

Abstract
Advancements in human genomics over the last two decades have shown that cancer is mediated by somatic aberration in the host genome. This discovery has incited enthusiasm among cancer researchers; many now use therapeutic approaches in genetic manipulation to improve cancer regression and find a potential cure for the disease. Such gene therapy includes transferring genetic material into a host cell through viral (or bacterial) and non-viral vectors, immunomodulation of tumor cells or the host immune system, and manipulation of the tumor microenvironment, to reduce tumor vasculature or to increase tumor antigenicity for better recognition by the host immune system. Overall, modest success has been achieved with relatively minimal side effects. Previous approaches to cancer treatment, such as retrovirus integration into the host genome with the risk of mutagenesis and second malignancies, immunogenicity against the virus and/or tumor, and resistance to treatment with disease relapse, have markedly decreased with the new generation of viral and non-viral vectors. Several tumor-specific antibodies and genetically modified immune cells and vaccines have been developed, yet few are presently commercially available, while many others are still ongoing in clinical trials. It is anticipated that gene therapy will play an important role in future cancer therapy as part of a multimodality treatment, in combination with, or following other forms of cancer therapy, such as surgery, radiation and chemotherapy. The type and mode of gene therapy will be determined based on an individual’s genomic constituents, as well as his or her tumor specifics, genetics, and host immune status, to design a multimodality treatment that is unique to each individual’s specific needs.
Keywords: Adenoviruses, Clinical trials, Electroporation, Gene silencing, Gene transfer technique, Immunomodulation, Molecular targeted therapy, Oncolytic viruses, Retroviruses, Suicide transgenes
Introduction
Over the last two decades, cancer research and genomics have experienced considerable advancements. In 2001, two independent draft versions of the human genome sequence and the concomitant identification of approximately 30,000 genes were published [1, 2]. As expected, this was followed by extensive research in molecular genetics with advanced tools used to dissect gene function and explore the biological processes involved in causing genetic aberration and malignant transformation [3]. The recent proliferation of knowledge of cancer has led to the development of novel therapeutic approaches in cancer management, particularly gene therapy.
Gene therapy implies any procedure intended to treat or alleviate a disease by genetically modifying the cell of a patient [4]. The material to be transferred into patient cells may be genes, gene segments, or oligonucleotides. Gene transfer therapy can be conducted either as in vivo or ex vivo approaches. In the in vivo approach, targeted cells are approached directly, such as the intradermal injection of a metastatic nodule, or intravesical therapy for superficial bladder cancer. In the ex vivo approach, targeted cells from a tumor are selected, then collected, grown in culture media at a controlled microenvironment, manipulated genetically by the insertion of a new gene or protein (transgene) in the cell genome, then introduced back into the host. The ex vivo approach is much simpler to achieve as it is easier to manipulate target cells externally.
Concerning cancer, initial efforts to deactivate oncogenes and replace non-functioning tumor suppressor genes were barely successful. Subsequently, new approaches have been developed to transfer genetic materials (transgenes) directly into target cells aiming to transiently or permanently change their phenotypes [5]. Target cells may be normal cells, cancerous cells, immune mediated cells, or pluripotent stem cells. Once the transgene enters a cancer cell, it can then assists in its death or restore normal cellular functions, whereas for normal cells, the transgene can protect them from drug-induced toxicities, or activate an immune cell to get rid of the cancer cell. Gene and vector-based molecular therapies for cancer comprise a wide range of treatment modalities to modify cancer cells, normal cells, and/or a tumor microenvironment [6].
History
The history of cancer therapy dates back to the eighteenth century, when surgery was the primary treatment for early stages of cancer, and patients suffered from frequent relapses [7]. Once the disease spread, patients were treated with herbal medications, castor oil, or arsenic. In 1895, radiation therapy was discovered, but resulted in few cures [8]. At that time, several cases of spontaneous cancer regression following bacterial infection were reported [9]. In 1868 a patient with soft tissue sarcoma went into remission following an erysipelas infection, but this regression lasted for a short duration [9]. In 1943, nitrogen mustard was used in the management of patients with lymphoma [10], and in 1948 folic acid antagonists led to transient remission in childhood leukemia [11]. Since then, there has been a dramatic advancement in chemotherapy treatment for cancer [7]. Viruses were also found to be effective in controlling malignancies in animal models, and subsequently in humans in 1956 [12]. Adenoviruses in particular have been studied more intensively in humans, with the subsequent development of gene therapy [13]. In 1987, immunotherapy was introduced in the management of cancer patients with subsequent FDA approval of rituximab antibodies in the treatment of patients with lymphoma (1997) [14].
The first FDA-approved gene therapy experiment in the United States occurred in 1990 for a patient with severe combined immunodeficiency disorder [15]. Since then, many clinical trials have been conducted for patients with cancer, using different approaches in gene therapy, with successful results reported in patients with chronic lymphocytic leukemia, acute lymphocytic leukemia, brain tumors, as well as others. Several commercially approved medications for gene therapy were released, including ONYX-15 (Onyx Pharmaceuticals) for refractory head and neck cancer (2005) [16]; human papilloma virus vaccine (Gardasil) (Merck Sharp & Dohme) for the prevention of cancer cervix (2006) [17]; and modified dendritic cells, sipuleucel-T (Provenge) (Dendreon Corporation, Seattle, WA), for minimally symptomatic, castration-resistant metastatic prostate cancer (2010) [18].
Methods of gene therapy
Gene therapy implies an approach that aims to modify, delete, or replace abnormal gene(s) at a target cell. Such target cells may be malignant primary or metastatic nodules, circulating tumor cells or dormant stem cells, and specific cells such as T-cell lymphocytes or dendritic cells. With the presence of over 20,000 active genes in human cells, exposed to numerous factors whether hereditary, environmental, infectious or spontaneous, unlimited possibilities for gene mutation, aberration, dysfunction or deletion have been expected, leading to clinical presentation of various medical disorders, including cancer. Furthermore, genomics of cancer evolve between primary and metastases. For example, estrogen receptor gene (ESR J) mutations in breast cancer were found in proportion of metastases but not in primary tumors [19, 20]. Whole-exome sequencing of metastatic samples reported among the top 17 mutated genes, only five were mutated in primary tumors [21]. The evolution from minority clone to lethal metastases follows branched evolution. Thus, tumors with high a level of intratumor heterogenicity and genomic instability could be more likely to escape from targeted therapies such as gene therapy, unless such a branched evolution is taken into consideration. Hence, gene therapy is somewhat difficult to achieve, with limited success. Presently, most approaches are for monogenic gene therapy, tackling one or more critical gene defects. Selection of the appropriate mode of gene therapy is based on the assessment of the immune status, and determination of the molecular nature of a patient’s disease. With the recent increases in knowledge of molecular biology of various medical disorders, a more advanced and comprehensive gene therapy approach will ultimately become available, with anticipated improved results.
Gene transfer delivery system
Several methods have been developed to facilitate the entry of genetic materials (transgenes) into target cells, using various vectors. They are broadly divided into two major categories: viral (or bacterial) and non-viral vectors [Table 1]. Viruses usually bind to target cells and introduce their genetic materials into the host cell as part of their replication process. As they enter target cells, they can carry a load of other genetic material called “transgenes”. For non-viral vectors, different approaches have been utilized, using physical, chemical, as well as other modes of genetic transfer. Transferring genetic material directly into cells is referred to as “transfection”, while moving them into cells carried by a viral or bacterial vector is termed “transduction”. Non-viral approaches have the advantage of safety and easy modifiability, but have a lower transfection efficiency compared to viral vectors [22].
Table 1.
Gene transfer and immunomodulation in cancer therapy
| Predominant action | Examples | Commercially available* | Clinical trials, Phases II,III,IV ** |
|---|---|---|---|
| Gene transfer | |||
| Non-Viral | Electroporation, nanoparticles, hydrodynamics, cationic liposomes, transposon, synthetic viruses | 18,1,0 | |
| Bacterial | Escherichia coli, Salmonella, Clostridium, Listeria, CEQ508 | 6,0,0 | |
| Viruses | |||
| ssDNA viruses | Adeno-Associated: Parvovirus | ||
| dsDNA viruses | Adenoviruses: Ad5-D24, CG870, Ad5-CD/TKrep, Recombinant H103, Gutless adenovirus, OBP-301 | ONYX-015 | 11,3,0 |
| dsDNA viruses | Herpetic viruses: Herpes simplex-1, TVEC | 42,10,0 | |
| ssRNA viruses | Lentiviruses: HIV-1, HIV-2, Simian IV, Feline IV. | 8,2,0 | |
| dsRNA viruses | Reoviruses | 9,1,0 | |
| Immunomodulation | |||
| Active immunotherapy | 41,3,0 | ||
| Single Tumor cell surface antigen vaccine | |||
| Antigen-specific plasmid-based vaccine: PSA, HER/2, Modified CEA vaccine. | |||
| Tumor cells, irradiated as vaccine | |||
| Genetically modified tumor cell vaccine: Using Poxvirus, Vaccinia virus, Recombinant fowlpox virus, Combination (TRICOM) (Prostvac-VF vaccine). | |||
| Passive immunotherapy | 219,29,2 | ||
| Antibodies against: | Rituximab | ||
| CD20 Protein on lymphoma cells | Rituximab | ||
| HER/2 receptor protein in breast cancer | Trastuzumab | ||
| CD52 Protein on CLL | Alemtuzumab | ||
| CD20 Protein on lymphoma cells | Ibritumomab | ||
| CD20 Protein on lymphoma cells | Tositumomab | ||
| EGFR Receptor on squamous CA | Cetuximab | ||
| EGFR Receptor on colorectal CA | Panitumumab | ||
| CD20 Protein on CLL | Ofatumumab | ||
| CD30 Protein on Hodgkin lymphoma cells | Brentuximab | ||
| HER/2 receptor protein in breast cancer | Pertuzumab | ||
| HER/2 receptor protein in breast cancer | Ado-Trastuzumab | ||
| CD20 Protein on CLL | Obinutzumab | ||
| Adoptive immunotherapy | 15,1,0 | ||
| Autologous activated T- lymphocytes | Sipuleucel-T | ||
| Genetically modified activated T-lymphocytes | |||
| Chimeric antigen receptor integrated T-lymphocytes | |||
| Activated dendritic cells | |||
| Genetically modified dendritic cells | |||
| Immune enhancement | 11,1,0 | ||
| Antibodies blocking CTLA-4 Inhibitors for malignant melanoma. | Ipilimumab | ||
| Microenvironment modification | |||
| Impact on vasculature | Humanized monoclonal antibodies against VEGFR-A | Bevacizumab | |
| Anti-angiogenic genes (against VEGFR-A): Endostatin, Angiostatin | 22,4,0 | ||
Abbreviations: CA Cancer; CEA carcinoembryonic antigen; CLL chronic lymphocytic leukemia; ds double stranded; CTLA-4 cytostatic T-lymphocyte antigen 4; DNA deoxy nucleic acid; EGFR epidermal growth factor receptor; FDA Food and Drug Administration in United States; HER/2 human epidermal growth factor receptor-2; HIV human immunodeficiency virus; PSA prostatic acid phosphatase antigen; RNA ribonucleic acid; ss single stranded; VEGF-A vascular endothelial growth factor A receptor.
*Commercially approved medications by FDA US as of July 1, 2014. ONYX-015 was previously approved by FDA China.
**Clinical trials: Number of active clinical trials on gene therapy for cancer (Phases-II, -III, and –IV) as of July 1, 2014 (http://www.clinicaltrials.gov).
Physical mediated gene transfer
DNA genetic material that is coated with nanoparticles from gold or other minerals, and with their kinetic energy supplemented by compressed air or fluid (gene gun), or using ultrasound, can force the genetic material into the target cell, followed by the release of DNA into its nucleus. They are best suited for gene delivery into tissue or in case of gene vaccination [23].
