

Abstract
A c-Met inhibitor tivantinib is a candidate anticancer agent for patients with hepatocellular carcinoma (HCC), and CYP2C19 is the key metabolic enzyme for tivantinib. Previous Japanese phase I studies in patients with solid tumors (except HCC) recommend 360 mg twice daily (BID) and 240 mg BID for CYP2C19 extensive metabolizers (EM) and poor metabolizers (PM), respectively. In this study, Japanese patients with HCC in whom sorafenib treatment has failed were enrolled to evaluate the safety, tolerability and pharmacokinetics of oral tivantinib as a single agent. The dose was escalated separately in EM and PM, from 120 mg BID to 240 mg BID, in both capsule and tablet formulations. A total of 28 patients (EM: 21, PM: 7) received tivantinib. At a dose of 120 mg BID, dose-limiting toxicities (DLT) did not develop in 12 EM (capsule: 6, tablet: 6) and 7 PM (capsule: 4, tablet: 3) during the DLT-observation period (for 29 days after first dosing). At this dose, the pharmacokinetic profiles of tivantinib (AUC0–12 and Cmax) did not remarkably differ between EM and PM. When treated with 240 mg BID, 5 of 9 EM (capsule: 4 of 6, tablet: 1 of 3) developed neutropenia-related DLT accompanying plasma tivantinib concentration higher than expected from the previous studies. Consequently, PM did not receive 240 mg BID. In conclusion, 120 mg BID of tivantinib is recommended among Japanese patients with HCC regardless of CYP2C19 phenotype.
This would be one of the first manuscripts demonstrating that the recommended dose of an anti-cancer agent for patients with hepatocellular carcinoma (HCC) could be distinct from its recommended dose for patients with other solid tumors.
Keywords: c-Met inhibitor, CYP2C19 polymorphisms, hepatocellular carcinoma, phase I study, tivantinib
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death in the world.1 Sorafenib is the only approved agent for medical treatment of HCC.2 A prespecified subgroup analysis from phase III study of sorafenib indicated that sorafenib was less effective in patients with high expression of hepatocyte growth factor (HGF) in tumor tissues.3 In addition, high expression of HGF and its receptor c-Met have been linked to poor outcomes in patients with HCC.4 The c-Met pathway may, therefore, be a therapeutic target in patients in whom sorafenib treatment has failed.
Tivantinib (ARQ 197) is a selective c-Met inhibitor being studied in many types of cancers.5–7 A placebo-controlled randomized phase II study in the USA and Europe (ARQ 197-215 study) demonstrated that monotherapy with tivantinib in a capsule formulation prolonged the time to progression in patients with HCC in whom one previous regimen of systemic therapy has failed.7 A prespecified subgroup analysis from the ARQ 197-215 study demonstrated that tivantinib was particularly effective in patients whose tumors showed high c-Met expression on immunohistochemical analysis (i.e. ≥50% of cells expressing 2+ or 3+).7 To confirm and extend these results, a phase III (METIV-HCC) study in patients with HCC expressing high levels of c-Met who failed sorafenib therapy is now ongoing in Western countries to evaluate the effectiveness of tivantinib in tablet formulation.8
CYP2C19 is the key metabolic enzyme of tivantinib. Genetic polymorphisms of CYP2C19 leading to enzymatic dysfunction are found in approximately 20% of Asians, but are rare in White and Black people.9 Several phase I trials of tivantinib have been performed in Asians with solid tumors, including non-small-cell lung cancer (NSCLC), to evaluate the safety and tolerability of tivantinib according to the CYP2C19 genotype of each patient.10,11 A previous phase I study in Japanese patients with solid tumors (ARQ 197-0701 study, HCC not included) revealed that the plasma concentration of tivantinib in CYP2C19 poor metabolizers (PM) was higher than that in extensive metabolizers (EM) receiving the same dose. Therefore, the recommended dose of tivantinib was based on the CYP2C19 phenotype: 360 mg twice daily (BID) for EM and 240 mg BID for PM.10 To date, however, clinical trials of tivantinib have not been performed in Asian patients with HCC. Here we report a phase I study in Japanese patients with HCC in whom sorafenib treatment has failed. Our main objectives were to evaluate the safety and tolerability of tivantinib as a single agent and to determine the recommended doses of tivantinib for CYP2C19 EM and PM, respectively.
