

Abstract
The fourth edition of the World Health Organization (WHO) classification of myeloid neoplasms refined the criteria for some previously described myeloid neoplasms and recognized several new entities based on recent elucidation of molecular pathogenesis, identification of new diagnostic and prognostic markers, and progress in clinical management. Protein tyrosine kinase abnormalities, including translocations or mutations involving ABL1, JAK2, MPL, KIT, PDGFRA, PDGFRB and FGFR1, have been used as the basis for classifying myeloproliferative neoplasms (MPN). Two new entities - refractory cytopenia with unilineage dysplasia and refractory cytopenia of childhood have been added to the group of myelodysplastic syndromes (MDS), and “refractory anemia with excess blasts-1” has been redefined to emphasize the prognostic significance of increased blasts in the peripheral blood. A list of cytogenetic abnormalities has been introduced as presumptive evidence of MDS in cases with refractory cytopenia but without morphologic evidence of dysplasia. The subgroup “acute myeloid leukemia (AML) with recurrent genetic abnormalities” has been expanded to include more molecular genetic aberrations. The entity “AML with multilineage dysplasia” specified in the 2001 WHO classification has been renamed “AML with myelodysplasia-related changes” to include not only cases with significant multilineage dysplasia, but also patients with a history of MDS or myelodysplasia-related cytogenetic abnormalities. The term “therapy-related myeloid neoplasms” is used to cover the spectrum of disorders previously known as t-AML, t-MDS, or t-MDS/MPN occurring as complications of cytotoxic chemotherapy and/or radiation therapy. In this review, we summarize many of these important changes and discuss some of the diagnostic challenges that remain.
Keywords: WHO 2008, myeloproliferative neoplasm, myelodysplastic/myeloprolierative neoplasm, myelodysplastic syndrome, acute myeloid leukemia
1. Introduction
In the fourth edition of the World Health Organization (WHO) classification of hematopoietic and lymphoid neoplasms, published in 2008, several important changes were made in the criteria for diagnosis and classification of myeloid neoplasms (Table 1, Vardiman JW et al, 2008). In this review, we highlight many of the recent advances in our understanding of the pathogenesis of myeloid neoplasms and the application of this knowledge to the diagnosis and classification of myeloid malignancies. As the current WHO classification is extensive, we have limited our comments to selected myeloid neoplasms in which there are significant advances or changes in diagnostic criteria.
Table 1.
The 2008 WHO classification of myeloid neoplasms
| Myeloproliferative Neoplasms |
| Chronic myelogenous leukaemia, BCR-ABL1 positive |
| Chronic neutrophilic leukaemia |
| Polycythaemia vera |
| Primary myelofibrosis |
| Essential thrombocythaemia |
| Chronic eosinophilic leukaemia, NOS |
| Mastocytosis |
| Myeloproliferative neoplasm, unclassifiable |
| Myeloid and Lymphoid Neoplasms with Eosinophilia and Abnormalities of PDGFRA PDGFRB or FGFR1 |
| Myelodyspastic/Myeloproliferative Neoplasms |
| Chronic myelomonocytic leukaemia |
| Atypical chronic myeloid leukaemia, BCR-ABL1 negative |
| Juvenile myelomonocytic leukaemia |
| Myelodysplastic/myeloproliferative neoplasm, unclassifiable |
| Refractory anaemia with ring sideroblasts associated with marked thrombocytosis |
| Myelodysplastic Syndromes |
| Refractory cytopenia with unilineage dysplasia |
| Refractory anaemia with ring sideroblasts |
| Refractory cytopenia with multilineage dysplasia |
| Refractory anaemia with excess blasts |
| Myelodysplastic syndrome associated with isolated del(5q) |
| Myelodysplastic syndrome, unclassifiable |
| Childhood myelodysplastic syndrome |
| Refarctory cytopenia of childhood |
| Acute Myeloid Leukaemia (AML) and Related Precursor Neoplasms |
| AML with recurrent genetic abnormalities |
| AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFβ-MYH11 |
| Acute promyelocytic leukemia with t(15;17)(q22;q12); PML-RARA |
| AML with t(9;11)(p22;q23); MLLT3-MLL |
| AML with t(6;9)(p23;q34); DEK-NUP214 |
| AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); RPN1-EVI1 |
| AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1 |
| Provisional entity: AML with mutated NPM1 |
| Provisional entity: AML with mutated CEBPA |
| AML with myelodysplasia-related changes |
| Therapy-related myeloid neoplasms |
| Acute myeloid leukaemia, not otherwise specified |
| AML with minimal differentiation |
| AML without maturation |
| AML with maturation |
| Acute myelomonocytic leukaemia |
| Acute monoblastic and monocytic leukaemia |
| Acute erythroid leukaemia |
| Acute megakaryoblastic leukaemia |
| Acute basophilic leukaemia |
| Acute panmyelosis with myelofibrosis |
| Myeloid sarcoma |
| Myeloid proliferations related to Down syndrome |
| Blastic plasmacytoid dendritic cell neoplasm |
2. Myeloproliferative neoplasms
In the fourth edition of the WHO classification, the term “chronic myeloproliferative disease” is replaced by “myeloproliferative neoplasm (MPN)” and the category now includes eight diseases. These include chronic myelogenous leukemia, BCR-ABL1+, chronic neutrophilic leukemia, polycythemia vera, primary myelofibrosis, essential thrombocythemia, chronic eosinophilic leukemia, not otherwise specified, myeloproliferative neoplasm, unclassifiable, and a new addition – mastocytosis (Table 2).
Table 2.
Gene mutations in myeloproliferative neoplasms with activated receptor tyrosine kinases
| Disease entity | Gene | Mechanism of activation | Main cell target |
|---|---|---|---|
| CML | BCR-ABL1 | t(9;22)(q34;q11) | all myeloid cells |
| PV | JAK2 | point mutation (V617F) | erythroid cells, granulocytes, megakaryocytes |
| ET | JAK2MPL | point mutation (V617F, W515L/K) | megakaryocytes |
| PMF | JAK2MPL | point mutation (V617F, W515L/K) | granulocytes, erythroid cells, megakaryocytes |
| Mastocytosis | KIT | point mutation (D816V) | mast cells |
| MPN with eosinophilia | PDGFRA-FIP1L1 | interstitial deletion at 4q12 | eosinophils, mast cells |
| MPN with eosinophilia | ETV6-PDGFRB | t(5;12)(q31–33;p13) | eosiniphils |
| MPN with eosinophilia | FGFR1-ZNF198 | t(8;13)(p11;q12) | eosinophils |
CML, chronic myelogenous leukemia; PV, polycythemia vera; ET, essential thrombocythemia; PNF, primary myelofibrosis; MPN, myeloproliferative neoplasm
2.1 Protein tyrosine kinases and chronic myelogenous leukemia
Protein tyrosine kinases (PTKs) play an important role in the pathogenesis of MPNs. Mutations and rearrangements of genes encoding PTKs cause constitutive activation of PTKs and its downstream signal transduction pathways, and provide cells with an advantage for proliferation and survival. Molecular abnormalities of PTKs have been used in the diagnosis, classification, prognosis prediction, and detection of minimal residual disease, as well as for targeted therapy in MPN. The best understood example is ABL1, activated as a result of t(9;22)(q34;q11)/BCR-ABL1 in chronic myelogenous leukemia (CML). BCR-ABL1 has been used to refine the diagnosis of CML. BCR-ABL1 is valuable for detection of minimal residual disease, and has been successfully targeted with specific therapies. Mutational analysis of ABL1 also has value in predicting response to therapy. The success of targeted therapy in patients with CML, changing a fatal disease into a chronic manageable disease, is a model for acquiring in-depth molecular understanding of all diseases, and thereby allowing for the rational treatment approaches to many myeloid diseases and cancer in general (Figure 1).
Figure 1.
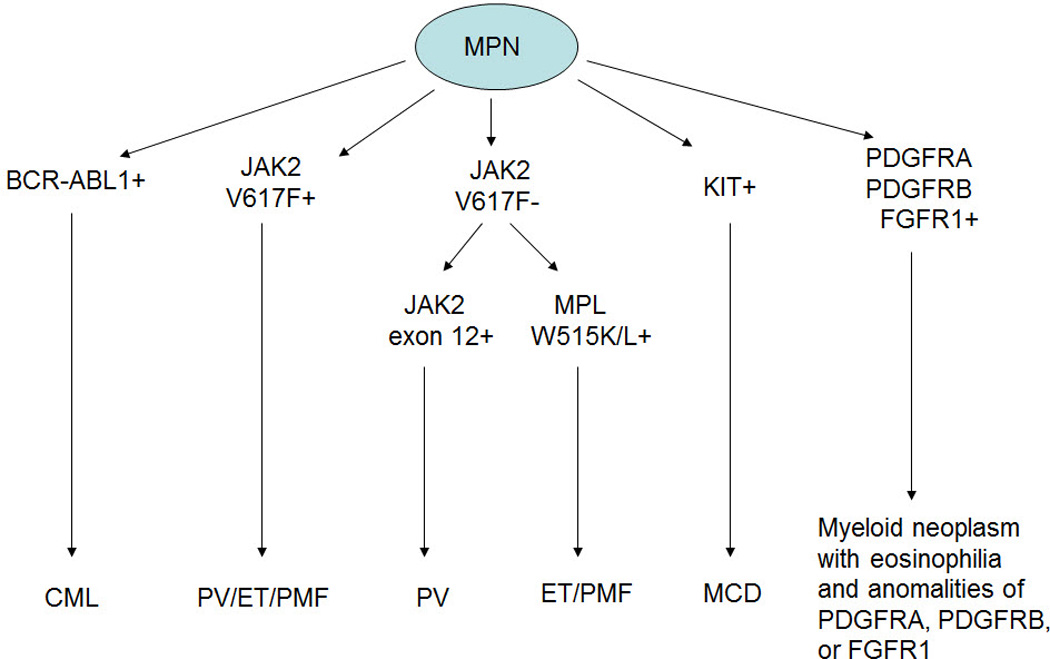
Diagnostic algorithm of myeloproliferative neoplasms associated with receptor tyrosine kinase activation.
