0.10 | – |
| MPCL570 | 73.3 ± 4.0 | 0.143 | −4.6 ± 0.3 | 92.0 ± 3.4 | 4.40 ± 0.17 | – |
Results are the mean ± SD (n = 3)
Cellular uptake
The amount of ETO accumulated in B16F10 cells after incubation with plain ETO and micellar formulations was determined by a reverse-phased HPLC method. Since it is important to know the effect of the surface density of peptide on the cellular internalization of micelles, the present study was conducted using micelles with various EILDV surface densities. EILDV conjugated on the surface of micelles with different surface densities (5%, 10%, and 20%) showed different cellular uptakes, as represented in Fig. 1. Plain ETO showed maximum cellular uptake of 75.0 ± 5.23% because of its solubilized state which helped in the rapid diffusion of the drug into the cell. Compared with plain ETO, micellar formulations showed cellular uptakes <16%. EILDV-conjugated micelles EPCL235 and EPCL570 exhibited higher cellular uptakes due to receptor-mediated uptake compared with MPCL235 and MPCL570. An increase in the cellular uptake of micelles was also observed upon increasing the surface density of EILDV from 5% to 20%. However, we observed a modest increase in cellular uptake upon increasing the surface density from 10% to 20%. Based on the results obtained in cellular uptake studies of EILDV-conjugated micelles, further experiments were conducted with micelles having 10% EILDV surface density.
Fig. 1.

Percent cell uptake of ETO after incubation of plain ETO and micellar formulations with B16F10 cells for 2 h. EPCL235 and EPCL570 micellar formulations used for cell uptake studies were with different EILDV surface densities (5%, 10%, and 20%). Results are the mean ± SD, n = 6 (*P < 0.05, **P < 0.01 compared with MPCL235; # P < 0.05, ## P < 0.01 compared with MPCL570)
Cytotoxicity assay
The cytotoxicity of EILDV-conjugated and non-conjugated micellar formulations were carried out on B16F10 cell line by the MTT assay with different drug concentrations and incubation times. Table 2 presents the IC50 values obtained after plotting a graph of percent cell viability vs. concentrations. After 24 h of drug incubation, plain ETO showed higher cytotoxicity at all concentrations compared with micellar formulations, and the IC50 value obtained was significantly lower (P < 0.001). After 48 h of drug incubation, an increase in the cytotoxicity of ETO was observed (Table 2), which was still higher compared with all micellar formulations. The micellar formulations showed an increase in cytotoxicity up to 40- to 50-fold after 48 h of drug incubation compared with 24 h of drug incubation, indicating the controlled-release properties of micelles after intracellular uptake. An increase in the drug incubation time up to 72 h resulted in no further increase in the cytotoxicity of plain ETO, while MPCL235 and MPCL570 micelles exhibited a cytotoxicity profile comparable to plain ETO. An increase in cytotoxicity to 1.72- and 1.38-fold was observed with EPCL235 (P < 0.01) and EPCL570 micelles compared with plain ETO. EILDV-conjugated placebo micellar formulations and plain EILDV peptide at a maximum concentration (350 μg/ml) showed no toxic effects against B16F10 cells when incubated for 72 h (data not shown).
Table 2.
Inhibitory concentrations (50%) of various formulations at different times of incubation with B16F10 cells
| Formulation code | IC50 (μM) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| ETO | 0.087 ± 0.010 | 0.050 ± 0.004 | 0.043 ± 0.003 |
| MPCL235 | 4.3 ± 0.61*** | 0.082 ± 0.011* | 0.034 ± 0.008 |
| EPCL235 | 2.5 ± 0.30** | 0.055 ± 0.007 | 0.025 ± 0.001** |
| MPCL570 | 5.0 ± 0.80*** | 0.120 ± 0.013*** | 0.040 ± 0.005 |
| EPCL570 | 3.1 ± 0.42*** | 0.075 ± 0.07 | 0.031 ± 0.002 |
Values are the mean ± SD (n = 6)
***P < 0.001, **P < 0.01, *P < 0.05 (compared with ETO at each time point)
Cell migration assay
The migration of B16F10 cells toward wound created was found to be inhibited upon treatment with a subtoxic dose of plain ETO and micellar formulations. Figure 2 presents the cell migration images of B16F10 cells in treated and untreated plates after fixing and staining, in which the maximum cell migration was observed in untreated control. Considering the cell migration of untreated control as 100%, plain ETO showed cell migration of 41.9%, while MPCL235 and MPCL570 micelle-treated cell showed cell migration of 32.3% and 33.2%, respectively (Fig. 3). B16F10 cells treated with EPCL235 and EPCL570 micelles exhibited maximum inhibition in cell migration of 28.9% and 21.7%, respectively, compared with plain ETO. Placebo EILDV-conjugated micelles EPCL235-P and EPCL570-P showed no inhibition in cell migration, and migration showed a resemblance to untreated control.
