

Abstract
Personalized medicine is to deliver the right drug to the right patient in the right dose. Pharmacogenomics (PGx) is to identify genetic variants that may affect drug efficacy and toxicity. The availability of a comprehensive and accurate PGx-specific drug-gene relationship knowledge base is important for personalized medicine. However, building a large-scale PGx-specific drug-gene knowledge base is a difficult task. In this study, we developed a bootstrapping, semi-supervised learning approach to iteratively extract and rank drug-gene pairs according to their relevance to drug pharmacogenomics. Starting with a single PGx-specific seed pair and 20 million MEDLINE abstracts, the extraction algorithm achieved a precision of 0.219, recall of 0.368 and F1 of 0.274 after two iterations, a significant improvement over the results of using non-PGx-specific seeds (precision: 0.011, recall: 0.018, and F1: 0.014) or co-occurrence (precision: 0.015, recall: 1.000, and F1: 0.030). After the extraction step, the ranking algorithm further improved the precision from 0.219 to 0.561 for top ranked pairs. By comparing to a dictionary-based approach with PGx-specific gene lexicon as input, we showed that the bootstrapping approach has better performance in terms of both precision and F1 (precision: 0.251 vs. 0.152, recall: 0.396 vs. 0.856 and F1: 0.292 vs. 0.254). By integrative analysis using a large drug adverse event database, we have shown that the extracted drug-gene pairs strongly correlate with drug adverse events. In conclusion, we developed a novel semi-supervised bootstrapping approach for effective PGx-specific drug-gene pair extraction from large number of MEDLINE articles with minimal human input.
Keywords: pharmacogenomics, text mining, information extraction, personalized medicine
Graphical abstract
Background
Pharmacogenomics and personalized medicine
Pharmacogenomics (PGx)is important for personalized medicine. Different patients respond differently to the same drug, with genetics accounting for 20 to 95 percent of the variability [1]. Pharmacogenomics is the study of how human genetic variations affect an individual’s response to drugs, with foci on drug metabolism, absorption, and distribution [2]. Pharmacogenomics plays an important role in identifying drug responders and non-responders, avoiding adverse events, and optimizing drug dose [3,4]. Recently, the U.S. Food and Drug Administration (FDA) has become a strong pharmacogenomics advocate in an effort to make drugs safer and more effective [5,6]. In order to improve the quality of already-marketed drugs, the FDA has updated certain drug labels to include PGx information. Currently, over one hundred FDA-approved drugs have PGx information on their labels that describe genes responsible for drug exposure, clinical response variability, and risk for adverse events1. One of the well-known PGx-specific drug-gene associations is warfarin-CYP2C9. Gene CYP2C9 encodes an important cytochrome P450 (CYP) enzyme that plays a major role in the metabolizing of more than 100 therapeutic drugs, one of which is warfarin. The genetic polymorphisms of CYP2C9 are associated with altered enzyme activity leading to toxicity at normal therapeutic doses of warfarin. Understanding how the genetic variants contribute to various drug responses is an essential step of personalized medicine [1, 7, 8]. The success of personalized drug treatment largely depends on the availability of accurate and comprehensive knowledge bases of PGx-specific drug-gene relationships, such as warfarin-CYP2C9 and irinotecan-UGT1A.
Automatic methods in extracting PGx-specific drug-gene pairs from literature
There are substantial research efforts in constructing PGx knowledge bases using both manual and automatic approaches. The Pharmacogenomics Knowledge Base (PharmGKB) is the largest manually created resource of information on how variations in human genetics lead to variations in drug response (http://www.pharmgkb.org) [9]. The PharmGKB project involves a large number of curators who read the literature and manually extract relationships among genes, drugs, and diseases from the pharmacogenetic literature. However, manually extracting PGx knowledge and other biomedical information in general from published literature and transforming it into machine-understandable knowledge is a difficult task because biomedical knowledge and terminology comprise huge, dynamic, and highly complicated fields. In addition, human curators are liable to error and subjective bias.
Development of automatic approaches to extract PGx-specific drug-gene relationships from published biomedical literature is an active research area. Both statistical and natural language processing (NLP) methods have been used [10–17]. Recently, we have developed a conditional approach to extract PGx-specific drug-gene pairs from 20 million MEDLINE abstracts using known drug-gene pairs available in PharmGKB as prior knowledge to implicitly classify sentences before relationship extraction. We have demonstrated that the conditional drug-gene relationship extraction approach significantly improves the precision and the F1 measure when compared with the unconditioned approach [18]. One common feature among above studies is that these drug-gene relationship extraction algorithms used either PGx-specific gene lexicons as input or PGx-related articles as the text corpus. These gene lexicons were either manually compiled or were derived from PharmGKB drug-gene pairs. PharmGKB is the largest pharmacogenomics knowledge, however the genes in this knowledge are often a mixture of non PGx-specific genes (e.g., IL2, VDR, EGFR, KRAS, ERBB2, and BRCA1) and PGx-specific genes (CYP2C9, VKORC1, ABCB1, UGT1A). Correspondingly, the drug-gene pairs in PharmGKB are also a mixture of non PGX-specific pairs. In addition, the recall of the PharmGKB gene lexicon is also limited. For example, there are total of 60 CYP (cytochrome P450) gene symbols approved by the HUGO Gene Nomenclature Committee (HGNC) (http://www.genenames.org/), but PharmGKB contains only 30 of them. Therefore, in order to increase the recall of extracted drug-gene pairs, we need to either compile a more comprehensive PGx-specific gene lexicon as done in [19], or start from all human genes and develop an algorithm to extract valid drug-gene pairs and classify them by their PGx-relevance.
Our semi-supervised iterative approach in extracting PGx-specific drug-gene pairs from literature
In this study, instead of using a precompiled PGx-specific gene lexicon, we use all human protein coding genes (total 19,055) as the underlying gene lexicon input to the drug-gene extraction algorithm. Since PGx-specific drug-gene pairs only account for a very small of portion of all drug-gene semantic pairs, using all human genes as the input gene lexicon makes the task of extracting PGx-specific drug-gene pairs more challenging and interesting. Therefore, it is critical to develop a ranking algorithm to rank extracted drug-gene pairs according to their PGx relevance. Another critical difference from our previous knowledge-driven approach [18] is that instead of using a significant portion of PharmGKB drug-gene pairs as prior knowledge, we use only one or a few known PGx-specific drug-gene pairs (eg. warfarin-CYP2C9, or caffeine-CYP1A2) as seeds to start the whole extraction process. Our previous conditional approach was guided by many known drug-gene pairs and therefore constituted a supervised learning approach. The method we present in this study is a semi-supervised approach since it depends on only a few seeds to start the whole learning process. Our study is based on the assumption that PGx-specific drug-gene pairs are often clustered together in a sentence. If we start with a known PGx-specific pair such as warfarin-CYP2C9, it is likely that sentences containing this pair are also PGx-specific. The other drug-gene pairs extracted from these PGx-related sentences are likely PGx-specific. The likelihood increases as the relatedness of the sentences increases, which depends on the relatedness of other drug-gene pairs in it. For example, using seed pair “warfarin-CYP2C9”, we retrieved the following sentence “Genetic factors (VKORC1, CYP2C9, EPHX1, and CYP4F2) are predictor variables for warfarin response in very elderly, frail inpatients.” (PMID19794411). Since this sentence contains a PGx-specific drug-gene pair warfarin-CYP2C9, the sentence itself is highly likely to be related to PGx. The other three drug-gene pairs (warfarin-VKORC1, warfarin-EPHX1, and warfain-CYP4F2) are likely to be PGx-specific pairs.