The electroporation gene therapy approach aims to achieve cellular membrane disruption with high-voltage electrical pulses, resulting in the formation of nanopores through which naked DNA, foreign genetic materials, and even chemotherapeutic agents can enter cells [23, 24]. This approach is best suited for plasmid DNA-based gene transfer therapy with the advantage of effectiveness in a vast array of cell types, ease of its administration, lack of genome integration with the risk of malignancy, as well as the low potential for unwanted immunogenicity [22]. Electroporation is presently being tested in several clinical trials, especially on patients with malignant melanoma, prostate cancer, colorectal cancer, and leukemia [22].
Chemical mediated gene transfer
Cationic liposomes are microscopic vesicles of synthetic phospholipids and cholesterol that can enter into cells by endocytosis [25], with the capability of carrying a variety of molecules such as drugs, nucleotides, proteins, plasmids and large genes [23]. Their advantage is selectivity to endothelial cells, a relatively high rate of gene transfer efficiency, a broad application as carriers for many genes, and the lack of severe side effects [26]. When combined with small interfering RNA (siRNA), cationic liposomes may lead to the inhibition of tumor proliferation, inducement of apoptosis, and enhancement of radiosensitivity to tumor cells [27].
Synthetic viruses have been developed to exploit the efficiency of viral vectors and the advantage of liposomes [28]. Once they enter the target cell, DNA is released from the endosome. This method has shown promising results in preclinical studies [29–32]. Transposons can also transport genetic material inside the cell as well as into the nucleus [33].
Bacterial mediated gene transfer
Some bacteria have the capability of specifically targeting tumor cells, leading to RNA interference (RNAi) and gene silencing with blockage of RNA functions, including cellular metabolism and protein synthesis. Examples include Escherichia coli, Salmonella typhimurium, Clostridium, and Listeria [34]. Bacterial vectors can deliver pro-drug-converting enzymes and cytotoxic agents into tumor cells, and can mediate the host immune response. They can be engineered to carry magnetic or fluorescent material to enhance the utility of diagnostic approaches in tumor localization, such as with magnetic resonance imaging (MRI) [35], and even in the development of cancer vaccines [36]. However, the outcome has been far less pronounced compared to other RNA interference silencing techniques. Overall, genetically engineered bacteria acting as vectors for RNA interference are relatively safe, effective, practical and cheaper to manufacture compared to viral vectors. They selectively colonize and grow within the tumor. They can also be administered orally, hence their use in the management of gastrointestinal disorders [34].
Viral mediated gene transfer
Viruses are small particles that contain either ribonucleic acid (RNA) or deoxyribonucleic acid (DNA), and may be single-stranded (ss) or double-stranded (ds). The viral structure consists of a genome surrounded by a protective protein coat (viral capsid) which helps the virus attach to host cell receptors, and prevents viral destruction by cell nuclease enzymes. Some viruses may also have a lipid bilayer envelope derived from the host cell’s membrane, and an outer layer of viral envelope made of glycoprotein. A complete viral particle (virion) by itself is unable to replicate. For propagation, the virus needs to insert its genetic material into a host cell, in order to acquire metabolic and biosynthetic products for viral transcription and replication.
Viruses can be modified genetically to be noninfectious. As they enter the cell, they can carry genetic material for delivery into a target cell’s cytoplasm, and subsequently into the nucleus. In monogenic gene therapy, virus vectors can carry a load of 2–10 kb of relevant genes. In high complex gene therapy, other supporting molecules can also be added, such as immune-stimulatory molecules to the virus’s DNA for subsequent release during viral replication. The advantage of viral vectors in gene therapy is the ease of purification into high titers, and prolonged gene expression with minimal side effects. Retroviruses including lentiviruses can integrate themselves into host cell genome at the nucleus, while adenoviruses and adeno-associated viruses predominantly persist as extrachromosomal episomes [24, 37].
RNA viruses comprise about 70% of all viruses, and vary greatly in genomic structures. They usually have a higher mutation rate with increased adaptation to attack different host cells. Single-stranded RNA viruses may have a viral reverse transcriptase enzyme in their genome, which helps in genetic transcription of the viral genome inside the host nucleus, into double-stranded pro-viral DNA. With viral integrase, the pro-viral DNA then integrates with the host DNA making the subsequent transcription of other parts of the virus possible, in order to give rise to a new retrovirus progeny. The proteins of the mature virion are then rearranged to form the new viral particles. Viral particles subsequently destroy the host cell, and release mature viruses to attack neighboring cells. Double-stranded DNA viruses enter the host cells by endocytosis through interaction between the virus and cell receptor. The virus then enters the nucleus through nuclear pores once they escape cellular endosome [38, 39]. The virus subsequently releases two gene products which bind to the retinoblastoma and p53 tumor suppressor genes, thus allowing viral replication. New viruses then cause cell lysis and the released viruses spread to attack neighboring cells [38].
Adenoviruses
Adenoviruses are double-stranded DNA viruses that usually cause mild respiratory, digestive and ocular infection in humans. In gene therapy, modified versions of adenovirus and adeno-associated viral vectors have been designed. Compared to wild-type, they are more potent in infecting cells, both dividing and non-dividing, replicate exclusively in tumor cells [40], and selectively target certain cellular receptors or molecular defects. They pose a very high transduction efficiency, which may approach 100%, with fewer tendencies for viral shedding and latent infection. They can easily be produced commercially in large quantities, and are capable of carrying pro-drug genes as well as others [41]. However, they have several pitfalls, including the tendency to develop genetic instability of carried genes. Subsequent chromosomal aberration may lead to the development of lymphoproliferative disorders. As nearly half of all humans have been exposed to these viruses during their lifetime, with the generation of neutralizing antibodies, they may lead to high immunogenicity, with shorter duration of adenoviral-mediated transgene expression [42].
Modified oncolytic adenoviruses are presently tested in different clinical trials, especially in patients with astrocytoma of the brain, in combination with radiation and/or temozolomide chemotherapy [43]. ONYX-015 (Onyx Pharmaceuticals) is a modified oncolytic adenovirus that was previously approved by the Chinese Food and Drug Administration (2005) in the management of refractory head and neck cancer in combination with cisplatinum [16]. It is presently being investigated in the management of other solid malignancies. Other oncolytic adenoviruses include Ad5-D24, recombinant H103, Ad5-CD/TKrep, CG7870, KH901, and OBP-301 (Telomelysin). The latest generation of adenoviral vectors is the gutless adenovirus; it has an impressive safety profile, less in vivo immune response and long-term sustained gene expression [24]. Most clinical trials using oncolytic adenoviruses rarely produce dramatic tumor response. However, when combined with other modalities of cancer therapy, good tumor regression has been reported [38, 42].
Adeno-associated virus
This represents small, single-stranded DNA viruses, which do not usually cause infection without co-infection of a helper virus, such as adenovirus, or herpes simplex virus. They have the advantage of broad host range, low level of immune response, and longer gene expression. One example is the Eukaryotic adeno-associated virus, which is a chimeric virus vector containing parvovirus and adenovirus [44]. It is capable of transfecting mitotic and quiescent cells, lacks immunogenicity and pathogenicity in humans, and integrates stably into the host DNA at a predictable location within a chromosome-19 in cell culture, but not in mammalian cells.
Herpes simplex virus
This is a large, enveloped double-stranded DNA virus (150 kb), naturally neurotropic (prefer nerve cells), that infects humans particularly at the oral and genital mucosa, but ultimately spreads to sensory nerves to replicate or become dormant at the sensory ganglions. Viral reactivation may lead to oral or genital ulcerations, skin rashes, or even encephalitis. Up to 80% of the population are seropositive to the virus [45, 46]. With genetic engineering, a modified oncolytic recombinant replication-selective herpes simplex virus has been developed, and has exhibited several advantages: it has broad tropism, potent in causing tumor cell lysis, it is non-integrating in targeting the cell genome (apart from nonessential genes), can evade the host immune system; and in case of toxicities, several effective antiviral therapies are presently available to control viral replication. Another advantage is its viral capability to carry a large load of transgenes, such as a pro-drug-activating gene thymidine kinase enzyme that enhances tumor lysis when ganciclovir medication is subsequently administered intravenously (suicide gene) [45]; therapeutic immunomodulatory transgenes that augment the antitumor immune response (such as talimogene laherparepvec) (OncoVEX GM-CSF) [47]; and antiangiogenic genes to suppress tumor vasculature [48]. Presently, modified oncolytic herpes simplex viruses such as Talimogene laherparepvec (TVEC) as well as others, are being tested in several clinical trials either as a monotherapy, or in association with surgery, radiation therapy or chemotherapy, particularly on patients with high-grade glioma. Currently, some success has been reported [45].
Lentivirus vector
Lentiviruses are retroviruses that infect bovine, equine, nonhuman primates and humans [49]. One of the most destructive human pathogens is human immunodeficiency virus infection (HIV). It constitutes a class of enveloped viruses that contain a single-stranded 9.2 kb RNA genome. The lentivirus carries a reverse transcriptase enzyme that transcribes RNA into double-stranded DNA once it enters the cytoplasm. It then integrates permanently into the nuclear genome of the target cells. Examples include lentiviral vectors derived from immunodeficiency viruses such as HIV-1, HIV-2. With genetic engineering, researchers have removed the infectious parts of the virus and added other parts from different viruses such as cytomegalovirus, generating a highly modified lentivirus [50]. Another genetic modification employed by previous researches created an integration-deficient lentiviruses that did not integrate into a host genome [51, 52], though with slightly lower transduction efficiency. Such a modified lentivirus has the advantage of being relatively safe, of having variable specificity to either a particular cell or is broad enough to infect all cells, and of having efficient transduction of both dividing and non-dividing cells. Modified viruses have low antivirus immunity, low potential for genotoxicity due to insertional mutagenesis, and the capability of carrying genes inside the nucleus [49]. Major disadvantages include inadequate immune responses as well as antitumor response, the risk of viral transformation into pathogenic HIV infection, especially in immunized individuals, and insertional mutagenesis of new cancer genes into the host genome, with the risk of second malignancy [49].
Reovirus
This is an oncolytic virus that usually infects animals. In humans, it rarely causes major illness except for respiratory and gastrointestinal symptoms. Nearly 100% of human adults are seropositive to the virus [53]. It is non-enveloped, double-stranded RNA (dsRNA), and its oncolytic activities are mainly through stimulation of the immune system, particularly through bystander immune activation. The release of tumor-associated antigens following cellular lysis further stimulates innate immunity against tumor cells. The virus is considered relatively benign, with good safety records, does not require genetic alterations to become an oncolytic virus, and is less expensive to be produced commercially. Because of its relative safety, the virus is presently being used in several clinical trials, as oncolytic reovirus monotherapy, administered intratumoral, intravenously, or intraperitoneally; or as polytherapy, in combination with radiation therapy or chemotherapy [53].
Gene therapy implementation
Once genetic materials are transferred into target cells and incorporated into nuclear genetic DNA, they may induce silencing, down-regulation, modification, or repair of the target cell genes. Depending on the intensity of the gene expression, it may lead to cell death and tumor necrosis (as with the suicide gene), or impaired cell growth with tumor regression (as with the silencing gene). Modification of the gene may improve the response from subsequent cancer therapy, such as chemotherapy, immunotherapy, or radiation. Repair of the target gene may help in preventing subsequent malignancy or cancer-related complications such as thrombosis. They may also be helpful in the future by preventing hereditary cancer syndromes.
