

Abstract
Silent information regulator 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase. In mice, mSirt1 deficiency causes the onset of fatty liver via regulation of the hepatic nutrient metabolism pathway. In this study, we demonstrate SIRT1 expression, activity and NAD+ regulation using noncancerous liver tissue specimens from hepatocellular carcinoma patients with non-B non-C (NBNC) hepatitis. SIRT1 expression levels were higher in NBNC patients than in healthy donors, while SIRT1 histone H3K9 deacetylation activity was suppressed in NBNC patients. In the liver of hepatitis patients, decreased NAD+ amounts and its regulatory enzyme nicotinamide phosphoribosyltransferase expression levels were observed, and this led to inhibition of SIRT1 activity. SIRT1 expression was associated with HIF1 protein accumulation in both the NBNC liver and liver cancer cell lines. These results may indicate that the NBNC hepatitis liver is exposed to hypoxic conditions. In HepG2 cells, hypoxia induced inflammatory chemokines, such as CXCL10 and MCP-1. These inductions were suppressed in rich NAD+ condition, and by SIRT1 activator treatment. In conclusion, hepatic SIRT1 activity was repressed in NBNC patients, and normalization of NAD+ amounts and activation of SIRT1 could improve the inflammatory condition in the liver of NBNC hepatitis patients.
Keywords: Nicotinamide adenine dinucleotide, nicotinamide phosphoribosyltransferase, non-alcoholic steatohepatitis, non-B non-C hepatitis, silent information regulator 1
In Japan, the incidence of infection with hepatitis B virus (HBV) and hepatitis C virus (HCV) is decreasing, and continued drug development is contributing to the control of viral hepatitis-induced hepatocellular carcinoma (HCC).1 However, the incidence of non-viral hepatitis (e.g. alcoholic steatohepatitis [ASH] and non-alcoholic steatohepatitis [NASH]-induced HCC) is increasing.1 Globally, the prevalence of non-alcoholic fatty liver disease ranges between 15 and 30%,2 and appears to be on the increase. Therefore, it is likely that there will be a continued increase in NASH patients. Unfortunately, the development of drugs to treat these types of hepatitis is yet to progress. ASH and NASH display similar pathological processes, such as steatosis, hepatic inflammation, liver fibrosis, cirrhosis and hepatocarcinogenesis.3 To manage ASH and NASH progression, regulation of the inflammatory response may be beneficial. Indeed, inflammatory chemokine, such as Cxcl10 or Mcp-1, disrupted animals are resistant to non-viral hepatitis.4,5 In the liver of ASH patients, CXC chemokine expression levels are upregulated.6
An animal model with HCC incidence through NASH-like pathology has not been fully developed. Recently, however, a NASH-HCC mouse model, named STAM mice, was developed using neonatal exposure to streptozotocin and continuous high-fat diet feeding.7 STAM mice display diabetes mellitus (DM) and progression of NASH, such as fatty liver, hepatitis, fibrosis and high incidences of HCC.7
Silent information regulator 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase. It deacetylates histones and several proteins.8,9 For example, SIRT1 deacetylates p53,10 NF-κB,11 FOXOs12 and PGC1-α.13 SIRT1 is able to either repress or activate the transcriptional activities of these targets, thereby regulating diverse cell cycle and inflammatory pathways. In the liver of mice, mSirt1 is expressed in the nucleus of periportal hepatocytes.14 SIRT1 deficiency causes the onset of fatty liver via regulation of the hepatic nutrient metabolism pathway.15 Researchers have reported that SIRT1 overexpression or activator treatment attenuates the progression of diet-induced fatty liver.14,16–18
SIRT1 activity is dependent on the amount of NAD+. The amount of NAD+ is regulated by its salvage pathway, which consists of nicotinamide nucleotide adenylyltransferase 1 (NMNAT1) and nicotinamide phosphoribosyltransferase (NAMPT).19 NAD+ metabolism is associated with cancer cell biology or metabolic syndromes.20,21. In the liver, NAMPT expression is downregulated as NASH progresses.22 Glucose and alcohol metabolism require NAD+.23,24 Therefore, SIRT1 activation may be restricted by intracellular NAD+ decreases.