Material and Methods
Patients
Patients with advanced HCC who were refractory to or intolerant to the previous sorafenib treatment and, also, patients who had refused to start a sorafenib treatment were eligible for the present study. Other inclusion criteria were: age ≥20 years, Eastern Cooperative Oncology Group performance status of 0 or 1, Child-Pugh class A, adequate organ functions (i.e. neutrophils ≥1500/μL, platelets ≥60 000/μL, hemoglobin ≥9.0 g/dL, total bilirubin ≥2.0 mg/dL, aspartate aminotransferase and alanine aminotransferase ≤5 times the institutional upper limit of normal (ULN), serum creatinine ≤1.5 times the ULN, prothrombin time/international normalized ratio ≥0.8 times to ≤ULN and albumin ≥2.8 g/dL) and a life expectancy of ≥3 months. Exclusion criteria were as follows: prior treatment with a c-Met inhibitor, treatment with anticancer agents (including investigational drugs) within 2 weeks before enrollment (within 4 weeks before enrollment if the anticancer agents were antibodies), locoregional therapies such as transcatheter arterial chemoembolization or hepatic arterial infusion chemotherapy within 4 weeks before enrollment, a history of liver transplantation, clinically serious infection of ≥Grade 3 according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 (excluding hepatitis B virus [HBV] and hepatitis C virus [HCV] infection), gastrointestinal disorders potentially affecting the absorption of tivantinib, positive tests for antibodies to human immunodeficiency virus, human T-cell lymphotropic virus type I, or both, ongoing interferon therapy for HBV/HCV, known symptomatic brain metastases and pregnancy.
Study design
This was a multicenter (six sites in Japan), open-label, dose-escalating phase I study. The study was conducted in accordance with institutional guidelines, Good Clinical Practice, and the Declaration of Helsinki. All patients provided written informed consent before undergoing any procedure related to this study. This study is registered at ClinicalTrials.gov: NCT01656265. Tivantinib was supplied by Kyowa Hakko Kirin (Tokyo, Japan) as 120 mg capsules and 120 mg tablets. Patients underwent the blood test for CYP2C19 genotyping before enrollment and were classified into CYP2C19 EM or PM. Patients with either the *2(G681A) or *3(G636A) allele were defined as PM, and others were defined as EM.
This study was originally designed to evaluate the capsule formulation of tivantinib. EM and PM were separately enrolled to respective cohorts testing the initial dose of 120 mg BID, which was escalated to up to 240 mg BID in accordance with the 3 + 3 rule. An EM cohort of six patients was scheduled to receive 240 mg BID. During the study, there were two major protocol amendments. First, after completing the capsule formulation cohort in EM (at both 120 mg BID and 240 mg BID), the protocol was amended to terminate assessment of 240 mg BID capsule formulation in the PM cohort and to add EM and PM cohorts given 120 mg BID tablet formulation, which is the putative commercial formulation. Second, after completion of the tablet formulation cohort in EM (120 mg BID), the protocol was amended to additionally study EM and PM cohorts given the tablet formulation at 240 mg BID. An EM cohort of six patients was scheduled to receive 240 mg BID.
On the first day of treatment (Day 1), tivantinib was orally administered only once, with or immediately after breakfast, to evaluate the pharmacokinetics, especially elimination from plasma. From Day 2 onward, tivantinib was administered twice daily, with or immediately after meals. Dose-limiting toxicity (DLT) was defined as any of the following drug-related adverse events (AE) occurring during the DLT-observation period (Day 1–29): Grade 4 neutropenia lasting for >7 days (administration of granulocyte colony-stimulating factor [G-CSF] allowed), Grade 4 thrombocytopenia lasting for >5 days, any thrombocytopenia requiring platelet transfusion, febrile neutropenia lasting for >2 days, Grade 4 anemia (blood transfusion allowed), Grade ≥3 non-hematologic toxicity (excluding nausea, vomiting, or diarrhea if controllable, and abnormal laboratory-test results not requiring treatment), any drug-related AE requiring interruption of tivantinib at the assigned dose a total of ≥15 times, or any drug-related AE considered DLT by the investigator. Patients were allowed to continue treatment until they met discontinuation criteria, which included disease progression and unacceptable toxicities. End-of-treatment examinations were performed 28 days after the day of discontinuation or within 7 days before the initiation of a post-study treatment, whichever came first in each patient.