Suicide gene
These are transgenes that make up products that can cause a cell to kill itself through apoptosis. Such gene products are usually transcribed by various factors (promoters) leading to cell death and necrosis. One example of such promoters is the human H19 RNA gene which is highly expressed in most fetal organs but rapidly cleared immediately after birth [54]. This gene has shown an abnormal expression in various types of cancer cells, and plays an important role in cancer cell proliferation, genetic instability, vascular angiogenesis, tumor metastases, multidrug resistance as well as cell survival despite hypoxia, with secondary tumor progression and dissemination [55]. Blocking H19 gene function leads to marked tumor regression, cellular death and necrosis. Another important promoter is human telomerase reverse transcriptase (hTERT), which is a critical factor for cell immortalization and tumorigenesis [56]. Its blockage with agents such as OBP-301 (Telomelysin) (Oncolys BioPharma) leads to cell necrosis and tumor regression (Figure 1). Other approaches to induce tumor cell death include the use of small-molecule drugs, monoclonal antibodies, and toxin gene therapy with agents such as Corynebacterium diphtheriae toxin-A chain (DTA-H19 therapy) [57].
Figure 1.
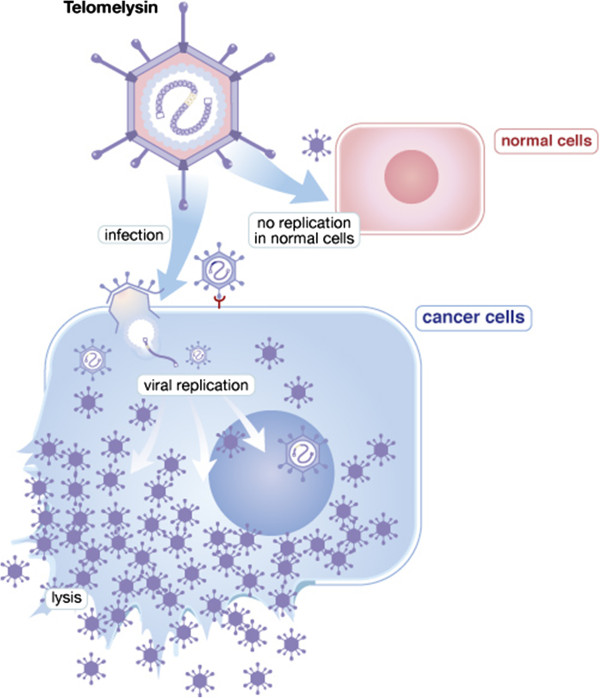
Genetically-modified adenovirus acting as a suicide gene. The above mode of action represents an example of a modified virus acting as a suicide gene, namely OBP-301 (Telomelysin) (Courtesy Oncolys BioPharma Company, Tokyo, Japan).
Gene silencing
This has been achieved through specific delivery of a small interfering double-stranded RNA (siRNA) into target cells, and subsequent duplex formation of RNA-induced silencing complex (RISC) that destroys messenger-RNA (mRNA), thus leading to interference with RNA functions and protein synthesis within the target cells [58]. Through the appropriate design of siRNA, it is theoretically possible to use the technology in silencing any gene in the body, providing a greater therapeutic potential in cancer therapy [59], as well as in the management of other medical disorders such as the hepatitis B virus, human papilloma virus, hypercholesterolemia and liver cirrhosis [59, 60]. As siRNA does not interact with chromosomal DNA, it does have a lower risk of inducing target cell gene alterations and possible mutagenesis. It is highly specific against target genes, with low systemic toxicities, and does not induce multidrug resistance. Furthermore, these genes can induce potent gene silencing of many cancer-related genes, leading to tumor regression, but do not abolish abnormal genes. siRNA therapy can be administered directly into tumors; however, for systemic administration, it is somewhat difficult as a naked siRNA protein is liable for host-mediated clearance by enzymatic degradation, renal filtration, and host cellular phagocytosis. Several delivery systems for siRNA have been developed to protect them from enzymatic degradation, and facilitate their effect in silencing specific genes. Examples of siRNA systemic delivery system presently in clinical trials include CALAA-01 (Calando Pharmaceuticals) for patients with malignant melanoma [61], and ALN-VSPOI (Alnylam Pharmaceuticals) for liver cancer and solid tumors [62]. However, limited success has been achieved mainly due to relatively high toxicity and low transfection efficiency [58, 59].
Gene modification
This may be helpful in improving cancer therapy results, such as with radiation therapy. Radiosensitizing gene therapy promotes transgene expression in tumor tissue, thus increasing tumor sensitivity to radiation with better tumor control [63–65]. In contrast, radioprotective gene therapy distributes transgenes and their products to surrounding normal tissue, thus limiting radiation induced toxicities to normal tissue [66]. The concept of combining both approaches is presently being investigated in several preclinical studies.
Gene repair
This can be achieved using zinc finger nuclease attached to the lentiviral vector. Once the viral vector enters the nucleus, it binds to a specific location in the double-stranded DNA, breaking it at specific location, with subsequent endogenous repair mechanisms, to create a newly edited double-stranded DNA [23]. Other technological approaches include transcription activator-like effector nucleases (TALENs) [67, 68], and clustered regularly interspaced short palindromic repeats (CRISPR) [69].
Gene therapy for mitochondria
Gene therapy may also be directed to cytoplasmic organelles such as mitochondria. The mammalian mitochondria are responsible for metabolic functions. Nearly 300 of the known mutations causing metabolic diseases are secondary to disorders affecting the mitochondrial genome [23]. Several approaches have been used to transfer genes successfully into cell mitochondria.
Immunomodulation
It has become apparent that the immune system is a crucial element in cancer regression or progression. There are two types of immune responses: humoral immunity and cellular immunity. Furthermore, the tumor microenvironment plays an important role in host immune effects against cancer cells [Table 1].
Humoral immunity is mediated by antibodies released by B-cells with a high-binding affinity to specific tumor antigens. The Food and Drug Administration in the United States (FDA) approved several antibodies against malignant cells, which include trastuzumab for breast cancer [70], rituximab for indolent lymphoma [71], cetuximab for lung cancer [72], and bevacizumab for various solid tumors [49, 73], and many others [74] [Table 2].
Table 2.
Monoclonal antibodies in cancer management
| Name | Class | Target | Approved initial indications | FDA Approved |
|---|---|---|---|---|
| Rituximab (Rituxan) | Chimeric IgG1 | CD20 | Non-Hodgkin lymphoma | 1997 |
| Trastuzumab (Herceptin) | Humanized IgG1 | HER2 | Breast cancer | 1998 |
| Alemtuzumab (Campath) | Humanized IgG1 | CD52 | B-cell CLL | 2001 |
| Ibritumomab tiuxetan (Zevalin) | Murine IgG1 | CD20 | Non-Hodgkin lymphoma | 2002 |
| Tositumomab (Bexxar) | Murine IgG2a | CD20 | Non-Hodgkin lymphoma | 2003 |
| Cetuximab (Erbitux) | Chimeric IgG1 | EGFR | Squamous cancer head & neck | 2004 |
| Bevacizumab (Avastin) | Humanized IgG1 | VEGF | Colorectal cancer | 2004 |
| Panitumumab (Vectibix) | Humanized IgG2 | EGFR | Colorectal cancer | 2006 |
| Ofatumumab (Arzerra) | Humanized IgG1 | CD20 | CLL | 2009 |
| Denosumab (Xgeva) | Humanized IgG2 | RANKL | Bone metastases | 2010 |
| Ipilimumab (Yervoy) | Humanized IgG1 | CTLA-4 | Metastatic melanoma | 2011 |
| Brentuximab vedotin (Adcetris) | Chimeric IgG1 | CD30 | Hodgkin lymphoma | 2011 |
| Pertuzumab (Perjeta) | Humanized IgG1 | HER2 | Breast cancer | 2012 |
| Trastuzumab emtansine (Kadcyla) | Humanized IgG1 | HER2 | Breast cancer | 2013 |
| Obinutzumab (Gazyva) | Humanized IgG1 | CD20 | B-cell CLL | 2013 |
| Name | Class | Target | Present indications | Clinical Trials** |
| Amatuximab | Chimeric IgG1 | mesothelin | mesothelioma | Phase-I |
| Elotuzumab | Humanized IgG1 | CS1 | Multiple myeloma | Phase-III |
| Farletuzumab | Humanized IgG1 | FRA | Ovarian and lung cancers | Phase-III |
| Inotuzumab ozogamicin | Humanized IgG4 | CD22 | ALL, Malignant lymphoma | D/C |
| Moxetumomab pasudotox | Murine Fv-CD22 | CD22 | Hairy cell Leukemia | Phase-III |
| Naptumomab estafenatox | Murine Fab | 5 T4 | Renal and solid malignancies | Phase-II |
| Necitumumab | Humanized IgG1 | EGFR | NSCL (Squamous cell) | Phase-III |
| Nivolumab | Humanized IgG4 | PDI | NSCL, Melanoma, Renal | Phase-III |
| Ontuximab | Humanized IgG1 | TEM1 | Solid tumors | Phase-I/II |
| Onartuzumab | Humanized IgG1 | c-Met | NSCL, Gastric | D/C |
| Racotumomab vaccine (Vaxira) | Murine | GM3 | NSCL | Phase-III |
| Rilotumumab | Humanized IgG2 | HGF/SF | Gastric, GEJ | Phase-III |
Abbreviations: 5 T4 Antigen expressed on several solid tumors; ALL acute lymphocytic leukemia; c-MET MNNG HOS proto-oncogene that encodes hepatocyte growth factor receptor; CLL chronic lymphocytic leukemia; CTLA-4 cytotoxic T lymphocyte inhibitors mediated by malignant cells; CS1 human CS1 antigen glycoprotein belonging to CD2 subset of the immunoglobulin superfamily; D/C clinical trials that were discontinued due to the lack of efficacy or excessive toxicities; EGFR, epidermal growth factor receptor; FDA Food and Drug Administration in United States; FRA folate receptor alpha; GM3 tumor antigen N-glycolil, a type of ganglioside present on the surface of breast and lung cancer cells; HGF human hepatocyte growth factor receptors; Mesothelin mesothelin is cell surface glycoprotein overexpressed in multiple malignancies such as mesothelioma; NSCL non-small cell lung cancer; PD-1 human cell surface receptor programed death-1, results in activation of T cell mediated immune responses; RANKL RANK ligand protein that acts as the primary signal for bone removal, loss, or destruction; TEM1 tumor endothelial Marker-1 and CD248 (Morphotek Inc, Exton, PA); VEGF vascular endothelial growth factor receptor.
**Active clinical trials on monoclonal antibodies in cancer management as of July 1st, 2014 (http://www.clinicaltrials.gov).
Cellular immunity is mediated by cell-to-cell contact that leads to antigen recognition and cell destruction of a target cell. Based on the intensity of tumor-associated antigens (TAAs) on the surface of tumor cells, they are recognized by the host immune system [75]. Dendritic cells are specialized in antigen recognition as well as mediation of immune responses against infectious agents or malignant cells, through direct stimulation or inhibition of immune effector cells such as T-cells, B-cells, and natural killer (NK) cells [76]. Dendritic cells are derived from the bone marrow and migrate to lymph nodes and distant tissue, looking for those foreign antigens [49].
Cancer cells can evade the immune system by secreting immunosuppressive cytokines that can downregulate major histocompatibility molecules, can recruit regulatory T-cells, and can kill reactive cytotoxic lymphocytes. Thus, the tumor microenvironment is highly immunosuppressive, which allows a tumor to grow and metastasize [77]. Several efforts have been undertaken to manipulate the tumor microenvironment in order to induce tumor regression.
Innate immunity
Most tumor cells express antigens that can mediate antitumor immune responses [78]. Earlier studies on antigens for therapeutic targeting were based on shared antigens that are expressed on self-tissue or peripheral cells, which can lead to immunologic tolerance for the interaction between antigen peptide, major histocompatibility complex (MHC), and T-cell antigen receptor (TCR). Generated immunologic responses were restricted with low therapeutic efficacy [78]. Recently, it has been found that neoantigens generated by point mutation in normal genes, which are unique to particular tumors, can result in much more potent antitumor T-cell response. Some cancers display hundreds or even thousands of mutations in coding exons, representing a large resource of potential targets for recognition by the immune system. However, despite such a plethora of antigens, most cancers progress and evade immune-system mediated destruction [78].