Recent studies in semi-supervised iterative learning approaches are motivated by the use of a very large collection of texts (web) [20] and the possibility of handling multiple entity types [21]. Semi-supervised pattern learning approaches are advantageous because they require minimal human intervention and no external domain knowledge. Therefore, semi-supervised information extraction systems are able to extract broad types of entities and relationships. Semi-supervised learning approaches have been used to extract information from the web [22–29]. Semi-supervised learning approaches depend on the regularity of language and the data redundancy. A big corpus such as MEDLINE (22 million articles as of the year 2012) is ideal for such tasks. However, the potential for semi-supervised approaches for biomedical information extraction was not fully explored until recently, when we developed semi-supervised pattern learning approaches for disease entity recognition [30] and medical intervention entity recognition [31], isa relationship extraction [32], and medical image retrieval from the web [33]. All iterative learning systems suffer from the inevitable problem of spurious patterns and instances introduced in the iterative process. We develop an iterative ranking algorithm to rank extracted drug-gene pairs according to their PGx-relatedness by combining the frequency of drug-gene pairs in MEDLINE with the PGx specificity of other co-occurred drug-gene pairs. The ranking algorithm is similar to the topic sensitive PageRank algorithm developed by Haveliwala [34]. Topic-Sensitive PageRank was based on the PageRank algorithm [35] in order to personalize search rankings using link analysis. Topic-sensitive PageRank computed a set of PageRank vectors, biased using a set of representative topics, in order to capture the importance with respect to a particular topic (details in Methods Section).
Data and Methods
Figure 1 depicts the iterative process of PGx-specific drug-gene extraction. The system consists of the following components: (1) build a local MEDLINE search engine; (2) iteratively extract drug-gene pairs; (3) rank extracted pairs; and (4) analyze extracted pairs.
Figure 1.
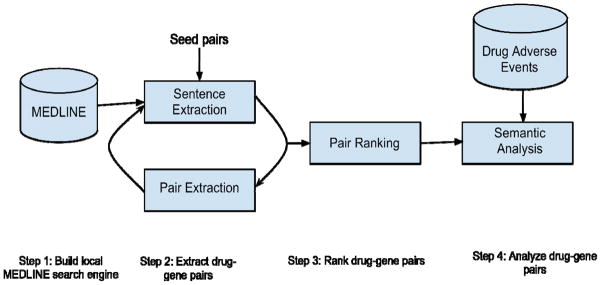
System diagram of iterative drug-gene extraction, pair ranking and semantic analysis process.
Build local MEDLINE search engine
We have used 20 million MEDLINE abstracts (roughly 100 million sentences) published from 1965 to 2010 as the text corpus for our task of PGx-specific drug-gene relationship extraction. The 2010 MEDLINE/PubMed baseline XML files were downloaded from NLM’s anonymous FTP server at ftp://ftp.nlm.nih.gov/nlmdata/.medleasebaseline/. The MEDLINE XML files were then parsed. The abstracts and PMID information from the XML files were extracted. Abstracts were subsequently split into sentences. We used the publicly available information retrieval library Lucene (http://lucene.apache.org) to create a local search engine with indexes for both sentences and abstracts. The drug lexicon was downloaded from DrugBank (http://www.drugbank.ca/) in 01/2012, and contains 6,516 drugs. The gene symbols were downloaded from www.genenames.org in 05/2012 and consisted of 19,055 human protein coding gene symbols.
Extract drug-gene pairs
The iterative process starts with a typical PGx-specific drug-gene seed pair such as “warfain-CYP2C9” or “caffeine-CYP1A2.” The program loops over a procedure that consists of a sentence extraction step and a pair extraction step (Fig. 1). In the sentence extraction step, the seed pair(s) are used as search queries to the local search engine. The sentences or abstracts containing the seed(s) are retrieved. In the pair extraction stage, we first find gene and drug entities from these returned sentences, and then extract the drug-gene co-occurrence pairs from the sentences. We use case-insensitive exact string matching for drug entity tagging, and case-sensitive extract string matching for gene symbol tagging. In the sentence extraction step in the subsequent iteration, the drug-gene pairs extracted from the previous iteration are used as queries to retrieve more sentences from which more drug-gene pairs are extracted. After each iteration, we rank the extracted drug-gene pairs and evaluate the precision and recall. The process was stopped after three iterations since further iterations did not improve the recall while precision decreased.
Rank drug-gene pairs
The ranking score of drug-gene pairs according to their PGx-specificity at a given iteration is given as the following:
The term on the left is the ranking score of a drug-gene pair being PGx-specific in iteration N, and the term within the summation on the right is the ranking score of its co-occurring drug-gene pairs (Pij) being PGx-specific in iteration N−1, weighted by co-occurrence frequency (Wij). K is the number of co-occurrence drug-gene pairs for pair Pi. Starting from a seed drug-gene pair, the ranking algorithm iteratively propagates its confidence score to its co-occurred pairs. The ranking score of each drug-gene pair is the sum of scores of its co-occurring pairs weighted by co-occurrence count. The confidence score of the seed pair was given a score of 1.0. For example, starting with the seed pair warfarin-CYP2C9, we extracted four additional genes P1, P2, P3 and P4 after first iteration, with the co-occurrence (with seed) counts 100, 20, 10, and 1, respectively. Then the weight of P1 after the first iteration is (100/131)* 1.0. At the second iteration, the ranking scores are calculated again based on the score vector from the first iteration. If more than one seed has been used, the initial scores of these seeds is one divided by the number of seeds. In one of our experiments in investigating the effect of number of seeds on the whole drug-gene extraction and ranking process, we used 68 drug-gene pairs extracted from FDA drug labels. The initial score for each of these 68 seeds was 1/68. The basic idea of our ranking algorithm is that if a sentence or abstract contains a known PGx-specific seed drug-gene pair, then the topic (or classification) of this sentence is related to PGx study. Other drug-gene pairs contained in the sentence are likely PGx-specific. The degree of PGx specificity a sentence confers to the unknown drug-gene pairs in it depends on the scores of the known drug-gene pairs in the same sentence. The difference from Topic Sensitive PageRank algorithm is that instead of continue the iteration process until rank stabilizes to within some threshhold, we evaluate the performance at each iteration and stop the iteration process after certain iterations.
