

Abstract
Bispecific antibodies play an important role in immunotherapy. They have received intense interest from pharmaceutical enterprises. The first antibody drug, OKT3 (muromonab-CD3), showed great performance in clinical treatment. We have successfully developed a single-chain variable fragment (ScFv) combination of anti-CD3 ScFv and anti-CD138 ScFv with the hIgG1 Fc (hIgFc) sequence. The novel bispecific T-cell engager (BiTE) with an additional hIgFc (BiTE-hIgFc, STL001) can target T cells, natural killer cells, and multiple myeloma cells (RPMI-8226 or U266). In addition, BiTE-hIgFc (STL001) has nanomolar-level affinity to recombinant human CD138 protein and shows more potent antitumor activity against RPMI-8226 cells than that of separate aCD3-ScFv-hIgFc and aCD138-ScFv-hIgFc, or the isotype mAb in vitro or in vivo.
Keywords: Bispecific antibodies, immunotherapy, multiple myeloma, natural killer cells, T cells
Multiple myeloma is a cancer of plasma cells, a type of white blood cell normally responsible for producing antibodies.1 It is the second most common hematological malignancy. The prevailing therapeutic strategy for MM is still chemotherapy. Some novel compounds and drugs have been approved for MM in recent years, such as bortezomib, thalidomide, PEGylated liposomal doxorubicin, and lenalidomide.2–5 Despite the development of chemotherapeutics, MM remains incurable, and all patients eventually relapse with this disease. This indicates that there is a great and urgent need to develop new therapeutic strategies for MM. One of the most promising approaches is immunotherapy that effectively harnesses the cellular immune response and circumvents mechanisms of tumor evasion. Moreover, therapeutic biologicals with potent clinical applicability are relatively straightforward to design and produce.
Immunotherapy represents one of the most promising cancer therapeutic strategies in this arena.6 The immune cells, specifically CTLs, play a vital role in an effective antitumor immune response.7–9 In prevailing immunotherapy treatments, the bispecific antibody therapeutic strategy has been shown to effectively engage and activate T cells by binding CD3 molecules and tumor antigens on the surface of target tumor cells. Novel Bispecific T-cell Engager (BiTE; Micromet, Munich, Germany) antibodies are designed to transiently connect T cells with cancer cells for initiation of redirected target cell lysis.10,11 Regular IgG1 antibodies cannot engage T cells because they lack Fcγ receptors, which are needed for interactions with antibodies. BiTE antibodies are composed of two flexibly linked single-chain antibodies, one binding to CD3 molecules on T cells and the other to a surface antigen on the target cells.12 Therefore, unlike the regular T-cell response, the APC, MHC-I/peptide complex, and co-stimulatory molecules are not required in BiTE-mediated cytotoxicity.
Many reports have analyzed the application of BiTE antibodies and have characterized antitumor activity in tumor cell lines and xenograft models. The BiTE principle of antibody-based T-cell engagement is one proof-of-concept strategy. Most studies characterized BiTE antibodies targeting CD19 on B-cell malignancies, or EpCAM on adenocarcinoma. The CD19/CD3-bispecific BiTE antibody blinatumomab (MT103) has shown high response rates at very low doses in patients with non-Hodgkin's lymphoma and B-precursor acute lymphoblastic leukemia.13–15 Some pharmaceutical organizations selected MM as the target for their pipelines, developing agents such as rituximab (CD20), elotuzumab (CS1), lucatumumab (CD40), milatuzumab (CD74), daratumumab (CD38), B-B4-DM1 (CD138), CD56, and CD317. Some have achieved powerful biological activities at the clinical research stage. Many studies have shown that CD138 may be an important target protein for MM treatment. In this immunotherapy study, the novel CD138×CD3 BiTE-hIgFc antibody combines the excellent anti-CD3 and anti-CD138 molecular modules from Micromet and Immunogen (Waltham, MA, USA), respectively. The anti-CD3 ScFv and the additional human IgG1 Fc are designed to target T cells and NK cells with the aim of substantially enhancing the cytotoxicity against MM tumor cells.
Materials and Methods
Molecular constructions
A mouse OKT3 ScFv gene sequence (US 7635472 B2; Micromet) with the addition of NcoI and BamHI (New England Biolabs, Ipswich, MA, USA) cloning sites consecutively at the 5′-end, and a BglII cloning site at the 3′-end was synthesized (Genewiz, South Plainfield, NJ, USA). We cloned the mouse OKT3 ScFv gene into pFUSE-hIgFc vector (pfuse-hg1fc1; InvivoGen, San Diego, CA, USA) between the NcoI(579) and BglII(587) (New England Biolabs) cloning sites. The hIgFc gene sequence followed the mouse OKT3 ScFv gene closely. The constructed OKT3-ScFv-hIgFc was used as a basic structure for subsequent BiTE construction. We also synthesized an anti-CD138 ScFv gene (US 2009/0169570-A1, nBT062; Immunogen) with an NcoI cloning site and a signal peptide (atggagacag acacaatcct gctatgggtg ctgctgctct gggttccagg ctccactggt) consecutively at the 5′-end, and a G4S linker and a BamHI cloning site consecutively added at the 3′-end. The anti-CD138 ScFv gene was cloned into the OKT3-ScFv-pFUSE-hIgFc vector between the NcoI and BamHI cloning sites. As a result, an anti-CD138-ScFv-OKT3-ScFv-pFUSE-hIgFc (named BiTE-hIgFc, STL001) construct was obtained. Based on BiTE-hIgFc (STL001) with NcoI, BamHI, and BglII cloning sites, the single OKT3-ScFv-hIgFc (named aCD3-ScFv-hIgFc) or anti-CD138-ScFv-hIgFc (named aCD138-ScFv-hIgFc) construct with additional signal peptide and hIgFc could be produced easily.
Recombinant antibody expression and purification
The aCD3-ScFv-hIgFc, aCD138-ScFv-hIgFc, and BiTE-hIgFc (STL001) plasmids were transformed into DH5α Escherichia coli cells using the heat-shock approach. Individual colonies of plasmid-transformed DH5α were incubated in LB medium (Life Technologies, Grand Island, NY, USA) at 37°C for 16 h. The three plasmids for transfection were prepared using the Endofree Plasmid Maxi Kit (Qiagen, Shanghai, China). The plasmid DNA was delivered with Lipofectamine 2000 (Life Technologies) DNA transfection reagent into HEK-293 cells per the manufacturer's protocol. The supernatant was purified using a protein-A affinity column.
