

Abstract
Systems approaches to analyzing disease phenotype networks in combination with protein functional interaction networks have great potential in illuminating disease pathophysiological mechanisms. While many genetic networks are readily available, disease phenotype networks remain largely incomplete. In this study, we built a large-scale Disease Manifestation Network (DMN) from 50,543 highly accurate disease-manifestation semantic relationships in the United Medical Language System (UMLS). Our new phenotype network contains 2305 nodes and 373,527 weighted edges to represent the disease phenotypic similarities. We first compared DMN with the networks representing genetic relationships among diseases, and demonstrated that the phenotype clustering in DMN reflects common disease genetics. Then we compared DMN with a widely-used disease phenotype network in previous gene discovery studies, called mimMiner, which was extracted from the textual descriptions in Online Mendelian Inheritance in Man (OMIM). We demonstrated that DMN contains different knowledge from the existing phenotype data source. Finally, a case study on Marfan syndrome further proved that DMN contains useful information and can provide leads to discover unknown disease causes. Integrating DMN in systems approaches with mimMiner and other data offers the opportunities to predict novel disease genetics. We made DMN publicly available at nlp/case.edu/public/data/DMN.
Keywords: Ontology, Disease phenotype network, Network analysis
1. Introduction
Linking complex human diseases to their genetic basis remains a challenging task. For computational strategies to discover candidate disease genes, incorporating new data may lead to new discoveries. Traditional methods prioritized genes for a disease if the genes have similar functions with the known disease genes [2,38,44,39,32,17,48]. Recent studies incorporate disease phenotype similarities in addition to the genomic data to increase the ability of identifying new disease genes [19,23,43,46,47,16,35,37], assuming that similar phenotypes and overlapping genetic causes are correlated [5,29,15,2,9,10].
However, the disease phenotype networks used in current gene prediction approaches remain largely incomplete. Most phenotype databases were constructed through mining textual phenotype descriptions [18,6]. For example, van Driel and the colleagues extracted disease-phenotype associations from OMIM through text mining, calculated the pairwise disease similarities, and stored them in the database called mimMiner [42], which is one of the most widely-used phenotype networks in recent disease gene discovery methods [23,43,33,36,16]. Combining different phenotype data has the potential to reduce the bias in each data source and improve the network-based prediction models [26,30]. Therefore, we explored new accurate and publicly accessible disease phenotype data in addition to the existing phenotype networks.
In this study, we created Disease Manifestation Network (DMN), using the highly accurate and structured clinical manifestation data from Unified Medical Language System (UMLS) [24,4,25]. Clinical manifestation captures a major aspect of disease phenotype and can predict disease causes [5]. For example, the Stickler syndrome, Marshall syndrome and Otospondylomegaepiphyseal dysplasia (OSMED) have highly similar manifestations and also involve mutations in interacting collagen genes COL2A1, COL11A2, and COL11A1, respectively [1]. The UMLS semantic network currently uses 50,543 disease-manifestation semantic relationships to explicitly link 2,305 diseases to their clinical manifestations. In this knowledge base, all disease and manifestation terms are formally represented by unified concepts and the semantic relationships between concepts were collected from multiple different ontologies.
We hypothesized that DMN not only reflects known disease-gene relationships, but also contains different phenotypic knowledge compared with mimMiner. We tested the hypothesis through network comparative analysis between DMN, mimMiner [42], and the two variants of human disease network (HDN) [12], which connects diseases if they share genes. The correlation between DMN and HDNs indicated that DMN reflects existing knowledge on genetic relationships among diseases. The comparison between DMN and mimMiner demonstrated that the two phenotype networks are largely complementary in nodes, edges and community structures. The overall analysis suggests that combining DMN with previous phenotype data sources, such as mimMiner, may potentially improve the data-driven methods for biomedical applications, such as disease gene discovery and drug repositioning.
2. Data and methods
Our study consists of the following steps (Fig. 1): (1) Constructed DMN using the disease-manifestation associations from UMLS; (2) compare phenotypic relationships in DMN and genetic relationships among diseases; (3) compared DMN with mimMiner [42]; and (4) conducted a case study on the phenotypic relationships of Marfan syndrome in DMN.
Fig. 1.
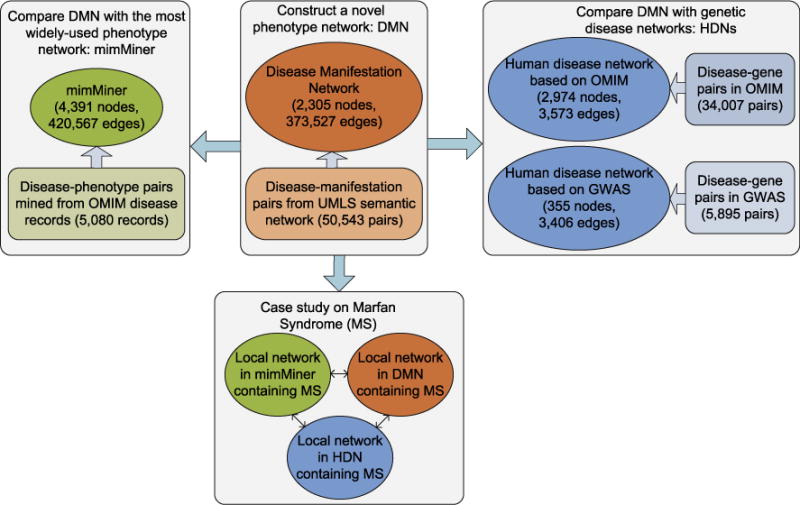
The four steps of network analysis for DMN.