Patient assessments
The baseline evaluation included medical history, physical examination, and laboratory tests, performed within 14 days before enrollment. During the observation period, AE were assessed and graded (CTCAE ver. 4.0) continuously on the basis of the results of physical examination, routine laboratory evaluations (including hematology, biochemical tests and urinalysis), vital signs, Child-Pugh scoring and electrocardiography. Tumor response was evaluated according to the Response Evaluation Criteria in Solid Tumors (ver. 1.1) with modification (i.e. imaging schedule: baseline, Day 29 and every 6 weeks thereafter).12 Patients who received at least one dose of tivantinib were included in both the safety analysis set and efficacy analysis set. The DLT analysis set was defined as the safety analysis set after excluding patients who missed ≥15 doses during the DLT observation period and had no DLT. For pharmacokinetic assessment, blood samples were collected before and 1, 2, 4, 6, 10, 12 and 24 h after drug administration on Day 1. Plasma samples were stored at −70°C until analysis by liquid chromatography/tandem mass spectrometry. Pharmacokinetic values were calculated using WinNonlin (Pharsight, Mountain View, CA, USA). If tumor tissues were provided under written consent, c-Met expression was evaluated immunohistochemically.
Results
Enrollment
A total of 61 Japanese patients with HCC were screened for CYP2C19 genotype. Fifty-four were classified as EM and 7 as PM. Twenty-eight of these patients (EM: 21, PM: 7) met the eligibility criteria and were enrolled in the respective cohorts (Table1). Owing to the much lower proportion of PM, genetic testing for CYP2C19 was continued to screen for PM, even after enrolling the specified number of patients in the EM cohorts. As shown in Table2, patient characteristics did not remarkably differ between the EM and PM. Incidence of each CYP2C19 genotype was mostly as anticipated.9
Table 1.
Patient enrollment
| Cohort | Tivantinib formulation | Dose | EM | PM |
|---|---|---|---|---|
| n = 21 | n = 7 | |||
| 1 | Capsule | 120 mg BID | 6 | 4† |
| 2 | Capsule | 240 mg BID | 6‡ | Not tested |
| 3 | Tablet | 120 mg BID | 6 | 3§ |
| 4 | Tablet | 240 mg BID | 3¶ | Not tested |
One patient was replaced because of withdrawal of consent early in the dose-limiting toxicities (DLT) observation period.
This cohort was planned to be started with six patients, based on the assumption from the ARQ 197-215 study that 240 mg twice daily (BID) as the capsule formulation would be tolerable.7
Adding three patients for confirmation was not planned, if none of the first three patients had DLT.
Adding three patients for confirmation was abandoned owing to the drug safety update from the METIV-HCC study, in which higher incidence of neutropenia was reported in patients with hepatocellular carcinoma (HCC) treated with 240 mg BID as the tablet formulation.13 EM, extensive metabolizers; PM, poor metabolizers.
Table 2.