Cell culture
The human MM cell line RPMI-8226 and the chronic myelogenous leukemia cell line K562 were maintained in Iscove's modified Dulbecco's medium (Hyclone, Logan, UT, USA) supplemented with 10% FBS in a 5% CO2 incubator at 37°C. In addition, we used RPMI-1640 media with 10% FBS for cell culture of the acute T-cell leukemia cell line Jurkat and the PBMCs, and with 20% FBS for the human MM cell line U266. Iscove's modified Dulbecco's medium (SH30228.01), RPMI-1640 medium (SH30027.01) and FBS (SH30401.01) were purchased from ThermoScientific HyClone (Thermo Fisher Scientific, Logan, UT, USA).
Enzyme-linked immunosorbent assay
Antibodies (aCD138-ScFv-hIgFc and BiTE-hIgFc (STL001), 100 μL per well) at an appropriate dilution were added to different wells in a 96-well ELISA plate that was coated with recombinant hCD138 (rCD138) protein (Sino Biological Inc., Beijing, China), and blocked with 0.5% BSA in PBS. A standard indirect ELISA procedure was followed with HRP-labeled goat anti-human IgG1-Fc antibody (Sigma, St. Louis, MO, USA) and signal development with 3,3’,5,5’-tetramethylbenzidine substrate (Dako, Hamburg, Germany) for 10 min. The absorbance was measured at 450 nm with a 96-well microplate reader (BioTek, Winooski, VT, USA).
To analyze the interaction of BiTE-hIgFc (STL001) and rCD138 antigen, the primary antibody solutions, at graded concentrations, harvested from the first 96-well ELISA plate, were pipetted into a second ELISA plate. The ELISA procedure was repeated as previously described. In addition, 0–300 nM rCD138 protein was used as a blocking antigen concentration to carry out a competitive ELISA using BiTE-hIgFc (STL001) antibody.
Western blot analysis
The harvested cell lysates (2–5 μg/lane) were separated by 8–12% SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skimmed milk for 1 h and incubated with unconjugated primary antibodies aCD138-ScFv-hIgFc, aCD3-ScFv-hIgFc, and BiTE-hIgFc (STL001) overnight at 4°C. Immune complexes were detected by incubating the PVDF membranes with HRP-conjugated anti-human IgG1-Fc antibody (Sigma) at room temperature for 1 h and developing the membranes using enhanced chemiluminescence reagents (Millipore) for different periods of time.
Flow cytometry analysis
RPMI-8226, U266, Jurkat, and K562 cells, as well as PBMCs, were harvested by centrifugation and washed twice with pH 7.4 PBS. After fixation with 4% formaldehyde and blocking with 0.5% BSA-PBS, the harvested cells were stained with aCD138-ScFv-hIgFc, aCD3-ScFv-hIgFc, and BiTE-hIgFc (STL001) at the appropriate dilution in the assay tubes at room temperature for 1 h. The cells were harvested again by centrifugation and washed twice with 0.5% BSA-PBS. Then a fluorochrome-conjugated anti-human IgG1-Fc antibody (Invitrogen, Grand Island, NY, USA) at the appropriate dilution was used to label the harvested cells at room temperature for 30 min. After centrifugation and washing twice, the cells were analyzed using a flow cytometer (BD Biosciences, San Jose, CA, USA).
Bio-layer interferometry to determine equilibrium dissociation constant KD
The equilibrium dissociation constant KD of aCD138-ScFv-hIgFc and BiTE-hIgFc (STL001) antibody against rCD138 antigen was determined by the ForteBio Octet-96 machine (Menlo Park, CA, USA) using a bio-layer interferometry approach. The rCD138 protein labeled with biotin was incubated with an SA biosensor in the Octet-96. For KD determination, aCD138-ScFv-hIgFc or BiTE-hIgFc (STL001) was diluted to the appropriate concentration using ForteBio's kinetic buffer. To confirm the specific binding of loaded rBiTE antibodies to rCD138 protein conjugated to the SA biosensor, blank kinetic buffer or overloaded rBiTE solution only was added to the rCD138-coated SA biosensor or blank SA biosensor, respectively. All data were analyzed using the Octet Data Analysis 7.0 software (ForteBio).
T cell activation assay
We used the standard Ficoll (GE Healthcare, Pittsburgh, PA, USA) density gradient centrifugation procedure to isolate human PBMCs from buffy coats provided by healthy donors from the First Affiliated Hospital of Soochow University (Suzhou, China). The harvested PBMCs were washed with PBS (pH 7.4) and resuspended in RPMI-1640 (SH30027.01) cell medium containing 10% FBS (SH30401.01). Cell culture for PBMCs was carried out as previously described.
On day 1, 100 μL cells were plated onto a 24-well plate at a density of 3 × 106 cells/mL per well. BiTE-hIgFc (STL001), a mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc, and isotype control human IgG1-Fc (Abcam, Cambridge, UK) were diluted to 100 ng/mL with PBS (pH 7.2). Anti-CD28 mAb (100 ng/mL; BD Biosciences) and IL-2 (10 U/mL; Roche, Basel, Switzerland) were added to different groups of the T cell activation assay. After T cell activation for 24 h, the CD4+ or CD8+ T cells were gated, and the activated T cell surface markers CD25 and CD69 were analyzed using anti-CD25 and anti-CD69 antibodies (BD Biosciences) by a flow cytometer (BD Biosciences).
Specific binding of NK cells by the Fc gamma receptor
Human PBMCs from healthy donors were harvested and cultured using the method described above. Isolated PBMCs were stimulated by the irradiated K562 cell lines (100 cGy) transfected with membrane-bound IL-21, IL-15, CD137L, and CD86 molecules. Interleukin-2 (60 U/mL) was added to enrich NK cell populations. The NK cells were gated with FITC-labeled anti-CD3 and APC-labeled anti-CD56 antibodies (BD Biosciences). The specific binding of BiTE-hIgFc (STL001) to NK cells by Fc and FcR, respectively, was analyzed by flow cytometry.