Antigens recognition by dendritic cells induce a T-cell inflamed reaction consisting of infiltrating T-cells, a broad chemokine profile, and type I interferon signature indicative of innate immune activation. Presence of excessive infiltration by CD8+ cells both within the tumor and in the peritumoral stroma (high immunoscore) had a favorable prognostic significance with improved survival, even in advanced cancer stages, compared to tumors with poor or no T-cell infiltration (low immunoscore) at an earlier stage of malignancy [79–81]. Hence, a heavy presence of activated CD8+ T-cells reflects good innate immune responses against such malignancy. Recent investigations have suggested two explanations for tumor escape recognition by host immune system, based primarily on cellular and molecular characteristics of the tumor microenvironment [78].
One explanation is that tumors resist immune attacks through inhibitory effects mediated by immune system suppressive pathways. This was evident in some tumors such as melanoma with high expression of PD-LI and indoleamine-2,3-dioxygenase (IDO) [82], leading to T-cell anergy and dysfunction with subsequent immune escape detection [83]. The presence of transcription factor forkhead box 03 proteins (Fox3) in the peritumoral microenvironment leads to the inhibition of tumor-infiltrating dendritic cell stimulatory functions [84]. The US FDA’s approval in 2011 of the anti-CTLA-4 monoclonal antibody ipilimumab for the treatment of patients with advanced malignant melanoma [85] represents the first-in-class strategy of uncoupling inhibitory pathways for initial antigen recognition by the host immune system [78] [Figures 2 and 3].
Figure 2.
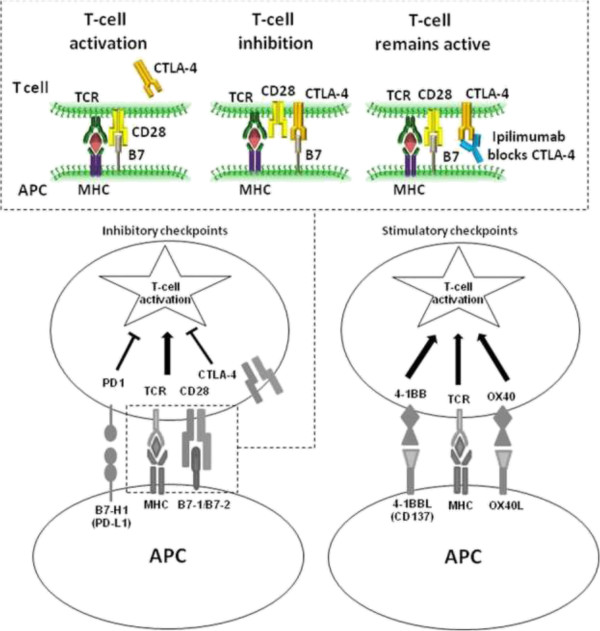
Mechanism of action of monoclonal antibody ipilimumab. Generation of an immune signal requires presentation of tumor antigen by major histocompatibility complex (MHC) class I or II molecules, on an antigen presenting cell (APC) such as dendritic cell. However, T-cell activation and proliferation requires a second signal, typically generated by CD28 antigen. When CD28 antigen on T-cell surface simultaneously binds to costimulatory B7-1/B7- ligand on the antigen presenting cell (APC), T-cell upregulate and translocate CTLA-4 receptor molecules to the surface, which binds B7 with a higher avidity than CD28, leading to suppressor effects, with T-cell inhibition, reduction of interleukin-2 (IL-2) secretion, and prevention of immune response against malignant cell. Ipilimumab blocks cytotoxic T-lymphocyte antigen-4 (CTLA-4) receptor, thus prevents such inhibitory effect, and allows T-cell to proliferate and mediate an immune reaction against malignant cells. Other regulatory checkpoints with the potential for modulation include the coinhibitory molecule PD-1, as well as costimulatory molecules such as OX40 and 4-1BB. Abbreviations: APC, antigen presenting cells; CTLA-4, cytotoxic T-lymphocyte antigen-4; MHC, major histocompatibility antigen; TCR, T-cell receptor. (Courtesy of Annals of New York Academy of Science and Wiley, Hoboken, New Jersey, Publisher) [86].
Figure 3.
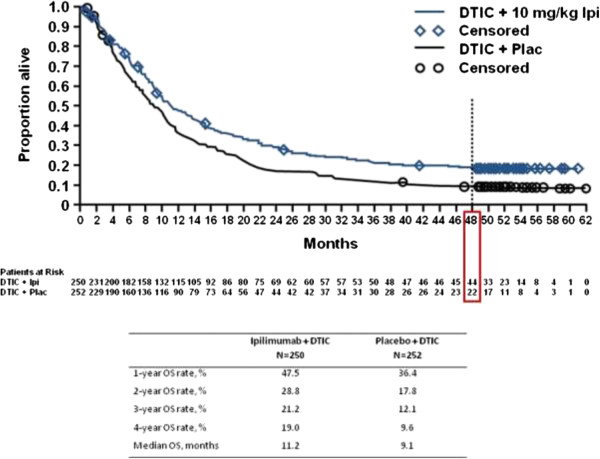
Analysis of overall survival comparing monoclonal antibody ipilimumab plus dacarbazine to placebo plus dacarbazine in metastatic melanoma patients. Kaplan–Meier analysis of overall survival in the phase III study CA184-024. Survival analysis of overall survival in treatment-naive patients with advanced melanoma who received ipilimumab at 10 mg/kg plus DTIC or placebo plus DTIC in the phase III trial, CA184-024. The survival curves reach a plateau beginning at approximately three years after initiation of treatment. Continued survival follow-up of more than four years demonstrates a long-term survival benefit that is consistent with the results of other ipilimumab studies. Abbreviations: DTIC, dacarbazine; Ipi, ipilimumab, Plac, placebo (Courtesy of Annals of New York Academy of Science and Wiley, Hoboken, New Jersey, Publisher) [86].
The other mechanism is immune system exclusion or ignorance with subsequent poor or no T-cell inflammatory reaction. Such tumors appear to lack a type I interferon signature and/or chemokines for recruitment of T-cells. Microenvironment vasculature may be nonpermissive for entry by T-cells, and the stromal element may prevent trafficking and/or function of T-cells. Radiations of tumors have shown to induce productions of interferon-beta and augment function of intratumoral dendritic cells with improved accumulation of T-cells leading to tumor regression [87]. Imatinib in gastrointestinal stromal cell tumors may cause down-modulation of IDO with improved antitumor response [88]. In patients with malignant melanoma, inhibition of R-Raf enzyme activity with vemurafenib can induce a T-cell infiltration within 1–2 weeks of therapy with some tumor responses [89]. It has been suggested that combination regimens consisting of strategies to improve innate immune system activation, T-cell trafficking in the tumor microenvironment, vaccination or adoptive T-cell transfer, and blockage of immune inhibitory pathways may be necessary to achieve clinical benefit in patients with a non-inflamed tumor phenotype. Such an approach is presently being tested in clinical trials [90, 91].
Immunomodulatory approaches in cancer therapy
Immunotherapy in cancer can be classified into four major categories [92]. Active immunotherapy includes strategies that directly sensitize the host immune system to tumor-specific antigens, exemplified as cancer vaccines. Passive immunotherapy utilizes humanized or chimeric antibodies to specifically target tumor antigens without direct activation of the immune system. Adoptive immunotherapy utilizes patients’ immune cells, whether T-cells or dendritic cells, stimulated or manipulated ex vivo, then infused back, to better react against tumor antigens. Immune enhancement therapy aims to augment co-stimulatory molecules or block inhibitory molecules. Immune-based therapy may include one or more of the above approaches, either as distinct immunotherapy treatment, or in combination with other modalities of cancer therapy [Table 1].
Autologous stimulated T-lymphocytes
Adoptive T-cell therapy has been shown to induce tumor regression in some patients with solid malignancies. In a recent study on patients with human papilloma virus (HPV)-induced metastatic cervical cancer who failed to respond to chemotherapy and radiation, and were selected for HPV-E6 and HPV-E7 reactivity, researchers collected tumor-infiltrating T-cell lymphocytes (TIL), and infused them back to patients. This was preceded by a non-myeloablative conditioning regimen and followed by a high-dose of bolus aldesleukin (interleukin-2). Three out of six patients with HPV reactivity achieved objective tumor responses, including two patients with metastatic disease that achieved complete tumor regression for 18 and 11 months after therapy. Side effects were minimal [93].
Autologous activated T-lymphocytes
Host T-cell lymphocytes have been found to be successful in controlling metastatic cancer with transient side effects. The first commercially available vaccine was modified dendritic cells, sipuleucel-T (Provenge) (Dendron Corporation), which was approved by the FDA for minimally symptomatic castration-resistant metastatic prostate cancer. CD54 T-cell lymphocytes were obtained from the patients using density gradient centrifugation, and then activated ex vivo with a prostatic specific antigen in addition to granulocyte macrophage colony stimulating factor (GM-CSF) to form sipuleucel-T. Autologous activated T-cell lymphocytes, at a dose of at least fifty million CD54 cells were infused back to the patient, intravenously over 60 minutes, every two weeks for three infusions. Premedications included acetaminophen and diphenhydramine. Side effects included transient fever, chills, fatigue, asthenia, backaches, and headaches. However, infusion-induced hypersensitivity reactions with cerebrovascular events have been reported in 3.5% of patients. Compared to a control group treated with a placebo, there were significant improvements in the survival of 20.5% versus 16.1% at four years [94].
Genetically modified activated T-lymphocytes
The adoptive transfer of lymphokine-activated lymphocytes can mediate the cellular immune response against cancer cells, which may lead to tumor regression. However, clinical trials have led to limited success. An alternative approach is to use genetically modified T-cells by altering their receptor for better recognition of tumor antigens. In such an approach, T-cells are collected from patient apheresis using density gradient centrifugation. As resting T-cell lymphocytes are non-dividing, refractory to gene therapy with lentiviral vectors, they need to be stimulated using cytokines such as interleukin-2. T-cells are then exposed to lentiviral vectors with the attached gene for 1–2 days of gene transfer. After transduction by the lentivirus, cells are then stimulated further to obtain a therapeutically effective number of cells. Genetically modified T-cell lymphocytes are then re-infused back into the patient [95]. The high-affinity of modified T-cells in detecting very low levels of tumor antigens is an extremely potent approach against tumor cells. However, it may also destroy normal cells. In another study using T-cell receptor gene-modified cells against melanoma differentiated antigens led to higher responses in patients with malignant melanoma [96]. It also destroyed normal melanocytes leading to vitiligo (skin depigmentation), uveitis, and hearing impairment [97].