Patients characteristics
| EM (n = 21) | PM (n = 7) | |||
|---|---|---|---|---|
| n (%) | n (%) | |||
| Gender | ||||
| Female | 5 (24) | 1 (14) | ||
| Male | 16 (76) | 6 (86) | ||
| Age (year) | ||||
| Mean | 65 | 67 | ||
| CYP2C19 genotype | ||||
| *1/*1 | 6 (29) | *2/*2 | 4 (57) | |
| *1/*2 | 11 (52) | *2/*3 | 3 (43) | |
| *1/*3 | 4 (19) | *3/*3 | 0 (0) | |
| BCLC stage at screening | ||||
| Stage A | 0 (0) | 0 (0) | ||
| Stage B | 1 (5) | 4 (57) | ||
| Stage C | 19 (91) | 3 (43) | ||
| Stage D | 1 (5) | 0 (0) | ||
| Epidemiology | ||||
| HBV | 5 (24) | 2 (29) | ||
| HCV | 5 (24) | 3 (43) | ||
| Alcoholic hepatitis | 6 (29) | 0 (0) | ||
| Non-alcoholic steatohepatitis | 2 (10) | 1 (14) | ||
| Other | 3 (14) | 1 (14) | ||
| ECOG PS | ||||
| 0 | 17 (81) | 5 (71) | ||
| 1 | 4 (19) | 2 (29) | ||
| History of locoregional therapy† | ||||
| Surgery | 14 (67) | 4 (57) | ||
| TACE | 7 (33) | 5 (71) | ||
| TAE | 5 (24) | 0 (0) | ||
| HAIC | 7 (33) | 5 (71) | ||
| RFA | 4 (19) | 3 (43) | ||
| PEIT | 1 (5) | 0 (0) | ||
| Others | 0 (0) | 1 (14) | ||
| Reason for discontinuation of prior sorafenib | ||||
| Disease progression | 18 (86) | 7 (100) | ||
| Toxicity | 2 (10) | 0 (0) | ||
| Subject's refusal | 1 (5) | 0 (0) | ||
Some of the patients had received multiple locoregional therapies. BCLC, Barcelona Clinic Liver Cancer; ECOG, Eastern Cooperative Oncology Group; HAIC: hepatic arterial infusion chemotherapy; PEIT, percutaneous ethanol injection therapy; RFA: radiofrequency ablation, TACE, transcatheter arterial chemoembolization; TAE, transcatheter arterial embolization.
Tolerability and safety
Extensive metabolizers who received 120 mg BID of tivantinib did not experience any DLT during the DLT observation period, irrespective of the capsule (n = 6) or tablet (n = 6) formulation. Consequently, 120 mg BID of tivantinib in both formulations was considered tolerable in Japanese EM with HCC. However, when the dose was increased to 240 mg BID of tivantinib, 4 of 6 EM given the capsule formulation and 1 of 3 EM given the tablet formulation experienced DLT, all of which were associated with neutropenia or febrile neutropenia (Table S1). In addition, during evaluation of 240 mg BID as the tablet formulation in the present study, the protocol of the ongoing Western phase III trial of tivantinib in advanced HCC (METIV-HCC) was amended to reduce the starting dose of tivantinib from 240 mg BID to 120 mg BID, because patients who received 240 mg BID as the tablet formulation had an increased incidence of neutropenia.13 Therefore, it was concluded that 240 mg BID of tivantinib was intolerable in Japanese EM with HCC, regardless of the formulation. All DLT in this study resolved after the interruption of tivantinib and treatment with G-CSF.
Poor metabolizers who received 120 mg BID of tivantinib did not experience any DLT during the DLT observation period, irrespective of capsule (n = 3) or tablet (n = 3) formulations. Per the study protocol, one PM given the capsule formulation was replaced owing to the short-period observation of DLT. The patient wished to withdraw informed consent after 11 days of treatment without developing any AE, which in the opinion of the investigator may interfere with continuation of this study. It was concluded that both formulations of tivantinib were tolerable at 120 mg BID in PM. Dose escalation to 240 mg BID in PM was terminated because this dose was not tolerated by the EM or the patients enrolled in the METIV-HCC trial.
Throughout the study, hematologic toxicities such as leukopenia, anemia, neutropenia, thrombocytopenia and lymphocytopenia were the most frequent AE in EM as well as PM (Table3). Besides hematologic toxicity, fatigue and anorexia were relatively common in both phenotypes. Hand-hoot syndrome, hypertension, bleeding and interstitial lung disease (ILD) were not reported during this study.
Table 3.