Evaluation
We used the drug-gene pairs from PharmGKB as the gold standard. As we discussed previously, the drug-gene pairs in PhamGKB are limited in both precision and recall. However, as long as the drug-gene pairs in PharmGKB are largely PGx-specific and not biased to certain sets of drugs or genes, using them as gold standard to compare different algorithms should give fair evaluation. We downloaded the PharmGKB databases in 04/2012. There are a total of 4,399 drug-gene pairs classified as “PD” (pharmacodynamics) and/or “PK” (pharmacokinetics). Among them, 1,561 pairs appeared in MEDLINE sentences and 2,083 in MEDLINE abstracts. We used the 1,561 pairs appearing in MEDLINE sentences as the gold standard for sentence-level drug-gene extraction evaluation. Similarly, we used the 2,083 pairs appearing in MEDLINE abstracts as the gold standard for abstract-level extraction evaluation. Precision, recall and F1 measures were used for both relationship extraction and ranking. Precision was calculated as the proportion of gold standard pairs amongst extracted and ranked pairs. The recall was estimated as the proportion of all gold standard pairs extracted. The F1 measure was defined as the harmonic mean of precision and recall: F1 = 2 * (precision *recall)/(precision + recall). The Student’s t-test was performed for significance evaluation. A comparison evaluation was determined as significant when the p value was less than 10E-7.
Analyze drug-gene pairs
Studies have shown that PGx-specific genes are responsible for drug toxicities [36]. To demonstrate the potential use of the extracted PGx-specific drug-gene pairs in drug adverse event prediction, we studied the correlations of these extracted drug-gene pairs with drug adverse events. We downloaded a total of 100,049 drug adverse event pairs from SIDER (Side Effect Resource), a side effect resource compiled from FDA package inserts using text-mining methods [37] (http://sideeffects.embl.de/download/, accessed 03/2012). These drug adverse event pairs contained 996 drugs. For drug-drug pairs that shared PGx-specific genes bases on the extracted drug-gene pairs, we calculated the average shared adverse events and compared them to that shared among all drug-drug pairs.
Results
Performance comparison of the extraction algorithms based on different types of seeds
We compared the performances of different types of drug-gene seeds on the overall drug-gene extraction algorithm. The seeds included PGx-specific drug-gene pairs (Warfarin-CYP2C9 or Caffeine-CYP1A2) and a non-PGx-specific pair (Captopril-ACE). We also compared the extraction algorithm using seed pairs to the one without using any seeds. We investigated the effects of the number of iterations (one, two, and three iterations) and document types (sentence-level vs. abstract level) on the overall extraction algorithm. As shown in Table 1, the drug-gene extractions using PGx-specific seeds (“Warfarin-CYP2C9” or “Caffeine-CYP1A2”) had significantly better precision, recall, and F1 values compared to those using the non-PGx-specific seed (“Captopril-ACE”) or pure co-occurrence (“None”). After two iterations, the algorithm extracted a total of 2,622 drug-gene pairs starting from the seed warfarin-CYP2C9, with a precision of 0.219, a recall of 0.368, and an F1 of 0.274. Similarly, starting with the seed caffeine-CYP1A2, the algorithm extracted a total of 2,578 drug-gene pairs, with a precision of 0.195, a recall of 0.322, and an F1 of 0.243. The overall precision and recall for these two PGx-specific seeds are similar. However, if a non-PGx-specific seed (“Captopril-ACE”) or no seed (“NONE”) was used, the performance measures were at least ten times lower (precision of 0.011, recall of 0.018, and F1 of 0.014 for “Captopril-ACE”, precision of 0.015, recall of 0.134, and F1 of 0.026 for “None”). Similar results were observed for the other non-PGx-specific seed “Gefitinib-EGFR” (data not shown). After one iteration, we extracted a total of 391 drug-gene pairs from sentences starting from seed warfarin-CYP2C9, with a precision of 0.340, a recall of 0.085, and an F1 of 0.136. After two iterations, the algorithm extracted 2,622 pairs, with lower precision (0.219) but higher recall (0.368) and F1 (0.274). As we ran the algorithm for another iteration (three iterations), the recall increased as expected, but both precision and F1 significantly decreased. The same trend was observed for the seed “Caffeine-CYP1A2.” By comparing sentence-level to abstract-level drug-gene relationship extractions, we have shown that sentence-level extraction in general had better precision and F1 value, but lower recall. For instance, the drug-gene pairs extracted from sentences after two iterations had a precision of 0.219, a recall of 0.368, and an F1 of 0.274. The ones extracted from abstracts had a lower precision of 0.085, a higher recall of 0.511, and a lower F1 of 0.145.
Table 1.
Comparison of drug-gene extraction using different types of seeds: PGx-specific seeds (“Warfarin-CYP2C9” and “Caffeine-CYP1A2”), non PGx-specific seed (“Captopril-ACE”) and no seeds (“None”)
| Document Type | Round | Seed | N | Precision | Recall | F1 |
|---|---|---|---|---|---|---|
| Sentence | 1 | Warfarin-CYP2C9 | 391 | 0.340 | 0.085 | 0.136 |
| 2 | 2,622 | 0.219 | 0.368 | 0.274 | ||
| 3 | 8,844 | 0.103 | 0.583 | 0.175 | ||
| 1 | Caffeine-CYP1A2 | 397 | 0.270 | 0.069 | 0.109 | |
| 2 | 2,578 | 0.195 | 0.322 | 0.243 | ||
| 3 | 8,534 | 0.097 | 0.529 | 0.164 | ||
| 1 | Captopril-ACE | 319 | 0.053 | 0.011 | 0.018 | |
| 2 | 2,545 | 0.011 | 0.018 | 0.014 | ||
| 3 | 14,278 | 0.015 | 0.134 | 0.026 | ||
| Co-occurrence | None | 102,916 | 0.015 | 1.000 | 0.030 | |
| Abstract | 1 | Warfarin-CYP2C9 | 1,262 | 0.222 | 0.134 | 0.167 |
| 2 | 12,580 | 0.085 | 0.511 | 0.145 | ||
| 3 | 68,415 | 0.025 | 0.823 | 0.049 | ||
| 1 | Caffeine-CYP1A2 | 1,028 | 0.198 | 0.098 | 0.131 | |
| 2 | 10,793 | 0.09 | 0.467 | 0.151 | ||
| 3 | 69,653 | 0.025 | 0.796 | 0.049 | ||
| 1 | Captopril-ACE | 897 | 0.025 | 0.011 | 0.015 | |
| 2 | 20,106 | 0.015 | 0.144 | 0.027 | ||
| 3 | 96,086 | 0.016 | 0.749 | 0.032 | ||
| Co-occurrence | None | 190,452 | 0.011 | 1.000 | 0.022 |
In summary, more than half of the PGx-specific drug-gene pairs were reachable from a PGx-specific seed in only three hops as shown by the greater than 0.5 recall values after three iterations. Starting from a non-PGx-specific seed, only around 13% (recall of 0.134) of all PGx-specific drug-genes proved reachable after three iterations. This demonstrated that PGx-specific drug-gene pairs are indeed clustered together in sentences or abstracts. Compared to drug-gene pairs extracted based on co-occurrence, the pairs extracted using PGx-specific seeds were highly enriched in PGx-specific pairs.