2.2 JAK2-mutated MPN
In 2005, several groups independently reported the presence of the JAK2 V617F mutation in approximately 95% of patients with polycythemia vera (PV), and 40–50% of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF) (Baxter EJ et al, 2005; James C et al, 2005; Jones AV et al, 2005; Levine RL et al, 2005; Kralovics R et al, 2005). Subsequently, JAK2 exon 12 mutations were identified in rare cases of PV without the JAK2 V617F mutation (Pardanani A et al, 2007; Scott LM et al; 2007), and W515L/K mutations in the thrombopoietin receptor (MPL) were identified in approximately 5% of patients with ET and PMF (Pardanani AD et al, 2006; Pikman Y et al, 2006). The overlapping clinical and biologic features of PV, ET, and PMF may now be explained by the presence of a JAK2 mutation. JAK2 mediates activation of receptors for erythropoietin, thrombopoietin, granulocyte-macrophage colony-simulating factor and granulocyte colony-stimulating factor. Therefore, the different clinical manifestations of PV, ET and PMF may reflect the stage of differentiation at which the JAK2 mutation occurs, other genetic events that evolve during disease progression, and differences in the genetic background of the patient. Knowledge of JAK2 mutation also suggests that JAK2-mutated PV, ET and PMF may be better considered as a spectrum of diseases with a common molecular origin. JAK2 and MPL mutation are now included as major diagnostic criteria for PV, ET and PMF.
However, at least for the present diagnostic distinctions between PV, ET, and PMF are still based on hematologic and clinical data, and bone marrow findings are also very helpful (Kvasnicka HM and Thiele J, 2010). It also should be remembered that JAK2 mutation is not an initiating event, and appears to be a late event in the molecular evolution of MPNs. Clearly, additional understanding of the molecular basis of these diseases is needed. The lateness of JAK2 mutation in evolution also has implications for therapy, as drugs that inhibit JAK2 may alleviate symptoms and not eradicate the monoclonal cell population.
A reasonable algorithm for the diagnosis of PV, ET and PMF is to initially test for JAK2 V617F, followed by assessment for either a JAK2 exon 12 mutation for patients with suspected PV, or an MPL W515L/K mutation for those with suspected ET or PMF. However, the appropriate specimen type and testing methodology are still subject to debate. It has been shown that JAK2 V617F mutation can be identified with the same high degree of specificity using either DNA or RNA when either fresh peripheral blood (PB) or bone marrow (BM) aspirate is used. The use of RNA is more sensitive and appears to be ideal for patients with low-tumor burden or for detection of minimal residual disease, whereas DNA is the preferred material when archived tissue is used (Gattenlohner S et al, 2007). Since JAK2-mutated progenitors are a fraction of all progenitor cells, some investigators have suggested using BM instead of PB to increase the detection rate, whereas others have shown that JAK2 mutation can be accurately and reliably detected, with concordant results, using either PB or BM (Mirza I et al, 2008). Plasma enriched with tumor-specific nucleic acid has been suggested as the sample of choice for JAK2 mutational analysis by one group (Ma W et al, 2008). Subsequent studies, however, have shown that granulocyte lysis during storage can affect accurate quantification of JAK2. Therefore, plasma may be better used for screening rather than as a measure for allelic burden (Salama ME et al, 2009). Granulocyte enrichment also has been proposed as a means to improve detection frequency. Sorting of granulocytes, however, is expensive and labor-intensive, with the potential for cell loss during sample procession. Testing of unfractionated samples with a sensitive assay is more practical (Stevenson WS et al, 2006). In fact, it has been demonstrated that the choice of a detection technique and the methods of results interpretation impact the detection rate.
Several qualitative and quantitative methods are currently available to assess for JAK2 mutation. Restriction fragment length polymorphism (RFLP) and direct Sanger sequencing are relatively insensitive (lower limit of sensitivity of 20%) and non-quantitative, and therefore not recommended. Pyrosequencing (lower limit 1%) or mutation-specific quantitative polymerase chain reaction (PCR) (lower limit 0.01%) provide better sensitivity and quantification to differentiate PV from ET and PMF, and are methods of choice (Yin CC and Jones D, 2010). Quantitifcation of JAK2 mutation is of value diagnostically. High levels of allele burden (consistent with homozygous mutation) are typically observed in patients with PV. By contrast, low allelic burdens (in the range of 30–50%) are more often seen in patients with ET. Mutation level is also helpful in distinguishing prodromal phase of PMF from ET (Kvasnicka HM and Thiele J, 2010).
2.3 Mastocytosis
Mastocytosis, an old and well known disease, is a new addition to the MPN category. KIT mutations at codon 816 in exon 17 (usually D816V) have been reported in 50–95% of adults with systemic mastocytosis (SM) and in 30–50% of pediatric patients with cutaneous mastocytosis (CM) (Lim KH et al, 2008). Rare sites of KIT mutation include D820G, E839K, F522C and V560G (Lim KH et al, 2008). The wide range in the reported frequency of KIT mutations may be explained by different diagnostic criteria, sampling issues owing to the focal nature of mast cell aggregates and BM fibrosis, whether aspirate or biopsy specimens are analyzed, and/or the differing sensitivity of detection methods. Pyrosequencing or mutation-specific quantitative PCR on DNA extracted from grossly-microdissected BM or skin biopsy specimens, or from mast cells sorted from BM aspirates are preferred methods (Zhao W et al, 2007). Direct Sanger sequencing is not recommended. The detection of KIT mutations in non-mast cell lineages at least partially explains the increased association of SM with other hematopoietic neoplasms (i.e., SM with associated clonal hematological non-mast cell lineage disease, SM-AHNMD) (Yavuz AS et al, 2002). It is noteworthy that although imatinib mesylate is active against the KIT kinase, the D816V mutation confers resistance, and other inhibitors (e.g. dasatinib) have been shown to be more effective (Akin C, 2006).
2.4 Issues not addressed by the revised WHO classification of MPNs
Several issues remain unaddressed in the fourth edition of the WHO classification of myeloproliferative neoplasms. For example, various diagnostic criteria have been used in the literature to define the accelerated phase and blast phase of CML. Table 4 summarizes the criteria used by the WHO classification and The University of Texas M. D. Anderson Cancer Center (MDACC). The fourth edition of the WHO classification continues to use the criteria for accelerated and blast phases that were introduced in the third edition on the basis of the published literature and the collective experience of a clinical advisory committee (Vardiman JW et al, 2002) without being tested in published trials. The criteria used at MDACC were, on the other hand, developed from a multivariate analysis of factors that independently predicted an outcome inferior to that seen in patients with chronic phase CML treated with imatinib mesylate (Cortes JE et al, 2006). Because appropriate staging of CML affects treatment and clinical outcome, it is highly desirable to establish a uniform, well-validated system for diagnosing accelerated and blast phases of CML, especially one that is adequately evaluated in the era of PTK inhibitor therapy.
Table 4.
Criteria of accelerated and blast phases of chronic myelogenous leukemia
| Phase | WHO | MDACC |
|---|---|---|
| Accelerated | ||
| Blasts | 10–19% | 15–29% |
| Blasts and promyelocytes | NA | ≥30% |
| Basophils | ≥20% | ≥20% |
| Platelets (x109/L) | <100 or >1000* | <100* |
| WBC (x109/L) | >10* | NA |
| Splenomegaly | increased | NA |
| Cytogenetics | CE | CE |
| Blast | ||
| Blasts | ≥20% | ≥30% |
| EMT | present | present |
| Other | large foci of blasts in BM biopsy | NA |
persistent and unrelated/unresponsive to therapy
WHO, World Health Organization; MDACC, M. D. Anderson Cancer Center; NA, not applicable; WBC, white blood cells; CE, clonal evolution
Another unaddressed issue is the problematic distinction between Philadelphia chromosome (Ph)-positive acute myeloid leukemia (AML) and blast phase of CML. It has been suggested that Ph+ AML simply represents CML presenting as myeloid blast phase in a patient without a symptomatic chronic phase. However, there is evidence to support the interpretation that Ph+ AML, although rare with an incidence of <1%, is indeed a distinct entity. Soupir et al. compared the clinical, morphologic, immunophenotypic and molecular cytogenetic features of Ph+ AML with those of blast phase CML in a large multi-institutional analysis (Soupir CP et al, 2007). They found that patients with Ph+ AML, compared with patients with blast phase CML, were less likely to have splenomegaly or PB and/or BM basophilia, had lower BM cellularity and myeloid:erythroid ratios, and less commonly had major additional cytogenetic abnormalities that are typically seen in blast phase of CML. Other evidence that favors a diagnosis of Ph+ AML includes the absence of a clinical history of a hematologic disorder, lack of evidence of a chronic or accelerated phase of CML after induction chemotherapy, and return to a normal karyotype after induction chemotherapy.