Immunofluorescence analysis
Human PBMCs from healthy donors were harvested and cultured using the method described above. Before biomembrane stimulation for T cell enrichment, 100 ng/mL anti-CD3 antibody (BD Biosciences) was added to 1 × 106 cells/mL PBMCs (for T cell group), and incubated in an incubator 5% CO2 at 37°C for 30 min. Subsequently, NK and T cells were enriched separately using the biomembrane stimulation method described above. Activated T cells were stained with 10 μM CFSE solution (Life Technologies) for 15 min at 37°C. Meanwhile, MM cells were seeded in 24-well microplate embedded coverslips. Multiple myeloma, activated NK, and T cells with 1 μg/mL BiTE-hIgFc (STL001) were mixed in a 24-well microplate for co-incubation in 5% CO2 incubator at 37°C for 2 h. After 4% paraformaldehyde fixation, and 2% BSA blocking, all cells reacted with Alexa Fluor 488-labeled mouse anti-human IgG (H+L) (Life Technologies). Finally, DAPI for cell nuclear staining and the mounting medium (Life Technologies) were added. Cell staining was analyzed by fluorescence microscopy (BZ-9000E; KEYENCE, Itasca, IL, USA).
BiTE-hIgFc-based cytotoxicity assay in vitro and in vivo
Tumor target cell RPMI-8226 and negative control K562 cells were stained with 10 μM CFSE at 37°C for 15 min. The PBMC effector cells (1.4 × 106) and tumor cells (2.0 × 105) (E:T ratio = 7:1) were mixed in a 96-well microtiter plate in a total volume of 200 μL in the presence or absence of BiTE-hIgFc (STL001), or the mixture of anti-CD138 and anti-CD3 antibodies, or isotype control hIgG1 mAb. Anti-CD28 mAb (100 ng/mL) and IL-2 (10 U/mL) were also added to each well. After incubation at 37°C in 5% CO2 for 24 h and 48 h, the mixed cells were stained with PE-Cy5-labeled 7-aminoactinomycin D solution (BD Biosciences). The cytotoxicity in vitro was estimated by flow cytometry.
Antitumor activity analysis of BiTE antibody was carried out in a human MM xenograft mouse model. RPMI-8226 tumor cells (8 × 106) resuspended in 200 μL PBS were inoculated s.c. into NOD/SCID mice (SLAC, Shanghai, China) to establish tumors. Unstimulated human naïve PBMCs at a ratio of 3:1(E:T), alone or in combination with 3 mg/kg BiTE-hIgFc (STL001) antibody and 2.5 × 106 U/kg rIL-2 (Roche) were i.v. injected through mouse lateral tail vein (day 0 after tumor cell inoculation). An isotype antibody was set as the negative control. In subsequent days, BiTE antibody along with rIL-2 were i.v. injected to mice every 3 days, six times in total. Tumor development was measured periodically and the tumor volume was determined by caliper measurement, and calculated using the formula [length (mm) × width (mm)2]/2. The animal procedures were approved by the Experimental Animal Ethics Committee of Soochow University.
Statistical analysis
Statistical analysis was carried out using Minitab 16 software (Minitab, State College, PA, USA). For statistical analyses, Student's t-test was used. P-values below 0.05 were considered statistically significant: *P < 0.05; **P < 0.01; ***P < 0.005. Correlation analyses were carried out using the correlation test with a confidence interval of 95%.
Results
Recombinant antibody expression, purification, and evaluation by Western blot and competitive ELISA
The synthesized aCD138-ScFv and aCD3-ScFv were successfully inserted into the pFUSE vector after two steps of enzyme digestion and ligation using NcoI, BamHI, and BglII cloning sites (Fig. S1). Based on the pFUSE-BiTE-hIgFc plasmid, pFUSE-aCD138-ScFv-hIgFc or pFUSE-aCD3-ScFv-hIgFc was obtained (data not shown). The plasmids aCD138-ScFv-hIgFc, aCD3-ScFv-hIgFc, and BiTE-hIgFc (STL001) were prepared using the Endofree Plasmid Maxi Kit and then transfected into 500 mL HEK-293 cells for 48 h. The protein A-purified antibodies were analyzed by SDS-PAGE and Western blot using HRP-labeled goat anti-human IgG1-Fc (Fig.1A). The predicted molecular weights of the three products are 52 kDa (aCD3-ScFv-hIgFc), 52 kDa (aCD138-ScFv-hIgFc), and 79 kDa (BiTE-hIgFc). The amino acid sequence of BiTE-hIgFc (STL001) is shown in Figure1(B). The orange letters represent the (G4S)3 or G4S linker.
Fig 1.

Expression of aCD138-ScFv-hIgFc, aCD3-ScFv-hIgFc, and bispecific T-cell engager (BiTE)-hIgFc antibodies. (A) Twelve percent reducing SDS-PAGE (3 μg/lane) and Western blot analysis of the supernatant of the three expression products showing molecular weights close to those predicted (52, 52, and 79 kDa). (B) Amino acid sequence of BiTE-hIgFc (STL001). The orange letters represent the (G4S)3 or G4S linker. In addition, the signal peptide and hIgG1-Fc sequence are shown at the NH2-terminus and COOH-terminus, respectively.
Interaction of BiTE-hIgFc (STL001) and rCD138 antigen was analyzed by competitive ELISA. The rCD138 coating concentration was 0.1 μg/mL. After incubation with 0.00, 0.03, 0.06, 0.13, 0.25, 0.50, or 1.00 μg/mL antibody, the primary antibody solution in the first 96-well ELISA plate (Fig. S2A, blue line) was pipetted into the second ELISA plate (Fig. S2A, red line). For the two plates, the ELISA was carried out following the same procedure as described in “Materials and Methods”. The coefficient of determination R2 demonstrated a good fit of linear regression for both plates (0.9794 and 0.9981). According to the formula (Slope1 − Slope2)/Slope1, the calculated bound ratio of the BiTE-hIgFc fraction in the first plate was 17.96%. With a higher rCD138 coating concentration, the bound ratio was much higher (data not shown). Therefore, 0.1 μg/mL was selected as the rCD138 coating concentration in the subsequent blocking experiment. A graded blocking concentration of rCD138 solution (0.0–300.0 nM) and 1.00 μg/mL primary BiTE-hIgFc (STL001) antibodies were used to determine the blocking ability of rCD138 against BiTE-hIgFc (STL001) by ELISA (Fig. S2B). The graph indicates a good blocking curve. A concentration of 9.38 nM rCD138 protein almost completely blocked BiTE-hIgFc (STL001).