Performance of extraction algorithm using different number of PGx-specific seeds
As we have shown in previous section, the drug-gene pairs extracted starting from one PGx-specific seed were significantly enriched with PGx-specific pairs (precision 0.219) compared to those extracted using a non-PGx-specific seed or based on pure co-occurrence (0.011 and 0.015, respectively). We further investigated whether or not we could improve both precision and recall by starting with more PGx-specific seeds. We experimented with the extraction algorithm starting with one seed (“Warfarin-CYP2C9” or “Caffeine-CYP1A2”), two seeds ((“Warfarin-CYP2C9” and “Caffeine-CYP1A2”), and 68 PGx-specific drug-gene pairs extracted from FDA drug labels. As shown in Table 2, there was a tradeoff in precision and recalls caused by using a different number of seeds. After two iterations, the algorithms with 68 seeds had significant better recall compared to that which had one seed (0.522 vs. 0.368); however the precision decreased from 0.219 to 0.171. The overall F1 values were similar among algorithms using one seed, two seeds, and 68 seeds. After three iterations, we had extracted the majority of the PGx-specific drug-gene pairs when 68 seeds were used (recall of 0.701 for sentence-level and, 0.876 for abstract-level); however the overall precision was low.
Table 2.
Comparison of drug-gene extraction using different number of seeds: none, one, two, and 68 PGx-specific seeds.
| Document Type | Round | Seed | N | Precision | Recall | F1 |
|---|---|---|---|---|---|---|
| Sentence | 1 | Warfarin-CYP2C9 | 391 | 0.340 | 0.085 | 0.136 |
| 2 | 2,622 | 0.219 | 0.368 | 0.274 | ||
| 3 | 8,844 | 0.103 | 0.583 | 0.175 | ||
| 1 | Warfarin-CYP2C9+Caffeine-CYP1A2 | 669 | 0.281 | 0.120 | 0.169 | |
| 2 | 3146 | 0.192 | 0.386 | 0.256 | ||
| 3 | 10811 | 0.087 | 0.603 | 0.152 | ||
| 1 | 68 FDA drug-gene pairs | 1524 | 0.278 | 0.272 | 0.275 | |
| 2 | 4767 | 0.171 | 0.522 | 0.258 | ||
| 3 | 15798 | 0.069 | 0.701 | 0.126 | ||
| Co-occurrence | None | 102,916 | 0.015 | 1.000 | 0.030 | |
| Abstract | 1 | Warfarin-CYP2C9 | 1,262 | 0.222 | 0.134 | 0.167 |
| 2 | 12,580 | 0.085 | 0.511 | 0.145 | ||
| 3 | 68,415 | 0.025 | 0.823 | 0.049 | ||
| 1 | Warfarin-CYP2C9+Caffeine-CYP1A2 | 1,879 | 0.185 | 0.167 | 0.175 | |
| 2 | 15,434 | 0.075 | 0.554 | 0.132 | ||
| 3 | 77,344 | 0.023 | 0.836 | 0.044 | ||
| 1 | 68 FDA drug-gene pairs | 3,886 | 0.165 | 0.308 | 0.215 | |
| 2 | 26,089 | 0.057 | 0.711 | 0.105 | ||
| 3 | 99,376 | 0.018 | 0.876 | 0.036 | ||
| Co-occurrence | None | 190,452 | 0.011 | 1.000 | 0.022 |
In summary, the extraction algorithm is effective in finding PGx-specific drug-gene pairs. However, the low precision demonstrated the need to develop a ranking algorithm to further rank PGx-specific pairs highly amongst the extracted pairs.
Performance comparison of different ranking algorithms
We developed three ranking methods to rank the extracted drug-gene pairs : (1) ranking based on the scores calculated using the formula defined in the Method section; (2) ranking based on MEDLINE frequency count. The intuition behind the MEDLINE frequency-based ranking is that if a drug-gene pair co-occurs in MEDLINE many times, it is likely that there is some semantic relationship between the drug and the gene; (3) random ranking. We used the 11-point interpolated average precision measures, often used for measuring ranked retrieval results for search engines [38], to evaluate the drug-gene ranking algorithms. The interpolated precision was measured at the 11 recall levels of 0.0, 0.1, 0.2, ..., 1.0. The interpolated precision pinterp at 11 recall cutoff values r is defined as the highest precision found for any recall value r′ >= r:
The precision-recall curves for both sentence- and abstract-level extractions after two iterations are shown in Fig. 2. The precision-recall curves after three iterations are shown in Fig. 3. In each of these figures, we plotted the precision-recall curves for pairs ranked by pure MEDLINE frequency (“Cooc_All”), by method 1 (“Warfarin_CYP2C9_ranked” and “FDA_PGx_ranked”), and for random ranking (“Warfarin_CYP2C9_random” and “FDA_PGx_random”). As shown in the figures, the precision for both random ranking and ranking by MEDLINE frequency were low at all cutoff recall values. On the other hand, the ranking algorithm we developed was able to rank PGx-specific drug-gene pairs highly amongst extracted pairs. For instance, the precision at recall of 0.10 was 0.881 for “Warfarin_CYP2C9_ranked”, compared to 0.26 for “Cooc_All” and 0.357 for “Warfarin_CYP2C9_random” (Fig. 2). Similar trends were seen for both sentence-level and abstract-level extraction after two or three iterations. This demonstrated that our ranking algorithm was robust and insensitive to the seeds used (warfarin-CYP2C9 vs. FDA PGx seeds), number of seeds used (one seed vs. 68 seeds), iterations (two or three iterations), or document types (sentences vs. abstracts).