Similarly, in some instances the term blast phase of MPN has not been used appropriately for a patient with a history of a Ph-negative MPN who subsequently develops AML. The incidence of AML at 10 years after initial presentation is 2–5% for PV, 1–2% for ET, and 5–30% for PMF (Yin CC and Jones D, 2010). AML is known to occur more frequently in patients treated with cytotoxic agents (e.g. hydroxyurea) (Thiele J et al, 2008) and JAK2 mutations are not identified in some of these AML cases (Theocharides A et al, 2007). These facts support the idea that some of these AMLs are more consistent with secondary leukemia rather than MPN progression. A careful investigation of clinical history (leukocytosis, thrombocytosis, basophilia, and splenomegaly), complete morphologic evaluation (background myeloid hyperplasia, eosinophilia and basophilia, marked megakaryocytic hyperplasia/dysplasia as highlighted by immunostain for CD61, and fibrosis and osteosclerosis as revealed by special stains for reticulum and collagen), routine cytogenetic and molecular testing (BCR-ABL1 fusion transcript, JAK2 V617F or exon 12 mutation, MPL mutation, and FIP1LI-PDGFRA rearrangement) may aid in the recognition of a de novo presentation of MPN in blast phase.
3 Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1
Diseases associated with eosinophilia are numerous and diagnostically challenging. Reactive cases secondary to aberrant release of cytokines or other proteins by reactive or neoplastic T-cells are much more common than are primary hematopoietic neoplasms such as chronic eosinophilic leukemia, acute myelomonocytic leukemia, CML, chronic myelomonocytic leukemia and mastocytosis. The revised WHO classification includes a new category “myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1”. This is an umbrella term that we believe includes three different diseases involving constitutive activation through chromosomal rearrangement of one of three PTKs: platelet derived growth factor receptor (PDGFR)A PDGFRB or fibroblast growth factor receptor 1 (FGFR1).. Multiple fusion gene partners have been reported, with the most common being FIP1L1, ETV6/TEL and ZNF198 for PDGFRA, PDGFRB and FGFR1, respectively (Table 3, Cross NC and Reiter A, 2008). However, considerable heterogeneity exists in the cell populations affected and in the clinical manifestations of these entities (Bain BJ et al, 2008; Cross NC and Reiter A, 2008). For patients with PDGFRA and PDGFRB rearrangements, imatinib mesylate has been used for treatment, but no kinase inhibitor is currently available for patients with FGFR1 abnormalities. The spectrum of diseases in patients with FGFR1 abnormalities is also broader as many patients with t(8;13)(p11;q12)/ZNF198-FGFR1 develop lymphoblastic lymphoma/leukemia, usually of T-cell lineage (Jackson CC et al, 2010).
Table 3.
Genes involved in the rearrangements of PDGFRAPDGFRB, and FGFR1
| Tyrosine kinase gene Chromosome | Partner gene | Chromosome | |
|---|---|---|---|
| PDGFRA | 4q12 | FIP1L1* | 4q12 |
| STRN | 2p24 | ||
| CDK5RAP2 | 9q33 | ||
| KIF5B | 10p11 | ||
| ETV6 | 12p13 | ||
| BCR | 22q11 | ||
| PDGFRB | 5q31–33 | ETV6/TEL* | 12p13 |
| TPM3 | 1q21 | ||
| PDE4DIP | 1q22 | ||
| WDR48 | 3p22 | ||
| GOLGA4 | 3p22 | ||
| PRKG2 | 4q21 | ||
| HIP1 | 7q11 | ||
| CCDC6/H4 | 10q21 | ||
| GPIAP1 | 11p13 | ||
| GIT2 | 12q24 | ||
| NIN | 14q24 | ||
| KIAA1509 | 14q32 | ||
| TRIP11/CEV14 | 14q32 | ||
| TP53BP1 | 15q22 | ||
| NDE1 | 16p13 | ||
| SPECC1 | 17p11 | ||
| RABEP1 | 17p13 | ||
| FGFR1 | 8p11 | ZNF198/RAMP/FIM* | 13q12 |
| FGFR1OP/FOP | 6q27 | ||
| TIF1 | 7q34 | ||
| CEP110 | 9q33 | ||
| FGFR1OP2 | 12p11 | ||
| MYO18A | 17q11 | ||
| LOC113386/HERV-K | 19q13 | ||
| BCR | 22q11 | ||
Most common partner gene
4. Myelodysplastic/myeloproliferative neoplasms
There are four diseases in this category: chronic myelomonocytic leukemia, atypical CML, BCR-ABL1 negative, juvenile myelomonocytic leukemia, and myelodysplastic/myeloproliferative neoplasm, unclassifiable.
4.1 Refractory anemia with ring sideroblasts and thrombocytosis
Refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) was initially proposed as a provisional entity. The current version of the WHO classification addresses this issue in the chapter designated “myelodysplastic/myeloproliferative neoplasm, unclassifiable” (Vardiman JW et al, 2008). In this chapter the authors of the WHO classification keep RARS-T as a provisional entity, yet they also question the merits of classifying RARS-T as a distinct entity. The criteria for RARS-T are similar, except that the threshold for diagnosing thrombocytosis has been lowered to 450 × 109/L (to be consistent with ET criteria), and the presence of megakaryocytes with morphologic features similar to those seen in ET or PMF is required.
The uniqueness of RARS-T as a distinct entity has been debated in the literature. Is RARS-T simply ET with increased ring sideroblasts or RARS with thrombocytosis? It was recently reported that patients with RARS-T had JAK2 V617F or MPL W515 mutation at a frequency similar to that seen in patients with ET (Boissinot M et al, 2006; Ceesay MM et al, 2006; Atallah E et al, 2008). A recent review of RARS-T summarized the similarities between RARS-T and ET: the prognosis for these two diseases was similar, the megakaryocyte morphology was identical (by definition), and they had JAK2 V617F and MPL W515 point mutations at a similar frequency (Wardrop D and Steensma DP, 2009). These authors concluded that ring sideroblasts were a non-specific finding and that the designation as RARS-T rather than ET gave no additional biologic insight and had no special treatment implications, other than the need to consider the effects of managing thrombocytosis and anemia. They further stated that molecular results should prompt reconsideration of RARS-T as a distinct category.
4.2 Atypical chronic myeloid leukemia, BCR-ABL1 negative
Although the designation “atypical chronic myeloid leukemia” has been used in the literature for many years, the term was first codified as a distinct entity under the umbrella term of “myelodysplastic/myeloproliferative diseases” in the third edition of the WHO classification. This entity was renamed “atypical chronic myeloid leukemia, BCR-ABL1 negative” (aCML) in the fourth edition, while the diagnostic criteria remained similar (Vardiman JW et al, 2008). aCML is an extremely rare leukemic disorder characterized by both myelodysplastic and myeloproliferative features at the time of initial diagnosis. Patients with aCML present with PB leukocytosis due to increased number of neutrophils and their precursors with prominent dysgranulopoiesis. No absolute basophilia or monocytosis is present. Anemia is very common and platelet counts are variable. The BM is typically hypercellular with granulocytic predominance often associated with multilineage dysplasia. Conventional cytogenetics commonly shows abnormalities, especially trisomy 8 and del(20q), but there is no evidence of the BCR-ABL1 fusion gene, or PDGFRA or PDGFRB rearrangement. JAK2 V617F, NRAS, and KRAS mutations have been reported.
To this point, we have simply summarized the fourth edition of the WHO classification. There is very little information on aCML in the literature (Bernett JM et al, 1994; Hernandez JM et al, 2000; Breccia M et al, 2006). Some investigators have used the term Ph-negative CML interchangeably (Kurzrock R et al, 2001). Although we agree that there are cases that, in part, resemble CML and yet lack BCR-ABL1 and are associated with dysplasia, it seems clear that aCML is a heterogeneous and poorly defined “entity”, if it indeed is an entity. In one study at our institution (Kurzrock 2001), a retrospective review of aCML cases showed that appropriately one-third of these cases were better classified as chronic neutrophilic leukemia. We suspect that some other cases may represent CMML, detected before the monocytic component is sufficiently high.
Furthermore, the term aCML is both unimaginative and confusing since it carries little molecular resemblance to Ph+ CML. Why not use a term that is at least self-explanatory, such as “chronic granulocytic proliferation, BCR-ABL1 negative, associated with dysplasia”? We admit this term is not catchy and alternative suggestions are welcome.