Fig. 2.

Microscopic images of migration of B16F10 cells after treatment with a subtoxic concentration of plain ETO and micellar formulations. Zero-hour plate was fixed immediately after wound creation. UC untreated control
Fig. 3.

Percent B16F10 cell migration after treatment with plain ETO and micellar formulations at IC25 concentrations. Results are the mean ± SD, n = 6 (***P < 0.001, **P < 0.01 compared with plain ETO)
Cell adhesion study
Percent adhesion of B16F10 cells treated with subtoxic doses (IC25, 48 h) of plain ETO and micellar formulations to substrate-coated plate was presented in Fig. 4. We observed a significant reduction in cell adhesion (P < 0.001) in plain ETO and micellar formulation-treated B16F10 cells compared with the untreated control. The adhesion of B16F10 cells treated with plain ETO was 66.3 ± 5.27%, while MPCL235 and MPCL570 showed 54.84 ± 6.37% and 61.96 ± 8.67%, respectively (Fig. 4). A maximum inhibition in cell adhesion was observed in EILDV-conjugated micelles EPCL235 and EPCL570 (P < 0.01), which exhibited 1.73- and 1.62-fold reductions in cell adhesion compared with plain ETO-treated B16F10 cells (Fig. 4). Placebo micellar formulations EPCL235-P and EPCL570-P also showed cell adhesions up to 81.8 ± 3.52% and 91.5 ± 2.57% toward the substrate-coated plates.
Fig. 4.

Percent adhesion of B16F10 cells toward substrate-coated plates after treatment with plain ETO and micellar formulations at IC25 (48 h). Results are the mean ± SD, n = 6 (**P < 0.01 compared with ETO)
Confocal microscopy
Qualitative uptake of 6-coumarin-loaded, EILDV-conjugated and non-conjugated micellar formulations by B16F10 cells were carried out after 1 h of incubation (Fig. 5). It was demonstrated that compared with MPCL235 and MPCL570 micelles, a more prominent fluorescence intensity was observed in EILDV-conjugated micelles, i.e., EPCL235- and EPCL570-treated B16F10 cells.
Fig. 5.

Confocal images of B16F10 cells after 1-h incubation with 6-coumarin-loaded micelles. Scale bar, 20 μm)
In vitro treatment of B16F10 melanoma cells and its effect on inhibition of lung metastasis (pretreatment)
In this study, B16F10 cells treated with a subtoxic concentration of plain ETO and ETO-loaded micellar formulations (IC25, 48 h) were injected to mice via the tail vein to examine the effect formulations on the inhibition of pulmonary metastatic nodule formation. The total number of metastatic nodules found in the untreated control group was 154.4 ± 26.8, and based on this, the percent inhibition in lung nodule formation of the formulation-treated group was calculated and presented in Fig. 6.
Fig. 6.

Percent inhibition of metastatic nodule formation after inoculation of in vitro-treated B16F10 cells to C57BL/6 mice via the tail vein. Results are the mean ± SD, n = 5 (***P < 0.001 compared with plain ETO)
The in vitro-treated B16F10 cells with plain ETO and ETO-loaded micellar formulations exhibited a reduction in the formation of pulmonary metastatic nodules, and the maximum effect was seen in peptide-conjugated micelles (P < 0.001; Fig. 6). B16F10 cells treated with plain ETO showed 47.92 ± 11.78% inhibition in metastatic nodules, which was lower compared with micellar formulations. MPCL235 and MPCL570 micellar formulations showed 54.91 ± 12.77% and 58.80 ± 3.50% inhibitions in pulmonary metastatic nodule formation, respectively. EILDV-conjugated micelles demonstrated close to 1.5-fold higher efficacies in the prevention of metastasis compared with plain ETO.