Flow cytometry analysis of BiTE-hIgFc (STL001), aCD138-ScFv-hIgFc, and aCD3-ScFv-hIgFc
RPMI-8226, U266, and Jurkat cells were stained using 10 μg/mL BiTE-hIgFc (STL001), aCD138-ScFv-hIgFc, or aCD3-ScFv-hIgFc. After incubation with FITC-labeled anti-hIgG1 Fc antibody, the cell samples were analyzed by flow cytometry (Fig.2). The staining percentage for different groups is summarized in Table S1. The stars indicate the degree of staining. As shown in Table S1 and Figure2, BiTE-hIgFc (STL001) and aCD138-ScFv-hIgFc antibodies showed a similar staining pattern (five stars) on MM cell lines RPMI-8226 (Fig.2A,E) and U266 (Fig.2B,F). However, BiTE-hIgFc (STL001) and aCD3-ScFv-hIgFc antibodies achieved the same pattern (four stars) on Jurkat (Fig.2C,K) and PBMCs (Fig.2D,L). Due to the bispecific characteristic of BiTE-hIgFc (STL001), targeting hCD138 and hCD3, the antibody could stain all four cell lines strongly (Fig.2A–D). However, aCD138-ScFv-hIgFc specifically stained the MM cell lines RPMI-8226 (Fig.2E) and U266 (Fig.2F) only, and aCD3-ScFv-hIgFc specifically stained CD3+ Jurkat (Fig.2K) and PBMCs (Fig.2L) only.
Fig 2.

Flow cytometry analysis of bispecific T-cell engager (BiTE)-hIgFc (STL001), aCD138-ScFv-hIgFc, and aCD3-ScFv-hIgFc. BiTE-hIgFc (STL001), aCD138-ScFv-hIgFc, and aCD3-ScFv-hIgFc (all 10 μg/mL) stained RPMI-8226 (A, E, I), U266 (B, F, J), and Jurkat (C, G, K) cells and peripheral blood mononuclear cells (D, H, L) for 1 h. Finally, FITC-labeled anti-human IgG1-Fc antibody at 1:5000 dilution was used to label the harvested cells at room temperature for 30 min. After centrifugation and being washed twice, the cells were analyzed using a flow cytometer. Data shown are representative of three independent experiments.
Measuring KD by bio-layer interferometry
The coating concentration of biotin-labeled rCD138 was 10 μg/mL in Octet Kinetic buffer, and the loading concentrations of aCD138-ScFv-hIgFc and BiTE-hIgFc (STL001) were set in increments of 500.0, 250.0, 125.0, and 62.5 nM, and 333.3, 166.7, 83.3, and 41.7 nM, respectively. As shown in Figure3, sensor E8 and G8 curves show that blank kinetic buffer or overloaded rBiTE solution cannot bind the rCD138-coated SA biosensor or blank SA biosensor, respectively, which verifies that the binding of loaded rBiTE to the coating rCD138 on the SA biosensor is specific. After 300 s of association and 1800 s of dissociation, the KD measurements of aCD138-ScFv-hIgFc and BiTE-hIgFc (STL001) against rCD138 were found to be 13.5 nM and 29.5 nM, respectively. It is clear that when the aCD138-ScFv molecule is modified to BiTE-hIgFc (STL001), the affinity KD remains at a nanomolar level.
Fig 3.
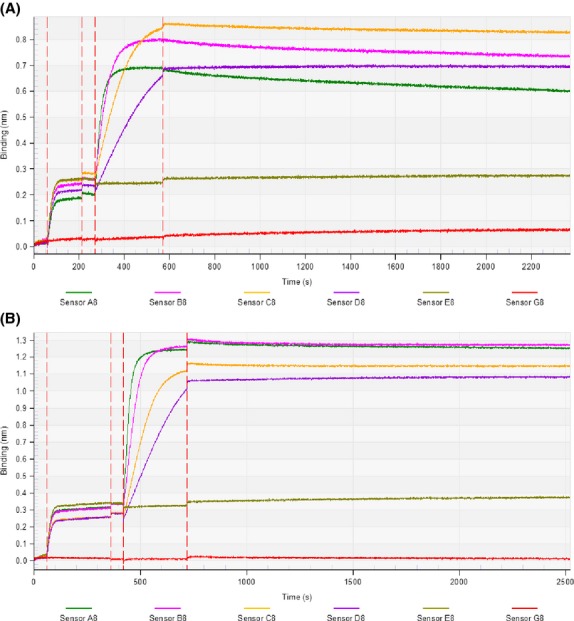
Determination of equilibrium dissociation constant (KD) of bispecific T-cell engager (BiTE)-hIgFc (STL001) and aCD138-ScFv-hIgFc. Biotin-labeled rCD138 (10 μg/mL) in Octet Kinetic buffer was loaded onto an streptavidin (SA) biosensor. For sensors A8–D8, 500.0, 250.0, 125.0, and 62.5 nM aCD138-ScFv-hIgFc (A) or 333.3, 166.7, 83.3, and 41.7 nM BiTE-hIgFc (STL001) (B) against the coated rCD138 protein were analyzed by the bio-layer interferometry approach in the KD determination experiment. The association and dissociation time was set to 300 and 1800 s, respectively. The KD of aCD138-ScFv-hIgFc and BiTE-hIgFc (STL001) against rCD138 protein were determined to be 13.5 and 29.5 nM, respectively. The blank kinetic buffer (Sensor E8 curve) or overloaded rBiTE solution (Sensor G8 curve) only was added to the rCD138-coated SA biosensor or blank SA biosensor, respectively, which was designed to verify that blank kinetic buffer or overloaded rBiTE solution cannot non-specifically bind the rCD138-coated SA biosensor or blank SA biosensor, respectively. Data shown are representative of three independent experiments.