2.1. Construct DMN using disease-manifestation associations in UMLS
We first extracted disease-manifestation relationships from the UMLS file MRREL.RRF (2013 version). The file contains 647 different kinds of semantic relationships between biomedical concepts. We collected the concepts pairs linked by the “has manifestation” relationship, and obtained 50,543 disease-manifestation pairs. The disease-manifestation relationships come from OMIM [14], Ultrasound Structured Attribute Reporting [3], and Minimal Standard Digestive Endoscopy Terminology [40]. OMIM is the major contributor among these data sources.
The manifestation terms vary greatly in abundance. For example, common manifestations such as “seizures” are associated with many diseases, while rare manifestations such as “Amegakaryocytic thrombocytopenia” are only associated with one disease. We used the information content (1) to weight each manifestation concept.
| (1) |
Variable wc is the weight of the manifestation concept c, nc is the number of diseases associated with manifestation c, and N is the total number of diseases. Then we modeled the manifestation similarity between disease x and y by the cosine of their feature vectors in (2), in which the feature vectors consist of manifestations xi and yi for disease x and y. The cosine similarity was used before [19,42] to quantify phenotype overlaps.
| (2) |
We constructed DMN as a weighted network with the manifestation similarities. The edges weights are in the range (0, 1].
2.2. Compare phenotypic relationships in DMN with genetic disease associations
We conducted two experiments to evaluate whether the phenotypic relationships in DMN reflect genetic associations among diseases. The first experiment is to calculate the correlation between the disease similarities in DMN and two quantified measures of genetic associations. We first ranked the edges (disease pairs) in DMN by their weights (disease similarities) from large to small. For top N disease pairs, we counted the percentage of disease pairs that share associated genes in OMIM and the average number of genes shared by the N disease pairs. Then we calculated the Pearson’s correlations between N and the genetic measures.
In the second experiment, we compared the network topologies between DMN and two genetic disease networks. A well-studied genetic disease network is HDN, in which diseases were connected if they share associated genes in OMIM and edges were weighted by the number of overlapping genes [12]. Here we inherited the network construction method of HDN, but used two different disease-gene association data: the updated data in OMIM (April, 2013) and GWAS catalog (August, 2013). We represented the disease terms in OMIM-based HDN and GWAS-based HDN with 2974 and 355 UMLS concept unique identifiers, respectively, to enable the comparison with DMN. The two genetic disease networks both contains rich information of disease genetics [20,22], but are largely different. The OMIM-based HDN mostly contains Mendelian diseases with strong genetic causes; the GWAS-based HDN mostly contains common complex diseases. The two networks only share 45 diseases.
We compared the edges and community structures between DMN and the two HDNs. Network community structure reveals the biological network properties and offered insights into cell functions, protein interactions, and disease dynamics [8,31,34]. We applied a widely-used community detection algorithm [28] and calculated the two-way similarities between community groups:
| (3) |
| (4) |
|X| and |Y| are the number of disease pairs that appear in the same community in DMN and HDN, respectively. |X∩Y| is the count of disease pairs that were grouped into one community in both networks.
We tested the significance of edge and community similarities between DMN and HDNs by creating a background distribution of similarities expected at random. We kept the number and size of communities in DMN, and randomly swapped the assignments of disease nodes into each community. Then we linked nodes inside a community with probability Pin, and those across communities with probability Pout. The Pin and Pout were estimated from the edge density within and between communities in DMN, respectively. We repeated 100 times of randomizing DMN, and compared each random network to HDNs to create the background signals. Finally, we compared the observed similarities with the background signals using Wilcoxon signed-rank test.
2.3. Compare DMN with the widely-used disease phenotype network mimMiner
DMN and mimMiner both contain phenotypic knowledge based on clinical observations. Here, we compared DMN with mimMiner to demonstrate that the two phenotype networks contain different knowledge, so that combining them in applications, such as disease gene discovery and drug repositioning, may potentially lead to improved performance. We first mapped the 5080 diseases in mimMiner from OMIM identifiers to UMLS concept unique identifiers to allow the comparison. Since text mining introduced false positive disease-phenotype relationships, we needed to tradeoff between the data coverage and accuracy in mimMiner. Based on previous analysis [42], we chose to connect two disease nodes if their similarities are above 0.3. The network of mimMiner contains 4,391 disease nodes after these processes. We then compared the node, edges and community structures between DMN with mimMiner.