5. Myelodysplastic syndromes
Several major changes have been made in the categorization of myelodysplastic syndromes (MDS) in the fourth edition of the WHO classification. These changes include incorporation of refractory cytopenia with unilineage dysplasia (RCUD) and refractory cytopenia of childhood (RCC) as separate entities. As a result, there are now seven diseases in this category. These diseases not already mentioned include refractory anemia with ringed sideroblasts, refractory cytopenia with multilineage dysplasia, refractory anemia with excess blasts (RAEB-1 and 2), MDS with isolated del(5q), and MDS unclassifiable. The category of RAEB-1 also has been redefined to include cases with 2–4% blasts in the PB to emphasize the prognostic significance of increased blasts in the PB. A list of cytogenetic abnormalities that are considered presumptive evidence of MDS in the presence of a refractory cytopenia but without morphologic evidence of dysplasia also has been introduced in the current WHO classification (Brunning RD et al, 2008).
5.1 Refractory cytopenia with unilineage dysplasia
Cases that are now recognized as RCUD were considered as refractory anemia (RA) or MDS, unclassifiable in the third edition of the WHO classification. Such cases encompass patients with unicytopenia or bicytopenia associated with unilineage dysplasia. The WHO classification also recommends criteria for defining cytopenia: <10 g/dL for hemoglobin, <1.8 × 109/L for absolute neutrophils, and <100 × 109/L for platelets. An advantage of this category is that one can further specify lineage: refractory anemia, neutropenia, or thrombocytopenia. This approach has more meaning than lumping these cases into RA as was done previously. Blasts are absent or very rare in the PB of RCUD patients. Patients with pancytopenia and unilineage dysplasia are still classified as having MDS, unclassifiable, owing to the uncertain clinical significance of these findings (Brunning RD et al, 2008).
5.2 Refractory cytopenia of childhood
A second important addition to MDS is the provisional entity, refractory cytopenia of childhood (RCC). This category encompasses children with MDS that have persistent cytopenia with less than 2% blasts in the PB and less than 5% blasts in the BM (Baumann I et al, 2008). Unlike adult MDS, pediatric patients often present with multilineage dysplasia and/or increased myeloblasts. Single lineage dysplasia, such as RA, is less common (McKenna RW, 2004). Ring sideroblasts (RS) are extremely rare in children, and their presence is highly suggestive of a mitochondrial disorder or disorder of heme synthesis (Bader-Meunier B et al 1994; Hasel H, 2007). Chromosome abnormalities are detected more frequently in pediatric than in adult MDS cases; monosomy 7 or del(7q) is most common. Monosomy 5 and del(5q), common aberrations in adult MDS, are not usually seen in children (McKenna RW, 2004). The most difficult diagnostic challenge in pediatric MDS occurs when the blast count is not elevated and clonality cannot be established. These cases must be extensively worked up for secondary causes of dyspoiesis, including nutritional deficiency, medications, toxins, metabolic diseases, infections, autoimmune diseases, growth factor therapy, and congenital disorders of hematopoiesis. In fact, approximately one-third of children with MDS have a predisposing genetic condition such as Fanconi anemia or Down syndrome (McKenna RW, 2004). Furthermore, a greater proportion of pediatric patients with MDS have hypocellular BM; therefore the distinction of MDS from acquired aplastic anemia (AA) and inherited BM failure syndromes is often difficult as discussed below. Myeloid leukemia in children with Down syndrome has unique clinical and biological features and is considered as a separate entity in the current WHO classification.
5.3 Hypoplastic MDS
Hypoplastic MDS accounts for approximately 10% of adult and 70% of pediatric cases of MDS, and is usually defined as BM less than 30% cellular at time of diagnosis in patients younger than 60 years of age, or less than 20% cellular in patients ≥60 years. Hypoplastic MDS represents a difficult diagnostic challenge, especially in distinguishing MDS from acquired AA. The degree of dysplasia and the blast counts appreciated on BM aspirate smears may overlap, and the hypocellularity renders interpretation difficult. Thus, an adequate core biopsy specimen with a touch imprint is necessary. The fact that some cases of AA demonstrate clonal cytogenetic abnormalities and knowledge that a subset of patients with AA progress to MDS further complicate the separation of these two disorders (Young NS, 1992; Maciejewski JP and Selleri C, 2004; Dolan MM et al, 2006; Konoplev S et al, 2007). It has been reported that some pediatric patients with AA may develop MDS with marked dysmegakaryopoiesis and monosomy 7 (Dolan MM et al, 2006). Another study showed that the use of growth factors might unmask hypoplastic MDS that mimicked AA (Konoplev S et al, 2007). Bennett and Orazi summarized the results of the studies by the French-American-British (FAB) Cooperative Leukemia Working Group, and recommended guidelines for distinguishing hypoplastic MDS from AA (Bennett JM and Orazi A, 2009). The presence of blasts in the PB, as well as an increased percentage of CD34-positive blasts and the tendency of these blasts to form aggregates in the abnormal central BM cavity was indicative of MDS. Thus, an effort should be made to count at least 100 cells in the PB and 200 cells in the BM to determine the percentage of blasts in spite of marked leukopenia. Dysplasia in granulocytes or megakaryocytes is abnormal and inconsistent with a diagnosis of AA. The presence of greater than 10% hypogranular or pseudo-Pelger-Huet neutrophils is indicative of MDS; smaller numbers raise the suspicion but are not definitive. In addition, the presence of easily identifiable micromegakaryocytes (as highlighted by CD61 immunostaining) in an architecturally disorganized BM favors MDS over AA. Erythroid dysplasia, if present alone, must be moderate or severe to support a diagnosis of MDS. The presence of ring sideroblasts (rare in pediatric cases) is considered as evidence of dyserythropoiesis in this setting. The presence of reticulin fibrosis also favors MDS over AA. On the basis of these guidelines, special stains for reticulum and iron, as well as immunohistochemical stains for CD34, CD61 and CD117 are very useful tools in evaluating hypocellular BM for MDS versus AA. In addition, flow cytometry immunophenotyping, cytogenetic and molecular analyses are valuable ancillary studies.
5.4 MDS with fibrosis
Moderate to marked BM fibrosis is observed in approximately 15% of MDS and has been identified as a distinct subgroup of MDS (MDS-F) that is associated with multilineage dysplasia, poor-risk cytogenetics, high transfusion requirement, and poor prognosis (Della Porta MG et al, 2009). MDS-F can be a diagnostic challenge because BM smears are generally of poor quality for assessment of dysplasia and blast percentage. Evaluation of a touch imprint prepared from an adequate core biopsy specimen is extremely helpful in this scenario. Histologic assessment of the core biopsy specimen and immunohistochemical analysis using antibodies for CD34, CD61 and CD117 are often mandatory, and correlation with the PB smear and complete blood count (CBC) findings is necessary for the diagnosis of MDS-F. Important entities in the differential diagnoses of MDS-F include acute panmyelosis with myelofibrosis, acute megakaryocytic leukemia, MPN with fibrosis, and MDS/MPN with fibrosis, as well as non-myeloid neoplasms, such as hairy cell leukemia and metastatic tumors. The diagnosis of MDS-F can usually be made when there are moderate to marked reticulin fibrosis, dysplasia in at least two cell lineages, and associated PB cytopenias. Organomegaly is not a prominent feature in patients with MDS-F. Unlike MPN and MDS/MPN with fibrosis, JAK2 V617F mutation is less frequent in MDS-F (Della Porta MG et al, 2009).
5.5 Idiopathic cytopenia of uncertain significance
Idiopathic cytopenia of uncertain significance (ICUS) is a term proposed for patients with persistent (≥ 6 months), unexplained cytopenia in one or more myeloid lineages (hemoglobin <11g/dL, absolute neutrophil count <1.5 × 109/L, platelet <100 × 109/L), and who do not fulfill diagnostic criteria for MDS (Valent P et al, 2007). ICUS is a diagnosis of exclusion and requires detailed history (e.g. nutritional deficiency, medications, toxin exposure, infections, etc), CBC with differential count, careful examination of PB and BM smears, iron stain and examination of BM biopsy specimen with immunohistochemistry, as well as ancillary studies including flow cytometry immunophenotyping, chromosome analysis (Table 5), and molecular analysis. An algorithm for evaluation of a patient with cytopenia is illustrated in Figure 2. Patients with ICUS should be closely followed with frequent repeat testing to confirm or exclude MDS. A recent abstract from the Mayo Clinic reported that ICUS is rare, with only 10 cases identified over 12 years; 60% of patients went on to develop MDS within 1 to 10 years (Hanson CA et al, 2010).
Table 5.
Presumptive karyotypic evidence of myelodysplastic syndrome*
| Type | Cytogenetic abnormalities |
|---|---|
| Balanced | t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1), t(1;3)(p36.3;q21.2), t(2;11)(p21;q23), inv(3)(q21q26.2), t(6;9)(p23;q34) |
| Unbalanced | −7 or del(7q), −5 or del(5q), i(17q) or t(17p), −13 or del(13q), del(11q), del(12p) or t(12p), del(9q), idic(X)(q13) |
These cytogenetic abnormalities can be used as presumptive evidence of myelodysplastic syndrome in the setting of persistent cytopenias of undertermined etiology without definite morphologic dysplasia
Figure 2.