BiTE-hIgFc (STL001) activated T cells and induced CD25 and CD69 secretion
CD4+ (Fig.4A–F) and CD8+ (Fig.4G–L) T cells were gated from PBMCs using aCD4-APC and aCD8-PE-Cy5 antibodies, respectively. After 24 h of T cell activation, flow cytometry analysis revealed that BiTE-hIgFc (STL001) (Fig.4A,B,G,H), or the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc (Fig.4C,D,I,J) showed higher activation efficiency compared with the hIgG1 isotype control group (Fig.4E,F,K,L). The histogram (Fig.4M) clearly indicates the significant activation difference of the sample group (Fig.4A and G, B and H, C and I, D and J) and the hIgG1 isotype control group (Fig.4E and K, F and L). In all 12 groups, BiTE-hIgFc (STL001), or the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc, showed much higher activation efficiency than the hIgG1 isotype control group. For the BiTE-hIgFc (STL001), the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc antibodies, and isotype hIgG1 mAb, six CD4+ T-cell groups showed similar activation patterns compared to six CD8+ T-cell groups. In addition, the absence (groups B and H, D and J, F and L) or presence (groups A and G, C and I, E and K) of co-stimulatory signal receptor aCD28 did not significantly affect T cell activation efficiency significantly.
Fig 4.
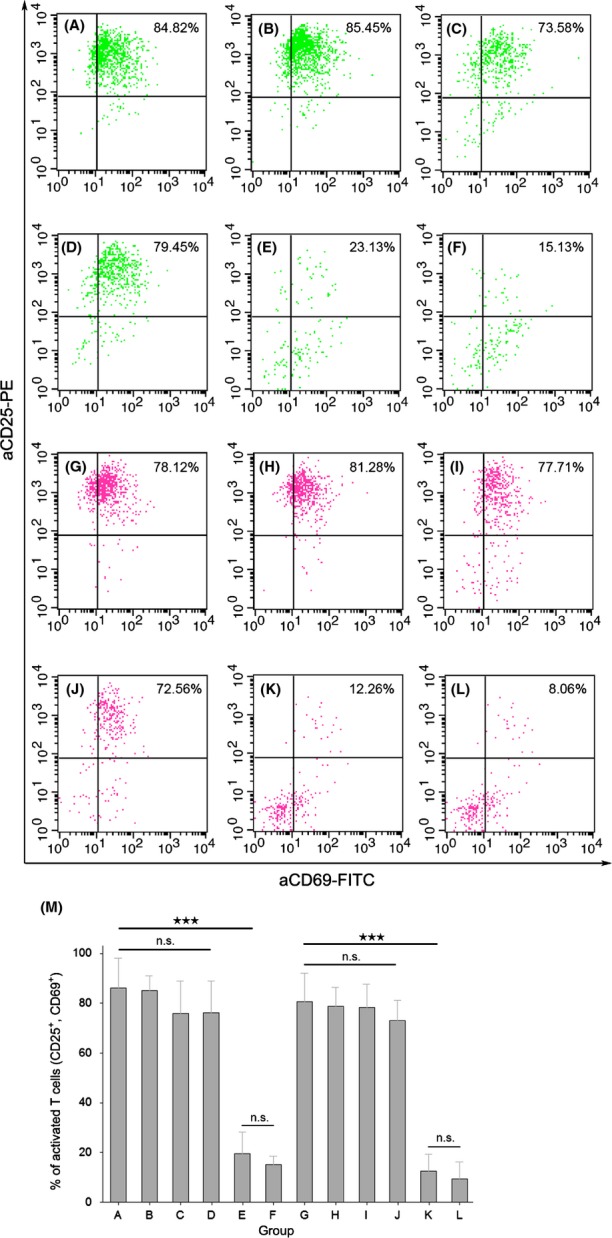
Bispecific T-cell engager (BiTE)-hIgFc (STL001) activated T cells and induced CD25 and CD69 secretion. CD4+ (A–F) and CD8+ (G–L) T cells were gated from peripheral blood mononuclear cells using aCD4-APC and aCD8-PE-Cy5 antibodies, respectively. T cell activation for 24 h with different antibody sets, BiTE-hIgFc (STL001) (A, B, G, H), the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc antibodies (C, D, I, J), and isotype hIgG1 mAb (E, F, K, L). The histogram (M) clearly shows the significant activation difference in sample groups 1 (A, G), 2 (B, H), 3 (C, I), and 4 (D, J), and hIgG1 isotype control groups 5 (E, K) and 6 (F, L). Interleukin-2 (10 U/mL) was added to all groups of the T-cell activation assay, but the additional anti-CD28 mAb (100 ng/mL) was added to groups 1, 3, and 5 only. Data shown in M are means ± SEM. ***P < 0.005, Student's t-test. n.s., not significant.
Specific binding of NK cells by Fc gamma receptor
After 2 weeks of PBMC stimulation and activation, enriched NK cells accounted for 95.51% of the total cell populations (shown in Fig. S3A). Compared with the negative control (Fig. S3B), the binding rate of NK cells by BiTE-hIgFc (STL001) through Fc was more than 96% of the total NK cells gated by CD3(−) and CD56(+) (Fig. S3C). This high staining percentage of BiTE-hIgFc (STL001) against NK cells reveals that BiTE-hIgFc (STL001) can specifically and strongly target FcR(+) immunological cells, such as NK cells and macrophages. It supports the notion that BiTE-hIgFc (STL001) also plays an important role in antibody-dependent cellular cytotoxicity.