2.4. Case study on Marfan syndrome
Marfan syndrome is a common inherited disorder of the connective tissue, occurring once in every 10,000 to 20,000 individuals [45]. About 75% of patients with Marfan syndrome have mutations in the FBN1 gene, which encodes brillin-1, a protein that provides strength and flexibility to connective tissue. Despite this well-defined mutation, gene FBN1 cannot always predict the wide variety of phenotypes in patients, and other unknown genetic factors that account for the diversified phenotypes of Marfan syndrome may exist. Here, we conducted a case study on Marfan syndrome and its phenotype relationships in DMN. We compared the corresponding subnetworks in DMN, mimMiner and HDN to show that the DMN contains different phenotypic knowledge and has the potential in deepening the understanding of MS pathogenesis.
3. Results
3.1. DMN network properties
DMN contains 2305 nodes and 373,527 edges. The network has a long-tail degree distribution and is robust to random removal of nodes. Removing the nodes with large degrees can quickly break down the network into small components (Fig. 2). Table 1 lists the network properties of DMN. To understand DMN better, we also showed the properties of three other disease networks, including OMIM-based HDN, GWAS-based HDN and mimMiner. DMN is denser than mimMiner, but the nodes tend to cluster into disjoint components. Both the phenotype networks are evidently different from the genetic networks: DMN and mimMiner are denser (higher network density), less cliquish (lower clustering coefficients) and more connective (less connected components) than HDNs. Fig. 3 shows example subnetworks from DMN, mimMiner, and HDNs containing randomly sampled nodes. In contrast to the densely-connected subnetworks of DMN and mimMiner, OMIM-based HDN mostly contains small components such as triangles and chains. GWAS-based HDN contains complex diseases, which are often associated with multiple genes, thus its edge density is higher than OMIM-based HDN, but still lower than DMN.
Fig. 2.
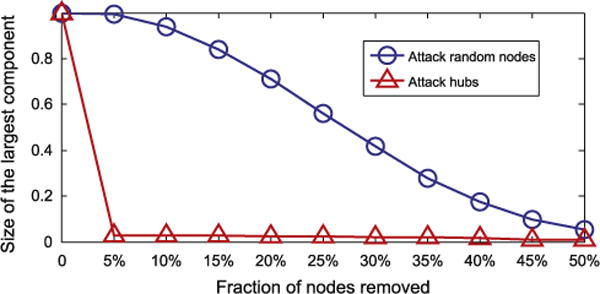
Robustness of DMN with respect to the removal of random nodes and hub nodes.
Table 1.
Global properties of DMN and the other disease networks, including HDNs (genetic disease networks) and mimMiner (widely-used phenotype network) based on OMIM text mining. The last three columns represent average shortest path, average cluster coefficient, and connected component, respectively.
| Disease network | Number of nodes | Network density | Network diameter | Avg. shortest path | Avg. clu. coeff. | Conn. comp. |
|---|---|---|---|---|---|---|
| DMN | 2305 | 0.14 | 6 | 2.042 | 0.649 | 6 |
| HDN(OMIM) | 2974 | 0.001 | 9 | 2.341 | 0.74 | 797 |
| HDN(GWAS) | 355 | 0.054 | 5 | 2.505 | 0.702 | 17 |
| MimMiner | 4391 | 0.044 | 7 | 2.445 | 0.421 | 1 |
Fig. 3.
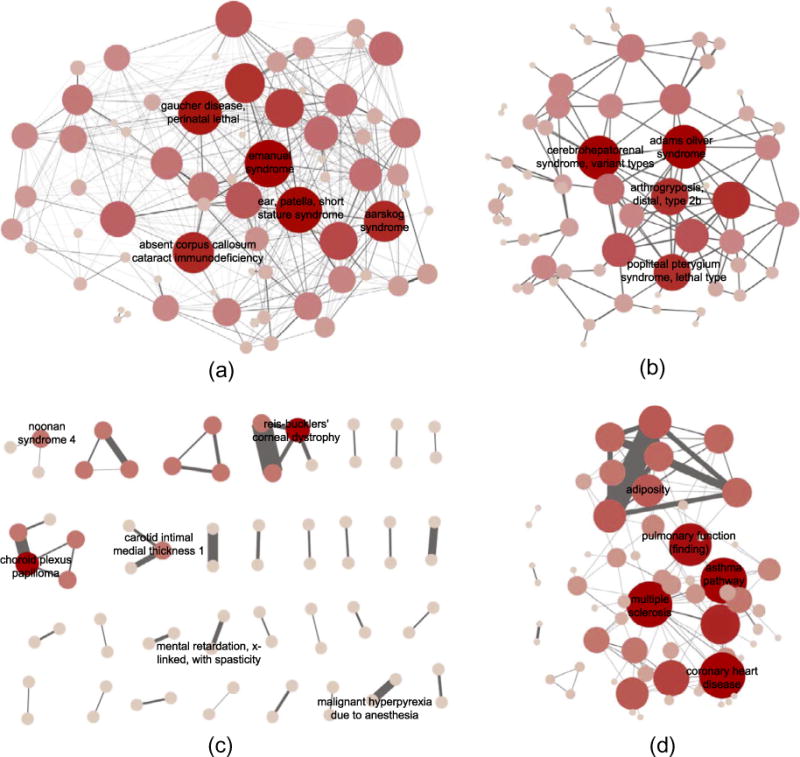
Randomly selected subgraphs of (a) DMN, (b) mimMiner, (c) OMIM-based HDN and (d) GWAS-based HDN. Only part of the node labels are shown in the figure due to space limit. In contrast to DMN and mimMiner, the sub-graphs in HDNs are less connective and cliquish.
