

Abstract
A new tertiary amine co-initiator (TUMA) containing three methacrylate-urethane groups was synthesized for application in dentin adhesives. The photopolymerization kinetics and leaching of unreacted components from methacrylate-based dental polymers formulated with this new co-initiator were determined. The newly synthesize co-initiator showed good chemical stability and decreased amine release from the initiator system. This study provides important information for the future development of biocompatible dentin adhesives/composites.
Keywords: Amine, Co-initiator, Degree of conversion, Dental restorative material, Photo-polymerization, Leachable
1. Introduction
Polymer-based composites have become the most common dental restorative material and are used twice as often as dental amalgam [1]. While composites exhibit many of the properties required for direct restorative materials, there are limitations that lead to a reduced clinical lifetime. One of the limitations of the dimethacrylate-based resins used in dental composites and adhesives is incomplete monomer to polymer conversion. Incomplete conversion of the methacrylates leads to the release of unreacted components. The release of methacrylic monomers together with components of the polymerization initiator system, including unreacted co-initiators[2] has been considered a source of adverse reactions, e.g. local and systemic toxicity, pulpal irritation, allergic and estrogenic effects[3–8].
The initiator system exerts considerable influence on the behavior of polymeric restorative materials in the wet, oral environment. A three-component photoinitiator system that is reactive upon exposure to visible light has been widely used in dental restoratives. The photoinitiator system generally contains a light absorbing photosensitizer (such as CQ), an elector donor (typically a tertiary amine compound as the co-initiator), and a third component (often a diaryliodonium or sulfonium salt). The co-initiator (amine compound) plays a critical role in the photoinitiation process [9]. The type of co-initiator as well as its ratio to the photosensitizer influence the quality of the polymerization [10]. Many of the available amine co-initiators cannot be used in dental restorative materials because of cytotoxicity concerns [2, 11].
Ethyl-4-(dimethylamino) benzoate (EDMAB), which is a very effective hydrogen donor, has been widely used in combination with CQ in dental resins formulations [12–15]. EDMAB may cause serious biocompatibility issues because it is an aromatic amine and it cannot be copolymerized into the polymer network. A coinitiator such as 2-(dimethylamino) ethyl methacrylate (DMAEMA) carrying a methacrylate group could provide improved biocompatibility by co-polymerizing with methacrylate-monomers. For this reason, CQ/DMAEMA is the most commonly used photoinitiator system in current photoactivated dental materials [12–15]. The stability of DMAEMA is, however, an unresolved problem. DMAEMA can be degraded up to 82% after 24 h aging in water [2]. The degradation products which contain organic amine compounds may cause tissue sensitivity and/or other problems related to biocompatibility [2]. Nie and colleagues synthesized and characterized a series of polymerizable amine-coinitiators with the goal of reducing the toxicity [5, 11, 16–19].
The leaching of co-initiators after extended aging has received limited attention, i.e. the majority of the studies have focused on elution of co-initiator from methacrylate based dental resins after 24 h [2, 20, 21]. The twofold objectives of this work were to synthesize a new tertiary amine co-initiator with three methacrylate-urethane groups and to characterize this new co-initiator in terms of photopolymerization kinetics and release of leachables from the polymers after extended aging, i.e. 60 days. It is hypothesized that (1) the DC with the newly synthesized amine-cointiator TUMA is comparable with that of EDMAB and DMAEMA and (2) amine leachables could be reduced by introducing this new co-initiator (TUMA).
2. Experimental
2.1 Materials
2,2-Bis[4-(2-hydroxy-3-methacryloxypropoxy) phenyl]-propane (BisGMA, Polysciences, Warrington, PA) and 2-hydroxyethylmethacrylate (HEMA, Acros Organics, NJ) were used as received without further purification, as monomers. Based on our previous studies, [14, 15, 22–25] camphorquinone (CQ), an amine co-initiator and diphenyliodonium hexafluorophosphate (DPIHP) were used as a three-component-photoinitiator system. CQ, DPIHP, ethyl-4-(dimethylamino) benzoate (EDMAB), 2-(dimethylamino) ethyl methacrylate (DMAEMA) were obtained from Aldrich (Milwaukee, WI, USA) and used without further purification. EDMAB, DMAEMA and TUMA were used as the co-initiators, respectively. TUMA was synthesized in our lab. 2-isocyantoethyl methacrylate (IEM), triisopropanolamine (TIPA), dibutyltin dilaurate (DBTL), ethanol, ethyl acetate, acetonitrile and all other chemicals were purchased from Sigma-Aldrich at reagent grade and used without further purification.