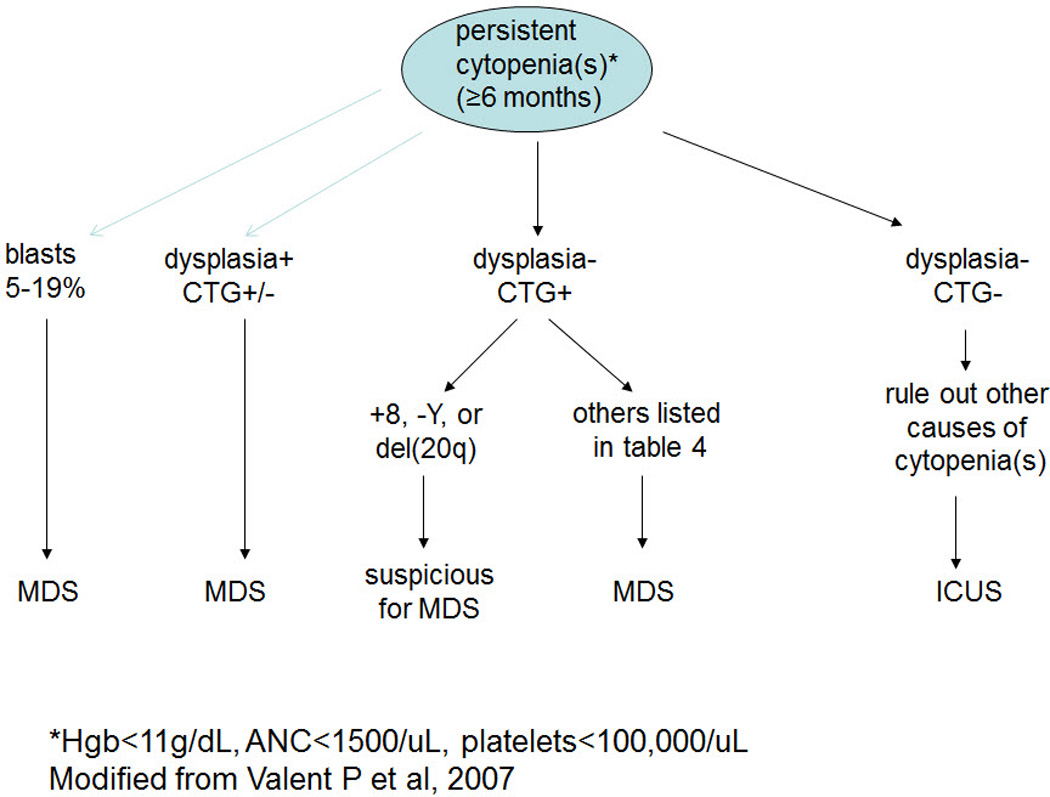
Diagnostic algorithm of cases with persistent cytopenia.
5.6 Refractory anemia with excess blasts in transformation – revisited
Refractory anemia with excess blasts in transformation (RAEB-T) is a high-grade MDS with increased blasts in the range of 20–29% as was defined in the FAB classification. This category was eliminated in the third edition of the WHO classification based on the fact that the survival of patients with RAEB-T was similar to those with a blast count of 30% or more. However, this decision has been somewhat controversial since the third edition was published in 2001, and the issues are not yet resolved currently. A number of studies have shown that many features of patients with RAEB-T, such as the frequency of poor-prognosis cytogenetic abnormalities and the presence of antecedent hematologic diseases, more resemble that of MDS than AML (Estey E et al, 1997; Albitar M et al, 2000; Huh YO et al, 2002). Furthermore, MDS is a disease in which ineffective hematopoiesis is thought to be attributable for the most part, to apoptosis unlike AML in which impaired differentiation and proliferation play greater roles. It has been shown that there are no significant differences in levels of apoptosis between RAEB-T and RAEB (Huh YO et al, 2002). Therefore, RAEB-T may be biologically closer to an advanced stage of MDS than it is to AML. To state this point another way, although the decision to change the cutoff may make sense using survival data, the historical cutoffs of 20% and 30% may reflect, in part, true biologic differences. Molecular markers are needed to further resolve this issue. Until they are developed, one should take into consideration the unique aspects of RAEB-T as we develop novel therapies that specifically target the biological and molecular abnormalities in leukemic cells. For the purpose of enrolling patients into certain treatment protocols, it may be helpful to classify such disease process as both AML based on the WHO classification and RAEB-T based on the FAB classification, as we do currently at our institution.
6. Acute myeloid leukemias
6.1 AML with recurrent genetic abnormalities
The group “AML with recurrent genetic abnormalities” has been expanded to include seven neoplasms associated with specific genetic abnormalities. Recent neoplasms included in this categoiry include AML associated with t(6;9)(p23;q34)/DEK-NUP214, AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2)/RPN1-EVI1, and AML with t(1;22)(p13;q13)/RBM15-MKL1. Cases with mutated NPM1 or mutated CEBPA are also listed as provisional entities.
In “acute promyelocytic leukemia (APL) with t(15;17)(q22;q21)/PML-RARA”, variant RARA translocations with other partner genes are recognized separately since not all have typical APL features and some are resistant to all-trans retinoic acid (ATRA). The former entity “AML with 11q23/MLL abnormalities” has been redefined as “AML with t(9;11)(p22;q23)/MLLT3-MLL”. Balanced translocations other than those involving MLLT3 should be specified in the diagnosis. Other abnormalities of MLL, such as partial tandem duplication, should not be placed under this entity (Arber DA et al, 2008). As a result of the rapid advances in the understanding of molecular leukemogenesis, this group is destined to undergo additional modifications in future classification revisions. AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2)/RPN1-EVI1 also may be more appropriately listed as “AML with EVI1 overexpression” because cases that overexpress EVI1 as a result of translocation with partners other than RPN1 have also been associated with aggressive clinical course (Lin P et al, 2006; Yin CC et al, 2006; Lennon PA et al, 2007). Other molecular abnormalities such as FLT-3 and KIT mutations are well-studied events that play important roles in leukemogenesis, whereas WT1 mutation and overexpression of BAALC, ERG and MN1 have more recently been shown to be associated with relatively poor clinical outcome (Mrozek K et al, 2007).
It should also be noted that the blast percentage cutoff for most types of AML with a recurrent genetic abnormalities remains 20%, but this is not true for AML with t(8;21)(q22;q22)/RUNX1-RUNXT1, inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB-MYH11, and t(15;17)(q22;q21)/PML-RARA. In these three entities recognition of the molecular abnormality defines the disease, even when the blast count in PB and BM is below 20%. The scientific basis for handling the blast count in this inconsistent manner for the other neoplasms in this group is unclear. Based on our experience, we have seen patients with t(8;21)(q22;q22)/RUNX1-RUNXT1, or inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB-MYH11, or t(15;17)(q22;q21)/PML-RARA and a blast count less than 20% in PB and BM. Follow-up showed a rapid rise in blast count supporting the diagnosis of AML based on the presence of the molecular abnormality and therefore we agree with the WHO approach for these neoplasm. It seems reasonable, in our opinion, to extend this approach to other AMLs associated with distinctive translocations, such as t(6;9)(p23;q34)/DEK-NUP214, as these patients in our experience also fair poorly (Oyarzo MP et al, 2004). We agree that the 20% blast cutoff should be retained for provisional AML categories based on gene mutations (e.g. AML with NPM1 mutation). We believe these issues need to be addressed in future revisions of the WHO classification.
6.2 AML with myelodysplasia-related changes
Acute myeloid leukemia with myelodysplasia-related changes (AML-MRC) is the term proposed by the fourth edition of the WHO classification to encompass all AML cases with a previous history of MDS or MDS/MPN, or with myelodysplasia-related cytogenetic abnormalities, or with morphologic evidence of dysplasia in 50% or more of the cells in two or more myeloid lineages (Arber DA et al, 2008). Even in the short time since publication, opinion differs regarding whether this category is truly a distinct entity. Earlier studies suggested that myelodysplasia-related morphologic abnormalities correlated with unfavorable cytogenetic abnormalities but had no independent impact on prognosis (Haferlach T et al, 2003; Wandt H et al, 2008). Weinberg et al. more recently compared patients with AML, NOS to patients with AML-MRC (Weinberg OK et al, 2009). Patients in the latter group were significantly older, presented with a lower hemoglobin level, exhibited a decreased frequency of CEBPA mutations, and had a significantly worse overall survival. The authors concluded that their data supported the clinical, morphologic, and cytogenetic criteria for AML-MRC. We believe that cases of AML-MRC differ from cases of AML, NOS. However, we wonder if the category of AML-MRC is too heterogeneous. In our opinion, patients with no morphologic evidence of MDS or history of MDS who present with a cytogenetic abnormality (e.g. del(7q) or monosomy 7) seem different from patients with overt MDS. It also seems likely that the pathogenic mechanisms attributable to the wide number of cytogenetic abnormalities in MDS are different. It is expected that AML-MRC is a term that will be substantially modified in future revisions of the WHO classification.