The differences in global structures between phenotype and genetic disease networks indicate that we may have not fully discovered the genes accounting for the observed phenotypic connections. Systematic studying the disease phenotype networks offers a chance to detect new disease genes, particularly for the disease whose genetic basis is completely unknown. Note that non-genetic factors, such as common environments and life styles, may also contribute to the overlapping phenotypes. To evaluated the potential of phenotype networks to predict disease genes, we show the correlation between phenotypic and genetic relationships in the next section.
3.2. DMN partially correlates with the genetic disease networks
In the first experiment, we found that the manifestation similarities in DMN have correlations with quantified measures of disease genetic associations. Fig. 4 (left) shows that the disease pairs with larger manifestation similarities (higher ranks) are more likely to share genes. The Pearson’s correlation between the ranks of manifestation similarities and the probabilities of sharing gene is −0.603 (p≪ E−8). Also, Fig. 4 (right) shows that diseases with larger manifestation similarities tend to share more genes. The Pearson’s correlation between the ranks of manifestation similarities and average number of shared genes is −0.647 (p≪ E−8).
Fig. 4.
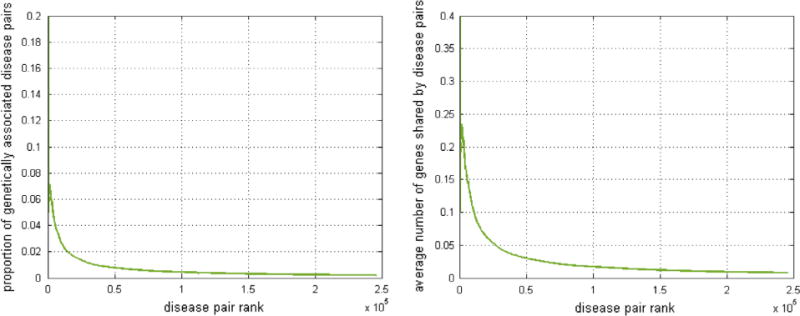
Correlation between manifestation similarities and genetic associations. Left: Correlation between proportion of genetically associated disease pairs (x-axis) and the phenotype similarity ranks (y-axis) in DMN. Right: Correlation between the average numbers of genes shared by disease pairs (x-axis) and the phenotype ranks (y-axis) in DMN. Diseases with larger phenotype similarity in DMN tend have stronger genetic association.
We found that only a small percentage of disease pairs share associated genes despite the significant correlations between phenotype similarities and genetic associations. For example, among the top five disease pairs with highest phenotype similarities, only one pair shared associated genes. This observation indicates that the overlapping manifestations may result from unknown genes, shared pathways, protein complexes, or common environment. Discovering unknown genetic factors responsible for overlapping phenotypes among diseases is one of the goals of studying the disease phenotype networks.
In the second experiment, we compared the edges and community structures of DMN with the genetic disease networks. Table 2 shows that the number of common edges between DMN and HDNs is significant higher than the random distribution. We found that mimMiner also contains 520 common edges with OMIM-based HDN and 14 with GWAS-based HDN. However, DMN and mimMiner share different disease connections with HDNs: 76 of 278 (27%) edge overlaps between DMN and OMIM-based HDN do not appear in mimMiner, and 5 of 6 edge overlaps between DMN and GWAS-based HDN do not appear in mimMiner.
Table 2.
Compare the edge overlaps N between DMN and the genetic disease networks. Network B′ represents the randomized graph that preserves the properties of Network B. Column N(A,B′) represents the average number of edge overlap comparing network A and the randomized networks.
| Network A | Network B | N(A,B) | N(A,B′) | P-value |
|---|---|---|---|---|
| HDN(OMIM) | DMN | 278 | 65.4 | ≪ E−8 |
| HDN(GWAS) | DMN | 6 | 2.93 | ≪ E−8 |
Table 3 lists the community structure similarities between DMN and HDNs. If two diseases are grouped together in OMIM-based HDN, they have over 60% chances to stay in one community in DMN. On the other hand, diseases in one community in DMN have 0.6% chance of being grouped together in OMIM-based HDN. The absolute values of community structure similarities may be biased: OMIM-based HDN mostly contains small size clusters, and the probability of two diseases share one cluster is naturally low. However, statistical test shows that the similarities in community partitions between DMN and HDN are significantly higher than the random distribution, indicating that the observed similarities reflect intrinsic correlations between the biological networks. The community structure correlation between DMN and GWAS-based HDN is also significant compared with random signals.
Table 3.