Chimeric antigen receptor integrated into T-lymphocytes
Elimination of malignant cells through host immune system depends largely on α β T-cell receptor that specifically recognize a cell target in the context of the major histocompatibility complex (MHC). Unfortunately, for many B-lineage leukemias and lymphomas, the resident immune system of patients remains incapable of controlling tumor growth, since autologous T-cells lack expression of the required receptors and tumor cells have adapted to evade immunological recognition [98]. It has been demonstrated that a chimeric antigen receptor (CAR) integrated into T-cells from patient (or even from healthy individuals), can directly recognize the CD19 molecule expressed on the cell surface of B-cell malignancies independent of major histocompatibility complex [99]. Recently, CD19-specific chimeric antigen receptor redirected T-lymphocytes have been utilized as gene therapy for patients with B-cell malignancies. One approach is to use a microelectroporator to achieve high throughput non-viral gene transfer of naked DNA plasmid, of in vitro transcribed CAR mRNA into human T cells that had been numerically expanded ex vivo using interleukin-2. After electroporation, a procedure that usually takes about 10 minutes, up to 80% of the passaged T-cells expressed the CD19-specific CAR, with redirected effector function of the genetically manipulated T-cells to specifically lyse CD19+ tumor cells. Preserved T-cells can then be re-infused into patient as an effector form of adoptive immunotherapy [98] [Figure 4]. Similar approaches have been used against other B-lineage restricted antigens such as CD20 in lymphoma, the light chain of human immunoglobulins, or CD30 expressed by Reed-Sternberg Cells in Hodgkin lymphoma [100]. Adding costimulatory endodomain within the chimeric receptors such as CD28, 4-1BB, or their combination, usually leads to enhancement of T-cell functions through the release of interleukin-2, interleukin-7 or interleukin-15 cytokines [100]. Excellent results in patients with B-cell malignancies have been reported [101–103]. CAR-modified allogeneic T-cells, such as those obtained from healthy individuals, have the potential to act as universal effector cells, which can be administered to any patient regardless of MHC type. Such universal effector cells could be used as an 'off-the-shelf' cell-mediated treatment for cancer [104, 105].
Figure 4.
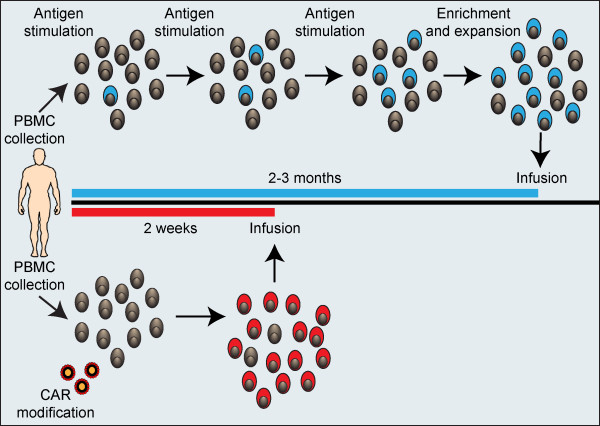
Chimeric antigen receptor modified T-lymphocyte therapy for B-cell malignancies. Generation of tumor-specific T cells by repeated antigen stimulation or genetic modification to express a tumor-targeting receptor. PBMC collected from a patient or healthy individual can be stimulated in vitro with tumor antigen at regular intervals to induce gradual enrichment of antigen-specific T cells (blue). Multiple stimulations followed by additional enrichment or expansion strategies are required to ensure sufficient antigen-specific T cells are generated. The entire process may take 2–3 months. In contrast, approaches that utilize genetic modification to redirect T cell specificity to a tumor antigen are much more rapid. PBMC can be collected from a patient or healthy donor and retrovirally or lentivirally transduced to express a tumor-reactive CAR (or TCR). The enriched CAR-modified tumor-reactive T cells (red) can be infused into the patient in as little as 1–2 weeks. Abbreviations: PBMC: Peripheral blood mononuclear cells, CAR: Chimeric antigen receptor modified T-lymphocytes. (Courtesy of The International Journal of Hematology, and Springer-Tokyo, Publisher) [102].
Genetically modified dendritic cells
Dendritic cells are the most powerful antigen-presenting cells (APCs) for antigen identification, T-cell costimulation and cytokine production by T lymphocytes [106]. Dendritic cells can be generated from monocytes or a CD34+ cell precursor [107], and can be harvested from peripheral blood using density gradient centrifugation. When cultured in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) plus interleukin-4, monocytes develop into immature dendritic cells in 3–5 days. Cells are then exposed to a variety of different stimuli over another 2 days in culture media to become mature dendritic cells, which are more effective in stimulating T-cells [108]. To target a specific tumor, mature dendritic cells are incubated with certain peptides, proteins, or irradiated tumor cells. An alternative approach is genetic modification of dendritic cells through viral and non-viral gene transfer vectors, resulting in dendritic cells that are more potent in antitumor immunity. Dendritic cell vaccines have a favorable safety profile, with toxicities limited to a local inflammatory reaction, flu-like symptoms, and vitiligo-like skin changes [109]. Despite numerous clinical trials, clinical outcomes have been modest. Emphasis has been shifted in using therapy on patients with a lower tumor burden, such as those after surgery, or following successful chemotherapy or radiation therapy. Indeed, significant synergy has been observed for chemotherapy and dendritic cell vaccines [107].
Genetically modified tumor cell vaccine
Previous attempts to use tumor cells or their products as a vaccine have not been successful. Subsequently, clinical trials have been conducted using methods to increase tumor antigenicity in order to enhance the immune-mediated tumor lysis by T-cells. One approach is to obtain tumor cells, infect them with a viral vector such as recombinant poxvirus that contains multiple costimulatory molecules to enhance the immunogenicity of tumor cells, and subsequently use those modified tumor cells as a vaccine. An alternative approach is to directly administer the poxviral vector into the tumor. Such an approach enhances tumor antigenicity and subsequent antigen-specific T-cell response, leading to an antitumor effect. Other viral vectors include vaccinia virus and recombinant nonreplicating fowlpox virus encoding GM-CSF (TRICOM) [110]. This has led to the development of the Prostvac-VF vaccine (Bavarian Nordic, Inc.) which employs a recombinant vaccinia vector as a primary vaccination, followed by multiple booster vaccinations employing a recombinant fowlpox vector. Both vectors contain the transgenes for prostate-specific antigen (PSA) and multiple T-cell costimulatory molecules (TRICOM). In a randomized clinical study, using the vaccine versus placebo, in 125 patients with metastatic castration resistant prostate cancer, there was an improved survival at 3 years of 30% for the vaccinated group compared to 17% for the placebo patients [18]. A Phase-III clinical trial is presently in progress.
Single-antigen plasmid-based vaccine
DNA plasmids containing a genetic sequence that encodes a desired antigen with other transcriptional elements have been considered as a mode of cancer vaccine. It readily accesses the nucleus of a transfected cell, transcribed into a peptide or protein, and may lead to cellular and humoral immune response [111]. The technique is considered to be relatively safe compared to viral or bacterial vectors, does not cause infection or autoimmune disorders, and is easy to develop and produce commercially [112]. However, its effectiveness wanes with time. Hence, the need for frequent booster immunizations. Examples of single-antigen plasmid-based vaccines include human prostatic acid phosphatase protein for patients with prostate cancer [113], human epidermal growth factor receptor-2 (HER-2/neu), protooncogene with low-doses of GM-CSF intradermally for patients with metastatic breast cancer [114], and modified carcinoembryonic antigen (CEA) gene fused to a promiscuous tetanus toxoid for colorectal cancer [115]. Although therapy was well tolerated, responses were minimal and transient. Using a multiple-antigens plasmid-based vaccine leads to broadly specific, long lasting, and multifunctional immune stimulation [116]. Improved results were noticed [117, 118].
Genetically modified microenvironment
The microenvironment around a tumor plays an important role in tumor progression and metastases. It includes stromal tissue, fibroblasts, and vascular endothelial cells. Interfering with such a microenvironment will lead to tumor regression. The most important target is angiogenesis, which is essential for tumor growth and metastases. It is mediated by tumor-derived pro-angiogenic cytokines, such as the vascular endothelial growth factor and fibroblast growth factor. These factors stimulate the proliferation of microvasculature around a tumor, with subsequent tumor progression and metastases. Compared to the recombinant antivascular endothelial factor antibody “bevacizumab”, gene therapy represents an attractive alternative to such drug therapy. Using an anti-angiogenic genes, such as angiostatin and endostatin, delivered by an adeno-associated virus vector, has led to tumor regression with minimal side effects [24].
Other gene therapy approaches in cancer management
As with other modes of cancer therapies, multimodality treatment frequently yields, better results compared to monotherapy. This is similarly true for gene therapy, and is evident when gene therapy is administered after maximum tumor load reduction following radical surgery or successful chemotherapy. Gene therapy has a synergistic effect when combined with chemotherapy, with higher tumor responses and lower therapy-related toxicities.
Gene directed enzyme prodrug therapy (GDEPT)
This is a new strategy in cancer management that aims to reduce the side effects of chemotherapy. With such an approach, a gene that expresses a nontoxic enzyme into cancer cells is first delivered to the cells, followed by the systemic administration of a pro-drug that can be converted into a toxic compound by the enzyme, leading to selective tumor cell death, with lower adverse effects on normal tissues [119]. Cell-to-cell diffusion of toxic metabolites may damage nearby and adjacent tumor cells (bystander effect) [120]. Release of tumor cell necrotic material in the circulation may activate the immune system in response to the tumor antigen, with subsequent regression of distant tumor cells, such as metastatic nodules (distant bystander effect) [121]. Examples include the use of a retroviral vector, such as suicide gene therapy and herpes simplex virus carrying the thymidine kinase enzyme, to the interior of tumor cells. The enzyme has a 1000-fold greater efficiency to selectively phosphorylate the acyclovir-derived pro-drug ganciclovir [120]. Following the systemic administration of ganciclovir, the drug is metabolized in tumor cells leading to cell death. As the efficacy of such a system is only about 10% of tumor cells, the extent of tumor regression is mainly mediated via bystander effects. The system has been tried in several clinical trials [122]. Replacing ganciclovir with a penciclovir drug, modified to generate radiolabeled analog, will also allow a closer follow-up of therapy results, using high-quality positron emission tomography imaging studies [123].
Cancer drug-resistance gene transfer
Several studies have used a gene transfer approach that aims to enhance chemotherapy and radiation effects against cancer cells, while protecting normal tissue against therapy mediated toxicities. Such gene transfer may also be used in the protection against HIV virus by making normal cells resistant to viral invasion, or correction of genetic disorders such as sickle cell anemia or metabolic disorders. However, incorporating a new gene into a host stem cell’s genome, for the life of an individual, may promote other oncogenes to develop malignant disorders, and may change other adjacent genes, thus creating other medical diseases. Hence, it is a risky approach in gene therapy. Few clinical trials have recently been conducted in this regards. One example is the multidrug-resistant protein-1, which is encoded by the human ABCBI gene named as MDR1 gene. It stimulates the cellular pump to remove cytotoxic drugs from normal cell cytoplasm to the outside, thus protecting normal cells from chemotherapy’s side effects, such as with vinca alkaloids, taxanes, epipodophyllotoxins and anthracyclines [124]. The MDR1 gene is minimally expressed in malignant cells; thus, chemotherapeutic medications entering the cytoplasm will remain at a higher concentration, leading to cell death. Other drug-resistant genes include methyl guanine methyltransferase (MGMT) for alkylating chemotherapy [125, 126], and glutathione transferase (GSTP1) for cisplatin, doxorubicin, and cyclophosphamide [127, 128, 124].
Theranostic approach
In a combined diagnostic and therapeutic system (theranostic), gene therapy may also be combined with other diagnostic measures to help diagnose, treat and monitor the response to therapy. For example, a small interfering double-stranded RNA (siRNA) delivery system can be labelled with imaging agents such as dextran-coated superparamagnetic nanoparticles for simultaneous noninvasive imaging of siRNA delivery to tumors, using magnetic resonance imaging (MRI) [59]. The siRNA delivery system can also be labeled with other imaging agents to closely monitor therapy, and may even predict the outcome of therapy long before any anatomical changes [129]. Such molecular diagnostic approaches have been evolving relatively fast in the last few years, and may become an important avenue in cancer diagnosis sometime in the near future [59].
Problems with gene therapy
The most frequent side effects following gene therapy include transient fever and flu-like symptoms [24]. A grade-3 hypersensitivity reaction following intravenous administration is usually transient and managed with the usual supportive measures. Leukocytopenia, and in particular, lymphopenia, may represent cellular redistribution of white blood cells to target tissue such as tumors. Mild transient anemia has also been reported [130]. However, toxicity, mutagenicity and immunogenicity associated with viral vector therapy have raised great concern [12].