Figure 2.

Ranked precision after two iterations for sentence-level (left) and abstract-level (right) drug-gene pair extraction.
Figure 3.
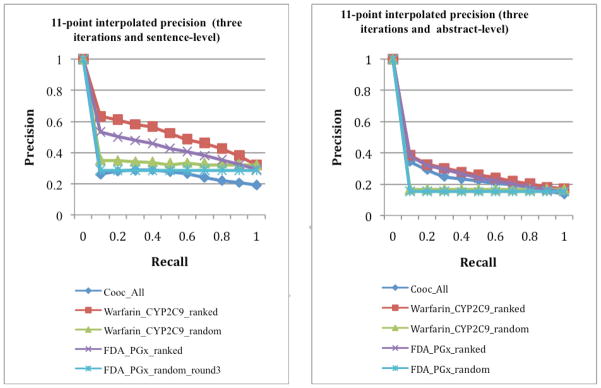
Ranked precision after three iterations for sentence-level (left) and abstract-level (right) drug-gene pair extraction
Ranked precision, recall and F1
We have demonstrated that starting from a few PGx-specific seeds, our extraction algorithm is able to effectively extract many other PGx-specific drug-gene pairs from either MEDLINE sentences or abstracts. We have also shown that the ranking algorithm is able to further rank PGx-specific drug-gene pairs highly among extracted pairs. Table 3 is the summary of precision, recall, and F1 at different ranking cutoffs for both PGx-specific and non-specific seeds. After two iterations, we extracted a total of 2,622 pairs with precision of 0.219, recall of 0.368, and F1 of 0.323. After ranking, the top 50% of ranked pairs had improved precision of 0.354 and F1 of 0.323 with decreased recall. The ranked F1 measures for Warfarin-CYP2C9 and 68 FDA drug-gene pairs are similar at all cutoffs. The pairs extracted with non-PGx-specific seed (“Captopril-ACE”) or based on pure co-occurrence without seeds (“None”) had significantly lower precision and F1 at all cutoffs.
Table 3.
Summary of ranked precision, recall and F1.
| Seed | Cutoff (%) | N | Precision | Recall | F1 |
|---|---|---|---|---|---|
| Warfarin–CYP2C9 | 10 | 262 | 0.561 | 0.094 | 0.161 |
| 25 | 655 | 0.472 | 0.198 | 0.279 | |
| 50 | 1,311 | 0.354 | 0.297 | 0.323 | |
| 75 | 1,966 | 0.273 | 0.343 | 0.304 | |
| 100 | 2,622 | 0.219 | 0.368 | 0.274 | |
| 68 FDA drug-gene pairs | 10 | 476 | 0.374 | 0.114 | 0.175 |
| 25 | 1,191 | 0.335 | 0.256 | 0.290 | |
| 50 | 2,383 | 0.260 | 0.397 | 0.314 | |
| 75 | 3,575 | 0.209 | 0.479 | 0.291 | |
| 100 | 4,767 | 0.171 | 0.522 | 0.258 | |
| Captopril-ACE | 10 | 254 | 0.063 | 0.010 | 0.018 |
| 25 | 636 | 0.033 | 0.013 | 0.019 | |
| 50 | 1,272 | 0.019 | 0.015 | 0.017 | |
| 75 | 1,908 | 0.013 | 0.016 | 0.014 | |
| 100 | 2,545 | 0.011 | 0.018 | 0.014 | |
| None | 10 | 10,291 | 0.054 | 0.359 | 0.094 |
| 25 | 25,729 | 0.035 | 0.575 | 0.066 | |
| 50 | 51,458 | 0.024 | 0.783 | 0.046 | |
| 75 | 77,187 | 0.018 | 0.896 | 0.036 | |
| 100 | 102,916 | 0.015 | 1.000 | 0.030 |
Comparison with a dictionary-based approach in extracting drug-gene pairs from MEDLINE sentences
A total of 4,399 PGx-specific (with subtype “PD” or “PK”) drug-gene pairs were extracted in PharmGKB and consist of 637 drugs and 859 genes. We used the 859 genes and the 637 drugs as the inputs to a dictionary-based PGx-specific drug-gene extraction algorithm. Since both of these genes and drugs are from PGx-specific drug-gene pairs, we assume that they are more PGx-specific than those not contained in PharmGKB. On the other hand, the bootstrapping approach used all 19,055 human protein coding genes and 6,516 drugs from DrugBank as inputs. We compared the dictionary-based approach to the bootstrapping approach (with seed “Warfarin-CYP2C9” or 68 FDA drug-gene pairs) in terms of precision, recall and F1. Among the 4,399 pairs from PharmGKB, 1561 pairs co-occurred in MEDLINE sentences. We used these 1561 pairs as goldstandard for both approaches in extracting drug-gene pairs from MEDLINE sentences. In fact, this evaluation dataset favors the dictionary-based approach towards high recall (recall of 1.0). It will underestimate precision of the bootstrapping approach when it extracts PGx-specific pairs that are not included in the goldstandard dataset. Using the dictionary-based approach, we extracted a total of 12,444 co-occurrence pairs from sentences. We then ranked the extracted pairs according to their co-occurrence frequency. We measured the precision, recall and F1 at two ranking cutoffs: top 50% and top 100% (all) pairs. As shown in Table 4, the dictionary-based approach achieved a precision of 0.178, a recall of 0.711 and a F1 of 0.285 at cutoff of 50%, and a precision of 0.125, a recall of 1.000 and a F1 of 0.223 at cutoff of 100%. It seems that ranking by co-occurrence frequencies can improve the precision. Compared to the dictionary-based approach, the bootstrapping approach has significantly higher precisions at all corresponding cutoffs: average precision of 0.152 for the dictionary-based approach vs. average precision of 0.251 for the bootstrapping approach. The recall of the dictionary approach (average of 0.856) is significantly higher than that of the bootstrapping approach (average of 0.396) partly because of the specific evaluation dataset we used. We used drug-gene pairs (with subtype “PD” or “PK”) from PharmGKB as evaluation set. Even though PharmGKB is a manually curated knowledge base, we found out that some drug-gene pairs in PharmGKB are non-PGx-specific pairs, such as cetuximab-EGFR, cisplatin-EGFR, caffeine-BRCA1, cisplatin-BRCA1, alendronate-VDR, and pioglitazone-IL6. In fact, by a quick search using term ‘IL’, we found that a total of 34 drug-gene pairs (with assigned subtype ‘PD’ or ‘PK’) are associated with interleukin ligand or receptor genes such as gefitinib-IL15, gefitinib-IL8, and gefitinib-IL8RA. Inclusion of these non-PGx-specific drug-gene pairs in the evaluation set will decrease the recall of the bootstrapping method since it may not retrieve these non-PGx-specific pairs. However, the average F1 of the dictionary-based approach is 0.254, which is lower than that of the bootstrapping approach (average F1: 0.292). Both approaches perform better than the co-occurrence approach with 19,055 genes and 6516 drugs as input (precision of 0.015, recall of 1.000 and F1 of 0.030 at cutoff of 100%).