Compare the community structures between DMN and the genetic disease networks. SA→B and SB→A represent the two-way the similarity in community partitions between network A and B.
| Network A | Network B | SA→B | SA→B′ | P-value | SB→A | SB→A′ | P-value |
|---|---|---|---|---|---|---|---|
| HDN(OMIM) | DMN | 0.655 | 0.281 | ≪ E−8 | 0.006 | 0.002 | ≪ E−8 |
| HDN(GWAS) | DMN | 0.611 | 0.279 | ≪ E−8 | 0.297 | 0.156 | ≪ E−8 |
In summary, DMN is partially correlated with the genetic disease networks in both edges and community structures. On the one hand, the phenotype relationships among diseases in DMN reflects shared genetic mechanisms. On the other hand, many disease-associated genes and pathways may have not been discovered yet. In addition, comparative analysis to HDNs also show that DMN and mimMiner contain different knowledge. The phenotype relationships in DMN have the potential to provide leads for discovering new disease genetics.
3.3. DMN contains knowledge different from mimMiner
We compared DMN with the widely-used phenotype network mimMiner to show their differences. Table 4 summarizes their differences in nodes, edges, and community structures. Though DMN shares 75% of the nodes with mimMiner, 295,975 edges (79.2%) are unique and do not appear in mimMiner. Examples of the unique edges are schizophrenia–myopia, autism–tuberous sclerosis, and familial mediterranean fever–alport syndrome. We extracted all unique disease pairs in DMN and made the data publicly accessible. In addition, the community structures of DMN and mimMiner are partly correlated. The community similarities in the two directions are comparable and both moderate, showing that we cannot completely predict the phenotype clusters in one network based on the other. Therefore, the knowledge captured in DMN and mim-Miner is complementary. Integrating these two networks is valuable for better prediction of candidate disease genes.
Table 4.
Compare DMN with mimMiner in nodes, edges and community structures.
| Network A | Network B | Unique node | Unique edge | WA→B | WB→A |
|---|---|---|---|---|---|
| mimMiner | DMN | 582 (25.2%) | 295,975 (79.2%) | 0.392 | 0.533 |
3.4. Case study of Marfan syndrome
We have demonstrated the difference between DMN and mimMiner through network comparison. In this section, we conducted a case study on Marfan syndrome (Ms) to further compare disease relationships in DMN and mimMiner. The direct neighbors of MS in DMN (665 nodes) and mimMiner (363 nodes) have overlaps (241 common neighbors), but are largely different. Fig. 5 shows the top twenty MS neighbors with the highest weights in DMN, mimMiner and OMIM-based HDN (GWAS-based HDN does not contain MS, therefore is not shown). The difference shows that the phenotype networks may contain new leads to discover the unknown causes of MS. The subgraphs of DMN (Fig. 5(a)) and mimMiner (Fig. 5(b)) share six of the twenty nodes. The edges in the DMN subgraph vary greatly in weights, while those in the mimMiner subgraph have almost uniform weights. Both the phenotype networks contain disease relationships that cannot be found in the other, hence are able to complement each other. For example, Lujan-Fryns syndrome (LFS) is among the top neighbors of MS in DMN, but is not connected to MS in mimMiner. Many literatures support the phenotype similarities between LFS and MS, such as tall stature, long limbs, and heart problems [11,41,27,13]. Inspired by these common phenotype features, a few studies looked for new genetic origins of MS and LFS [21,7].
Fig. 5.
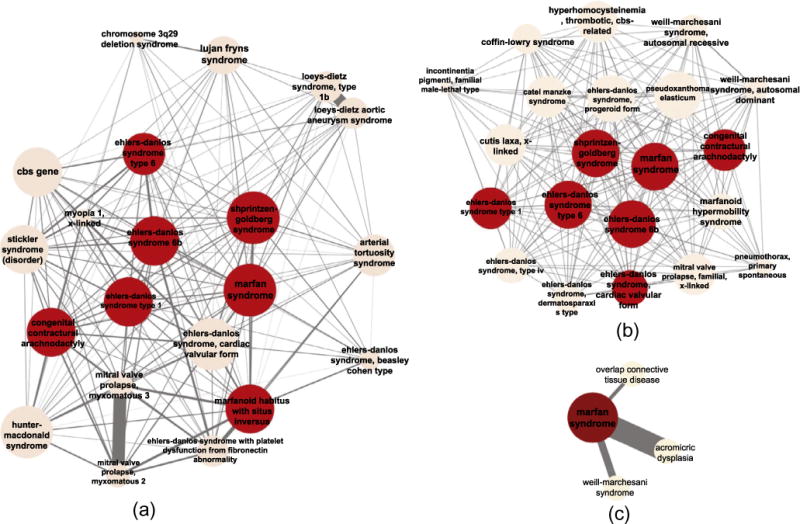
Top 20 nodes directly connected to Marfan syndrome with the highest weights in (a) DMN, (b) mimMiner, and (c) HDN. The common nodes among the three subnetworks are highlighted. The thickness of edges represents the weights.
We manually traced the data sources to explain the different connections between LFS and MS in the two phenotype networks. For DMN, we extracted disease-manifestation associations from UMLS ontologies and found seven common manifestations between LFS and MS, such as “Contracture of joint,” “Congenital funnel chest,” and “Aneurysm of ascending aorta.” For mimMiner, we manually curated both full text and clinical synopsis fields from OMIM disease records for both diseases, but only found three out of the seven common manifestations in UMLS shared by the two diseases. In addition, and text mining approaches introduced false signals when extracting disease-phenotype associations. As a result, the LFS-MS connection has a weight below the threshold in mimMiner.