2.2 Synthesis of TUMA
2-isocyantoethyl methacrylate (IEM, 18.2 g, 0.117 mol) with 30 mL dry ethyl acetate (EA) was added dropwise to a three-neck flask containing triisopropanolamine (TIPA, 10 g, 0.052 mol), dibutyltin dilaurate (DBTL, 0.03 g) and EA (50 mL). The solution was stirred continuously at 25°C throughout the process. Following complete addition of IEM, the reaction was allowed to continue at room temperature for another 24 h. The reaction was monitored by thin layer chromatography (mobile phase: dichloromethane). After reaction completion, the product-containing solution was purified by washing with distilled water and ethyl acetate until the solution was clear. The solution was dried over anhydrous MgSO4 and the solvent was removed with a rotary evaporator at 35–40 °C. The yield of this synthesis of TUMA, in its colorless, viscous state, was 95%. The scheme for the TUMA synthesis is shown in Figure 1.
Fig. 1.
Reaction scheme for TUMA.
2.3 Preparation of resin formulations
The procedure to prepare the resin formulations has been reported[26–28]. The formulations consisted of HEMA and BisGMA with a mass ratio of 45/55, which is similar to widely used commercial dentin adhesives. Camphorquinone (CQ, 0.5 wt %), amine co-initiator (0.5 wt % or 1.75 wt %) and diphenyliodonium hexafluorophosphate (DPIHP, 1.0 wt %) with respect to the total amount of monomers, were used as a three-component-photoinitiator system. Three different amine co-initiators were used: EDMAB (0.5 wt %), DMAEMA (0.5 wt %), TUMA (0.5 and 1.75 wt %). It should be noted that the mole concentration is the same for co-initiator with EDMAB at 0.5 wt % and TUMA at 1.75 wt %. The resin mixtures were prepared in brown glass vials and stirred for 48 h on an orbital shaker to form a homogeneous solution.
2.4 Real-time conversion and maximal polymerization rate
Real-time in situ monitoring of the photopolymerization of the resin formulations was performed using an infrared spectrometer (Spectrum 400 Fourier transform infrared spectrophotometer, Perkin-Elmer, Waltham, MA) at a resolution of 4 cm−1 [13, 15]. One drop of adhesive solution was placed on the diamond crystal top-plate of an attenuated total reflectance (ATR) accessory (Pike, GladiATR, Pike Technology, Madison, WI) and covered with a mylar film. A 40-s exposure to the commercial visible-light-polymerization unit (Spectrum 800®, Dentsply, Milford, DE, ~480–490 nm[25]) at an intensity of 550 mW cm−2 was initiated after 50 spectra had been recorded. Real-time IR spectra were recorded continuously for 600 s after light curing began. A time-resolved spectrum collector (Spectrum TimeBase, Perkin-Elmer) was used for continuous and automatic collection of spectra during polymerization. Three replicates were obtained for each adhesive formulation.
The change of the band ratio profile (1637 cm−1(C=C)/1608 cm−1(phenyl)) was monitored for calibrating the DC of the methacrylate groups. DC was calculated using the following equation, which is based on the decrease in the absorption intensity band ratios before and after light curing. The average of the last 50 values of time-based data points is reported as the DC value at 10 minutes.
The kinetic data were converted to Rp/[M]0 by taking the first derivative of the time versus conversion curve [12, 29, 30], where Rp and [M]0 are the rate of polymerization and the initial monomer concentration, respectively.
2.5 Nuclear magnetic resonance (NMR) characterization
The purity of synthesized TUMA was characterized by analyzing its 1H NMR and 13C NMR spectra, which were obtained on a Bruker Avance DRX 500 spectrometer equipped with a broadband probe. Deuterated chloroform (CDCl3) was used as the solvent.
The chemical stability of three amine co-initiators (EDMAB, DMAEMA and TUMA) was characterized by 1H NMR spectroscopy. Deuterated dimethyl sufoxide (DMSO-d6) /deionized water (80/20 wt/wt) was used as the solvent. The NMR spectra of 0.04 M amine samples were monitored as a function of storage time at 25 °C.