Immunofluorescence analysis of BiTE forming CD138+ MM cells, CD3+ T cells, and FcR+ NK cells
The MM cells and activated T and NK cells were validated by FITC-labeled anti-CD138, FITC-labeled anti-CD3, and APC-labeled anti-CD56 antibodies, respectively. Compared with the isotype negative control (Fig.5A–C, upper panels), the positive staining ratio of MM (CD138+), T (CD3+), and NK (CD56+) cells was 97.59%, 89.60%, and 90.94%, respectively (Fig.5A–C, lower panels). The presence of the complexes forming CD138+ MM cells, CD3+ T cells, and FcR+ NK cells was observed (Fig. 5D) after CFSE 17 staining for T cells (Fig. 5E), BiTE membrane staining (Fig. 5F), and DAPI nuclear staining (Fig. 5G) for MM/T/NK cells. As shown in Figure5(G), MM nuclei were obviously disrupted, which may have resulted from T/NK cell-mediated cytotoxicity on MM cells through the 2-h short incubation period. The immunofluorescence data was consistent with other evaluation data shown in this article.
Fig 5.
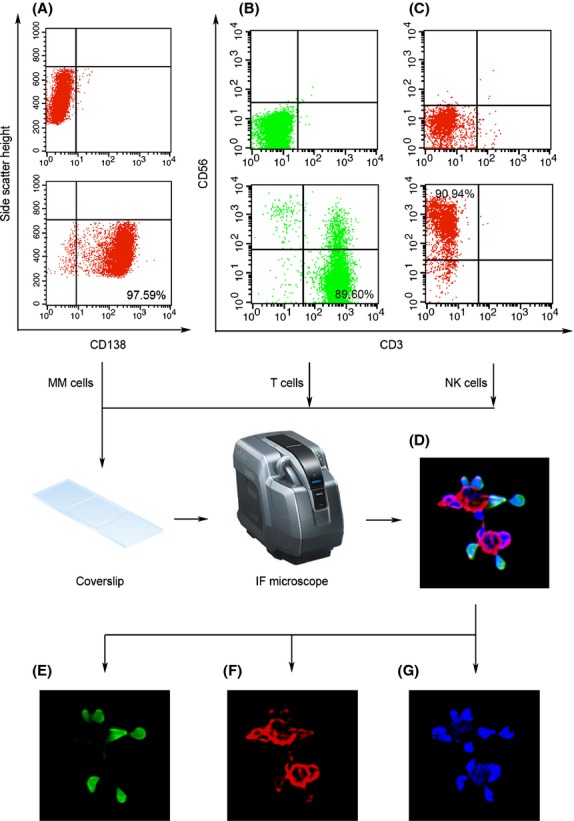
Immunofluorescence analysis of bispecific T-cell engager (BiTE) forming CD138+ multiple myeloma (MM) cells, CD3+ T cells, and Fc receptor (FcR)+ natural killer (NK) cells. Both T and NK cells were enriched using the biomembrane stimulation approach. The MM cells and activated T and NK cells were validated by FITC-labeled anti-CD138, FITC-labeled anti-CD3, and APC-labeled anti-CD56 antibodies, respectively. Compared with the isotype negative control (A–C, upper panels), the positive staining rates of MM (CD138+), T (CD3+), and NK (CD56+) cells were 97.59%, 89.60%, and 90.94%, respectively (A–C, lower panels). After carboxyfluorescein succinimidyl ester staining for T cells (E), BiTE membrane staining for MM/T/NK cells (F), and DAPI nuclear staining for MM/T/NK cells (G), the presence of the complexes forming CD138+ MM cells, CD3+ T cells, and FcR+ NK cells were observed (D). The BiTE staining was imaged using Alexa Fluor 488-labeled mouse anti-human IgG (H+L) antibody. Cell staining was analyzed by fluorescence microscopy.
BiTE-hIgFc (STL001) showed potent cytotoxicity against RPMI-8226 cells in vitro and in vivo
BiTE-hIgFc-mediated cytotoxicity against RPMI-8226 cells in vitro was evaluated (Fig. 6) After 24 h of incubation of PBMCs and RPMI-8226 cells, BiTE-hIgFc (STL001) (27.5%; Fig.6A), or the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc (20.3%; Fig.6B), showed stronger cell lysis of RPMI-8226 than that of K562 control groups (16.9%, Fig.6K; 12.8%, Fig.6L). Additionally, after 48 h of incubation, BiTE-hIgFc (STL001) (90.1%; Fig.6F) and the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc (70.5%; Fig.6G) showed outstanding cytotoxicity on RPMI-8226 cells compared with K562 control groups (13.8%, Fig.6P; 12.3%, Fig.6Q). However, all of the hIgG1 isotypes or blank control groups (Fig.6C–E,H–J,M–O,R–T) elicited low cytotoxicity. Meanwhile, BiTE-hIgFc (STL001) also showed stronger cell lysis of RPMI-8226 than that of single aCD138-ScFv-hIgFc, and BiTE-hIgFc (STL001) itself showed little cytotoxicity on MM cells if PBMCs were absent (Fig. S4).
Fig 6.

Bispecific T-cell engager (BiTE)-hIgFc-mediated cytotoxicity against RPMI-8226 multiple myeloma tumor cells. RPMI-8226 and negative control K562 cells were stained with 10 μM carboxyfluorescein succinimidyl ester solution at 37°C for 15 min. Peripheral blood mononuclear effector cells (1.4 × 106) and tumor cells (2.0 × 105) (effector:target = 7:1) were mixed in a 96-well microtiter plate in a total volume of 200 μL. Anti-CD28 mAb (100 ng/mL) and interleukin-2 (10 U/mL) were also added to each group, except for groups E, J, O, and T. In the RPMI-8226 group, after 24 h (A–E, K–O) or 48 h (F–J, P–T) incubation of peripheral blood mononuclear cells and multiple myeloma cells with the antibody mixture, BiTE-hIgFc (STL001) (A, F), or the mixture of aCD138-ScFv-hIgFc and aCD3-ScFv-hIgFc (B, G) showed stronger cell lysis of RPMI-8226 than that of K562 control groups (K, P and L, Q, respectively). However, the hIgG1 isotype control groups or blank control groups for RPMI-8226 (C, H or D, I, E, J, respectively) and K562 (M, R or N, S, O, T, respectively) showed much lower cytotoxicity. Before cytotoxicity analysis by flow cytometry, the mixed cells were stained with PE-Cy5-labeled 7-aminoactinomycin D (7-AAD) reagent. Data shown in U are means ± SEM. *P < 0.05, ***P < 0.005, Student's t-test. n.s., not significant.