In vivo treatment of B16F10 melanoma and its effect on inhibition of lung metastasis (post-treatment)
In the post-treatment method, plain ETO and ETO-loaded micellar formulations were injected by intravenous route on the second day of B16F10 cell inoculation to C57BL/6 mice. The percent inhibition in metastatic nodule formation after the 21st day of cell inoculation is represented in Fig. 7. It was observed that all formulations showed their potential toward inhibition in metastatic nodule formation. The percent inhibition in nodule formation observed with plain ETO was 36.72 ± 7.85%, while MPCL235 and MPCL570 showed 48.60 ± 10.04% and 53.99 ± 9.42% nodule formations, respectively (Fig. 7). EILDV-conjugated micelles compared with plain ETO and non-conjugated micelles showed close to 1.75- and 1.40-fold higher inhibitions in metastatic nodule formation, respectively.
Fig. 7.

Percent inhibition of metastatic nodule formation in B16F10-injected C56BL/6 mice after treatment with plain ETO and micellar formulations at a dose of 2 mg/kg. Results are the mean ± SD, n = 5 (**P < 0.01, ***P < 0.001 compared with ETO)
Histopathology
The histological examinations of lung sections are shown in Fig. 8, in which the untreated control exhibited small- to large-sized tumor islands which covered the entire lung parenchyma and also blocked the lumen of the medium and large blood vessels. Moreover, tumor cells penetrated the middle coat of blood vessels and lodged in several blood vessels. In the case of ETO, smaller-sized tumor islands with a lesser extent of multiplicity were observed compared with control. The MPCL235- and MPCL570-treated groups showed a reduction in the number of tumor islands with smaller sizes compared with the control. A greater extent of reduction in size as well as the number of tumor islands was observed in EILDV-conjugated micelles. In addition to this, the blood vessels were found unblocked in EILDV-conjugated micelles.
Fig. 8.

Histology section of lungs (pretreatment method) stained with hematoxilin and eosin. Black arrow indicates pulmonary metastatic nodule formation
Discussion
One approach for increasing the site specificity of a nanoparticulate carrier is to conjugate it with a ligand which can interact with an overexpressed cell receptor. Various molecules like folic acid, monoclonal antibodies, and peptides have been reported as ligands for tumor targeting of nanoparticles and liposomes (Sutton et al. 2007; Torchilin 2007). Despite promising outcomes showed by the folate-conjugated nanocarrier for tumor targeting, overexpression of folate receptors in normal tissues like placenta and kidney was reported by immunochemistry studies to impede the possible clinical translation of folate bioconjugates for human use (Alexis et al. 2008). On the other end, monoclonal antibodies possess two major drawbacks as ligands for tumor targeting: their larger size and nonspecific uptake by reticuloendothelial system (RES; Aina et al. 2002). Compared with these, cell surface binding peptides are considerably smaller in size, low immunogenic, low toxic, and generally do not bind to RES systems (Aina et al. 2002; Lopez-Barcons et al. 2004). In the present study, we showed that the conjugation of EILDV to polymeric micelles resulted into a receptor-mediated uptake, which is helpful in the treatment of metastasis.