Summary of drug-related adverse events occurring in ≥2 patients in either CYP2C19 EM or PM
| CYP2C19 extensive metabolizers | CYP2C19 poor metabolizers | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Capsule | Tablet | Overall | Capsule | Tablet | Overall | |||||||||||
| 120 mg BID | 240 mg BID | 120 mg BID | 240 mg BID | 120 mg BID | 120 mg BID | |||||||||||
| n = 6 | n = 6 | n = 6 | n = 3 | n = 21 | n = 4 | n = 3 | n = 7 | |||||||||
| All grade | Gr.≧3 | All grade | Gr.≧3 | All grade | Gr.≧3 | All grade | Gr.≧3 | All grade | Gr.≧3 | All grade | Gr.≧3 | All grade | Gr.≧3 | All grade | Gr.≧3 | |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Hematological toxicity | ||||||||||||||||
| Leukopenia | 3 (50) | 2 (33) | 6 (100) | 5 (83) | 1 (17) | 0 (0) | 2 (67) | 1 (33) | 12 (57) | 8 (38) | 2 (50) | 0 (0) | 1 (33) | 1 (33) | 3 (43) | 1 (14) |
| Anemia | 4 (67) | 2 (33) | 5 (83) | 4 (67) | 2 (33) | 0 (0) | 1 (33) | 0 (0) | 12 (57) | 6 (29) | 2 (50) | 2 (50) | 1 (33) | 0 (0) | 3 (43) | 2 (29) |
| Neutropenia | 3 (50) | 2 (33) | 4 (67) | 4 (67) | 2 (33) | 0 (0) | 1 (33) | 1 (33) | 10 (48) | 7 (33) | 1 (25) | 0 (0) | 2 (67) | 1 (33) | 3 (43) | 1 (14) |
| Thrombocytopenia | 0 (0) | 3 (50) | 4 (67) | 0 (0) | 0 (0) | 3 (50) | 1 (33) | 0 (0) | 5 (24) | 6 (29) | 2 (50) | 0 (0) | 0 (0) | 0 (0) | 2 (29) | 0 (0) |
| Lymphocytopenia | 2 (33) | 3 (50) | 5 (83) | 5 (83) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 7 (33) | 0 (0) | 2 (50) | 1 (25) | 1 (33) | 1 (33) | 3 (43) | 2 (29) |
| Non-hematological toxicity | ||||||||||||||||
| Fatigue | 3 (50) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 2 (67) | 0 (0) | 7 (33) | 0 (0) | 2 (50) | 0 (0) | 0 (0) | 0 (0) | 2 (29) | 0 (0) |
| Anorexia | 1 (17) | 0 (0) | 3 (50) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 5 (24) | 0 (0) | 1 (25) | 0 (0) | 1 (33) | 0 (0) | 2 (29) | 0 (0) |
| Diarrhea | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 1 (33) | 0 (0) | 4 (19) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 1 (14) | 0 (0) |
| Cough | 2 (33) | 1 (17) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (19) | 1 (5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Alopecia | 1 (17) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 4 (19) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 1 (14) | 0 (0) |
| Malaise | 1 (17) | 0 (0) | 1 (17) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 3 (14) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Rash | 1 (17) | 0 (0) | 1 (17) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 3 (14) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Bradycardia | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Vomiting | 1 (17) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Nausea | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Fever | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 1 (14) | 0 (0) |
| Edema | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Hypoalbuminemia | 0 (0) | 0 (0) | 3 (50) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (14) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Hypophosphatemia | 0 (0) | 0 (0) | 2 (33) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Insomnia | 1 (17) | 0 (0) | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Dyspnea | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (33) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Febrile neutropenia | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| ALP elevation | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (25) | 0 (0) | 1 (33) | 0 (0) | 2 (29) | 0 (0) |
EM, extensive metabolizers; PM, poor metabolizers.
A total of seven serious AE (SAE) related to tivantinib were reported in seven patients during the study. The SAE were neutropenia (3 EM), febrile neutropenia (2 EM), cough (1 EM) and fatigue (1 PM). One patient who was an EM died on Day 127 due to the hepatic failure associated with disease progression. Simultaneously, a drug-related febrile neutropenia was developed in the patient on Day 120, but causal relationship was not established between the febrile neutropenia and the death.
Pharmacokinetic analysis
Figure1 shows plasma concentration-time profiles of tivantinib after oral administration on Day 1. Although inter-individual differences in plasma tivantinib concentrations were appreciable even in the same cohort, plasma exposure was clearly higher in patients given 240 mg than in those given 120 mg (Fig.1). The maximum plasma tivantinib concentration (Cmax) exceeded 4000 ng/mL in five of the eight patients given 240 mg of tivantinib (Fig.1), and there was no clear difference among *1/*1, *1/*2 and *1/*3 (data not shown).
Fig 1.