Alcohol consumption induces a disruption of the hepatic microenvironment, such as sinusoid narrowing or edema formation, and leads to inhibition of oxygenation of the liver. Oxygen consumption is increased in alcohol dehydrogenation by CYP2E1. In the NASH liver, hepatocyte ballooning by lipid deposition impairs the peripheral microenvironment, which could cause oxygenation to decrease. Therefore, a hypoxic condition would be induced in hepatocytes of ASH and NASH patients. Hypoxia inducible factor (HIF) has been reported as accumulating in the liver of alcohol-fed mice.25 Hypoxia enhances inflammatory chemokine expression.26 Therefore, a hepatic hypoxic condition would exacerbate ASH and NASH. To identify a novel therapeutic target for non-viral hepatitis-induced tumorigenesis, we analyzed SIRT1 gene expression and activity in livers of patients with non-B non-C (NBNC) hepatitis (either ASH or NASH). Activation of SIRT1 may result in an improvement to the inflammatory condition in the liver of NBNC hepatitis patients.
Materials and Methods
Human tissue specimens
We retrospectively analyzed mRNA expression using noncancerous liver tissue from 28 patients with NBNC hepatitis, 20 HBsAg positive patients as HBV patients, and 73 patients with chronic HCV infection. Histone acetylation and NAD+/NADH ratio were analyzed using tissues from 8 NBNC, 9 HBV and 14 HCV patients, whose tissue samples were available enough for those analyses, among the patients in this study. Patients in the present study had undergone a liver resection for HCC between 2003 and 2010 at our institute. For healthy control tissue, we analyzed liver samples taken from the living donors of the liver transplantation. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki. Samples were collected following an established protocol approved by the Ethics Committee of Kyushu University after patients had given their informed consent. The data does not contain any information that could lead to the identification of patients.
Measurement of mRNA expression using real-time RT-PCR
Total RNA was extracted from resected liver tissue or cells using reagents for RNA extraction, including ISOGEN and Ethachinmate (Nippon Gene, Tokyo, Japan). Synthesis of first-strand cDNA was performed using the SuperScript III First-Strand synthesis system for qRT-PCR (Life Technologies, Tokyo, Japan) according to the manufacturer's protocol. Real-time RT-PCR was performed using Taqman reagents (Life Technologies). Gene expression assays were purchased from Life Technologies.
Immunohistochemistry
Formalin-fixed paraffin-embedded 3-μm sections were deparaffinized in xylene, rehydrated through graded ethanol, and rinsed in PBS. Heat-induced epitope retrieval was performed in 10 mM citrate buffer, pH 6.0, with 1 mM EDTA, at 125°C for 4 min in a pressure boiler. Endogenous peroxidase activity was blocked by incubation with 0.3% H2O2 for 10 min. Nonspecific antibody binding was blocked by incubating the sections with normal goat serum (Dako, Glostrup, Denmark) for 10 min. The sections were then incubated with anti-SIRT1 (1:200, Sigma-Aldrich, Tokyo, Japan) or HIF1 rabbit polyclonal antibodies (1:50, Sigma-Aldrich) for 30 min and labeled using the Envision Detection System (Dako) for 30 min at room temperature. Sections were then developed with 3,3′-diaminobenzidine tetrahydrochloride (DAB plus; Dako) and counterstained with 10% Mayer's hematoxylin, dehydrated and mounted.
Western blotting
Cells were lysed in Cell Lysis Buffer (Cell Signaling Technology, Tokyo, Japan) containing Protease Inhibitor Cocktail (Sigma-Aldrich). Rabbit polyclonal antibodies against HIF1 were purchased from Cell Signaling Technology. Rabbit polyclonal antibodies against SIRT1 were purchased from Epitomics (Burlingame, CA, USA). Mouse monoclonal antibodies against β-actin (AC-15) were purchased from Sigma-Aldrich. Proteins were detected using an ECL Plus Western Blotting Detection System (GE Healthcare, Tokyo, Japan).
Cell culture and reagents
HepG2 and Hep3B cells were cultured in DMEM (Life Technologies) supplemented with 10% FBS. Cells were subjected to 1% O2 to create the hypoxic condition. For iron deficiency or iron-rich conditions, HepG2 cells were incubated with 100 μM deferoxamine (DFX) (Sigma-Aldrich) or 50 μM transferrin (Sigma-Aldrich) for 24 h. SIRT1 activator, SRT1720, was purchased from Selleck Chemicals (Houston, TX, USA). Recombinant TNF protein was obtained from R&D Systems (Minneapolis, MN, USA). For the high glucose medium, a DMEM containing 25 mM glucose (Life Technologies) was used.