One disadvantage of mimMiner is that we need to control the false positive disease-phenotype associations introduced by text mining. Practical applications, such as candidate disease gene prediction tasks, chose stringent threshold for disease similarities, which is often higher than 0.3 [19,23]. However, one threshold hardly fits all diseases. Directly removing disease pairs with small similarities may cause the miss of true disease connections such as LFS-MS. The disease-phenotype associations in UMLS were observations rather than the result of text mining, hence do not require the users to control the false positive signals. Therefore, combining DMN with mimMiner may improve the quality of disease phenotype network.
4. Discussion
We have constructed a phenotype network using the clinical manifestation data from the biomedical ontologies, and demonstrated the correlation between the manifestations based phenotype relationships and genetic associations among diseases. We have also compared DMN with another phenotype network that has been widely-used in candidate gene selection. Results show that the two phenotype networks are largely complementary.
Our work has a few limitations and can be improved in future studies. First, the manifestation data in UMLS is highly accurate but is limited in size. Though we have used 50,543 disease-manifestation pairs to construct DMN, the number of nodes in DMN is smaller than that in mimMiner. To increase the coverage of diseases, we need to integrate DMN and mimMiner and obtain a more complete phenotype network. In addition, many nodes in DMN are syndromes and rare diseases. Phenotype data obtained from other sources, such as literature, contains information of more common diseases and can greatly complement DMN. Currently, we are developing approaches to integrate heterogeneous phenotype networks, including DMN, mimMiner, and the network constructed from the disease-manifestation relationships based on literature mining [49].
Second, the comparison in network community indicate differences in the community structures between DMN and HDNs. One possible reason is that many disease associated genes may have not been discovered yet and the community structure of the genetic networks may largely bias towards known knowledge. In the future, we plan to discover new disease-gene associations by analyzing the observed phenotype similarities. Also, since similar manifestations between diseases may be caused by common functional modules or pathways, we will integrate gene functional relationships with the phenotype network in detecting new disease mechanisms.
Third, phenotypic data is high-dimensional, containing not only manifestations, but also other aspects on levels from genes, cells to organisms. Though our network uses highly accurate manifestation data, it can only reflect one aspect of the phenotype associations. In the future, we will integrate the DMN with multiple other kind of phenotype data, and incorporate the comprehensive phenotype network in disease gene discovering methods.
Finally, the scope of this study is to demonstrate the potential of DMN to predict unknown disease mechanisms. Our analysis showed the significant positive correlation between manifestation similarities and genetic overlaps among diseases through comparative analysis. To use DMN in discovering new disease mechanisms, however, we still need to develop systems approaches and exploit other network characteristics, such as network local structures, which are not discussed in this study.
5. Conclusions
Systems approaches in studying disease phenotype networks have great potential in discovering unknown disease mechanisms. Currently, disease phenotype networks remain largely incomplete. Clinical manifestation is an important aspect of the phenotype data. In this study, we built a disease phenotype network, DMN, using the high quality disease-manifestation semantic relationship data from ontologies in the UMLS. Phenotype-genotype correlation analysis based on network comparison have demonstrated that the phenotype relationships in DMN reflects overlapping genetic mechanisms of diseases, but also contains new leads to discover genetic disease causes. Also, we have shown that DMN and a widely-used phenotype network are complementary. With the integration of phenotype data from other sources, our network could strengthen current candidate disease-gene selection methods.
Acknowledgments
YC, GZ and RX are funded by Case Western Reserve University/Cleveland Clinic CTSA Grant (UL1TR000439). XZ are partially supported by US National Science Foundation IIS-1162374 and IIS-1218036. We would like to thank our funding and the reviewers for their invaluable comments and suggestions.
Footnotes
Author’s contributions
RX conceived the study. YC and RX designed the experiment. YC performed the experiment and wrote the manuscript. All authors have participated study discussion and manuscript preparation.