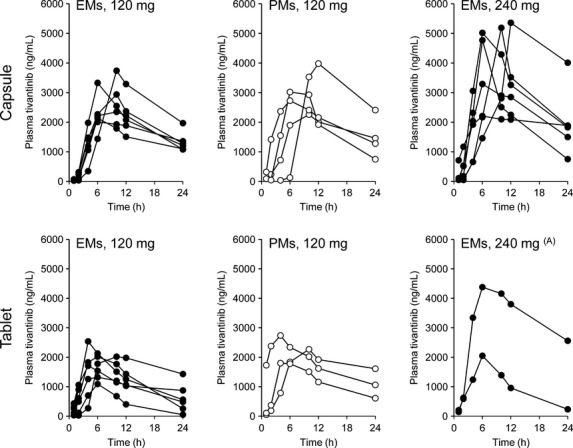
Plasma concentration–time profiles of tivantinib. Plasma concentrations after a single dose of tivantinib on Day 1 are presented for each patient treated with the indicated dose of tivantinib. Upper and lower panels indicate the profiles of the capsule formulation and tablet formulation, respectively. Closed circles indicate the profiles in CYP2C19 EM, and open circles indicate the profiles in CYP2C19 PM. (A) Data from one patient are missing because of a human error. EM, extensive metabolizers; PM, poor metabolizers.
After treatment with 120 mg, plasma exposure in PM was similar to, or slightly higher than, that in EM with both formulations. In addition, plasma tivantinib concentrations were somewhat higher in patients given the capsule formulation than in those given the tablet formulation. However, these differences were considered unremarkable, because the ranges of plasma tivantinib concentrations were generally similar in the cohorts given 120 mg. As shown in Figure2, the AUC0–24 in patients with HCC who received 120 mg was distributed in a similar range as historical AUC0–24 data from a previous study in which patients with NSCLC who had CYP2C19 EM or PM phenotypes received tivantinib as a single agent in a dose of 360 or 240 mg, respectively.11
Fig 2.
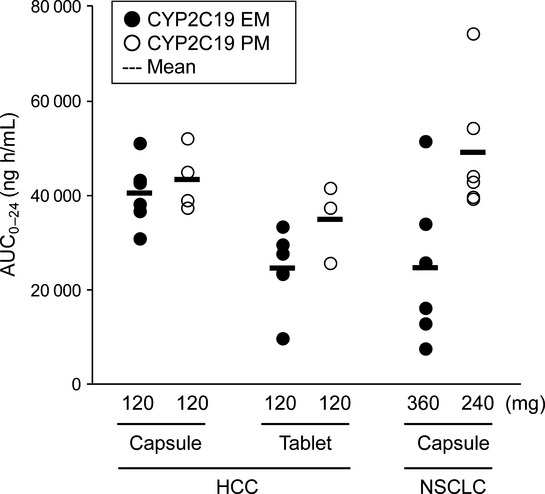
Comparison of AUC0–24 between patients with hepatocellular carcinoma (HCC) and patients with non-small-cell lung cancer (NSCLC). The results in HCC patients were from this study, while those in NSCLC patients were obtained from a previous study (ARQ 197-003/00511). Each dot indicates the AUC0–24 on Day 1 for individual patients treated with tivantinib monotherapy as indicated. Closed and open circles indicate the profiles in CYP2C19 EM and PM, respectively. EM, extensive metabolizers; PM, poor metabolizers.
Efficacy
Tumor response was assessable in all 28 patients. There were no complete or partial responses. Stable disease was reported in 17 of 21 EM (81.0%) and in 3 of 7 PM (42.9%). Progression-free survival (median and 95% confidence interval) according to phenotype was 113.0 days [69.0–154.0] for EM and 30.0 days [21.0–70.0] for PM, whereas overall survival data was immature at data cutoff. A total of four patients continued tivantinib treatment for more than 6 months, including one patient who received tivantinib for 407 days until disease progression. Tumor specimens obtained from 13 patients before enrollment in this study underwent immunohistochemical analysis for c-Met. High c-Met expression was found in 2 of the 13 patients. However, there was no apparent relation between c-Met expression and the response to tivantinib in this study (Table S2).