Table 4.
Comparison of bootstrapping approach with the dictionary-based approach with drugs and genes from PharmGKB PGx-specific drug-gene pairs as input.
| Method | Cutoff (%) | N | Precision | Recall | F1 |
|---|---|---|---|---|---|
| Bootstrapping (Seed “Warfarin–CYP2C9”) | 50 | 1,311 | 0.354 | 0.297 | 0.323 |
| 100 | 2,622 | 0.219 | 0.368 | 0.274 | |
| Bootstrapping (Seeds 68 FDA drug-gene pairs) | 50 | 2,383 | 0.260 | 0.397 | 0.314 |
| 100 | 4,767 | 0.171 | 0.522 | 0.258 | |
| Dictionary-based Approach | 50 | 6,222 | 0.178 | 0.711 | 0.285 |
| 100 | 12,444 | 0.125 | 1.000 | 0.223 | |
| None (co-occurrence with 19,055 genes and 6,516 drugs) | 50 | 51,458 | 0.024 | 0.783 | 0.046 |
| 100 | 102,916 | 0.015 | 1.000 | 0.030 |
By examining the errors (false positives) for the dictionary-based approach, we found that many of these errors are caused by inclusion of non-PGx-specific genes in the input gene lexicon. For example, we extracted 288 drug-AR pairs, 396 drug-VDR pairs and 127 drug-EGFR pair since gene symbols ‘AR’, ‘VDR’ and ‘EGFR’ are included in the gene lexicon. Among the 4,399 PGx drug-gene pairs extracted from PharmGKB, there are 11 drug-EGFR pairs such as cetuximab-EGFR and cisplatin-EGFR, 18 drug-VDR pairs such as alendronate-VDR and calcium-VDR and 3 drug-AR pairs such as ethanol-AR and paroxetine-AR. On the other hand, using the bootstrapping approach (with seed warfain-CYP2C9), we extracted only 11 drug-EGFR pairs, 0 drug-AR pairs and 0 drug-VDR pairs since PGx-specific drug-gene pairs such as warfarin-CYP2C9 often don’t appear togther with non-PGx-specific drug-gene pairs such as drug-AR or drug-VDR pairs.
One of the limitations inherent in this comparison is that we only implemented the most primitive dictionary-based method: co-occurrence ranked by frequency. More advanced implementation will have better performance than that shown here. The take-home message is that the bootstrapping approach by itself may not be sufficient as a standalone drug-gene relationship extraction method, but it effectively removed many false positives. Its output is enriched with PGx-specific drug-gene pairs compared to the dictionary-based co-occurrence method and can be used as input for more sophisticated methods to further improve the performance.
Analyze the correlations between the extracted PGx-specific drug-gene pairs and drug adverse events
A total of 100,049 drug adverse event pairs were downloaded from SIDER. For the 996 drugs contained in these drug-gene pairs, a total of 495,510 drug-drug combination pairs (order ignored) are possible. The average number of shared adverse events for these pairs is 20.4. Among the 996 drugs, 557 of them were mapped to the 2,622 drug-gene pairs (extracted using seed warfarin-CYP2C9 after two iterations). A total of 154,846 drug-drug pairs are possible for these 557 drugs. The average number of shared adverse events for these drug-drug pairs is 28.2. The number of shared side effects increases to 37.4 for drug-drug pairs sharing at least one gene (Fig. 4). The same is true for drug-gene pairs extracted using other PGx-specific seeds (“Caffeine_CYP1A2” and “FDA_PGx”). The number of shared adverse events increases from 28.2 to 41.7 as the number of share PGx-specific genes increases from 0 to 5. The correlation between drug-gene pairs extracted based on pure co-occurrence (“Cooc_All”) and drug adverse event associations is less significant. The strong positive correlation between the extracted PGx-specific drug-gene pairs and drug adverse event pairs demonstrated that these pairs may have potential in computational approaches in studying drug adverse events for personalized medicine.
Figure 4.

Correlation between extracted PGx-specific drug-gene pairs (using different seeds) with known drug adverse event associations.
Discussion
We developed a large-scale, semi-supervised relationship algorithm to extract PGx-specific drug-gene pairs from 20 million published MEDLINE abstracts. Starting with a few seeds, our extraction algorithm achieved a precision of 0.219, a recall of 0.368, and an F1 of 0.274 after two iterations, marking a significant improvement over methods using non-PGx-specific seeds (precision: 0.011, recall: 0.018, and F1: 0.014) or no seeds (precision: 0.015, recall: 1.000, and F1: 0.030). The ranking algorithm effectively ranked PGx-specific pairs highly and further significantly improved the precision from 0.219 for all extracted pairs to 0.561 for the highest ranked pairs. However, there are several limitations to our study.
First of all, even though our algorithm has significantly improved precision over methods using non-PGx-specific seed or without using any seed (0.219 vs. 0.015) and the ranking algorithm was able to further rank PGx-specific drug-gene pairs highly, the overall precision is still low. Starting from a PGx-specific seed, the algorithm implicitly classifies sentences into PGx-related or non-related. However, if n drugs and m genes co-occur in a PGx-specific sentences, the algorithm will extraction all n*m possible drug-gene pairs. The ranking algorithm then ranks each of these n*m drug-gene pairs based on the ranking scores of other co-occurring pairs. Both extraction and ranking algorithm are probabilistic and do not take into account of the syntactic relationships between drug entities and gene entities in sentences. Consider the following sentence: “Fifteen healthy subjects were administered single oral doses of caffeine (CYP1A2), warfarin (CYP2C9), omeprazole (CYP2C19), dextromethorphan (CYP2D6), and midazolam (CYP3A)” (PMID 16638741). There are five drugs and five genes in the sentence. Our algorithm will extract total of 5*5 = 25 drug-gene pairs from this sentence. In this case, manual curation, deep syntactic analysis, or prior knowledge are necessary. Even though our algorithm cannot decide the exact drug-gene correspondences in this case, it is able to find this PGx-related sentence out of millions of non-PGx-specific sentences in MEDLINE and filtering out many non-PGx-specific drug-gene pairs. Currently, we are developing a semi-automatic method to construct an accurate and comprehensive PGx-specific drug-gene knowledge base from published literature. We will use the semi-supervised method to first implicitly classify MEDLINE sentences into PGx-related or non-related. We will then manually extract drug-gene pairs from these classified PGx-related sentences. The newly extracted drug-gene pairs will immediately be fed into the extraction and ranking algorithm in a positive feedback loop and newly ranked sentences and drug-gene pairs will be presented to curators for confirmation or rejection. Both accepted and rejected pairs are used in the automatic learning process.