6.3 Therapy-related myeloid neoplasms
The fourth edition of the WHO classification uses the term “therapy-related myeloid neoplasms” (t-MN) to cover the spectrum of disorders previously known as t-AML, t-MDS, or t-MDS/MPN occurring as late complications of cytotoxic chemotherapy and/or radiation therapy (Vardiman JW et al, 2008). Excluded from this category is blast crisis of an underlying MPN since it is often not possible to determine if leukemic transformation is a result of disease evolution or is therapy-related. It should be noted that the term “therapy-related” is based on the patient’s history of prior exposure to cytotoxic agents and/or radiation, but the causal relationship remains to be proven, and the etiology and specific factors that predispose patients to t-MN is still largely elusive. Most cases show multilineage dysplasia, complex cytogenetic aberrations, and a poor prognosis (Yin CC et al, 2005). Approximately 75% of cases of t-MN develop after exposure to alkylating agents and/or radiation, and are characterized by a relatively long latency interval (5–10 years after exposure), presentation with t-MDS, and loss of chromosomes 5 and/or 7. The remaining patients with t-MN usually present in a relatively short interval (1–5 years) after therapy with topoisomerase II inhibitors. This subset of patients tends to develop overt AML without an antecedent MDS, and is associated with balanced chromosomal translocations involving 11q23 (MLL gene) or 21q22 (RUNX1 gene). The similarities in clinicopathologic and molecular genetic features between t-MDS and t-AML including the presence of multilineage dysplasia, similar chromosomal aberrations, as well as rapid progression from t-MDS to t-AML in most cases, in our opinion, justifies combining t-MDS and t-AML into one category. The newly proposed name, t-MN, is reasonable although the term t-MDS/AML also seems reasonable.
The development of t-MN is a result of a complex interplay of a number of factors including direct mutational effect of cytotoxic agents and/or radiation, the effect of an ineffective BM microenvironment due to injury to vascular supply or increasing fibrosis (caused by prior treatment), genetic instability likely driving BM dysplasia, immunosuppression from prior or current therapy, host genetic predisposition, and high frequency of unfavorable cytogenetic abnormalities. Various genetic pathways involving chromosomal rearrangements and mutations in multiple genes (e.g. FLT-3, RAS, KIT, RUNX1, MLL, TP53, CBF, NPM1, CEBPA) have been implicated. Based on cytogenetic abnormalities at initial presentation, Pedersen-Bjergaard et al.proposed eight different genetic pathways for the multistep development of t-MN (Pedersen-Bjergaard J et al, 1995), which have been modified subsequently with the discovery of more molecular genetic aberrations (Pedersen-Bjergaard J et al, 2006). These data raise the possibility that the category of t-MN is likely to be subdivided in future classification systems.
6.4 Acute erythroid leukemia
The category of acute erythroid leukemia (AEL) has substantially evolved as it is defined in the current WHO classification, in large part as a result of the new entity “AML-MRC” as previously discussed. Many cases of AML with blasts ≥20% of all BM cells and erythroid predominance that used to be classified as AEL are now classified as AML-MRC because they are associated with dysplasia. Cases with blasts <20% of all BM cells but ≥20% of the non-erythroid cells are classified as AEL, and cases with blasts <20% of the non-erythroid cells are classified as MDS (Figure 3). Using the criteria of the 2008 WHO classification, the distinction between AEL and AML-MRC or MDS with erythroid hyperplasia is based solely on the percentage of blasts. In fact, similarities exist among these entities, in particular, the high frequency of preceding MDS or the presence of multilineage dysplasia, as well as common cytogenetic abnormalities (Hasserjian RP et al, 2009; Santos FP et al, 2009). Hasserjian et al studied the clinicopathologic and cytogenetic features of 124 patients with AEL as defined using the 2008 WHO classification in comparison with patients with AML-MRC or MDS associated with erythroid hyperplasia, and concluded that AEL was part of a continuum with AML-MRC and MDS with erythroid hyperplasia, where karyotype rather than an arbitrary blast cutoff was the most important prognostic factor (Hasserjian RP et al, 2009). Another recent study also showed that AEL, as currently defined in the WHO classification, is both substantially less common than the reported frequency of 5% of AML and nevertheless remains heterogeneous. One subset of AEL patients had relatively low blast count and was diploid with a relatively good prognosis. The other subset of patients had cytogenetic abnormalities similar to those seen in patients with MDS and was associated with a poor prognosis (Kasyan A et al, in press).
Figure 3.
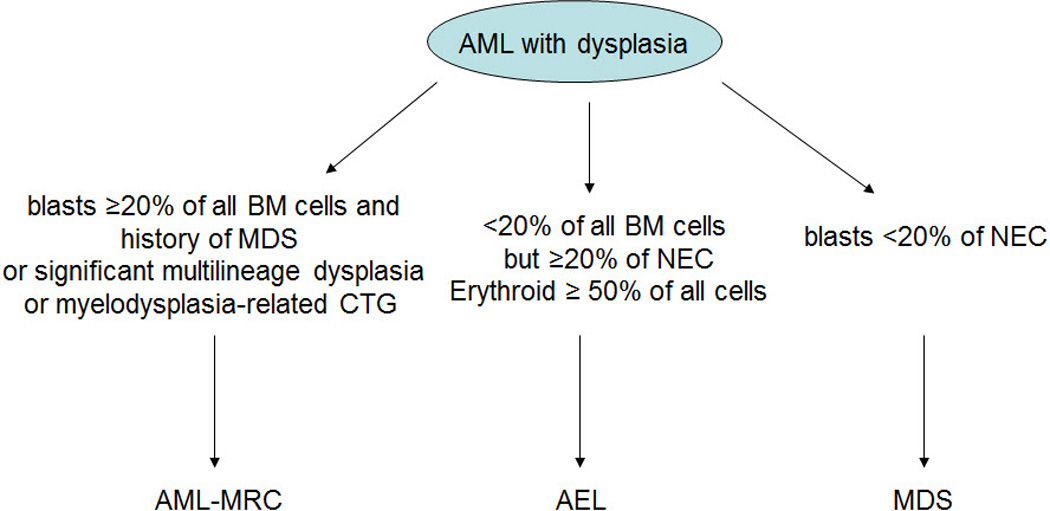
Diagnostic algorithm of AML with myelodysplasia and erythroid hyperplasia.
6.5 Acute megakaryoblastic leukemia
Acute megakaryoblastic leukemia is defined as an acute leukemia with 20% or more blasts of which at least 50% are of megakaryocytic lineage. Similar to AEL, acute megakaryoblastic leukemia has become an even more rare entity using the criteria of the fourth edition of the WHO classification (Oki Y et al, 2006). Cases previously classified in this category should now be placed in the appropriate genetic category if they are associated with inv(3)(q21q26.2) or t(3;3)(q21;q26.2)/RPN1-EVI1 or t(1;22)(p13;q13)/RBM15-MKL1. Those cases with myelodysplasia-related cytogenetic abnormalities should be re-classified as AML-MRC. Down syndrome-related cases are also excluded from this category and designated separately.
6.6 A role for the FAB classification in AML?
The FAB classification of AML, last updated in 1985, was based predominantly on traditional morphologic and cytochemical criteria. Although the current WHO classification of AML, NOS is essentially a modified FAB classification, there is little mention of the FAB, not even as synonyms for the various categories. Although we agree that it is best to define myeloid diseases on the basis of well-defined molecular alterations, the FAB classification, in our opinion, still has some value and the clinicians at our institution continue to ask us to provide both FAB and WHO terminology for AML cases. There are some advantages to this approach. First, a FAB designation can be provided rapidly after morphological examination and assessment of cytochemical stains (mostly myeloperoxidase and butyrate esterase). We then include the WHO terminology once the results of cytogenetics and molecular tests are available. Second, some of the FAB categories such as M3 and M4Eo highly correlate with the t(15;17)(q22;q21)/PML-RARA or inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB-MYH11, respectively. Lastly, the terminology of the FAB classification is convenient. For example, M0 is much easier to say or write than AML with minimal differentiation.
7. Conclusions
In summary, the fourth edition of the WHO classification of myeloid neoplasms clearly manifests the progress made in deciphering the molecular pathogenesis of myeloid neoplasms, as well as the development of targeted therapy since 2001, the year the third edition was published. It is expected that additional updates and revisions to this classification will be needed as progress continues to occur rapidly. It is hoped that the time to publication of the future fifth edition will be less than the seven-year interval between the third and the fourth edition.
Acknowledgement
We thank Karen Phillips from Scientific Publication at The University of Texas M. D. Anderson Cancer Center for editorial review of the manuscript.