MPCL570 and EPCL570 micelles showed higher percent drug loading and particle size compared with MPCL235 and EPCL235 micelles due to the influence of higher molecular weight. The results obtained indicate that the drug encapsulation efficiency depends on the copolymer composition and the particle size. However, the percent drug loading can be modulated by varying the hydrophilic-to-hydrophobic block ratio (Kim and Lee 2001). Moreover, we found lower percent drug loading in EILDV-conjugated micelles EPCL235 and EPCL570 due to loss of the surface-bound drug during the conjugation process. Up to this date, very few studies have reported the effect of different surface densities of ligands (cRGD, folate, and aptamers) on the intracellular uptake of nanocarriers (Gu et al. 2008; Nasongkla et al. 2004; Zhao and Yung 2008). These studies found that the surface density of ligands plays an important role in optimum cellular uptake mechanism. Pang et al. (2008) have reported the use of monoclonal antibody OX-26 for brain delivery of PEG–PCL micelles with various surface densities of OX-26. They found that maximum blood–brain barrier permeability surface area product (PS) and percentage of injected dose per gram of brain were dependent on the OX26 densities on the surface of polymersomes. On the effect of folate content on targeting efficiency studied on KB cells, it was observed that the maximum cellular uptake peaked at 65% folate content (Zhao and Yung 2008). It has also been pointed out that the high intracellular folate concentration may lead to saturation and shut off folate receptor uptake, leading to reduced intracellular uptake of micelles. However, in the case of peptide-conjugated micelles, it is difficult to deduce the same thing in terms of concentration, but the saturation and regeneration of receptors may play an important role. Cellular uptake studies conducted on B16F10 cell lines with various surface densities of EILDV demonstrated similar kinds of observations, and we found that upon increasing the density up to 20%, a minor increase in cellular uptake compared with the cellular uptake found with 10% surface density of EILDV was shown. MTT assay at three different time points (24, 48 and 72 h) showed time- and concentration-dependent cytotoxicity. Plain ETO, due to its solubilized state, leads to a rapid diffusion through the cell membrane and showed higher cytotoxicity at 24- and 48-h incubation periods (Li et al. 2008; Zhao and Yung 2008). Compared with plain ETO, micellar formulations showed a controlled-release behavior and showed much lesser toxicity at the initial time point of incubation, i.e., 24 h. EILDV-conjugated micelles exhibited the highest cytotoxicity at 72-h incubation compared with plain ETO and non-conjugated micellar formulations. This phenomenon appeared to correspond reasonably well to the receptor-medicated cellular uptake efficiency and is in agreement with reports of previous studies and cellular uptake studies carried out in the present work (Yang et al. 2008; Yoo and Park 2004; Zhao and Yung 2008).
It is known that migrating cells are less sensitive to standard chemotherapy due to their decreased proliferation rate (Douma et al. 2004; Lefranc et al. 2005). Therefore, therapeutic drugs or drug-loaded carrier which can target cell motility and invasion may appear to be important in decreasing the mortality and mobility rates among cancer patients. B16F10 cells after treatment with EILDV-conjugated micelles showed greater inhibition in migration compared with plain ETO and non-conjugated micelles at a similar inhibitory concentration, i.e., IC25. These might be due to the effect of ETO on the changes in cell cytoskeleton of B16F10 cells after drug exposure. It was reported that ETO promoted the changes in the distribution of tubulin in HL-60 cells with significant changes in cell morphology (Grzanka and Grzanka 2003). We supposed that the alteration in cell morphology due to ETO resulted into inhibition in the migration of B16F10 cells.
EILDV, after conjugation on the surface of micelles, showed a significant reduction in the adhesion of B16F10 cells toward substrate-coated plates, implying no alternation in biological activity after conjugation. In addition, we also found a reduction in adhesion of B16F10 cells treated with plain ETO and non-conjugated micellar formulations, which suggests that ETO might have some role in the prevention of adhesion of B16F10 cells. Exposure of ETO to the cells leads to apoptosis and gross changes in cell shape Kook et al. (2000). Moreover, a decline in focal adhesion sites was also seen in ETO-treated Rat-1 cells due to cleavage of the Crk-associated substrate by caspase-3. It was believed that the effect of ETO on the reduction in focal adhesion sites might have prevented the adhesion of B16F10 cells to substrate-coated plates. EPCL235-P and EPCL570-P micelles also showed a reduction in cell adhesion in the absence of ETO, which further supports the role of EILDV in the prevention of cell adhesion. Confocal microscopy analysis of 6-coumarin-loaded micelles demonstrated an enhanced uptake of EPCL235 and EPCL570 micelles by B16F10 cells compared with non-conjugated micelles, MPCL235 and MPCL570, attributing greater intracellular delivery due to receptor-mediated endocytosis.