Nicotinamide adenine dinucleotide/NADH quantification
Nicotinamide adenine dinucleotide and NADH were extracted from frozen liver tissue and quantified using a NAD/NADH Quantitation Colorimetric Kit (BioVision, Milpitas, CA, USA) according to the manufacturer's protocol. The NAD+/NADH ratio was calculated.
Total histone H3 and acetylated-histone H3K9 quantification
Histones were extracted from frozen liver tissue using an EpiQuik Total Histone Extraction Kit according to the manufacturer's protocol. Total histone H3 and acetylated-histone H3K9 were quantified using EpiQuik Total Histone H3 Quantification and EpiQuik Global Acetyl Histone H3K9 Quantification Kits, respectively. The acetylated-H3K9 value was normalized using the amount of total histone H3. These kits were purchased from Epigentek (Farmingdale, NY, USA).
STAM mice study
STAM mice liver tissue was obtained from Stelic Institute (Tokyo, Japan).7 Briefly, pathogen-free 14-day pregnant C57BL/6J mice were purchased from CLEA Japan (Tokyo, Japan). NASH-HCC was induced in male mice by a single s.c. injection of 200 μg STZ (Sigma-Aldrich) at 2 days after birth and fed with HFD32 (CLEA Japan) ad libitum after 4 weeks of age.
All animal procedures were performed in accordance with the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23, revised 1985).
Total RNA extractions and mRNA quantifications were performed, similar to those for human liver tissues.
Statistical analysis
Pearson's χ2-test was used for qualitative variables. Non-parametric Wilcoxon and Student's t-tests were used for quantitative variables.
Results
Silent information regulator 1 expression and histone acetylation in livers of non-B non-C patients
Table1 displays the patients’ characteristics in this study. Body mass index and DM complication rates in NBNC patients were significantly higher than in viral-hepatitis patients. ALT and the activity grade in HCV patients were higher than in other patients. We measured SIRT1 mRNA expression levels using RT-PCR, and compared expression levels between healthy donors, NBNC, HBV and HCV patients. SIRT1 mRNA expression levels were significantly higher in the livers of patients with NBNC hepatitis than in HCV infection patients (P < 0.001) and healthy controls (P = 0.001) (Fig.1a). Immunohistochemistry (IHC) using anti-SIRT1 antibodies showed staining to mainly hepatic parenchymal cellular nuclei in the perivascular area. Representations of positive sections (Fig.1b, left panel) and negative sections (Fig.1b, right panel) are shown. More vessels showed staining in NBNC hepatitis patients than in HCV infection patients (P = 0.005) (Fig.1c).
Table 1.
Patient characteristics
| Factor | NBNC (n = 28) | HBV (n = 20) | HCV (n = 73) | P-value (univariate) |
|---|---|---|---|---|
| Sex, male/female | 23/5 | 15/5 | 58/15 | 0.833 |
| Age, mean years (range) | 71 (34–86) | 59 (36–81) | 70 (55–87) | <0.001 |
| BMI (kg/m2), mean ± SD | 25 ± 4 | 23 ± 3 | 22 ± 3 | 0.004 |
| ALT (IU/L), mean ± SD | 35 ± 25 | 38 ± 22 | 60 ± 42 | 0.003 |
| Albumin (g/dL), mean ± SD | 3.9 ± 0.4 | 3.9 ± 0.4 | 3.9 ± 0.4 | 0.777 |
| Cholesterol (mg/dL), mean ± SD | 179 ± 35 | 188 ± 38 | 154 ± 31 | <0.001 |
| Total-bilirubin (mg/dL), mean ± SD | 0.8 ± 0.3 | 0.8 ± 0.4 | 0.8 ± 0.4 | 0.659 |
| DM complication (%) | 62 | 16 | 26 | 0.001 |
| Fibrosis stage, F0/1/2/3/4 | 7/6/4/5/6 | 3/4/3/3/7 | 5/13/15/19/21 | 0.414 |
| Activity grade, A0/1/2/3 | 4/19/5/0 | 1/14/5/0 | 1/18/38/16 | <0.001 |
Fibrosis stage and activity grade were classified according to the New Inuyama Classification. ALT, alanine aminotransferase; BMI, body mass index; DM, diabetes mellitus.