Discussion
In our study, Japanese patients with HCC received 120 mg BID and 240 mg BID of tivantinib in both capsule and tablet formulations. At a dose of 120 mg BID, no EM or PM experienced DLT with either formulation. In contrast, a high proportion of EM (5 of 9) who received 240 mg BID in either formulation developed DLT associated with neutropenia. At the same time, pharmacokinetic analysis revealed that tivantinib Cmax exceeded 4000 ng/mL in 5 of 8 EM who received 240 mg of tivantinib. This finding supports the results of a previous phase I study in Japanese patients with solid tumors (ARQ 197-0701 study), suggesting that a Cmax value exceeding 4000 ng/mL carries a high risk of severe neutropenia.10 Moreover, during our study, an ongoing Western phase III (METIV-HCC) trial indicated that treatment of tivantinib at the dose of 240 mg BID in tablet formulation was poorly tolerated.13 Taken together, it was suggested that 120 mg BID in either the capsule or tablet formulation is close to the maximum tolerable dose of tivantinib in Asian patients with HCC, irrespective of CYP2C19 phenotype.
Even at a relatively low dose of 120 mg of tivantinib, the AUC0–24 distribution in our patients with HCC, regardless of CYP2C19 phenotype, was similar to that in patients with NSCLC who received 360 mg (EM) or 240 mg (PM) of tivantinib in a previous study (Fig.2). The AUC0–24 values in patients with NSCLC were derived from the previous ARQ 197-003/005 study, in which patients with NSCLC received tivantinib in a similar manner to the present study (i.e. a crystalline formulation of tivantinib and administration with meals).11 It is still unclear why sufficient plasma exposure was achieved in patients with HCC even after administration of tivantinib at a dose lower than the recommended dose for patients with other solid tumors. However, it is supposed that the lower recommended dose in HCC would be the consequence of both HCC by itself and underlying cirrhosis, which might lead to the low neutrophil count at baseline, decreased activity of metabolic enzymes, and so on.14,15 On the basis of this concept and supported by the findings in this study, we concluded that 120 mg BID of tivantinib in either capsule or tablet formulation is the recommended dose for Japanese patients with HCC regardless of CYP2C19 genotype.
It was noteworthy that the same dose of tivantinib was recommended for both EM and PM among Japanese patients with HCC, although the recommended dose levels of this agent differ for patients with other solid tumors. For Asian patients with other solid tumors, the recommended dose of tivantinib is based on the results of CYP2C19 genetic testing.10,11 A previous study by Frye et al.15 suggests that this is a valid assumption. Their findings indicated that CYP2C19 is impaired more rapidly than other cytochrome P450 species, such as CYP1A2 or CYP2D6, during the development of hepatic functional impairment.15 Therefore, all patients with HCC, including those classified as EM on the basis of CYP2C19 genotype, are likely to have metabolic activity similar to or less than that associated with the CYP2C19 PM phenotype in patients with other solid tumors.
Hematologic toxicities were the major type of toxicity associated with tivantinib in Japanese patients with advanced HCC. It was implied that neutropenia would be easily exacerbated in HCC patients, because basal neutrophil counts are usually modest in HCC patients. The results of our small trial provided no evidence of a novel type of toxicity specific to patients with HCC or to EM or PM.7,10 In addition, the toxicity profile of tivantinib distinctly differed from that of vascular endothelial growth factor receptor (VEGFR) inhibitors, frequently associated with AE such as hypertension, proteinurea, hand-foot skin reaction and bleeding.16 Therefore, tivantinib may be a useful treatment option for patients with HCC who cannot tolerate VEGFR inhibitors such as sorafenib. As for the risk of ILD, Japanese patients with HCC who receive tivantinib as a single agent are probably at a much lower risk than Japanese patients with NSCLC who receive tivantinib plus erlotinib.17 ILD was one of the reasons for terminating a previous phase III study in which Asian patients with NSCLC were given a combination of erlotinib and tivantinib.17 However, there was no case of ILD in our study, consistent with the results of other Asian studies of tivantinib as a single agent.6,10
In this study, the efficacy of tivantinib was not prominent, but this remains inconclusive owing to the small number of patients. The ARQ 197-215 study reported that tivantinib significantly prolonged survival in the subgroup of patients who had tumors with high c-Met expression.7 Further studies, including the ongoing phase III studies in Japan (NCT02029157) and in Western countries (METIV-HCC), are evaluating the efficacy of tivantinib in the biomarker-defined population of HCC patients with high levels of c-Met.8
In conclusion, the same dose of tivantinib (120 mg BID) is recommended for both EM and PM among Japanese patients with HCC. This differs from the recommended dose for Asian patients with other solid tumors (i.e. 360 mg BID for EM and 240 mg BID for PM). A placebo-controlled phase III trial to evaluate the efficacy of tivantinib is ongoing in Japanese patients with advanced HCC who have tumors with high c-Met expression.