Experimental metastasis study conducted using highly metastatic B16F10 cells was reported to produce pulmonary metastatic nodules in lungs after intravenous injection of cells (Cameron et al. 2000; Gude et al. 1996). In actual metastatic process which is observed in subcutaneously developed tumor, several processes such as primary invasion, transport into circulation, survival in circulation, arrest in the capillary bed of an organ, extravasation into the parenchyma, and several other steps are involved. Compared with these, many of these steps are not seen in the experimental metastasis model. Although, this model is very useful in investigating the process of organ specificity of tumor cells involving homing to the lung and the subsequent cell–cell interaction. As metastatic cells need to attach themselves to lung basement membrane before they grow into nodules, cell adhesion plays a vital part in the metastatic spreading of B16 melanoma (Gude et al. 1996). Experimental metastasis conducted by two different techniques, i.e., pretreatment and post-treatment models, showed the effectiveness of micellar formulations over plain drug to prevent the colonization of B16F10 cells in the lung. The results obtained implies that ETO and ETO-loaded micellar formulations might have interacted directly or indirectly with tumor cell (B16F10 cell) surface receptors involved in adhesion to lung basement membrane (Gude et al. 1996). EILDV-conjugated micelles EPCL235 and EPCL570 exhibited higher inhibition in the formation of pulmonary metastatic nodules compared with plain ETO and non-conjugated micelles due to its interaction with cell surface receptors present on B16F10 cells. Pretreatment method exhibited higher inhibition in pulmonary metastatic nodule formation compared with the post-treatment method. However, it is difficult to correlate each other due to the difference in treatment and the dose of drug given in the intravenous route. Both pretreatment and post-treatment methods justify that two different situations exist in cancer patients. The pretreatment method describes the situation in which a patient is detected with primary tumor and receives chemotherapy, while the post-treatment method entails a situation in which metastasis has already been detected and then chemotherapy has started. Furthermore, liposomal pentoxifyline and niosomal cisplatin demonstrated higher antimetastatic activity compared with plain drug, indicating the usefulness of nanocarrier in treatment of metastasis (Gude et al. 2002; Sant et al. 2000). The market-available DOXIL® (pegylated doxorubicin liposomes) which is used in metastasis was not that much defined by clinical trials due to a poor understanding of its role in the prevention of metastasis. It is believed that the antimetastasis effect was rather due to the inhibition in growth of the primary tumor (Zhang et al. 2008). It was also suggested that for complete eradication of pulmonary metastasis, surgical removal of primary tumor plus multiple injections of pegylated doxorubicin liposomes are needed. In this situation, micelles conjugated with antimetastatic peptides like EILDV can play a major role in recognition of tumor cells which have been already detached from the primary tumor site and under process of metastasis.
Conclusion
In conclusion, EILDV-conjugated micelle was more effective in the treatment of metastasis compared with plain ETO. The in vitro cell line studies conducted on highly metastatic B16F10 cells demonstrated the superior antimigratory and antiadhesive activity of EILDV-conjugated micelles by virtue of their receptor-mediated endocytosis mechanism. Furthermore, experimental metastasis assay elucidated the potential of EILDV-conjugated micelles in the prevention and cure of metastasis, a condition which is considered difficult to eradicate completely by conventional chemotherapy. Overall, EILDV-conjugated micelles loaded with the anticancer drug ETO could open new opportunities for the effective treatment of metastasis.
Acknowledgment
Mr. Mukesh Ukawala is grateful to ICMR, New Delhi, for providing financial assistance (SRF). The authors are thankful to Mr. Manohar Parkar and Mr. Yuvaraj Nikam for their kind assistance in cell line and animal studies conducted at ACTREC, respectively.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Contributor Information
R. S. R. Murthy, Phone: +91-265-2794051, FAX: +91-265-2418927, Email: m_rsr@rediffmail.com
Rajiv Gude, Phone: +91-22-27405084, FAX: +91-22-27412894, Email: rgude@actrec.gov.in.