References
- Akin C. Molecular diagnosis of mast cell disorders: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2006;8:412–419. doi: 10.2353/jmoldx.2006.060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albitar M, Beran M, O'Brien S, Kantarjian H, Frieriech E, Keating M, Estey E. Differences between refractory anemia with excess blasts in transformation and acute myeloid leukemia. Blood. 2000;96:372–733. [PubMed] [Google Scholar]
- Arber DA, Brunning RD, Le Beau MM, Falini B, Vardiman JW, Porwit A, Thiele J, Bloomfield CD. Acute myeloid leukaemia with recurrent genetic abnormalities. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 110–123. [Google Scholar]
- Arber DA, Brunning RD, Orazi A, Bain BJ, Porwit A, Vardiman JW, Le Beau MM, Greenberg PL. Acute myeloid leukaemia with myelodysplasia-related changes. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 124–126. [Google Scholar]
- Atallah E, Nussenzveig R, Yin CC, Bueso-Ramos C, Tam C, Manshouri T, Pierce S, Kantarjian H, Verstovsek S. Prognostic interaction between thrombocytosis and JAK2 V617F mutation in the WHO subcategories of myelodysplastic/myeloproliferative disease-unclassifiable and refractory anemia with ringed sideroblasts and marked thrombocytosis. Leukemia. 2008;22:1295–1298. doi: 10.1038/sj.leu.2405054. [DOI] [PubMed] [Google Scholar]
- Bader-Meunier B, Rotig A, Meilot F, Lavergne JM, Croisille L, Rustin P, Landrieu P, Dommergues JP, Munnich A, Tchernia G. Refractory anaemia and mitochondrial cytopathy in childhood. Br J Haematol. 1994;87:381–385. doi: 10.1111/j.1365-2141.1994.tb04926.x. [DOI] [PubMed] [Google Scholar]
- Bain BJ, Gilliland DG, Horny HP, Vardiman JW. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB or FGFR1. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 68–73. [Google Scholar]
- Baumann I, Niemeyer CM, Bennett JM, Shannon K. Childhood myelodyspastic syndrome. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 104–107. [Google Scholar]
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, Sultan C, Cox C. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol. 1994;87:746–754. doi: 10.1111/j.1365-2141.1994.tb06734.x. [DOI] [PubMed] [Google Scholar]
- Bennett JM, Orazi A. Diagnostic criteria to distinguish hypocellular acute myeloid leukemia from hypocellular myelodysplastic syndromes and aplastic anemia: recommendations for a standardized approach. Haematologica. 2009;94:264–268. doi: 10.3324/haematol.13755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot M, Garand R, Hamidou M, Hermouet S. The JAK2-V617F mutation and essential thrombocythemia features in a subset of patients with refractory anaemia with ring sideroblasts (RARS) Blood. 2006;108:1781–1782. doi: 10.1182/blood-2006-03-008227. [DOI] [PubMed] [Google Scholar]
- Breccia M, Biondo F, Latagliata R, Carmosino I, Mandelli F, Alimena G. Identification of risk factors in atypical chronic myeloid leukemia. Hematologica. 2006;91:1568–1570. [PubMed] [Google Scholar]
- Brunning RD, Orazi A, Germing U, Le Beau MM, Porwit A, Baumann I, Vardiman JW, Hellstrom-Lindberg E. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 88–93. [Google Scholar]
- Brunning RD, Hasserjian RP, Porwit A, Bennett JM, Orazi A, Thiele J, Hellstrom-Lindberg E. Refractory cytopenia with unilineage dysplasia. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 94–95. [Google Scholar]
- Ceesay MM, Lea NC, Ingram W, Westwood NB, Gaken J, Mohamedali A, Cervera J, Germing U, Gattermann N, Giagounidis A, Garcia-Casado Z, Sanz G, Mufti GJ. The JAK2 V617F mutation is rare in RARS but common in RARS-T. Leukemia. 2006;20:2060–2061. doi: 10.1038/sj.leu.2404373. [DOI] [PubMed] [Google Scholar]
- Cortes JE, Talpaz M, O’Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, Verstovsek S, Rios MB, Shan J, Kantarjian HM. Staging of chronic myeloid leukemia in the imatinib era. An evaluation of the World Health Organization proposal. Cancer. 2006;106:1306–1315. doi: 10.1002/cncr.21756. [DOI] [PubMed] [Google Scholar]
- Cross NC, Reiter A. Fibroblast growth factor receptor and platelet-derived growth factor receptor abnormalities in eosinophilic myeloproliferative disorders. Acta Haematol. 2008;119:199–206. doi: 10.1159/000140631. [DOI] [PubMed] [Google Scholar]
- Della Porta MG, Malcovati L, Boveri E, Travaglino E, Pietra D, Pascutto C, Passamonti F, Invernizzi R, Castello A, Magrini U, Lazzarino M, Cazzola M. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J Clin Oncol. 2009;27:754–762. doi: 10.1200/JCO.2008.18.2246. [DOI] [PubMed] [Google Scholar]
- Dolan MM, Singleton TP, Neglia J, Cioc A. Aplastic anemia and monosomy 7-associated dysmegakaryocytopoiesis. Am J Clin Pathol. 2006;126:925–930. doi: 10.1309/50GWDKVWU3VWL5XW. [DOI] [PubMed] [Google Scholar]
- Estey E, Thall P, Beran M, Kantarjian H, Pierce S, Keating M. Effect of diagnosis (refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, or acute myeloid leukemia [AML]) on outcome of AML-type chemotherapy. Blood. 1997;90:2969–2977. [PubMed] [Google Scholar]
- Gattenlohner S, Peter C, Bonengel M, Einsele H, Bargou R, Muller-Hermelink HK, Marx A. Detecting the JAK2 V167F mutation in fresh and ‘historic’ blood and bone marrow. Leukemia. 2007;21:1599–1602. doi: 10.1038/sj.leu.2404701. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Schoch C, Löffler H, Gassmann W, Kern W, Schnittger S, Fonatsch C, Ludwig WD, Wuchter C, Schlegelberger B, Staib P, Reichle A, Kubica U, Eimermacher H, Balleisen L, Grüneisen A, Haase D, Aul C, Karow J, Lengfelder E, Wörmann B, Heinecke A, Sauerland MC, Büchner T, Hiddemann W. Morphologic dysplasia in de novo acute myeloid leukemia (AML) is related to unfavorable cytogenetics but has no independent prognostic relevance under the conditions of intensive induction therapy: results of a multiparameter analysis from the German AML Cooperative Group studies. J Clin Oncol. 2003;21:256–265. doi: 10.1200/JCO.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Hanson CA, Durnick DK, Hoyer JD, Hodnefield JM, Ketterling RP, Steensma DP. Idiopathic cytopenia of undetermined significance (ICUS): the importance of clinical, morphologic, and cytogenetic evaluation. Mod Pathol. 2010;23(Suppl 1):300A. [Google Scholar]
- Hasel H. Myelodysplastic and myeloproliferative disorders in children. Curr Opin Pediatr. 2007;19:1–8. doi: 10.1097/MOP.0b013e3280128ce8. [DOI] [PubMed] [Google Scholar]
- Hasserjian RP, Zuo Z, Garcia C, Tang G, Kasyan A, Luthra R, Abruzzo LV, Kantarjian HM, Medeiros LJ, Wang SA. Acute erythroid leukemia: a reassessment using criteria refined in the 2008 WHO classification. Blood. 2009 doi: 10.1182/blood-2009-09-243964. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez JM, del Canizo MC, Cuneo A, Garcia JL, Gutierrez NC, Gonzalez M, Castoldi G, San Miguel JF. Clinical hematological and cytogenetic characteristics of atypical chronic myeloid leukemia. Ann Oncol. 2000;11:441–444. doi: 10.1023/a:1008393002748. [DOI] [PubMed] [Google Scholar]
- Huh YO, Jilani I, Estey E, Giles F, Kantarjian H, Freireich E, Albitar M. More cell death in refractory anemia with excess blasts in transformation than in acute myeloid leukemia. Leukemia. 2002;16:2249–2252. doi: 10.1038/sj.leu.2402704. [DOI] [PubMed] [Google Scholar]
- Jackson CC, Medeiros LJ, Miranda RN. 8p11 myeloproliferative syndrome: a review. Hum Pathol. 2010;41:461–476. doi: 10.1016/j.humpath.2009.11.003. [DOI] [PubMed] [Google Scholar]
- James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, White H, Zoi C, Loukopoulos D, Terpos E, Vervessou EC, Schultheis B, Emig M, Emst T, Lengfelder E, Hehlmann R, Hochhaus A, Oscier D, Silver RT, Reiter A, Cross NC. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–2168. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- Kasyan A, Medeiros LJ, Zuo Z, Santos FP, Ravandi-Kashani F, Miranda R, Vadhan-Raj S, Koeppen H, Bueso-Ramos CE. Acute erythroid leukemia as defined in the World Health Organization Classification is a rare and pathogenetically heterogeneous disease. Mod Pathol. in press doi: 10.1038/modpathol.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konoplev S, Medeiros LJ, Lennon PA, Prajapati S, Kanungo A, Lin P. Therapy may unmask hypoplastic myelodysplastic syndrome that mimics aplastic anemia. Cancer. 2007;110:1520–1526. doi: 10.1002/cncr.22935. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- Kurzrock R, Bueso-Ramos CE, Kantarjian H, Freireich E, Tucker SL, Siciliano M, Pilat S, Talpaz M. BCR rearrangement-negative chronic myelogenous leukemia revisited. J Clin Oncol. 2001;19:2915–2926. doi: 10.1200/JCO.2001.19.11.2915. [DOI] [PubMed] [Google Scholar]