Human RPMI-8226 cells were inoculated s.c. into NOD/SCID mice to develop tumors. The unstimulated human naïve PBMCs from healthy human donors in the presence or absence of BiTE-hIgFc (STL001) antibody were injected i.v. into mice. As shown in Figure7, compared with the isotype control group, 3 mg/kg (each dose) of BiTE antibody effectively engaging T and NK cells to inoculated tumor cells, significantly inhibited tumor growth in NOD/SCID mice, especially in days 0–32. In total, the differences between BiTE antibody and the control were significant (P < 0.005; n = 3).
Fig 7.
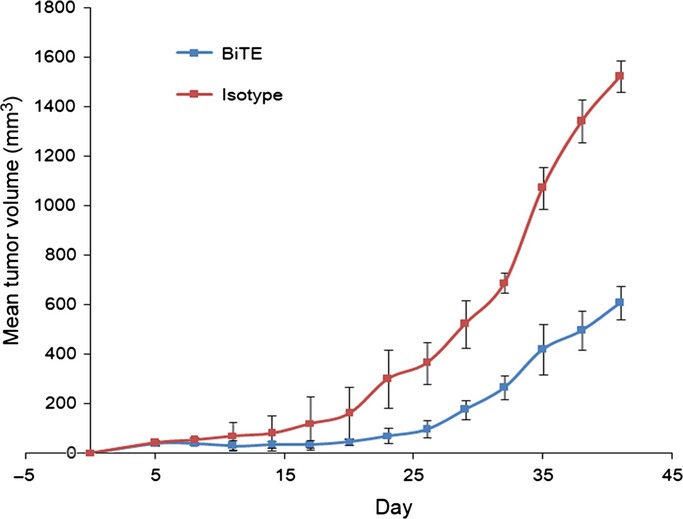
Bispecific T-cell engager (BiTE)-hIgFc reveals antitumor activity of BiTE antibody in a xenograft multiple myeloma model. RPMI-8226 tumor cells (8 × 106) were inoculated s.c. into NOD/SCID mice. Human naïve peripheral blood mononuclear cells at a ratio of 3:1 (effector:target) with BiTE antibody and rIL-2 were i.v. injected through mouse lateral tail vein (day 0). An isotype antibody was set as the negative control. BiTE antibody along with rIL-2 were injected every 3 days, six times in total. Tumor volume was determined using the formula [length (mm) × width (mm)2]/2. The mean tumor volume ± SD (n = 3) is shown. The differences between BiTE antibody and the control are significant (P < 0.005; n = 3).
Discussion
Compared with single aCD138 or aCD3 recombinants, the produced BiTE-hIgFc (STL001) showed an identical staining pattern on MM cells, Jurkat cells, and PBMCs. Importantly, BiTE-hIgFc (STL001) showed nanomolar level affinity to rCD138 antigen, and could activate T cells effectively. The specific binding ability of the Fc molecule in BiTE to NK cells has also been verified, which supports our future bispecific NK engager study. Moreover, BiTE-hIgFc (STL001) achieved potent cytotoxicity against MM tumor cells in vitro and in vivo. In combination with the analysis of T cell activation and cytotoxicity assays, these results indicate that BiTE-hIgFc (STL001) is a potential therapeutic approach to MM disease. All the effects of BiTE-hIgFc suggest this novel immunotherapy strategy toward MM treatment works well as an in vitro or in vivo agent.
However, based on current therapeutic principles, any single biomolecule is unlikely to achieve complete clinical effects, and they are always combined with other biomolecules or compounds to achieve excellent therapeutic performance. As the data show in the present study, despite its potent targeting ability and cytotoxicity, BiTE-hIgFc (STL001) may be unable to completely kill MM cells and prevent MM relapse in vitro or in vivo. BiTE-hIgFc (STL001) (90.1%; Figs6F,7) shows outstanding cytotoxicity in MM. Some studies have shown that MM stem cells may exist and can result in relapse of the disease.16 However, the origins of MM stem cells still seem to be controversial and different among published reports, including those by Matsui et al. (CD19+/CD20+/CD27+/CD138−,17,18 Yaccoby and Epstein and Kim et al. (CD38+/CD45−),19,20, and Pilarski and Belch (CD34+/CD45low).21 For example, the CD19 positivity in myeloma stem cells suggested by Matsui et al.17,18 could not be reproduced by other researchers. In the current situation, it is not easy for us to introduce one or multiple MM stem cell markers into our future BiTE strategy. However, besides MM stem cells, there is another strategy for combining BiTE-hIgFc with compounds such as bortezomib, thalidomide, PEGylated liposomal doxorubicin, and lenalidomide. BiTE-hIgFc would be more effective to eradicate the tumor if small-molecular compounds could be integrated into our proof-of-concept BiTE-hIgFc strategy in our future study.
To our knowledge, this BiTE-hIgFc (STL001) is the first construct to target CD138, CD3, and FcR, which are highly expressed on the surface of MM cells, T cells, and NK cells, respectively. The molecule backbone is shown in Figure S5. One could easily replace the antibody fragment (green or blue module) with antibodies against other tumor cells and produce versatile BiTE-hIgFc recombinants with potent antitumor activity by engaging T cells in vitro and in vivo. BiTE antibodies may contribute in the future to cancer immunotherapy by redirecting the vast number of existing T-cell clones in patients while ignoring many of the immune escape mechanisms that otherwise limit specific antitumor responses of T-cell clones.11 Taking EpCAM/CD3 and CD19/CD3 BiTE as examples, Klaus Brischwein and colleagues have developed a novel single-chain EpCAM/CD3–bispecific antibody construct, designated MT110. It appears to be an attractive bispecific antibody candidate for treatment of human EpCAM-overexpressing carcinomas.14 Additionally, clinical studies with a CD19/CD3–bispecific BiTE antibody suggest that this therapeutic paradigm is finally showing promise for treatment of both bulky and minimal residual disease.22
Another important question is how to produce antibody genes. Should the gene be produced from a mouse hybridoma, a camelid VHH domain (a single-domain antibody called Nanobody [Ablynx, Ghent, Belgium]), the Human Combinatorial Antibody Library (HuCAL; MorphoSys, Planegg, Germany), or others? Pharmaceutical enterprises are focusing on a range of different formats. Current antibody candidates are showing outstanding preclinical or clinical performance, and some have entered the medical marketplace. Hence, MM tumors may be eventually treated by deploying rationally designed immunotherapeutic agents.