References
- Aina OH, Sroka TC, Chen M, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184–199. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- Alexis F, Rhee JW, Richie JP, Radovic-Moreno AF, Langer R, Farokhzad OC. New frontiers in nanotechnology for cancer treatment. Urol Oncol Semin Ori. 2008;26:74–85. doi: 10.1016/j.urolonc.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Aliabadi HM, Mahmud A, Sharifabadi AD, Lavasanifar A. Micelles of methoxy poly(ethylene oxide)-b-poly(ε-caprolactone) as vehicles for the solubilization and controlled delivery of cyclosporine A. J Control Release. 2005;104:301–311. doi: 10.1016/j.jconrel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Bagi CM. Cancer cell metastases. J Musculoskel Neuron Interact. 2002;2:565–566. [PubMed] [Google Scholar]
- Bidard FC, Pierga JY, Vincent-Salomon A, Poupon MF. A “class action” against the microenvironment: do cancer cells cooperate in metastasis? Cancer Metastasis Rev. 2008;27:5–10. doi: 10.1007/s10555-007-9103-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SA, Lomax-Browne HJ, Carter TM, Kinch CE, Hall DMS. Molecular interactions in cancer cell metastasis. Acta Histochemica. 2010;112:3–25. doi: 10.1016/j.acthis.2008.11.022. [DOI] [PubMed] [Google Scholar]
- Cameron DM, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000;60:2541–2546. [PubMed] [Google Scholar]
- Chamber AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature Reviews Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Dua P, Gude RP. Antiproliferative and antiproteolytic activity of pentoxifylline in cultures of B16F10 melanoma cells. Cancer Chemother Pharmacol. 2006;58:195–202. doi: 10.1007/s00280-005-0155-8. [DOI] [PubMed] [Google Scholar]
- Dua P, Gude RP. Pentoxifylline impedes migration in B16F10 melanoma by modulating Rho GTPase activity and actin organization. Eur J Cancer. 2008;44:1587–1595. doi: 10.1016/j.ejca.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Dua P, Ingle A, Gude RP. Suramin augments the antitumor and antimetastatic activity of pentoxifylline in B16F10 melanoma. Int J Cancer. 2007;121:1600–1608. doi: 10.1002/ijc.22843. [DOI] [PubMed] [Google Scholar]
- Garmy-Susini B, Avraamides CJ, Schmid MC, Foubert P, Ellies LG, Barnes L, Feral C, Papayannopoulou T, Lowy A, Blair SL, Cheresh D, Ginsberg M, Varner JA. Integrin α4β1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res. 2010;70:3042–3051. doi: 10.1158/0008-5472.CAN-09-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzanka A, Grzanka D (2003) The influence of etoposide on the distribution of tubulin in human leukemia cell line HL-6. Med Sci Monit 9:BR66-69 [PubMed]
- Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci USA. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gude RP, Ingle AD, Rao SGA. Inhibition of lung homing of B16F10 by pentoxifylline, a microfilament depolymerizing agent. Cancer Lett. 1996;106:171–176. doi: 10.1016/0304-3835(96)04311-X. [DOI] [PubMed] [Google Scholar]
- Gude RP, Jadhav MG, Rao SGA, Jagtap AG. Effects of niosomal cisplatin and combination of the same with theophyline and with activated macrophages in murine B16F10 melanoma model. Cancer Biother Radiopharm. 2002;17:183–192. doi: 10.1089/108497802753773801. [DOI] [PubMed] [Google Scholar]
- Habeeb AFSA. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal Biochem. 1966;14:328–336. doi: 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- Janes SM, Watt FM. New roles for integrins in squamous cell carcinoma. Nature Reviews, Cancer. 2006;6:175–183. doi: 10.1038/nrc1817. [DOI] [PubMed] [Google Scholar]
- Kaneda Y, Yamamoto Y, Okada N, Tsutsumi Y, Nakagawa S, Kakiuch M, Maeda M, Kawasaki K, Mayumi T. Antimetastatic effect of synthetic Glu-Ile-Leu-Asp-Val peptide derivatives containing D-amino acids. Anticancer Drugs. 1997;8:702–707. doi: 10.1097/00001813-199708000-00009. [DOI] [PubMed] [Google Scholar]