Retroviral (such as lentiviruses) mediated gene therapy leads to viral integration into host genome, thus, it may cause mutagenic events with possible second malignancies. This was reported in earlier studies on the murine leukemia retrovirus vector in the treatment of patients with severe combined immunodeficiency and five out of 30 cases developed leukemia [131], though, no second malignancy has been reported so far, in gene therapy for cancer. Such mutagenicity depends on the site of viral insertion. For this reason, the FDA has required all clinical trials involving genomic integrated viral vectors to report and analyzes viral vector insertion sites. Initial methodology was linear amplification mediated polymerase chain reaction [132], but lately, high-throughput DNA sequencing methods have been used [133, 134]. Clinical trials that initially or subsequently show evidence of higher mutagenicity are usually discontinued. Information obtained from such studies is of major significance in designing new and much safer therapeutic approaches [58].
Another major problem with gene therapy for cancer is the resistance to treatment with subsequent tumor recurrences and shorter survival. A potential mechanism is intrinsic, and possibly acquired, tumor cell resistance to therapy-induced cell death (apoptosis) by dysregulation and release of anti-apoptotic inhibitor of apoptosis protein or Bcl-2 proteins [24]. Recently, some pharmaceutical companies have developed several medications such as Novartis-LBH589, cIAP1, and cIAP2 which inhibit the Bcl-2 protein, thus promoting cell death (apoptosis) and tumor regression, prevent or delay tumor resistance, and prolong remission following gene therapy. These medications are presently in clinical trials [24, 135].
Review, Conclusions
Gene therapy for cancer has evolved relatively fast in the last two decades, and presently, few drugs are commercially available while others are still in clinical trials. Most reports on gene therapy have shown good safety profiles with transient tolerable toxicities. The lack of success in several clinical trials may partly be attributed to patient selection. Similar to initial chemotherapy outcomes thirty years ago, patients with advanced and therapy-resistant malignancies are presently enrolled in gene therapy trials. Perhaps, gene therapy maybe much more successful in patients with earlier stages of malignancies, or in those who have a lower tumor burden. Alternatively, gene therapy may better be used after successful cancer therapy with maximum tumor load reduction, such as after radical surgery, following radiation therapy, or after successful chemotherapy.
In the future, the wide use of patient and tumor genomic analysis as well as the assessment of host humoral and cellular immunity, will facilitate a better selection of the most appropriate gene therapy per patient. Recent progress in developing safe and effective vectors for gene transfer, such as with synthetic viruses and non-viral methods, as well as the success in using autologous and allogenic chimeric antigen receptor integrated T-lymphocytes, even from healthy individuals, as universal effector cells in mediating adoptive immunotherapy, will increase the effectiveness and safety profile of gene therapy. Furthermore, with the advancement in biological research, much cheaper gene vectors will become commercially available, which will make gene therapy readily available to the majority of cancer patients, worldwide. This will transform the future of cancer therapy, from generalized cancer treatment strategies, based on tumor size, nature and location, to a more tailored, individualized cancer therapy, based on the patient’s specific genomic constituents, host immune status, and genetic profile of the underlying malignancy. Treatment is expected to be fast, effective, relatively less toxic and inexpensive, with higher cure rates, and may even, cancer prevention.
Authors’ information
The author is an internist and medical oncologist at a regional cancer center in the United States, with higher qualification from United Kingdom, Canada, and United States, previous academic title at Ohio State University, over thirty five years practice as a medical oncologist, and authored or co-authored over twenty five peer-reviewed papers on cancer management and research.
Abbreviations
- APCs
Antigen-presenting cells
- CEA
Carcinoembryonic antigen
- DNA
Deoxyribonucleic acid
- DTA-H19
Corynebacterium diphtheriae toxin-A chain
- EGFR
Epidermal growth factor receptor
- Fox3
Forkhead box 03 transcription factor protein
- FDA
The Food and Drug Administration in the United States
- GDEPT
Gene directed enzyme prodrug therapy
- GM-CSF
Granulocyte macrophage colony stimulating factor
- HER-2/neu
Human epidermal growth factor receptor-2
- HIV
Human immunodeficiency virus infection
- hTERT
Human telomerase reverse transcriptase
- IDO
Indoleamine-2,3-dioxygenase
- MHC
Major histocompatibility complex
- MDR1
Multidrug-resistant protein-1
- mRNA
Messenger-RNA
- NK
Natural killer cell lymphocytes
- PSA
Prostatic acid phosphatase antigen
- RISC
Ribonucleic acid induced silencing complex
- RNA
Ribonucleic acid
- RNAi
Ribonucleic acid interference
- siRNA
Small interfering double-stranded RNA
- TAAs
Tumor-associated antigens
- TCR
T cell antigen receptor
- TRICOM
Multiple T-cell costimulatory molecules
- VEGF-A
Vascular endothelial growth factor A receptor.
Footnotes
Competing interests
The author declare that he has no financial and non-financial competing interests.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, et al. The sequence of the human genome. Science (New York, NY) 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Dulbecco R. A turning point in cancer research: sequencing the human genome. Science (New York, NY) 1986;231(4742):1055–1056. doi: 10.1126/science.3945817. [DOI] [PubMed] [Google Scholar]
- 4.Strachan T, Read A. Gene therapy and other molecular genetic-based therapeutic approaches. In: Strachan T, Read AP, editors. Human Molecular Genetics. 2. New York: Wiley-Liss; 1999. [Google Scholar]
- 5.Miller AD. Retroviral vectors. Curr Top Microbiol Immunol. 1992;158:1–24. doi: 10.1007/BF02122002. [DOI] [PubMed] [Google Scholar]
- 6.Weichselbaum RR, Kufe D. Gene therapy of cancer. Lancet. 1997;349(Suppl 2):SII10–2. doi: 10.1016/S0140-6736(97)90013-1. [DOI] [PubMed] [Google Scholar]
- 7.DeVita VT, Jr, Rosenberg SA. Two hundred years of cancer research. N Engl J Med. 2012;366(23):2207–2214. doi: 10.1056/NEJMra1204479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curie P, Curie M, Bémont G. On a new, strongly radioactive substance contained in pitchblende. CR (East Lansing, Mich) 1898;127:1215–1217. [Google Scholar]
- 9.Lage H. Bacterial delivery of RNAi effectors. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 67–75. [Google Scholar]
- 10.Goodman LS, Wintrobe MM, Dameshek W, Goodman MJ, Gilman A, McLennan MT. Landmark article Sept. 21, 1946: nitrogen mustard therapy: use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodgkin's disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders by Louis S. Goodman, Maxwell M. Wintrobe, William Dameshek, Morton J. Goodman, Alfred Gilman and Margaret T. McLennan. JAMA. 1984;251(17):2255–2261. doi: 10.1001/jama.1984.03340410063036. [DOI] [PubMed] [Google Scholar]
- 11.Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. 1948;238(23):787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 12.Hemminki O, Hemminki A. Oncolytic adenoviruses in the treatment of cancer in humans. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 153–170. [Google Scholar]
- 13.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 14.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood. 1997;90(6):2188–2195. [PubMed] [Google Scholar]
- 15.Sheridan C. Gene therapy finds its niche. Nat Biotechnol. 2011;29(2):121–128. doi: 10.1038/nbt.1769. [DOI] [PubMed] [Google Scholar]
- 16.Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, Kracher J, Grossman SA, Fisher JD, Carson K, Rosenblum M, Mikkelsen T, Olson J, Markert J, Rosenfeld S, Nabors LB, Brem S, Phuphanich S, Freeman S, Kaplan R, Zwiebel J. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958–966. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 17.Block SL, Nolan T, Sattler C, Barr E, Giacoletti KE, Marchant CD, Castellsague X, Rusche SA, Lukac S, Bryan JT, Cavanaugh PF, Jr, Reisinger KS, Protocol 016 Study Group Comparison of the immunogenicity and reactogenicity of a prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in male and female adolescents and young adult women. Pediatrics. 2006;118(5):2135–2145. doi: 10.1542/peds.2006-0461. [DOI] [PubMed] [Google Scholar]
- 18.Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J, Dahut WL, Arlen PM, Gulley JL, Godfrey WR. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, Wilson RK, Ally A, Balasundaram M, Butterfield YS, Carlsen R, Carter C, Chu A, Chuah E, Chun HJ, Coope RJ, Dhalla N, Guin R, Hirst C, Hirst M, Holt RA, Lee D, Li HI, Mayo M, Moore RA, Mungall AJ, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shirley S, Heller R, Heller L. Electroporation gene therapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 93–106. [Google Scholar]
- 23.Baranyi L, Slepushkin V, Dropulic B. Ex vivo gene therapy: utilization of genetic vectors for the generation of genetically modified cell products for therapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 3–18. [Google Scholar]
- 24.Yuan Z, Pastoriza J, Quinn T, Libutti S. Targeting tumor vasculature using adeno-associated virus page vector coding tumor necrosis factor-a. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 19–33. [Google Scholar]
- 25.Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270(32):18997–19007. doi: 10.1074/jbc.270.32.18997. [DOI] [PubMed] [Google Scholar]
- 26.Tagami T, Suzuki T, Matsunaga M, Nakamura K, Moriyoshi N, Ishida T, Kiwada H. Anti-angiogenic therapy via cationic liposome-mediated systemic siRNA delivery. Int J Pharm. 2012;422(1–2):280–289. doi: 10.1016/j.ijpharm.2011.10.059. [DOI] [PubMed] [Google Scholar]
- 27.Yang W, Sun T, Cao J, Liu F. Survivin downregulation by siRNA/cationic liposome complex radiosensitises human hepatoma cells in vitro and in vivo. Int J Radiat Biol. 2010;86(6):445–457. doi: 10.3109/09553001003668006. [DOI] [PubMed] [Google Scholar]
- 28.Wagner E, Plank C, Zatloukal K, Cotten M, Birnstiel ML. Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle. Proc Natl Acad Sci U S A. 1992;89(17):7934–7938. doi: 10.1073/pnas.89.17.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soltani F, Sankian M, Hatefi A, Ramezani M. Development of a novel histone H1-based recombinant fusion peptide for targeted non-viral gene delivery. Int J Pharm. 2013;441(1–2):307–315. doi: 10.1016/j.ijpharm.2012.11.027. [DOI] [PubMed] [Google Scholar]
- 30.Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, Cantafio ME, Gulla A, Conforti F, Morelli E, Tomaino V, Rossi M, Negrini M, Ferrarini M, Caraglia M, Shammas MA, Munshi NC, Anderson KC, Neri A, Tagliaferri P, Tassone P. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res. 2012;18(22):6260–6270. doi: 10.1158/1078-0432.CCR-12-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soliman M, Nasanit R, Abulateefeh SR, Allen S, Davies MC, Briggs SS, Seymour LW, Preece JA, Grabowska AM, Watson SA, Alexander C. Multicomponent synthetic polymers with viral-mimetic chemistry for nucleic acid delivery. Mol Pharm. 2012;9(1):1–13. doi: 10.1021/mp200108q. [DOI] [PubMed] [Google Scholar]