Second, the precision and recall calculation of the algorithm largely depend on the precision and recall of the gold standard. As we discussed previously, the drug-gene pairs in PhamGKB are limited in both precision and recall. The drug-gene pairs in PharmGKB are largely PGx-specific and not biased to certain sets of drugs or genes; therefore using them as gold standard to compare different algorithms should give fair evaluation. However, the precision and recall may not reflect the real measures of the algorithms. We will need to manually create gold standard datasets for MEDLINE-based drug-gene relationship extraction. For example, for a given drug or gene, we can find all the sentences and abstracts wherein it appears. We can then extract PGx-specific drug-gene pairs from these retrieved sentences and abstracts to create MEDLINE-based PGx-specific drug-gene relationship extraction gold standards.
Third, the entire drug-gene relationship extraction algorithm starts with 19,055 human protein-coding genes, 6,707 drugs, 20 million MEDLINE abstracts, and a few PGx-specific drug-gene pairs as seeds. During the extraction and ranking process, many non-PGx-specific MEDLINE sentences were automatically excluded. However, if we can rank the 19,055 human protein-coding genes, most of which are not PGx-related, according to their PGx specificity, we can further improve the precision of the relationship extraction algorithm. We recently developed a gene-prioritizing algorithm to rank all human genes according to their relevance in drug metabolism [129]. In our future study, we will incorporate the gene ranking score into the pair-ranking algorithm to further improve the precision.
Fourth, we compared the performance of our drug-gene extraction algorithm to that of baseline method instead of state-of-art approaches. The reason is that most of the state-of-art PGx-specific drug-gene approaches used either PGx-specific gene lexicons as input or limited set of PGx-related articles as the text corpus, which already have much better baseline in terms of precision. On the other hand, we started with all 19,055 human protein-coding genes, 6,707 drugs, 20 million MEDLINE abstracts, and a few PGx-specific drug-gene pairs as seeds. In order for algorithms to be comparable to each other, the algorithms should take the same inputs: gene lexicon, drug lexicon and text corpus. It will be interesting to compare the overall performance when other algorithms take the same inputs we used in this study.
Conclusions
We developed a bootstrapping, semi-supervised learning approach to iteratively extract and rank drug-gene pairs according to their relevance to drug pharmacogenomics. Starting with 19,055 human protein-coding genes, 6,707 drugs, 20 million MEDLINE abstracts, and a few PGx-specific drug-gene pairs as seeds, the extraction algorithm achieved a precision of 0.219, a recall of 0.368, and an F1 of 0.274 after two iterations, which is a significant improvement over the results of using any seeds (precision: 0.015, recall: 1.000, and F1: 0.030). The ranking algorithm effectively ranked PGx-specific pairs highly and further improved the precision from 0.219 to 0.561. By comparing to a dictionary-based approach with PGx-specific gene lexicon as input, we showed that the bootstrapping approach has better performance in terms of precision and F1 (precision: 0.251 vs. 0.152, recall: 0.396 vs. 0.856 and F1: 0.292 vs. 0.254). To demonstrate the connection between drug metabolism and drug adverse events, we show that drug-drug pairs sharing at least one PGx-specific gene also share significant more adverse events (28.2 for all pairs vs. 37.4 for pairs sharing at least one PGx-specific gene). In the future, we will develop automatic and semi-automatic methods to further improve both precision and recall of the extracted drug-gene pairs and use the extracted pairs in drug adverse event prediction.
Knowledge of PGx-specific drug-gene relationships is important for personalized medicine.
Automatic PGx-specific drug-gene relationship extraction from free text is difficult.
We develop a semi-supervised relationship extraction method requiring minimal prior domain knowledge.
We develop a ranking algorithm to rank PGx-specific drug-gene pairs highly among extracted pairs.
The extracted PGx-specific drug-gene pairs have strong correlations with drug adverse events.
Acknowledgments
RX is funded by Case Western Reserve University/Cleveland Clinic Foundation CTSA Grant (UL1 RR024989) and Case Western Reserve University Provost Starting Grant. QW is funded by ThinTek LLC. RX and QW together have conceived the idea, designed and implemented the algorithm, and prepared the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis JC, Furstenthal L, Desai AA, Norris T, Sutaria S, Fleming E, Ma P. The microeconomics of personalized medicine: today’s challenge and tomorrow’s promise. Nature reviews Drug discovery. 2009;8:279–286. doi: 10.1038/nrd2825. [DOI] [PubMed] [Google Scholar]
- 2.Evans WE, McLeod HL. Pharmacogenomics: Drug disposition, drug Targets, and side effects. N Engl J Med. 2003;348:538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 3.Weinshilboum RM, Wang L. Pharmacogenetics and pharmacogenomics: development, science, and translation. Annu Rev Genomics Hum Genet. 2006;7:223–245. doi: 10.1146/annurev.genom.6.080604.162315. [DOI] [PubMed] [Google Scholar]
- 4.Roden DM, Tyndale RF. Pharmacogenomics at the tipping point: challenges and opportunities. Clin Pharmacol Ther. 2011;89:323–327. doi: 10.1038/clpt.2010.340. [DOI] [PubMed] [Google Scholar]
- 5.Frueh FW, Amur S, Mummaneni P, Epstein RS, Aubert RE, DeLuca TM, Verbrugge RR, Burckart GJ, Lesko LJ. Pharmacogenomic biomarker information in drug labels approved by the United States Food and Drug Administration: prevalence of related drug use. Pharmacotherapy. 2008;28:992–998. doi: 10.1592/phco.28.8.992. [DOI] [PubMed] [Google Scholar]
- 6.Lesko LJ, Zineh I. DNA, drugs and chariots: on a decade of pharmacogenomics at the US FDA. Pharmacogenomics. 2010;11:507–512. doi: 10.2217/pgs.10.16. [DOI] [PubMed] [Google Scholar]
- 7.Weiss ST. Creating and evaluating genetic tests predictive of drug response. Nat Rev Drug Discov. 2008;7:568–74. doi: 10.1038/nrd2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott SA. Personalizing medicine with clinical pharmacogenetics. Genet Med. 2011 Dec;13(12):987–95. doi: 10.1097/GIM.0b013e318238b38c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein TE, Chang JT, Cho MK, Easton KL, Fergerson R, Hewett M, Lin Z, Liu Y, Liu S, Oliver DE, Rubin DL, Shafa F, Stuart JM, Altman RB. Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base. The Pharmacogenomics Journal. 2001;1:167–170. doi: 10.1038/sj.tpj.6500035. [DOI] [PubMed] [Google Scholar]