- Kang YH, Lee E, Youk HJ, Kim SH, Lee HJ, Park YG, Lim SJ. Potentiation by alpha-tocopheryl succinate of the etoposide response in multidrug resistance protein 1-expressing glioblastoma cells. Cancer Lett. 2005;217:181–190. doi: 10.1016/j.canlet.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Kim SY, Lee YM. Taxol-loaded block copolymer nanospheres composed of methoxy poly(ethylene glycol) and poly(ε-caprolactone) as novel anticancer drug carriers. Biomaterials. 2001;22:1697–1704. doi: 10.1016/S0142-9612(00)00292-1. [DOI] [PubMed] [Google Scholar]
- Kim MS, Seo KS, Khang G, Cho SH, Lee HB. Preparation of poly(ethylene glycol) block-poly(ε-caprolactone) copolymers and their applications as thermo-sensitive materials. J Biomed Mater Res. 2004;70A:154–158. doi: 10.1002/jbm.a.30049. [DOI] [PubMed] [Google Scholar]
- Kook S, Shim SR, Choi SJ, Ahnn J, Kim JI, Eom SH, Jung YK, Paik YG, Song WK. Caspase-mediated cleavage of p130cas in etoposide induced apoptotic Rat-1 cells. Mol Biol Cell. 2000;11:929–939. doi: 10.1091/mbc.11.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuphal S, Bauer R, Bosserhoff AK. Integrin signaling in malignant melanoma. Cancer and Metastasis Reviews. 2005;24:195–222. doi: 10.1007/s10555-005-1572-1. [DOI] [PubMed] [Google Scholar]
- Leber MF, Efferth T. Molecular principles of cancer invasion and metastasis. Int J Oncol. 2009;34:881–895. doi: 10.3892/ijo_00000214. [DOI] [PubMed] [Google Scholar]
- Lee H, Hu M, Reilly RM, Allen C. Apoptotic epidermal growth factor (EGF)-conjugated block copolymer micelles as a nanotechnology platform for targeted combination therapy. Mol Pharmaceutics. 2007;4:769–781. doi: 10.1021/mp0700559. [DOI] [PubMed] [Google Scholar]
- Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol. 2005;23:2411–2422. doi: 10.1200/JCO.2005.03.089. [DOI] [PubMed] [Google Scholar]
- Li X, Li R, Qian X, Ding Y, Tu Y, Guo R, Hu Y, Jiang X, Guo W, Liu B. Superior antitumor efficiency of cisplatin-loaded nanoparticles by intratumoral delivery with decreased tumor metabolism rate. Eur J Pharm Biopharm. 2008;70:726–734. doi: 10.1016/j.ejpb.2008.06.016. [DOI] [PubMed] [Google Scholar]
- Lopez-Barcons LA, Polo D, Reig F, Fabra A. Pentapeptide YIGSR-mediated HT-1080 fibrosarcoma cells targeting of adriamycin encapsulated in sterically stabilized liposomes. J Biomed Mater Res. 2004;69A:155–163. doi: 10.1002/jbm.a.20235. [DOI] [PubMed] [Google Scholar]
- Maeda M, Izuno Y, Kawasaki K, Kaneda Y, Mu Y, Tsutsumi Y, Lem KW, Mayumi T. Amino acids and peptides XXXII: a bifunctional poly(ethylene glycol) hybrid of fibronectin-related peptides. Biochem Biophys Res Commun. 1997;241:595–598. doi: 10.1006/bbrc.1997.7855. [DOI] [PubMed] [Google Scholar]
- Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. PSEBM. 1999;122:124–138. doi: 10.1046/j.1525-1373.1999.d01-122.x. [DOI] [PubMed] [Google Scholar]
- Mould AP, Askari JA, Craig SE, Garratt AN, Clement J, Humphries MJ. Integrin α4β1-mediated melanoma cell adhesion and migration on vascular cell adhesion molecule-1(V CAM-1) and the alternatively spliced IIICS region of fibronectin. J Biol Chem. 1994;269:27224–27230. [PubMed] [Google Scholar]
- Mu Y, Kamada H, Kaneda Y, Yamamoto Y, Kodaira H, Tsunoda S, Tsutsumi Y, Maeda M, Kawasaki K, Nomizu M, Yamada Y, Mayumi T. Bioconjugation of laminin peptide YIGSR with poly(styrene co-maleic acid) increases its antimetastatic effect on lung metastasis of B16-BL6 melanoma cells. Biochem Biophys Res Commun. 1999;255:75–79. doi: 10.1006/bbrc.1999.9930. [DOI] [PubMed] [Google Scholar]
- Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem. 2004;116:6483–6487. doi: 10.1002/ange.200460800. [DOI] [PubMed] [Google Scholar]
- Oba M, Fukushima S, Kanayama N, Aoyagi K, Nishiyama N, Koyama H, Kataoka K. Cyclic RGD peptide-conjugated polyplex micelles as a targetable gene delivery system directed to cells possessing αvβ3 and αvβ5 integrins. Bioconjug Chem. 2007;18:1415–1423. doi: 10.1021/bc0700133. [DOI] [PubMed] [Google Scholar]
- Pang Z, Lu W, Gao H, Hu K, Chen J, Zhang C, Gao X, Jiang X, Zhu C. Preparation and brain delivery property of biodegradable polymersomes conjugated with OX26. J Control Release. 2008;128:120–127. doi: 10.1016/j.jconrel.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetwar V. Fluorescence and electron microscopy probes for cellular and tissue uptake of poly(d,l-lactide-co-glycolide) nanoparticles. Int J Pharm. 2003;262:1–11. doi: 10.1016/S0378-5173(03)00295-3. [DOI] [PubMed] [Google Scholar]