- 32.Nie Y, Schaffert D, Rodl W, Ogris M, Wagner E, Gunther M. Dual-targeted polyplexes: one step towards a synthetic virus for cancer gene therapy. J Control Release. 2011;152(1):127–134. doi: 10.1016/j.jconrel.2011.02.028. [DOI] [PubMed] [Google Scholar]
- 33.Hackett PB, Largaespada DA, Switzer KC, Cooper LJ. Evaluating risks of insertional mutagenesis by DNA transposons in gene therapy. Transl Res. 2013;161(4):265–283. doi: 10.1016/j.trsl.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon S, Min J. Genetically engineered Salmonella typhimurium for targeted cancer therapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 443–452. [Google Scholar]
- 35.Benoit MR, Mayer D, Barak Y, Chen IY, Hu W, Cheng Z, Wang SX, Spielman DM, Gambhir SS, Matin A. Visualizing implanted tumors in mice with magnetic resonance imaging using magnetotactic bacteria. Clin Cancer Res. 2009;15(16):5170–5177. doi: 10.1158/1078-0432.CCR-08-3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baban CK, Cronin M, O'Hanlon D, O'Sullivan GC, Tangney M. Bacteria as vectors for gene therapy of cancer. Bioeng Bugs. 2010;1(6):385–394. doi: 10.4161/bbug.1.6.13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 38.Kuroda S, Kagawa S, Fujiwara T. Selectively replicating oncolytic adenoviruses combined with chemotherapy, radiotherapy, or molecular targeted therapy for treatment of human cancers. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 171–183. [Google Scholar]
- 39.Wu E, Nemerow GR. Virus yoga: the role of flexibility in virus host cell recognition. Trends Microbiol. 2004;12(4):162–169. doi: 10.1016/j.tim.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Mathis JM, Stoff-Khalili MA, Curiel DT. Oncolytic adenoviruses - selective retargeting to tumor cells. Oncogene. 2005;24(52):7775–7791. doi: 10.1038/sj.onc.1209044. [DOI] [PubMed] [Google Scholar]
- 41.Zemp FJ, Corredor JC, Lun X, Muruve DA, Forsyth PA. Oncolytic viruses as experimental treatments for malignant gliomas: using a scourge to treat a devil. Cytokine Growth Factor Rev. 2010;21(2–3):103–117. doi: 10.1016/j.cytogfr.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 42.Balvers R, Gomez-Manzano C, Jiang H, Piya S, Klein S, Lamfers M, Driven C, Fueyo J. Advances on oncolytic virotherapy for brain tumors. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 137–151. [Google Scholar]
- 43.Kaliberova LN, Krendelchtchikova V, Harmon DK, Stockard CR, Petersen AS, Markert JM, Gillespie GY, Grizzle WE, Buchsbaum DJ, Kaliberov SA. CRAdRGDflt-IL24 virotherapy in combination with chemotherapy of experimental glioma. Cancer Gene Ther. 2009;16(10):794–805. doi: 10.1038/cgt.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nuesch JP, Lacroix J, Marchini A, Rommelaere J. Molecular pathways: rodent parvoviruses–mechanisms of oncolysis and prospects for clinical cancer treatment. Clin Cancer Res. 2012;18(13):3516–3523. doi: 10.1158/1078-0432.CCR-11-2325. [DOI] [PubMed] [Google Scholar]
- 45.White E, Gill S. Selectively replicating Herpes simplex viral vectors. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 199–211. [Google Scholar]
- 46.Sharp PM. Origins of human virus diversity. Cell. 2002;108(3):305–312. doi: 10.1016/S0092-8674(02)00639-6. [DOI] [PubMed] [Google Scholar]
- 47.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I, Medley LC, Michael A, Nutting CM, Pandha HS, Shorrock CA, Simpson J, Steiner J, Steven NM, Wright D, Coombes RC. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 48.Liu TC, Zhang T, Fukuhara H, Kuroda T, Todo T, Canron X, Bikfalvi A, Martuza RL, Kurtz A, Rabkin SD. Dominant-negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin Cancer Res. 2006;12(22):6791–6799. doi: 10.1158/1078-0432.CCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 49.Bryson P, Wang P. Lentivector vaccines. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 345–361. [Google Scholar]
- 50.Breckpot K, Aerts JL, Thielemans K. Lentiviral vectors for cancer immunotherapy: transforming infectious particles into therapeutics. Gene Ther. 2007;14(11):847–862. doi: 10.1038/sj.gt.3302947. [DOI] [PubMed] [Google Scholar]
- 51.Hu B, Yang H, Dai B, Tai A, Wang P. Nonintegrating lentiviral vectors can effectively deliver ovalbumin antigen for induction of antitumor immunity. Hum Gene Ther. 2009;20(12):1652–1664. doi: 10.1089/hum.2009.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michelini Z, Negri DR, Baroncelli S, Spada M, Leone P, Bona R, Klotman ME, Cara A. Development and use of SIV-based Integrase defective lentiviral vector for immunization. Vaccine. 2009;27(34):4622–4629. doi: 10.1016/j.vaccine.2009.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Comins C, Simpson G, Relph K, Harrington K, Melcher A, Pandha H. Reoviral therapy for cancer: strategies for improving antitumor efficacy using radio- and chemotherapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 185–198. [Google Scholar]
- 54.Pachnis V, Brannan CI, Tilghman SM. The structure and expression of a novel gene activated in early mouse embryogenesis. EMBO J. 1988;7(3):673–681. doi: 10.1002/j.1460-2075.1988.tb02862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matouk I, Raveh E, Ohana P, Lail RA, Gershtain E, Gilon M, De Groot N, Czerniak A, Hochberg A. The increasing complexity of the oncofetal h19 gene locus: functional dissection and therapeutic intervention. Int J Mol Sci. 2013;14(2):4298–4316. doi: 10.3390/ijms14024298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohana P, Matouk I, Amit D, Gilon M, Hochberg A. Toxin-based cancer gene therapy under the control of oncofetal H19 regulatory sequences. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 107–122. [Google Scholar]
- 57.Li Y, McCadden J, Ferrer F, Kruszewski M, Carducci M, Simons J, Rodriguez R. Prostate-specific expression of the diphtheria toxin A chain (DT-A): studies of inducibility and specificity of expression of prostate-specific antigen promoter-driven DT-A adenoviral-mediated gene transfer. Cancer Res. 2002;62(9):2576–2582. [PubMed] [Google Scholar]
- 58.Fung H, Gersson S. Viral insertion site detection and analysis in cancer gene therapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 35–46. [Google Scholar]
- 59.Gujrati M, Lu Z. Targeted systemic delivery of therapeutic siRNA. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 47–65. [Google Scholar]
- 60.Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, Takimoto R, Takada K, Miyanishi K, Matsunaga T, Takayama T, Niitsu Y. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26(4):431–442. doi: 10.1038/nbt1396. [DOI] [PubMed] [Google Scholar]
- 61.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6(3):659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 62.Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, Paz-Ares L, Cho DC, Infante JR, Alsina M, Gounder MM, Falzone R, Harrop J, White AC, Toudjarska I, Bumcrot D, Meyers RE, Hinkle G, Svrzikapa N, Hutabarat RM, Clausen VA, Cehelsky J, Nochur SV, Gamba-Vitalo C, Vaishnaw AK, Sah DW, Gollob JA, Burris HA., 3rd First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3(4):406–417. doi: 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- 63.Geng J, Xiao S, Zhang S, Liu C, Li Y, Ji J, Fang Z, Sun Y, Su X, Cai Y, Xu G, Li D, Zhu G, Xu B: Clinical effectiveness of recombinant adenovirus-p53 combined with radiotherapy in advanced soft tissue sarcoma: a report of 37 cases.J Clin Oncol 2014.,32(suppl; abstr e21514):
- 64.Pan JJ, Zhang SW, Chen CB, Xiao SW, Sun Y, Liu CQ, Su X, Li DM, Xu G, Xu B, Lu YY. Effect of recombinant adenovirus-p53 combined with radiotherapy on long-term prognosis of advanced nasopharyngeal carcinoma. J Clin Oncol. 2009;27(5):799–804. doi: 10.1200/JCO.2008.18.9670. [DOI] [PubMed] [Google Scholar]
- 65.Lai SY, Koppikar P, Thomas SM, Childs EE, Egloff AM, Seethala RR, Branstetter BF, Gooding WE, Muthukrishnan A, Mountz JM, Lui VW, Shin DM, Agarwala SS, Johnson R, Couture LA, Myers EN, Johnson JT, Mills G, Argiris A, Grandis JR. Intratumoral epidermal growth factor receptor antisense DNA therapy in head and neck cancer: first human application and potential antitumor mechanisms. J Clin Oncol. 2009;27(8):1235–1242. doi: 10.1200/JCO.2008.17.8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greenberger JS. Radioprotection. In Vivo. 2009;23(2):323–336. [PMC free article] [PubMed] [Google Scholar]
- 67.Cai Y, Bak RO, Mikkelsen JG. Targeted genome editing by lentiviral protein transduction of zinc-finger and TAL-effector nucleases. eLife. 2014;3:e01911. doi: 10.7554/eLife.01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uhde-Stone C, Sarkar N, Antes T, Otoc N, Kim Y, Jiang YJ, Lu B. A TALEN-based strategy for efficient bi-allelic miRNA ablation in human cells. RNA (New York, NY) 2014;20(6):948–955. doi: 10.1261/rna.042010.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sakuma T, Woltjen K. Nuclease-mediated genome editing: at the front-line of functional genomics technology. Develop Growth Differ. 2014;56(1):2–13. doi: 10.1111/dgd.12111. [DOI] [PubMed] [Google Scholar]
- 70.Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–2648. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 71.McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16(8):2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 72.Foon KA, Yang XD, Weiner LM, Belldegrun AS, Figlin RA, Crawford J, Rowinsky EK, Dutcher JP, Vogelzang NJ, Gollub J, Thompson JA, Schwartz G, Bukowski RM, Roskos LK, Schwab GM. Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int J Radiat Oncol Biol Phys. 2004;58(3):984–990. doi: 10.1016/j.ijrobp.2003.09.098. [DOI] [PubMed] [Google Scholar]
- 73.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 74.Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. 2014;11(1):20–33. doi: 10.7497/j.issn.2095-3941.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boon T, Van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183(3):725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449(7161):419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 77.De Souza AP, Bonorino C. Tumor immunosuppressive environment: effects on tumor-specific and nontumor antigen immune responses. Expert Rev Anticancer Ther. 2009;9(9):1317–1332. doi: 10.1586/era.09.88. [DOI] [PubMed] [Google Scholar]
- 78.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, Bruneval P, Trajanoski Z, Fridman WH, Pages F, Galon J. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29(6):610–618. doi: 10.1200/JCO.2010.30.5425. [DOI] [PubMed] [Google Scholar]
- 80.Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. 2011;29(15):1949–1955. doi: 10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- 81.Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, Vimond N, Concha A, Garrido F, Isambert N, Chaigneau L, Le Brun-Ly V, Dubreuil P, Cremer I, Caignard A, Poirier-Colame V, Chaba K, Flament C, Halama N, Jäger D, Eggermont A, Bonvalot S, Commo F, Terrier P, Opolon P, Emile JF, Coindre JM, Kroemer G, Chaput N, Le Cesne A, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013;73(12):3499–3510. doi: 10.1158/0008-5472.CAN-13-0371. [DOI] [PubMed] [Google Scholar]
- 82.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21(2):233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177(7):4521–4529. doi: 10.4049/jimmunol.177.7.4521. [DOI] [PubMed] [Google Scholar]