In conclusion, the present study shows the basic features of a BiTE combination of CD138- and CD3-specific antibodies. Moreover, the innovative BiTE structure can be replaced, supplemented, or conjugated easily with other antibodies or molecular fragments to deliver specific biological functions for development of a promising therapeutic antibody.
Acknowledgments
We thank Dr. Chun Wu and Qianjin Ke for expert technical assistance in the molecular construction. Dr. John Mountzouris is acknowledged for proofreading and editing of manuscript. This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the National Natural Science Foundation of China (Grant No. 31471283), and the Science and Technology Support Program Project of Jiangsu Province (Grant Nos. BE2010649 and BE2011682).
Glossary
Abbreviations
- APC
antigen-presenting cell
- BiTE
bispecific T-cell engager
- CFSE
carboxyfluorescein succinimidyl ester
- EpCAM
epithelial cell adhesion molecule
- E:T
effector:target
- FcR
Fc receptor
- IL
interleukin
- MM
multiple myeloma
- NK
natural killer
- PBMC
peripheral blood mononuclear cells
- SA
streptavidin
- ScFv
single-chain variable
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Design of BiTE-hIgFc molecule construction.
Fig. S2. Competitive ELISA analysis of rCD138 and BiTE-hIgFc (STL001).
Fig. S3. Specific binding of natural killer cells by Fc gamma receptor.
Fig. S4. Anti-tumor effect analysis of anti-CD138-ScFv-hIgFc and BiTE-hIgFc (STL001).
Fig. S5. Backbone of the BiTE-hIgFc (STL001) molecule.
Table S1. Staining percentages of different antibodies targeting RPMI-8226, U266, and Jurkat cells and peripheral blood mononuclear cells
References
- Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma. Lancet. 2009;374:324–39. doi: 10.1016/S0140-6736(09)60221-X. [DOI] [PubMed] [Google Scholar]
- Blum W, Schwind S, Tarighat SS, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119:6025–31. doi: 10.1182/blood-2012-03-413898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366:1770–81. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan ST, Keating GM. Pegylated liposomal doxorubicin: a review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi's sarcoma. Drugs. 2011;71:2531–58. doi: 10.2165/11207510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Mariz JM, Esteves GV. Review of therapy for relapsed/refractory multiple myeloma: focus on lenalidomide. Curr Opin Oncol. 2012;24(Suppl 2):S3–11. doi: 10.1097/01.cco.0000410243.84074.dc. [DOI] [PubMed] [Google Scholar]
- Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanagiri T, Shigematsu Y, Kuroda K, et al. Anti-tumor activity of human γδ T cells transducted with CD8 and with T cell receptors of tumor-specific cytotoxic T lymphocytes. Cancer Sci. 2012;103:1414–9. doi: 10.1111/j.1349-7006.2012.02337.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Seki S, Nakashima H, Nakashima M, Kinoshita M. Antitumor immunity produced by the liver Kupffer cells, NK cells, NKT cells, and CD8 CD122 T cells. Clin Dev Immunol. 2011;2011:868345. doi: 10.1155/2011/868345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum LG. T cell-based immunotherapy for cancer: a virtual reality? CA Cancer J Clin. 1999;49:74–100. doi: 10.3322/canjclin.49.2.74. [DOI] [PubMed] [Google Scholar]
- Choi BD, Cai M, Bigner DD, et al. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther. 2011;11:843–53. doi: 10.1517/14712598.2011.572874. [DOI] [PubMed] [Google Scholar]
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009;69:4941–4. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- Lutterbuese R, Raum T, Kischel R, et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A. 2010;107:12605–10. doi: 10.1073/pnas.1000976107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier T, Baeuerle PA, Fichtner I, et al. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3-bispecific single-chain antibody construct. J Immunol. 2003;170:4397–402. doi: 10.4049/jimmunol.170.8.4397. [DOI] [PubMed] [Google Scholar]
- Brischwein K, Schlereth B, Guller B, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–43. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Nagorsen D, Bargou R, Ruttinger D, Kufer P, Baeuerle PA, Zugmaier G. Immunotherapy of lymphoma and leukemia with T-cell engaging BiTE antibody blinatumomab. Leuk Lymphoma. 2009;50:886–91. doi: 10.1080/10428190902943077. [DOI] [PubMed] [Google Scholar]
- Huff CA, Matsui W. Multiple myeloma cancer stem cells. J Clin Oncol. 2008;26:2895–900. doi: 10.1200/JCO.2007.15.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui W, Huff CA, Wang Q, et al. Characterization of clonogenic multiple myeloma cells. Blood. 2004;103:2332–6. doi: 10.1182/blood-2003-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–7. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94:3576–82. [PubMed] [Google Scholar]
- Kim D, Park CY, Medeiros BC, Weissman IL. CD19-CD45 low/- CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia. 2012;26:2530–7. doi: 10.1038/leu.2012.140. [DOI] [PubMed] [Google Scholar]
- Pilarski LM, Belch AR. Clonotypic myeloma cells able to xenograft myeloma to nonobese diabetic severe combined immunodeficient mice copurify with CD34 (+) hematopoietic progenitors. Clin Cancer Res. 2002;8:3198–204. [PubMed] [Google Scholar]
- Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–7. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Design of BiTE-hIgFc molecule construction.
Fig. S2. Competitive ELISA analysis of rCD138 and BiTE-hIgFc (STL001).
Fig. S3. Specific binding of natural killer cells by Fc gamma receptor.
Fig. S4. Anti-tumor effect analysis of anti-CD138-ScFv-hIgFc and BiTE-hIgFc (STL001).
Fig. S5. Backbone of the BiTE-hIgFc (STL001) molecule.
Table S1. Staining percentages of different antibodies targeting RPMI-8226, U266, and Jurkat cells and peripheral blood mononuclear cells