- Ratheesh A, Ingle A, Gude RP. Pentoxifylline modulates cell surface integrin expression and integrin mediated adhesion of B16F10 cells to extracellular matrix components. Cancer Biol Ther. 2007;6:1743–1752. doi: 10.4161/cbt.6.11.4833. [DOI] [PubMed] [Google Scholar]
- Sant VP, Nagarsenkar MS, Rao SGA, Gude RP. Enhancement of anti-metastatic activity of pentoxifylline by encapsulation in conventional liposomes and sterically stabilized liposomes in murine experimental B16F10 melanoma model. J Pharm Pharmacol. 2000;52:1461–1466. doi: 10.1211/0022357001777667. [DOI] [PubMed] [Google Scholar]
- Shen S, Guo S, Lu C. Degradation behaviors of monomethoxy poly(ethylene glycol)-b-poly(ε-caprolactone) nanoparticles in aqueous solution. Polym Adv Technol. 2008;19:66–72. doi: 10.1002/pat.975. [DOI] [Google Scholar]
- Singleton C, Menino AR. Effects of inhibitor of integrin binding on cellular outgrowth from bovine inner cell masses in vitro. In Vitro Cell Dev Biol Animal. 2005;40:29–37. doi: 10.1290/0407054.1. [DOI] [PubMed] [Google Scholar]
- Snyder SL, Sobocinski PZ. An improved 2,4,6-trinitrobenzenesulfonic acid method for the determination of amines. Anal Biochem. 1975;64:284–288. doi: 10.1016/0003-2697(75)90431-5. [DOI] [PubMed] [Google Scholar]
- Sutton D, Nasongkla N, Blanco E, Gao J. Functionalized micellar systems for cancer targeted drug delivery. Pharm Res. 2007;24:1029–1046. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- Torchilin VP. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007;11:E128–E147. doi: 10.1208/aapsj0902015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard V, Kayuzhniy O, Riccio O, Potekhin S, Melnik TN, Kajava AV, RUegg U, Corradin G. Synthetic RGD-containing α-helical coiled coil peptides promote integrin-dependent cell adhesion. J Peptide Sci. 2006;12:206–212. doi: 10.1002/psc.707. [DOI] [PubMed] [Google Scholar]
- Wan X, Kim SY, Guenther LM, Mendoza A, Briggs J, Yeung C, Currier D, Zhang H, Mackall C, Li WJ, Tuan RS, Deyrup AT, Khanna C, Helman L. Beta 4 integrin promotes osteosarcoma metastasis and interacts with ezrin. Oncogene. 2009;24:3401–3411. doi: 10.1038/onc.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Fields AJ, Kapteijn BK, McDonald JA. The role of α4β1 integrin in cell motility and fibronectin matrix assembly. J Cell Sci. 1995;108:821–829. doi: 10.1242/jcs.108.2.821. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Kaneda Y, Okada N, Nakagawa S, Kubo K, Inoue S, Maeda M, Yamashiro Y, Kawasaki K, Mayumi T. Antimetastatic effects of synthetic peptides containing the core sequence of the type III connecting segment domain (IIICS) of fibronectin. Anticancer Drugs. 1994;5:424–428. doi: 10.1097/00001813-199408000-00006. [DOI] [PubMed] [Google Scholar]
- Yang X, Deng W, Fu L, Blanco E, Gao J, Quan D, Shuai X. Folate-functionalized polymeric micelles for tumor targeted delivery of a potent multidrug-resistance modulator FG020326. J Biomed Mater Res. 2008;86A:48–60. doi: 10.1002/jbm.a.31537. [DOI] [PubMed] [Google Scholar]
- Yoo HS, Park TG. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J Control Release. 2004;96:273–283. doi: 10.1016/j.jconrel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Yuan H, Miao J, Du YZ, You J, Hu F, Zeng S. Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int J Pharm. 2008;348:137–145. doi: 10.1016/j.ijpharm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li A, Wang Z, Han Z, He J, Ma J. Antimetastatic activities of pegylated liposomal doxorubicin in a murine metastatic lung cancer model. J Drug Target. 2008;16:679–687. doi: 10.1080/10611860802201340. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yung LYL. Selectivity of folate conjugated polymer micelles against different tumor cells. Int J Pharm. 2008;349:256–268. doi: 10.1016/j.ijpharm.2007.07.040. [DOI] [PubMed] [Google Scholar]