- Kvasnicka HM, Thiele J. Prodromal myeloproliferative neoplasms: the 2008 WHO classification. Am J Hematol. 2010;85:62–69. doi: 10.1002/ajh.21543. [DOI] [PubMed] [Google Scholar]
- Lennon PA, Abruzzo LV, Medeiros LJ, Cromwell C, Zhang X, Yin CC, Kornblau SM, Konopieva M, Lin P. Aberrant EVI1 expression in acute myeloid leukemias associated with the t(3;8)(q26;q24) Cancer Genet Cytogenet. 2007;177:37–42. doi: 10.1016/j.cancergencyto.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Lim KH, Pardanani A, Tefferi A. KIT and mastocytosis. Acta Haematol. 2008;119:194–198. doi: 10.1159/000140630. [DOI] [PubMed] [Google Scholar]
- Lin P, Medeiros LJ, Yin CC, Abruzzo LV. Translocation (3;8)(q26;q24): a recurrent chromosomal abnormality in myelodysplastic syndrome and acute myeloid leukemia. Cancer Genet Cytogenet. 2006;166:82–85. doi: 10.1016/j.cancergencyto.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Ma W, Kantarjian H, Zhang X, Sun W, Buller AM, Jilani I, Schwartz JG, Giles F, Albitar M. Higher detection rate of JAK2 mutation using plasma. Blood. 2008;111:3906–3907. doi: 10.1182/blood-2008-02-139188. [DOI] [PubMed] [Google Scholar]
- Maciejewski JP, Selleri C. Evolution of clonal cytogenetic abnormalities in aplastic anemia. Leuk Lymph. 2004;45:433–440. doi: 10.1080/10428190310001602363. [DOI] [PubMed] [Google Scholar]
- McKenna RW. Myelodysplasia and myeloproliferative disorders in children. Am J Clin Pathol. 2004;122(Suppl 1):S58–S69. doi: 10.1309/63C2VQ4GAN2C18RQ. [DOI] [PubMed] [Google Scholar]
- Mirza I, Sekora D, Frantz C. Testing for JAK2 V617F mutation across specimen types yields concordant results. J Clin Pathol. 2008;61:975. doi: 10.1136/jcp.2007.054379. [DOI] [PubMed] [Google Scholar]
- Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki Y, Kantarjian HM, Zhou X, Cortes J, Faderl S, Verstovsek S, O'Brien S, Koller C, Beran M, Bekele BN, Pierce S, Thomas D, Ravandi F, Wierda WG, Giles F, Ferrajoli A, Jabbour E, Keating MJ, Bueso-Ramos CE, Estey E, Garcia-Manero G. Adult acute megakaryocytic leukemia: an analysis of 37 patients treated at M.D. Anderson Cancer Center. Blood. 2006;107:880–884. doi: 10.1182/blood-2005-06-2450. [DOI] [PubMed] [Google Scholar]
- Oyarzo MP, Lin P, Glassman A, Bueso-Ramos CE, Luthra R, Medeiros LJ. Acute myeloid leukemia with t(6;9)(p23;q34) is associated with dysplasia and a high frequency of flt3 gene mutations. Am J Clin Pathol. 2004;122:348–358. doi: 10.1309/5DGB-59KQ-A527-PD47. [DOI] [PubMed] [Google Scholar]
- Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F–negative polycythemia vera. Leukemia. 2007;21:1960–1963. doi: 10.1038/sj.leu.2404810. [DOI] [PubMed] [Google Scholar]
- Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, Steensma DP, Elliott MA, Wolanskyj AP, Hogan WJ, McClure RF, Litxow MR, Gilliland DG, Tefferi A. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- Pedersen-Bjergaard J, Christiansen DH, Desta F, Andersen MK. Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2006;20:1943–1949. doi: 10.1038/sj.leu.2404381. [DOI] [PubMed] [Google Scholar]
- Pedersen-Bjergaard J, Pedersen M, Roulston D, Philip P. Different genetic pathways in leukemogenesis for patients presenting with therapy-related myelodysplasia and therapy-related acute myeloid leukemia. Blood. 1995;86:3542–3552. [PubMed] [Google Scholar]
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, DeAngelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama ME, Swierczek SI, Hickman K, Wilson A, Prchal JT. Plasma quantitation of JAK2 mutation is not suitable as a clinical test: an artifact of storage. Blood. 2009;114:223–224. doi: 10.1182/blood-2009-03-209593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos FP, Faderl S, Garcia-Manero G, Koller C, Beran M, O’Brien S, Pierce S, Freireich EJ, Huang X, Borthakur G, Bueso-Ramos C, de Lima M, Keating M, Cortes J, Kantarjian H, Ravandi F. Adult acute erythroleukemia: an analysis of 91 patients treated at a single institution. Leukemia. 2009;23:2275–2280. doi: 10.1038/leu.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupir CP, Vergilio JA, Dal Cin P, Muzikansky A, Karntarjian H, Jones D, Hasserjian RP. Philadelphia chromosome-positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol. 2007;127:642–650. doi: 10.1309/B4NVER1AJJ84CTUU. [DOI] [PubMed] [Google Scholar]
- Stevenson WS, Hoyt R, Bell A, Guipponi M, Juneja S, Grigg AP, Curtis DJ, Scott HS, Szer J, Alexander WS, Tuckfield A, Roberts AW. Genetic heterogeneity of granulocytes for the JAK2 V617F mutation in essential thrombocythaemia: implications for mutation detection in peripheral blood. Pathology. 2006;38:336–342. doi: 10.1080/00313020600820906. [DOI] [PubMed] [Google Scholar]
- Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, Talmant P, Tichelli A, Hermouet S, Skoda RC. Leukemic blasts in transformed JAK2-V617F–positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110:375–379. doi: 10.1182/blood-2006-12-062125. [DOI] [PubMed] [Google Scholar]
- Thiele J, Kvasnicka HM, Tefferi A, Barosi G, Orazi A, Vardiman JW. Primary myelofibrosis. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 44–47. [Google Scholar]
- Vardiman JW, Brunning RD, Arber DA, Le Beau MM, Porwit A, Tefferi A, Bloodfield CD, Thiele J. Introduction and overview of the classification of the myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 18–30. [Google Scholar]
- Vardiman JW, Bennett JM, Bain BJ, Brunning RD, Thiele J. Atypical chronic myeloid leukaemia, BCR-ABL1 negative. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 80–81. [Google Scholar]
- Vardiman JW, Bennett JM, Bain BJ, Baumann I, Thiele J, Orazi A. Myelodysplastic/myeloproliferative neoplasm, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 85–86. [Google Scholar]
- Vardiman JW, Arber DA, Brunning RD, Larson RA, Matutes E, Baumann I, Thiele J. Therapy-related myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 127–129. [Google Scholar]
- Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- Valent P, Horny HP, Bennett JM, Fonatsch C, Germing U, Greenberg P, Haferlach T, Haase D, Kolb HJ, Krieger O, Loken M, van de Loosdrecht A, Ogata K, Orfao A, Pfeilstocker M, Ruter B, Sperr WR, Stauder R, Wells DA. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: Consensus statements and report from a working conference. Leukemia Res. 2007;31:727–736. doi: 10.1016/j.leukres.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Wandt H, Schäkel U, Kroschinsky F, Prange-Krex G, Mohr B, Thiede C, Pascheberg U, Soucek S, Schaich M, Ehninger G. MLD according to the WHO classification in AML has no correlation with age and no independent prognostic relevance as analyzed in 1766 patients. Blood. 2008;111:1855–1861. doi: 10.1182/blood-2007-08-101162. [DOI] [PubMed] [Google Scholar]
- Wardrop D, Steensma DP. Is refractory anaemia with ring sideroblasts and thrombocytosis (RARS-T) a necessary or useful diagnostic category? Br J Haematol. 2009;144:411–419. doi: 10.1111/j.1365-2141.2008.07526.x. [DOI] [PubMed] [Google Scholar]
- Weinberg OK, Seetharam M, Ren L, Seo K, Ma L, Merker JD, Gotlib J, Zehnder JL, Arber DA. Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood. 2009;113:1906–1908. doi: 10.1182/blood-2008-10-182782. [DOI] [PubMed] [Google Scholar]
- Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661–665. doi: 10.1182/blood-2002-01-0203. [DOI] [PubMed] [Google Scholar]
- Yin CC, Cortes J, Barkoh B, Hayes K, Kantarjian H, Jones D. t(3;21)(q26;q22) in myeloid leukemia: an aggressive syndrome of blast transformation associated with hydroxyurea or antimetabolite therapy. Cancer. 2006;106:1730–1738. doi: 10.1002/cncr.21797. [DOI] [PubMed] [Google Scholar]
- Yin CC, Glassman AB, Lin P, Valbuena JR, Jones D, Luthra R, Medeiros LJ. Morphologic, cytogenetic, and molecular abnormalities in therapy-related acute promyelocytic leukemia. Am J Clin Pathol. 2005;123:840–848. doi: 10.1309/TJFF-K819-RPCL-FKJ0. [DOI] [PubMed] [Google Scholar]
- Yin CC, Jones D. Myeloproliferative neoplasms. In: Jones D, editor. Neoplastic Hematology. Humana Press; 2010. pp. 177–192. 2010. [Google Scholar]
- Young NS. The problem of clonality in aplastic anemia: Dr. Dameshek’s riddle, restated. Blood. 1992;79:1385–1392. [PubMed] [Google Scholar]
- Zhao W, Bueso-Ramos CE, Verstovsek S, Barkoh BA, Khitamy AA, Jones D. Quantitative profiling of codon 816 KIT mutations can aid in the classification of systemic mast cell disease. Leukemia. 2007;21:1574–1576. doi: 10.1038/sj.leu.2404680. [DOI] [PubMed] [Google Scholar]