Fig 1.

Silent information regulator 1 (SIRT1) expression and histone acetylation in the livers. (a) Comparison of hepatic SIRT1 mRNA expression levels between healthy donors, non-B non-C (NBNC), hepatitis B virus (HBV) and hepatitis C virus (HCV) patients. (b) Hepatic SIRT1 protein expression in NBNC patients determined by immunohistochemistry (IHC). Representative images of positive sections (left panels) and negative sections (right panel). (c) Comparison of SIRT1 positive tissue rates by IHC in liver between NBNC and HCV. (d) Comparison of intrahepatic H3K9 relative acetylation levels between healthy donors, NBNC, HBV and HCV patients. H3K9 relative acetylation is determined by calculating the acetylated-histone H3K9/total histone H3 ratio.
To evaluate SIRT1 activity, we detected a SIRT1-targeted deacetylation site, lysine 9 of histone H3 (H3K9),27 using total H3 and acetylated-H3K9 specific ELISA. Interestingly, H3K9 relative acetylation levels were significantly higher in patients with hepatitis than in the healthy donors (NBNC, P = 0.005; HBV, P = 0.007; HCV, P = 0.03) (Fig.1d).
Impairment of nicotinamide adenine dinucleotide salvage pathway in the liver with hepatitis
In NBNC patients, H3K9 acetylation levels were increased; however, SIRT1 expression levels were higher than levels in healthy donors. Therefore, the NAD+ salvage pathway and the amount of NAD+ in the liver were analyzed to clarify the regulation of SIRT1 activity in the NBNC liver. NAMPT mRNA expression levels were significantly lower in patients with hepatitis than those levels in healthy donors (NBNC, P < 0.001; HBV, P < 0.001; HCV, P < 0.001). In particular, NAMPT expression in the NBNC liver was significantly lower than in HBV (P = 0.004) or HCV (P = 0.04) patients (Fig.2a). NMNAT1 mRNA expression levels of healthy donors and NBNC livers were similar (Fig.2b). The NAD+/NADH ratio was measured. The NAD+/NADH ratio was significantly lower in the hepatitic liver than the ratio for healthy donor livers (Fig.2c). NAD+ and NADH amounts could not be measured in some tissues. The analysis of the NAD+/NADH ratio revealed a relationship, but not significantly, between healthy donor and NBNC livers. (Fig. S1) A decrease in NAD+ levels would impair SIRT1 activity and result in an increase of H3K9 acetylation. These results indicate a disruption of the NAD+ salvage pathway in the hepatitis liver.
Fig 2.
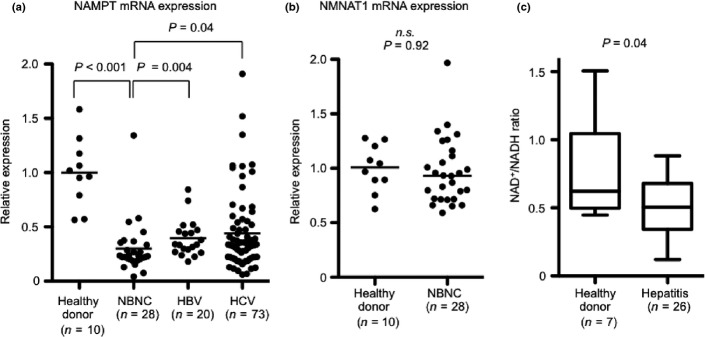
Suppression of nicotinamide phosphoribosyltransferase (NAMPT) expression and NAD/NADH ratio in liver of hepatitis. (a) Comparison of hepatic NAMPT mRNA expression levels between healthy donors, non-B non-C (NBNC), hepatitis B virus (HBV) and hepatitis C virus (HCV) patients. (b) Comparison of hepatic NMNAT1 mRNA expression levels between healthy donors and NBNC patients. (c) Comparison of the intrahepatic NAD+/NADH ratios between healthy donors and hepatitis patients, including NBNC (n = 6), HBV (n = 8) and HCV (n = 2). n.s., Not significant.