2.6 Leachables study by high performance liquid chromatography (HPLC)
Disc samples, 4 mm diameter and 1 mm thick, were used for the leachable study. These samples were prepared by injecting the resin formulations into hermetic lids (TA instruments, T 120110, USA) and covering with a glass coverslip (Ted pella, inc., Prod No. 26023). Five specimens were prepared for each formulation. The samples were light polymerized with a 40-s exposure to the commercial visible-light-polymerization unit (Spectrum®, Dentsply, Milford, DE) at an intensity of 550 mW cm−2. The polymerized samples were stored in the dark at room temperature for 48 h to provide adequate time for post-cure polymerization. The samples were extracted and the mass was recorded. The samples were stored in 1 mL ethanol (200 proof) at 25 °C. The solution was replaced with fresh ethanol and reserved for HPLC at fixed time intervals. The samples were centrifuged, and the supernatant was collected and analyzed by HPLC.
Co-initiators and monomers released from the polymers were assayed by reverse phase high performance liquid chromatography (RP-HPLC) as described by our group [31]. This technique allows for the separation, identification, and quantification of individual molecules in complex mixtures. A Shimadzu LC-HTC HPLC system equipped with a SPD-M20A photodiode array detector (PDA), an autosampler with EZStart chromatography software as a controller and also for data processing, was used for this study (Shimadzu, Columbia, MD). Separation was performed on a reverse-phase column- A phenomenex Luna 5 μm C18 4.6 × 250 nm (Phenomenex, Torance, CA) column and security guard cartridge were used to isolate the products. The mobile phase consisted of a mixture of a 10 mM potassium phosphate buffer and acetonitrile (CH3CN). The pH of the buffer was adjusted to 7.0 with sodium hydroxide. The elution was started at a constant flow rate of 0.2 mL/min with CH3CN: 10 mM potassium phosphate buffer (65:35, v/v) for 25 min and then ramped to 100% CH3CN in 1 s and kept constant for 5 min. Twenty microliters of the supernatant were injected onto the HPLC column with a constant column temperature of 40 °C. UV spectrum at 208 nm wavelength was used to detect the leachables. Leached monomers and amine co-initiators were identified and quantified by comparison with reference compounds of known composition. These reference compounds were measured under the same HPLC conditions (linear regression analysis for all leachables: R2=0.99). In addition, amounts of co-initiators or co-monomers released from 1 gram polymer samples were measured by calculation of the released mass values divided by mass values of the polymer samples. The results were obtained in μg/mL and were also reported in μg/g, relative to the weighted adhesives for data normalization.
2.7 Statistical analysis
For all experimental groups, the differences were evaluated using one-way analysis of variance (ANOVA), together with Tukey’s test at α= 0.05 to identify significant differences (Microcal Origin Version 8.0, Microcal Software Inc., Northampton, MA).
3. Results
3.1 Characterization of the synthesized tertiary amine co-initiator (TUMA)
The structure of the newly synthesized amine co-initiator (TUMA) was identified using FTIR (Fig. 2) and 1H NMR/ 13C NMR spectroscopies (Fig. 3). The characteristic FTIR peaks for TUMA are: 3351 cm−1 (NH stretching on -CONH), 1637 cm−1 (C=C bending on methacrylate groups). The disappearance of the –NCO of IEM stretching band at 2259 cm−1 confirmed the formation of the new co-initiator (Fig. 2). The 1H NMR/ 13C NMR spectra of TUMA (Fig. 3) show the chemical shifts, which confirm the desired structures. The methacrylate groups are supported by the presence of two singlets (δ = 6.15 and 5.63 ppm) on the 1H-NMR spectrum and by the peaks at 135.8 and 125.8 ppm in the 13C-NMR, assignable to the double bond of a methacrylate group.
Fig. 2.

FTIR spectra of TIPA, IEM (starting material) and TUMA.
Fig 3.

1H-NMR (A) and 13C-NMR (B) spectra of synthesized TUMA.