- 84.Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KE, Sklavos MM, Ambs S, Yagita H, Hurwitz AA. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest. 2011;121(4):1361–1372. doi: 10.1172/JCI44325. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O'Day SJ, Hoos A, Humphrey R, Berman DM, Lonberg N, Korman AJ. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Annals of the New York Academy of Sciences. 2013;1291:1–13. doi: 10.1111/nyas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71(7):2488–2496. doi: 10.1158/0008-5472.CAN-10-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, Taguchi T, Yuan J, Wolchok JD, Allison JP, DeMatteo RP. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–1100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, Hodi FS, Overwijk WW, Lizée G, Murphy GF, Hwu P, Flaherty KT, Fisher DE, Wargo JA. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liang H, Deng L, Chmura S, Burnette B, Liadis N, Darga T, Beckett MA, Lingen MW, Witt M, Weichselbaum RR, Fu Y. Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J Immunol. 2013;190(11):5874–5881. doi: 10.4049/jimmunol.1202612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, Hammers HJ, Mathios D, Tyler B, Brem H, Tran PT, Pardoll D, Drake CG, Lim M. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reardon DA, Wucherpfennig KW, Freeman G, Wu CJ, Chiocca EA, Wen PY, Curry WT, Jr, Mitchell DA, Fecci PE, Sampson JH, Dranoff G. An update on vaccine therapy and other immunotherapeutic approaches for glioblastoma. Expert Rev Vaccines. 2013;12(6):597–615. doi: 10.1586/erv.13.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hinrichs CS, Stevanovic S, Draper L, Somerville R, Wunderlich J, Restifo NP, Sherry R, Giao PQ, Kammula US, Yang JC, Rosenberg SA: HPV-targeted tumor-infiltrating lymphocytes for cervical cancer.J Clin Oncol 2014.,32(suppl; abstr LBA3008):
- 94.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 95.Varela-Rohena A, Carpenito C, Perez EE, Richardson M, Parry RV, Milone M, Scholler J, Hao X, Mexas A, Carroll RG, June CH, Riley JL. Genetic engineering of T cells for adoptive immunotherapy. Immunol Res. 2008;42(1–3):166–181. doi: 10.1007/s12026-008-8057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Johnson LA, Heemskerk B, Powell DJ, Jr, Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. JImmunol. 2006;177(9):6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang X, Nishimura M. Genetically engineered (T cell receptor) T cells for adoptive therapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 259–271. [Google Scholar]
- 98.Choi Y, Yuen C, Maiti SN, Olivares S, Gibbons H, Huls H, Raphael R, Killian TC, Stark DJ, Lee DA, Torikai H, Monticello D, Kelly SS, Kebriaei P, Champlin RE, Biswal SL, Cooper LJ. A high throughput microelectroporation device to introduce a chimeric antigen receptor to redirect the specificity of human T cells. Biomed Microdevices. 2010;12(5):855–863. doi: 10.1007/s10544-010-9440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, Hackett PB, Kohn DB, Shpall EJ, Champlin RE, Cooper LJ. Redirecting specificity of T-cell populations for CD19 using the sleeping beauty system. Cancer Res. 2008;68(8):2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, Liu H, Creighton CJ, Gee AP, Heslop HE, Rooney CM, Savoldo B, Dotti G. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Turtle CJ. Chimeric antigen receptor modified T cell therapy for B cell malignancies. Int J Hematol. 2014;99(2):132–140. doi: 10.1007/s12185-013-1490-x. [DOI] [PubMed] [Google Scholar]
- 103.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marcus A, Eshhar Z. Allogeneic chimeric antigen receptor-modified cells for adoptive cell therapy of cancer. Expert Opin Biol Ther. 2014;14(7):947–954. doi: 10.1517/14712598.2014.900540. [DOI] [PubMed] [Google Scholar]
- 105.Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovitch B, Lee DA, Champlin RE, Bonini C, Naldini L, Rebar EJ, Gregory PD, Holmes MC, Cooper LJ. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119(24):5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boudreau JE, Bonehill A, Thielemans K, Wan Y. Engineering dendritic cells to enhance cancer immunotherapy. Mol Ther. 2011;19(5):841–853. doi: 10.1038/mt.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pinder-Schenck M, Antonia S. Genetically modified dendritic cell vaccines for solid tumors. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 273–282. [Google Scholar]
- 108.Jonuleit H, Giesecke-Tuettenberg A, Tuting T, Thurner-Schuler B, Stuge TB, Paragnik L, Kandemir A, Lee PP, Schuler G, Knop J, Enk AH. A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int J Cancer. 2001;93(2):243–251. doi: 10.1002/ijc.1323. [DOI] [PubMed] [Google Scholar]
- 109.Tsao H, Millman P, Linette GP, Hodi FS, Sober AJ, Goldberg MA, Haluska FG. Hypopigmentation associated with an adenovirus-mediated gp100/MART-1-transduced dendritic cell vaccine for metastatic melanoma. Arch Dermatol. 2002;138(6):799–802. doi: 10.1001/archderm.138.6.799. [DOI] [PubMed] [Google Scholar]
- 110.Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, Hodge JW, Doren S, Grosenbach DW, Hwang J, Fox E, Odogwu L, Park S, Panicali D, Schlom J. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23(4):720–731. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 111.Wolff JA, Budker V. The mechanism of naked DNA uptake and expression. Adv Genet. 2005;54:3–20. doi: 10.1016/S0065-2660(05)54001-X. [DOI] [PubMed] [Google Scholar]
- 112.Kutzler MA, Weiner DB. DNA vaccines: ready for prime time? Nat Rev Genet. 2008;9(10):776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McNeel DG, Dunphy EJ, Davies JG, Frye TP, Johnson LE, Staab MJ, Horvath DL, Straus J, Alberti D, Marnocha R, Liu G, Eickhoff JC, Wilding G. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009;27(25):4047–4054. doi: 10.1200/JCO.2008.19.9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Norell H, Poschke I, Charo J, Wei WZ, Erskine C, Piechocki MP, Knutson KL, Bergh J, Lidbrink E, Kiessling R. Vaccination with a plasmid DNA encoding HER-2/neu together with low doses of GM-CSF and IL-2 in patients with metastatic breast carcinoma: a pilot clinical trial. J Transl Med. 2010;8:53. doi: 10.1186/1479-5876-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Staff C, Mozaffari F, Haller BK, Wahren B, Liljefors M. A Phase I safety study of plasmid DNA immunization targeting carcinoembryonic antigen in colorectal cancer patients. Vaccine. 2011;29(39):6817–6822. doi: 10.1016/j.vaccine.2010.12.063. [DOI] [PubMed] [Google Scholar]
- 116.Rosa DS, Ribeiro SP, Almeida RR, Mairena EC, Postol E, Kalil J, Cunha-Neto E. A DNA vaccine encoding multiple HIV CD4 epitopes elicits vigorous polyfunctional, long-lived CD4+ and CD8+ T cell responses. PLoS One. 2011;6(2):e16921. doi: 10.1371/journal.pone.0016921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia F, Petry KU, Muderspach L, Gold MA, Braly P, Crum CP, Magill M, Silverman M, Urban RG, Hedley ML, Beach KJ. ZYC101a for treatment of high-grade cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2004;103(2):317–326. doi: 10.1097/01.AOG.0000110246.93627.17. [DOI] [PubMed] [Google Scholar]
- 118.Auci D, Cecil D, Herendeen D, Broussard E, Liao J, Holt G, Disis M. Clinical application of plasmid-based cancer vaccines. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 335–343. [Google Scholar]
- 119.Karjoo Z, Ganapathy V, Hatefi A. Gene-directed enzyme prodrug cancer therapy. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 77–91. [Google Scholar]
- 120.Nicholas TW, Read SB, Burrows FJ, Kruse CA. Suicide gene therapy with Herpes simplex virus thymidine kinase and ganciclovir is enhanced with connexins to improve gap junctions and bystander effects. Histol Histopathol. 2003;18(2):495–507. doi: 10.14670/HH-18.495. [DOI] [PubMed] [Google Scholar]
- 121.Agard C, Ligeza C, Dupas B, Izembart A, El Kouri C, Moullier P, Ferry N. Immune-dependent distant bystander effect after adenovirus-mediated suicide gene transfer in a rat model of liver colorectal metastasis. Cancer Gene Ther. 2001;8(2):128–136. doi: 10.1038/sj.cgt.7700281. [DOI] [PubMed] [Google Scholar]
- 122.Edelstein ML, Abedi MR, Wixon J, Edelstein RM. Gene therapy clinical trials worldwide 1989-2004-an overview. J Gene Med. 2004;6(6):597–602. doi: 10.1002/jgm.619. [DOI] [PubMed] [Google Scholar]
- 123.Alauddin MM, Shahinian A, Gordon EM, Bading JR, Conti PS. Preclinical evaluation of the penciclovir analog 9-(4-[(18)F]fluoro-3-hydroxymethylbutyl)guanine for in vivo measurement of suicide gene expression with PET. J Nucl Med. 2001;42(11):1682–1690. [PubMed] [Google Scholar]
- 124.Lin Y, Gerson S. Clinical trials using LB-P140K-MGMT for gliomas. In: Lattime EC, Gerson SL, editors. Gene Therapy of Cancer. 3. San Diego (CA): Elsevier; 2013. pp. 379–391. [Google Scholar]
- 125.Maier P, Herskind C, Fleckenstein K, Spier I, Laufs S, Zeller WJ, Fruehauf S, Wenz F. MDR1 gene transfer using a lentiviral SIN vector confers radioprotection to human CD34+ hematopoietic progenitor cells. Radiat Res. 2008;169(3):301–310. doi: 10.1667/RR1067.1. [DOI] [PubMed] [Google Scholar]
- 126.Maier P, Spier I, Laufs S, Veldwijk MR, Fruehauf S, Wenz F, Zeller WJ. Chemoprotection of human hematopoietic stem cells by simultaneous lentiviral overexpression of multidrug resistance 1 and O(6)-methylguanine-DNA methyltransferase(P140K) Gene Ther. 2010;17(3):389–399. doi: 10.1038/gt.2009.133. [DOI] [PubMed] [Google Scholar]
- 127.Arai T, Miyoshi Y, Kim SJ, Akazawa K, Maruyama N, Taguchi T, Tamaki Y, Noguchi S. Association of GSTP1 expression with resistance to docetaxel and paclitaxel in human breast cancers. Eur J Surg Oncol. 2008;34(7):734–738. doi: 10.1016/j.ejso.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 128.Oguri T, Fujiwara Y, Katoh O, Daga H, Ishikawa N, Fujitaka K, Yamasaki M, Yokozaki M, Isobe T, Ishioka S, Yamakido M. Glutathione S-transferase-pi gene expression and platinum drug exposure in human lung cancer. Cancer Lett. 2000;156(1):93–99. doi: 10.1016/S0304-3835(00)00447-X. [DOI] [PubMed] [Google Scholar]
- 129.Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123(4):607–620. doi: 10.1016/j.cell.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 130.Seiradake E, Henaff D, Wodrich H, Billet O, Perreau M, Hippert C, Mennechet F, Schoehn G, Lortat-Jacob H, Dreja H, Ibanes S, Kalatzis V, Wang JP, Finberg RW, Cusack S, Kremer EJ. The cell adhesion molecule "CAR" and sialic acid on human erythrocytes influence adenovirus in vivo biodistribution. PLoS Pathog. 2009;5(1):e1000277. doi: 10.1371/journal.ppat.1000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schmidt M, Zickler P, Hoffmann G, Haas S, Wissler M, Muessig A, Tisdale JF, Kuramoto K, Andrews RG, Wu T, Kiem HP, Dunbar CE, von Kalle C. Polyclonal long-term repopulating stem cell clones in a primate model. Blood. 2002;100(8):2737–2743. doi: 10.1182/blood-2002-02-0407. [DOI] [PubMed] [Google Scholar]
- 133.Bartholomae CC, Glimm H, Von Kalle C, Schmidt M. Insertion site pattern: global approach by linear amplification-mediated PCR and mass sequencing. Methods Mol Biol. 2012;859:255–265. doi: 10.1007/978-1-61779-603-6_15. [DOI] [PubMed] [Google Scholar]
- 134.Gillet NA, Malani N, Melamed A, Gormley N, Carter R, Bentley D, Berry C, Bushman FD, Taylor GP, Bangham CR. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. 2011;117(11):3113–3122. doi: 10.1182/blood-2010-10-312926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Novartis . Investigational New Drug Development Program. 2014. [Google Scholar]