Induction of silent information regulator 1 expression in the hypoxic liver
To assess the state of the liver in NBNC patients, we explored the trigger that induced SIRT1 expression. We cultured human hepatoma HepG2 cells in the medium with H2O2 as a reactive oxygen species, high glucose, transferrin, tumor necrosis factor (TNF), transforming growth factor-β, or SIRT1 inhibitor. These treatments did not induce SIRT1 mRNA and protein expression. Chen et al.28 reported that hypoxia induces SIRT1 expression in Hep3B cells. In addition to Hep3B cells, we confirmed SIRT1 expression in hypoxic condition or by iron-chelator treatment in another liver cell line, HepG2 cells.
An iron chelator, DFX, induce both SIRT1 protein and mRNA expression (Fig.3a and data not shown). DFX also induced HIF1 protein accumulation (Fig.3a, left panel); therefore, we examined whether the hypoxic condition induces SIRT1 expression.
Fig 3.
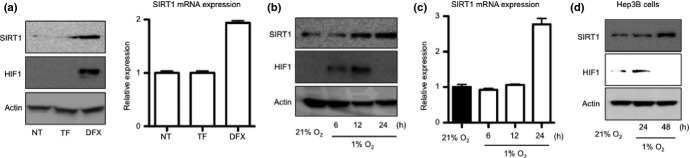
Silent information regulator 1 (SIRT1) expression in hypoxic condition. (a) HepG2 cells were treated with 50 μM transferrin (TF) or 100 μM deferoxamine (DFX) for 24 h. SIRT1 and HIF1α protein expression levels were detected by western blotting (left panels). Total RNA was extracted and SIRT1 mRNA was measured by real-time RT-PCR (right graph). Relative expression of non-treated cells (NT) is shown. (b, c) HepG2 cells were cultured in the hypoxic condition (1% O2) for 6, 12 and 24 h. (b) SIRT1 and HIF1α protein expression levels in cells were detected by western blotting. (c) Total RNA was extracted and SIRT1 mRNA was measured by real-time RT-PCR. Relative expression of normoxia-cultured (21% O2) cells is shown. (d) Hep3B cells were cultured in the hypoxic condition for 24 and 48 h. SIRT1 and HIF1α protein expression levels in cells were detected by western blotting.
SIRT1 expression was induced in HepG2 cells cultured in 1% O2 (Fig.3b,c). The hypoxic condition also induced SIRT1 expression in another hepatome cell, Hep3B (Fig.3d). We then detected HIF1 protein in liver tissue using IHC. HIF1 was observed to accumulate in the nuclei of hepatic parenchymal cells in the perivascular area (Fig. S2a, upper panels).
HIF1 positive cells were also stained with SIRT1 antibodies (Fig. S2a, lower panels) in comparison with the serial sections. All HIF1 positive tissue sections were also positive for SIRT1, and 12 sections of the 22 HIF1 negative sections were stained with SIRT1 antibodies (Fig. S2b).
These results suggest that SIRT1 expression is associated with the accumulation of HIF1 in the liver of NBNC hepatitis patients.
Impact of nicotinamide adenine dinucleotide and silent information regulator 1 on chemokine expression in the hypoxic condition
CXCL10 deficiency or neutralization antibodies attenuate non-viral hepatitis in mice.4 Therefore, we investigated whether CXCL10 increases in the livers of NBNC patients in this study. CXCL10 expression levels in some of the healthy donors were under the detection limit. Expression levels were higher in livers of NBNC patients than in livers of healthy donors (P = 0.04) (Fig.4a).
Fig 4.

Silent information regulator 1 (SIRT1) activation and hypoxia-induced chemokine expression. (a) Comparison of hepatic CXCL10 mRNA expression levels between healthy donors and non-B non-C (NBNC) patients. (b) HepG2 cells were cultured in the hypoxic condition in high glucose (25 mM) and low glucose (5 mM) medium for 24 h. The intracellular NAD+/NADH ratio is shown. (c) Hypoxia-induced CXCL10 expression in HepG2 cells is shown. Cells were cultured in the hypoxic condition in high glucose, low glucose or low glucose with 1 μM SRT1720 containing medium for 24 or 48 h. (d) Tumor necrosis factor (TNF)-induced CXCL10 expression levels in HepG2 cells is shown. Cells were cultured in 1 ng/mL TNF or TNF with 1 μM SRT1720 containing medium for 24 h. (e) Hypoxia-induced MCP-1 expression in HepG2 cells is shown. Cells were cultured in the hypoxic condition in high glucose, low glucose or low glucose with 1 μM SRT1720 containing medium for 24 or 48 h. n.s., Not significant.