3.2 Chemical stability of three amine co-initiators
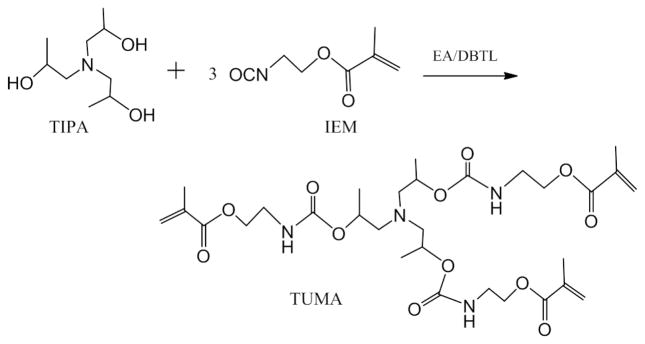
Fig. 4 shows the degradation results for the three amine co-initiators in water containing DMSO-d6 (H2O: 20 wt %). There is a rapid degradation rate for DMAEMA, with 37.6% degraded at 100 days. No degradation was detected for either EDMAB or TUMA after 100 days storage at 25 °C.
Fig 4.
Stability of three different amine co-initiators in DMSO-d6 with 20 wt% H2O at the concentration of 0.04 M.
3.3 Degree of conversion and maximum polymerization rate for resin formulations with different co-initiators
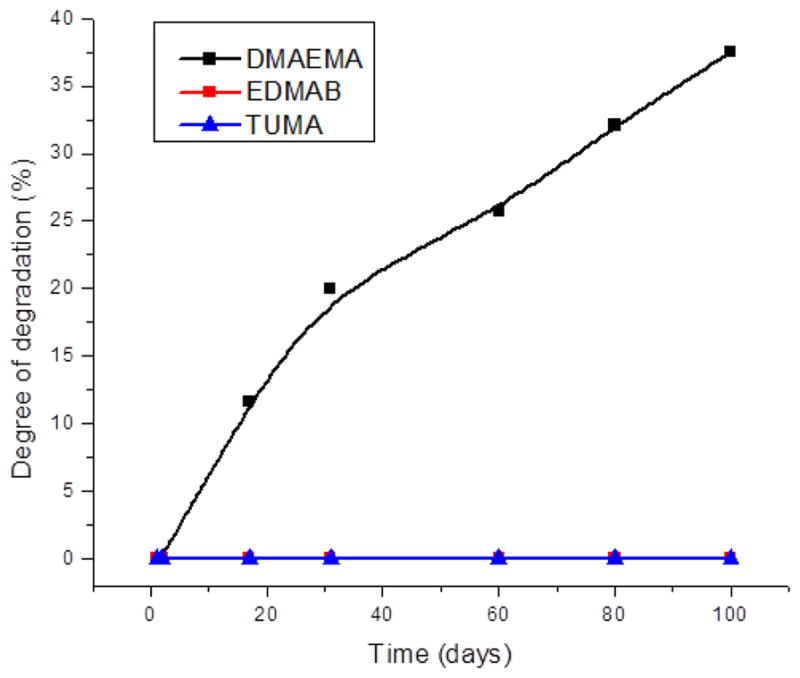
The kinetics of the photopolymerization of resin formulations are shown in Fig. 5. When EDMAB, DMAEMA and TUMA were used as the co-initiators with 0.5 wt %, the DC after 600s was 68.2%, 69.9% and 65.2%, respectively. When TUMA was used at 1.75 wt %, the DC increased to 70.1% (Fig. 5A). The maximum polymerization rates for EDMAB, DMAEMA and TUMA at 0.5 wt % concentration were 0.25, 0.21 and 0.12 s−1 respectively. The polymerization rate (Rp/[M]0) was 0.17 s−1 for TUMA at 1.75 wt% (Fig. 5B).
Fig 5.
Real-time conversion (A) and the maximum polymerization rate (B) of adhesive resins with different co-initiator. The adhesives were light-cured for 40 sec at room temperature using a commercial visible-light-curing unit (Spectrum® 800, Dentsply, Milford, DE. Intensity is 550 mW/cm−2). Real-time IR spectra were continuously recorded for 600 s after light activation began. N= 3 ± SD. * Significantly (p <0.05) different from the EDMAB-0.5. The values of 0.5 and 1.75 shown in the symbols mean there are 0.5 or 1.75 wt% of co-initiator in the formulations.