References
- 1.Annunen S, Körkkö J, Czarny M, Warman ML, Brunner HG, Kääriäinen H, et al. Splicing mutations of 54-bp exons in the col11a1 gene cause marshall syndrome, but other mutations cause overlapping marshall/stickler phenotypes. Am J Human Genet. 1999;65(4):974–83. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bell DS, Greenes R, Doubilet P. Proceedings of the annual symposium on computer application in medical care. American Medical Informatics Association; 1992. Form-based clinical input from a structured vocabulary: initial application in ultrasound reporting; p. 789. [PMC free article] [PubMed] [Google Scholar]
- 4.Bodenreider O. The unified medical language system (umls): integrating biomedical terminology. Nucl Acids Res. 2004;32(Suppl. 1):D267–70. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunner HG, van Driel MA. From syndrome families to functional genomics. Nat Rev Genet. 2004;5(7):545–51. doi: 10.1038/nrg1383. [DOI] [PubMed] [Google Scholar]
- 6.Butte AJ, Kohane IS. Creation and implications of a phenome-genome network. Nat Biotechnol. 2006;24(1):55–62. doi: 10.1038/nbt1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callier P, Aral B, Hanna N, Lambert S, Dindy H, Ragon C, et al. Systematic molecular and cytogenetic screening of 100 patients with marfanoid syndromes and intellectual disability. Clin Genet. 2013 doi: 10.1111/cge.12094. [DOI] [PubMed] [Google Scholar]
- 8.Caretta-Cartozo C, De Los Rios P, Piazza F, Liò P. Bottleneck genes and community structure in the cell cycle network of s. pombe. PLoS Comput Biol. 2007;3(6):e103. doi: 10.1371/journal.pcbi.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang H, Gough J. A disease-drug-phenotype matrix inferred by walking on a functional domain network. Mol BioSyst. 2013;9(7):1686–96. doi: 10.1039/c3mb25495j. [DOI] [PubMed] [Google Scholar]
- 10.Fang H, Gough J. A domain-centric solution to functional genomics via dcgo predictor. BMC Bioinform. 2013;14(Suppl. 3):S9. doi: 10.1186/1471-2105-14-S3-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fryns J-P, Buttiens M, Opitz JM, Reynolds JF. X-linked mental retardation with marfanoid habitus. Am J Med Genet. 1987;28(2):267–74. doi: 10.1002/ajmg.1320280202. [DOI] [PubMed] [Google Scholar]
- 12.Goh K-I, Cusick ME, Valle D, Childs B, Vidal M, Barabási A-L. The human disease network. Proc Natl Acad Sci. 2007;104(21):8685–90. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grahame R, Hakim AJ. Arachnodactyly-a key to diagnosing heritable disorders of connective tissue. Nat Rev Rheumatol. 2013;9(6):358–64. doi: 10.1038/nrrheum.2013.24. [DOI] [PubMed] [Google Scholar]
- 14.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online mendelian inheritance in man (omim), a knowledgebase of human genes and genetic disorders. Nucl Acids Res. 2005;33(Suppl. 1):D514–7. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houle D, Govindaraju DR, Omholt S. Phenomics: the next challenge. Nat Rev Genet. 2010;11(12):855–66. doi: 10.1038/nrg2897. [DOI] [PubMed] [Google Scholar]
- 16.Hwang T, Atluri G, Xie M, Dey S, Hong C, Kumar V, et al. Co-clustering phenome–genome for phenotype classification and disease gene discovery. Nucl Acids Res. 2012;40(19):e146–e146. doi: 10.1093/nar/gks615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Köhler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Human Genet. 2008;82(4):949–58. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korbel JO, Doerks T, Jensen LJ, Perez-Iratxeta C, Kaczanowski S, Hooper SD, et al. Systematic association of genes to phenotypes by genome and literature mining. PLoS Biol. 2005;3(5):e134. doi: 10.1371/journal.pbio.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lage K, Karlberg EO, Størling ZM, Olason PI, Pedersen AG, Rigina O, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol. 2007;25(3):309–16. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, Li H, Li J, Rebman E, Achour I, Regan KE, et al. Network models of genome-wide association studies uncover the topological centrality of protein interactions in complex diseases. J Am Med Inform Assoc. 2013;20(4):619–29. doi: 10.1136/amiajnl-2012-001519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerma-Carrillo I, Molina JD, Cuevas-Duran T, Julve-Correcher C, Espejo-Saavedra JM, Andrade-Rosa C, et al. Psychopathology in the lujan–fryns syndrome: report of two patients and review. Am J Med Genet Part A. 2006;140(24):2807–11. doi: 10.1002/ajmg.a.31503. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Lee Y, Chen JL, Rebman E, Li J, Lussier YA. Complex-disease networks of trait-associated single-nucleotide polymorphisms (snps) unveiled by information theory. J Am Med Inform Assoc. 2012;19(2):295–305. doi: 10.1136/amiajnl-2011-000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Patra JC. Genome-wide inferring gene–phenotype relationship by walking on the heterogeneous network. Bioinformatics. 2010;26(9):1219–24. doi: 10.1093/bioinformatics/btq108. [DOI] [PubMed] [Google Scholar]