HepG2 cells were then incubated in 1% O2. The medium included either high or low glucose. Intracellular NAD+ and CXCL10 expression levels were measured. The NAD+/NADH ratio showed a decrease in the 1% O2 condition with high glucose (Fig.4b). CXCL10 expression levels showed a decrease in the 1% O2 condition with low glucose (rich NAD+ condition). SIRT1 activator, SRT1720,29,30 inhibited CXCL10 induction most effectively (Fig.4c). SRT1720 treatment also inhibited TNF-induced CXCL10 expression in HepG2 cells (Fig.4d).
Another hepatitis-related chemokine, Mcp-1, was induced in the liver of STAM mice (Fig. S3a) and alcohol-fed mice.25 Therefore, MCP-1 expression in HepG2 cells incubated in the hypoxic condition was analyzed. MCP-1 expression was more strongly induced in the high glucose medium than in the low glucose medium, as with CXCL10 expression (Fig.4e). SRT1720 treatment also attenuated MCP-1 expression induced by hypoxia (Fig.4e). Normalization of the NAD+/NADH ratio and activation of upregulated SIRT1 could improve the inflammatory condition in the liver of NBNC hepatitis patients.
Discussion
Several previous studies have reported an association between SIRT1 and liver disease. For example, in mice studies, it has been reported that liver-specific mSirt1 deficiency promotes fatty liver development;15 and mSirt1 forced-expression improves fatty liver progression.14 However, the role of SIRT1 in the liver with non-viral hepatitis is still unknown.
In this present study, we demonstrate SIRT1 expression, activity and NAD+ regulation using noncancerous liver tissue specimens from NBNC patients. We observed that hepatic SIRT1 expression levels in NBNC patients increased compared with healthy donors or in case of HCV infection (Fig.1a). Recently, in vitro studies have shown that SIRT1 expression is repressed by HCV replication, and SIRT1 inhibition contribute to HCV replication.31,32 HCV replication may also be associated with low expression levels of SIRT1 in clinical liver tissue.
SIRT1 expression is upregulated and, therefore, we hypothesized that SIRT1 contributes to hepatic inflammation or HCC incidence. To clarify whether SIRT1 activity is upregulated, histone H3K9, the SIRT1-targeted deacetylation site27 and acetylation levels were determined. Unexpectedly, histone H3K9 acetylation levels were higher in NBNC patients than levels observed in healthy donors (Fig.1d). This indicates a repression of SIRT1 activity in NBNC patients.
NAMPT expression (that converts nicotinamide [NAM] into nicotinamide mononucleotide)19 levels were significantly lower in hepatitis patients than in healthy donors (Fig.2a). Therefore, we could not measure the NAM amount in the liver tissue, although the NAM amount in the hepatitic liver would be increased. The amount of NAD+ was also significantly lower (Fig.2c). NAD+ is indispensable in SIRT1 activity, and NAM is a SIRT1 inhibitor.33 SIRT1 inactivation in the hepatitic liver would be caused by NAD+ shortage and NAM accumulation.
We measured hepatic mNampt and mSirt1 expression levels using a NASH animal model (STAM mice)7 to analyze whether or not there is an association between the expression of mNampt and mSirt1 and disease progression. Mcp-1 and Col1a1 expression levels, inflammation and fibrosis progression markers in this model7, showed an increase compared with control mice (Fig. S3a). mNampt expression showed a significant decrease in the livers of 12-week-old STAM mice (P = 0.02) (Fig. S3a). These results indicate that NASH-like hepatitis progression is associated with a reduction in mNampt expression. mSirt1 expression was also measured in STAM mice. A slight but insignificant increase in 16- and 20-week-old mice was observed (Fig. S3b). SIRT1 static expression in STAM mice would be an interesting observation. The cause of SIRT1 induction might be independent of the cause of NAMPT decrease. Analysis using another NASH model with HCC incidence may be needed to address the SIRT1 induction mechanism in vivo. As an alternative hypothesis, decreased levels of mNampt expression could be followed by increased levels of SIRT1 expression to compensate for the impairment of SIRT1 activity.