Acknowledgments
We thank the patients, their families and caregivers, and all of the personnel who contributed to patient care and data collection for this study of tivantinib. We also thank the members of the Safety Review Committee; Dr Kazuo Tamura, Dr Masatoshi Kudo and Dr Kiyohiko Hatake. This work was supported by Kyowa Hakko Kirin Co., Ltd.
Disclosure Statement
JF is a consultant for Kyowa Hakko Kirin Co., Ltd. TS and YI are employees of Kyowa Hakko Kirin Co., Ltd. All remaining authors have declared no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Table S1. Dose-limiting toxicities occurring in this study.
Table S2. Relationship between c-Met expression and responses.
References
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Peña CE, Lathia CD, et al. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2012;18:2290–300. doi: 10.1158/1078-0432.CCR-11-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Ojima H, Tsuda H, et al. Clinical impact of c-Met expression and its gene amplification in hepatocellular carcinoma. Int J Clin Oncol. 2013;18:207–13. doi: 10.1007/s10147-011-0361-9. [DOI] [PubMed] [Google Scholar]
- Adjei AA, Schwartz B, Garmey E. Early clinical development of ARQ 197, a selective, non-ATP-competitive inhibitor targeting MET tyrosine kinase for the treatment of advanced cancers. Oncologist. 2011;16:788–99. doi: 10.1634/theoncologist.2010-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YK, Muro K, Ryu MH, et al. A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer. Invest New Drugs. 2014;32:355–61. doi: 10.1007/s10637-013-0057-2. [DOI] [PubMed] [Google Scholar]
- Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14:55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
- Fornaro L, Vivaldi C, Caparello C, et al. Dissecting signaling pathways in hepatocellular carcinoma: new perspectives in medical therapy. Future Oncol. 2014;10:285–304. doi: 10.2217/fon.13.181. [DOI] [PubMed] [Google Scholar]
- Kubota T, Chiba K, Iga T. Frequency distribution of CYP2C19, CYP2D6, and CYP2C9 mutant-alleles in several different populations. Xenobio Metab Dispos. 2001;16:69–74. [Google Scholar]
- Yamamoto N, Murakami H, Nishina T, et al. The effect of CYP2C19 polymorphism on the safety, tolerability, and pharmacokinetics of tivantinib (ARQ 197): results from a phase I trial in advanced solid tumors. Ann Oncol. 2013;24:1653–9. doi: 10.1093/annonc/mdt014. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Murakami H, Hayashi H, et al. CYP2C19 genotype-based phase I studies of a c-Met inhibitor tivantinib in combination with erlotinib, in advanced/metastatic non-small cell lung cancer. Br J Cancer. 2013;109:2803–9. doi: 10.1038/bjc.2013.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Press release from ArQule Inc. (19 Presidential Way, Woburn, MA, USA). [Cited 16 Jan 2014.] Available from URL: http://investors.arqule.com/releasedetail.cfm?ReleaseID=819847.
- Marrero JA, Fontana RJ, Barrat A, et al. Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology. 2005;41:707–16. doi: 10.1002/hep.20636. [DOI] [PubMed] [Google Scholar]
- Frye RF, Zgheib NK, Matzke GR, et al. Liver disease selectively modulates cytochrome P450-mediated metabolism. Clin Pharmacol Ther. 2006;80:235–45. doi: 10.1016/j.clpt.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Cohen RB, Oudard S. Antiangiogenic therapy for advanced renal cell carcinoma: management of treatment-related toxicities. Invest New Drugs. 2012;30:2066–79. doi: 10.1007/s10637-012-9796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma K, Yoshioka H, Yamamoto N, et al. Tivantinib plus erlotinib vs. placebo plus erlotinib in Asian patients with previously treated non-squamous NSCLC with wild-type EGFR: first report of a Phase III ATTENTION trial. J Clin Oncol. 2014;32:5s. (abstract 8044) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Dose-limiting toxicities occurring in this study.
Table S2. Relationship between c-Met expression and responses.