- 24.Lindberg DA, Humphreys BL, McCray AT. The unified medical language system. Methods Inform Med. 1993;32(4):281–91. doi: 10.1055/s-0038-1634945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCray AT. An upper-level ontology for the biomedical domain. Compar Funct Genom. 2003;4(1):80–4. doi: 10.1002/cfg.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mestres J, Gregori-Puigjané E, Valverde S, Sole RV. Data completeness-the achilles heel of drug-target networks. Nat Biotechnol. 2008;26(9):983–4. doi: 10.1038/nbt0908-983. [DOI] [PubMed] [Google Scholar]
- 27.Murphy-Ryan M, Psychogios A, Lindor NM. Hereditary disorders of connective tissue: a guide to the emerging differential diagnosis. Genet Med. 2010;12(6):344–54. doi: 10.1097/GIM.0b013e3181e074f0. [DOI] [PubMed] [Google Scholar]
- 28.Newman ME, Girvan M. Finding and evaluating community structure in networks. Phys Rev E. 2004;69(2):026113. doi: 10.1103/PhysRevE.69.026113. [DOI] [PubMed] [Google Scholar]
- 29.Oti M, Huynen MA, Brunner HG. Phenome connections. Trends Genet. 2008;24(3):103–6. doi: 10.1016/j.tig.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Oti M, Huynen MA, Brunner HG. The biological coherence of human phenome databases. Am J Human Genet. 2009;85(6):801–8. doi: 10.1016/j.ajhg.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palla G, Derényi I, Farkas I, Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435(7043):814–8. doi: 10.1038/nature03607. [DOI] [PubMed] [Google Scholar]
- 32.Piro RM, Di Cunto F. Computational approaches to disease-gene prediction: rationale, classification and successes. FEBS J. 2012;279(5):678–96. doi: 10.1111/j.1742-4658.2012.08471.x. [DOI] [PubMed] [Google Scholar]
- 33.Piro RM, Molineris I, Di Cunto F, Eils R, König R. Disease-gene discovery by integration of 3d gene expression and transcription factor binding affinities. Bioinformatics. 2013;29(4):468–75. doi: 10.1093/bioinformatics/bts720. [DOI] [PubMed] [Google Scholar]
- 34.Salathé M, Jones JH. Dynamics and control of diseases in networks with community structure. PLoS Comput Biol. 2010;6(4):e1000736. doi: 10.1371/journal.pcbi.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sifrim A, Popovic D, Tranchevent L-C, Ardeshirdavani A, Sakai R, Konings P, et al. Extasy: variant prioritization by genomic data fusion. Nat Methods. 2013;10(11):1083–4. doi: 10.1038/nmeth.2656. [DOI] [PubMed] [Google Scholar]
- 36.Singh-Blom UM, Natarajan N, Tewari A, Woods JO, Dhillon IS, Marcotte EM. Prediction and validation of gene-disease associations using methods inspired by social network analyses. PloS One. 2013;8(5):e58977. doi: 10.1371/journal.pone.0058977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singleton MV, Guthery SL, Voelkerding KV, Chen K, Kennedy B, Margraf RL, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Human Genet. 2014;94(4):599–610. doi: 10.1016/j.ajhg.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiffin N, Andrade-Navarro MA, Perez-Iratxeta C. Linking genes to diseases: it’s all in the data. Genome Med. 2009;1(8):77. doi: 10.1186/gm77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tranchevent L-C, Capdevila FB, Nitsch D, De Moor B, De Causmaecker P, Moreau Y. A guide to web tools to prioritize candidate genes. Brief Bioinform. 2011;12(1):22–32. doi: 10.1093/bib/bbq007. [DOI] [PubMed] [Google Scholar]
- 40.Tringali M, Hole WT, Srinivasan S. Proceedings of the AMIA symposium. American Medical Informatics Association; 2002. Integration of a standard gastrointestinal endoscopy terminology in the umls metathesaurus; p. 801. [PMC free article] [PubMed] [Google Scholar]
- 41.Van Buggenhout G, JP F. Lujan-fryns syndrome (mental retardation, x-linked, marfanoid habitus) Orphanet J Rare Dis. 2006;1(26):325. doi: 10.1186/1750-1172-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Driel MA, Bruggeman J, Vriend G, Brunner HG, Leunissen JA. A text-mining analysis of the human phenome. Eur J Human Genet. 2006;14(5):535–42. doi: 10.1038/sj.ejhg.5201585. [DOI] [PubMed] [Google Scholar]
- 43.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010;6(1):e1000641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Gulbahce N, Yu H. Network-based methods for human disease gene prediction. Brief Funct Genom. 2011;10(5):280–93. doi: 10.1093/bfgp/elr024. [DOI] [PubMed] [Google Scholar]
- 45.Webb GD, Smallhorn JF, Therrien J. Congenital heart disease. In: Bonow RO, Man DL, Zipes DP, editors. Heart disease: a textbook of cardiovascular medicine. 9. Vol. 2. Saunders Elsevier; 2011. pp. 53–76. [Google Scholar]
- 46.Wu X, Jiang R, Zhang MQ, Li S. Network-based global inference of human disease genes. Mol Syst Biol. 2008;4(1) doi: 10.1038/msb.2008.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu X, Liu Q, Jiang R. Align human interactome with phenome to identify causative genes and networks underlying disease families. Bioinformatics. 2009;25(1):98–104. doi: 10.1093/bioinformatics/btn593. [DOI] [PubMed] [Google Scholar]
- 48.Xu J, Li Y. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics. 2006;22(22):2800–5. doi: 10.1093/bioinformatics/btl467. [DOI] [PubMed] [Google Scholar]
- 49.Xu R, Li L, Wang Q. Towards building a disease-phenotype knowledge base: extracting disease-manifestation relationship from literature. Bioinformatics. 2013;29(17):2186–94. doi: 10.1093/bioinformatics/btt359. [DOI] [PMC free article] [PubMed] [Google Scholar]