We then explored how SIRT1 induction occurred in the NBNC liver. We treated HepG2 cells with transferrin or DFX. DFX induced SIRT1 expression (Fig.3a). The hypoxic condition induced SIRT1 expression (Fig.3b–d). Therefore, we focused on HIF1 protein, because accumulation of HIF1 protein was observed in DFX-treated and hypoxia-exposed cells. Several liver sections from NBNC patients were stained with HIF1 antibodies. HIF1 positive blood vessels were also positive for SIRT1 expression (Fig. S2). In HepG2 cells, DNA damage inducers moderately increased SIRT1 expression (data not shown). DNA damage or other stimuli may associate with SIRT1 expression in HIF1 negative tissues.
In the ASH and NASH liver the microenvironment is impaired and oxygenation is decreased. This results in a hypoxic condition with HIF1 protein accumulating in hepatocytes of NBNC patients. Therefore, HepG2 cells were exposed to hypoxic and low NAD+ conditions (Fig.4b) and CXCL10 and MCP-1 expression were induced (Fig.4c,e). Improvement in the NAD+/NADH ratio was achieved by culturing cells in low glucose which suppressed the expression of these chemokines. Treatment with SIRT1 activator SRT1720 even more significantly reduced expression (Fig.4c,e). In the alcohol-induced hypoxic liver, a HIF-1 activation in hepatocytes results in liver abnormalities, and liver-specific deletion of HIF-1 contributes to hepatocytes protection from alcohol-induced liver injury.25 SIRT1 suppresses HIF-1 activity via a HIF-1 deacetylation34 and, therefore, SIRT1 expression would be induced to maintain the liver function in response to hypoxic condition. However, SIRT1 activity is inhibited by the impairment of the NAD+/NADH ratio due to excess alcohol or nutrient metabolism in NBNC hepatitis, and that may lead to the exacerbation of hepatic inflammation or abnormalities. CXCL10 expression was higher in livers of NBNC patients than in livers of healthy donors (Fig.4a). CXCL10 deficiency or neutralization antibodies attenuate non-viral hepatitis in mice.4 Mcp-1 inhibition also improves the hepatitic condition.5 These results indicate that SIRT1 activation by normalization of the NAD+/NADH ratio and SIRT1 activator could improve the inflammatory condition of the liver in NBNC hepatitis patients.
A recent report suggests that the SIRT1 activator resveratrol inhibited HBV X protein-induced hepatocarcinogenesis.35 The present study suggests that NBNC hepatitis and hepatitis B have in common SIRT1 expression, activity and NAD+ regulation (Figs1a,d,2a). NAD+ normalization may suppress NBNC hepatitis and hepatitis B exacerbation; however, the NAD+ salvage pathway would be impaired in hepatitis. Therefore, regulation of other NAD+ metabolize enzymes would be needed. Other NAD+ase, CD38, inhibitor administration increased NAD+ levels, and improved glucose and lipid metabolism in obese mice.36 NAD+/NADH ratio was disrupted in NBNC hepatitis patients; therefore, CD38 inhibition could be a pharmacological target to treat such liver diseases through SIRT1 activation. Further in vivo studies are required to determine SIRT1 functions in non-viral hepatitis. SIRT1 regulation may have the potential to be developed as a novel therapeutic target to prevent non-viral hepatitis-induced hepatocellular carcinogenesis.
Acknowledgments
This study was supported by JSPS KAKENHI Grant Number 25861197, and a Grant-in-Aid from the Ministry of Health, Labour and Welfare, Japan (H23-kannen-003).
Disclosure Statement
H.K. is an employee of Chugai Pharmaceutical Co., Ltd. The other authors have no coflict of interest to declare.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Comparison of the intrahepatic NAD+/NADH ratios between healthy donors, non-B non-C (NBNC), hepatitis B virus (HBV) and hepatitis C virus (HCV) patients.
Fig. S2. Hepatic HIF1 and silent information regulator 1 (SIRT1) protein expression determined by immunohistochemistry (IHC).