- 10.Garten Y, Coulet A, Altman RB. Recent progress in automatically extracting information from the pharmacogenomic literature. Pharmacogenomics. 2010;11:1467–1489. doi: 10.2217/pgs.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang JT, Altman RB. Extracting and characterizing gene-drug relationships from the literature. Pharmacogenetics. 2004;14:577–586. doi: 10.1097/00008571-200409000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Garten Y, Altman RB. Pharmspresso: a text-mining tool for extraction of pharmacogenomic concepts and relationships from full text. BMC Bioinformatics. 2009;10:S6. doi: 10.1186/1471-2105-10-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theobald M, Shah N, Shrager J. Extraction of conditional probabilities of the relationships between drugs, diseases, and genes from PubMed guided by relationships in PharmGKB. AMIA Summit on Translational Bioinformatics. 2009:124–128. [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen NI, Brunak S, Altman RB. Generating genome-scale candidate gene lists for pharmacogenomics. Clin Pharmacol Ther. 2009;86:183–189. doi: 10.1038/clpt.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Liu M, Zheng W, Zhao Z, Xu H. Ranking gene-drug relationships in biomedical literature using latent dirichlet allocation. Pacific Symposium on Biocomputing. 2012 [PMC free article] [PubMed] [Google Scholar]
- 16.Ahlers CB, Fiszman M, Demner-fushman D, Lang F, Rindesch TC. Extracting semantic predications from MEDLINE citations for pharmacogenomics. Pac Symp Biocomput. 2007:209–220. [PubMed] [Google Scholar]
- 17.Coulet AM, Shah N, Garten Y, Musen M, Altman RB. Using text to build semantic networks for pharmacogenomics. J Biomed Inform. 2010;43:1009–1019. doi: 10.1016/j.jbi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu R, Wang Q. A knowledge-driven conditional approach to extract pharmacogenomics specific drug-gene relationships from free text. J of Biomedical Informatics. 2012 doi: 10.1016/j.jbi.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu R, Wang Q. An iterative searching and ranking algorithm for prioritizing pharmacogenomics genes. International Journal of Computational Biology and Drug Design. 2012 doi: 10.1504/IJCBDD.2013.052199. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etzioni O, Cafarella M, Downey D, Popescu AM, Shaked T, Soderland S, Weld D, Yates A. Unsupervised named-entity extraction from the web: an experimental study. Artificial Intelligence. 2005;165(1):91–134. [Google Scholar]
- 21.Nadeau D, Turney P, Matwin S. Unsupervised named entity recognition: Generating gazetteers and resolving ambiguity. Advances in Artificial Intelligence. 2006:266–277. [Google Scholar]
- 22.Agichtein E, Gravano L. Snowball: extracting relations from large plain-text collections. Proceedings of the fifth ACM conference on Digital libraries (DL); pp. 85–94.pp. 2000 [Google Scholar]
- 23.Brin S. Extracting patterns and relations from the World Wide Web. The World Wide Web and Databases. 1999:172–183. [Google Scholar]
- 24.Carlson A, Betteridge J, Kisiel B, Settles B, Hruschka ER, Jr, Mitchell TM. Toward an architecture for never-ending language learning. Proceedings of the Twenty-Fourth Conference on Artificial Intelligence. 2010;2:3–3. [Google Scholar]
- 25.Caprosaso JG, Baumgartner WA, Randolph DA, Cohen KB, Hunter L. Rapid pattern development for concept recognition systems: application to point mutations. Journal of Bioinformatics and Computational Biology. 2007;5(06):1233–1259. doi: 10.1142/s0219720007003144. [DOI] [PubMed] [Google Scholar]
- 26.Nakashole N, Theobald M, Weikum G. Scalable knowledge harvesting with high precision and high recall. In Proceedings of the fourth ACM international conference on Web search and data mining. 2012:227–236. [Google Scholar]
- 27.Pasca M, Lin D, Bigham J, Lifchits A, Jain A. Organizing and searching the world wide web of facts-step one: the one-million fact extraction challenge. In Proceedings of the 21st National Conference on Artificial Intelligence (AAAI-06) 2006;21(2):1400. [Google Scholar]
- 28.Riloff E, Jones R. Learning dictionaries for information extraction by multi-level bootstrapping. In Proceedings of the National Conference on Artificial Intelligence (AAAI-99) 1999:474–479. [Google Scholar]
- 29.Snow E, Jurafsky D, Ng A. Learning syntactic patterns for automatic hypernym discovery. In Proceedings of the 17th Conference on Advances in Neural Information Processing Systems (NIPS) 2005 [Google Scholar]
- 30.Xu R, Supekar K, Morgan A, Das A, Garber AM. Unsupervised Method for Automatic Construction of a Disease Dictionary from a Large Free Text Collection. Annual American Medical Informatics Association Symposium. 2008:820–824. [PMC free article] [PubMed] [Google Scholar]
- 31.Xu R, Morgan A, Das A, Garber AM. Investigation of Unsupervised Pattern Learning Techniques for Bootstrap Construction of a Medical Treatment Lexicon. Association for Computational Linguistics BioNLP Workshop. 2009:63–70. [Google Scholar]
- 32.Xu R, Das A, Garber AM. Unsupervised Method for Extracting Machine Understandable Medical Knowledge from a Large Free Text Collection. Annual American Medical Informatics Association Symposium. 2009:709–713. [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Zhang GQ, Xu R. Semi-supervised Image Classification for Automatic Construction of a Health Image Library. Proceedings of the 2nd ACM SIGHIT International Health Informatics Symposium; 2012. pp. 2111–120. [Google Scholar]
- 34.Haveliwala T. Topic-Sensitive PageRank: A Context-Sensitive Ranking Algorithm for Web Search. IEEE Transactions on Knowledge and Data Engineering. 2003 [Google Scholar]
- 35.Page L. Stanford Digital Library Project, talk. Aug 18, 1997. PageRank: Bringing Order to the Web. (archived 2002) [Google Scholar]
- 36.Chiang AP, Butte AJ. Data-driven methods to discover molecular determinants of serious adverse drug events. Clin Pharmacol Ther. 2009;85:259–268. doi: 10.1038/clpt.2008.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol. 2010 doi: 10.1038/msb.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning CD, Schutze H. Introduction to Information Retrieval. Cambridge University Press; 2008. [Google Scholar]