Fig. S3. Comparison of hepatic mMcp-1, mCol1a1 mNampt and mSirt1 mRNA expression.
References
- Okanoue T, Umemura A, Yasui K, Itoh Y. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in Japan. J Gastroenterol Hepatol. 2011;26(Suppl 1):153–62. doi: 10.1111/j.1440-1746.2010.06547.x. [DOI] [PubMed] [Google Scholar]
- Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–84. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Scaglioni F, Ciccia S, Marino M, Bedogni G, Bellentani S. ASH and NASH. Dig Dis. 2011;29:202–10. doi: 10.1159/000323886. [DOI] [PubMed] [Google Scholar]
- Hintermann E, Bayer M, Pfeilschifter JM, Luster AD, Christen U. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J Autoimmun. 2010;35:424–35. doi: 10.1016/j.jaut.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez M, Miquel R, Colmenero J, et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–50. doi: 10.1053/j.gastro.2009.01.056. [DOI] [PubMed] [Google Scholar]
- Fujii M, Shibazaki Y, Wakamatsu K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46:141–52. doi: 10.1007/s00795-013-0016-1. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Nat Acad Sci USA. 2000;97:5807–11. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu S, Giles A, et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011;25:1664–79. doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–38. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Nat Acad Sci USA. 2008;105:9793–8. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–42. doi: 10.1152/ajpgi.90358.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor RK, Baur JA, Gomes AP, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Frizzell KM, et al. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem. 2009;284:20408–17. doi: 10.1074/jbc.M109.016469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome–A key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–52. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8:287–96. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- Dahl TB, Haukeland JW, Yndestad A, et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2010;95:3039–47. doi: 10.1210/jc.2009-2148. [DOI] [PubMed] [Google Scholar]
- Ganapathy V, Thangaraju M, Prasad PD. Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009;121:29–40. doi: 10.1016/j.pharmthera.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- Nath B, Levin I, Csak T, et al. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology. 2011;53:1526–37. doi: 10.1002/hep.24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savransky V, Bevans S, Nanayakkara AMS, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G871–7. doi: 10.1152/ajpgi.00145.2007. [DOI] [PubMed] [Google Scholar]
- Suter MA, Chen A, Burdine MS, et al. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J. 2012;26:5106–14. doi: 10.1096/fj.12-212878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Dioum EM, Hogg RT, Gerard RD, Garcia JA. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J Biol Chem. 2011;286:13869–78. doi: 10.1074/jbc.M110.175414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Chung S, Hwang JW, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122:2032–45. doi: 10.1172/JCI60132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard BP, Gomes AP, Dai H, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339:1216–9. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LJ, Li SC, Zhao YH, Yu JW, Kang P, Yan BZ. Silent information regulator 1 inhibition induces lipid metabolism disorders of hepatocytes and enhances hepatitis C virus replication. Hepatol Res. 2013;43:1343–51. doi: 10.1111/hepr.12089. [DOI] [PubMed] [Google Scholar]
- Yu JW, Sun LJ, Liu W, Zhao YH, Kang P, Yan BZ. Hepatitis C virus core protein induces hepatic metabolism disorders through down-regulation of the SIRT1-AMPK signaling pathway. Int J Infect Dis. 2013;17:539–45. doi: 10.1016/j.ijid.2013.01.027. [DOI] [PubMed] [Google Scholar]
- Peck B, Chen CY, Ho KK, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9:844–55. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38:864–78. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Lin HC, Chen YF, Hsu WH, Yang CW, Kao CH, Tsai TF. Resveratrol helps recovery from fatty liver and protects against hepatocellular carcinoma induced by hepatitis B virus X protein in a mouse model. Cancer Prev Res (Phila) 2012;5:952–62. doi: 10.1158/1940-6207.CAPR-12-0001. [DOI] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013;62:1084–93. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Comparison of the intrahepatic NAD+/NADH ratios between healthy donors, non-B non-C (NBNC), hepatitis B virus (HBV) and hepatitis C virus (HCV) patients.
Fig. S2. Hepatic HIF1 and silent information regulator 1 (SIRT1) protein expression determined by immunohistochemistry (IHC).
Fig. S3. Comparison of hepatic mMcp-1, mCol1a1 mNampt and mSirt1 mRNA expression.