3.4 Leachables from polymers study by HPLC
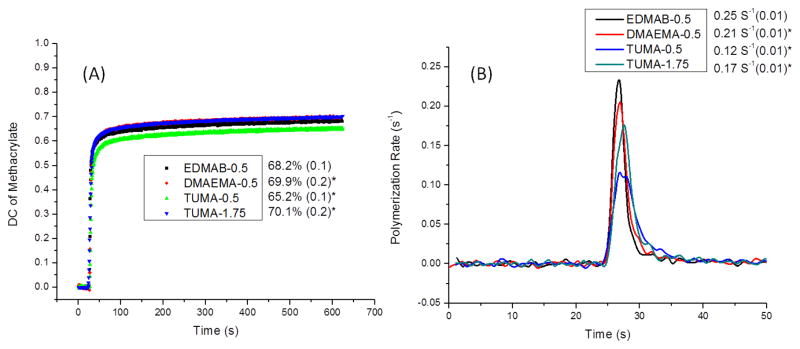
The retention time for EDMAB, DMAEMA and TUMA is 13.3, 11.3 and 13.5 min. Fig. 6A shows the results of cumulative amine co-initiators release from polymers as a function of incubation time in ethanol at 25 °C. The cumulative release of EDMAB (EDMAB-0.5: 53.6±4.2 μg/mL) after 60 days is significant higher (p<0.05) than that of DMAEMA (DMAEMA-0.5: 11.2±3.0 μg/mL). Moreover, no TUMA was detected by HPLC when TUMA was used as co-initiator at 0.5 or 1.75 wt%. In addition, there would be 2.68±0.21 mg EDMAB released from 1 gram polymer samples by calculation of the released mass values divided by mass values of polymer samples (Fig. 6B), and 0.56±0.15 mg of DMAEMA would be released from 1 gram polymer samples (Fig. 6B).
Fig 6.
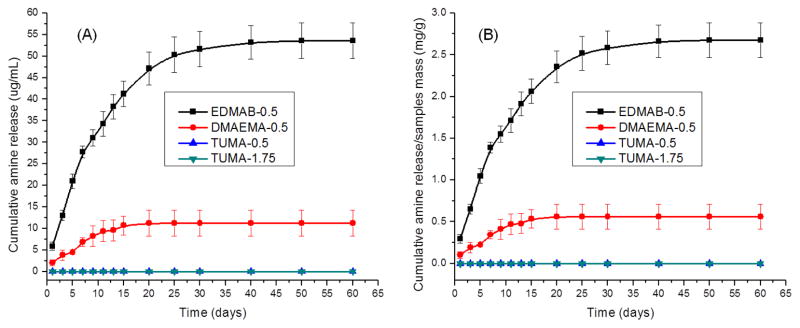
(A) Cumulative amine co-initiators release from dentin adhesives as a function of incubation time in ethanol. (B) Calculation was done by dividing the mass values of released compounds by the mass values of polymer samples. N= 3 ± SD. The values of 0.5 and 1.75 shown in the symbols mean there are 0.5 or 1.75 wt% of co-initiator in the formulations.
Fig. 7A shows the cumulative HEMA release as a function of time (The retention time for HEMA is 6.4 min). The cumulative HEMA release at 60 days with 0.5 wt % of TUMA (TUMA-0.5: 460.9±17.6 μg/mL) is significant higher (p<0.05) than 1.75 wt% TUMA, 0.5 wt% EDMAB, and 0.5 wt% DMAEMA. When TUMA was increased to 1.75 wt % (TUMA-1.75: 314.0±11.0 μg/mL), the release of HEMA was similar with that of EDMAB (EDMAB-0.5: 295.0±13.0 μg/mL) and slightly lower than that of DMAEMA (DMAEMA-0.5: 347.3±19.3 μg/mL). Based on the results in Fig. 7A, there would be 23.04±0.88, 17.36±0.96, and 14.75±0.65 mg of HEMA released from 1 gram polymer samples after 60 days (Fig. 7B) with TUMA, DMAEMA, and EDMAB as co-initiators (0.5 wt %), respectively. When TUMA is increased to 1.75 wt%, HEMA release from 1 gram polymer samples after 60 days is 15.70±0.55 mg.
Fig 7.
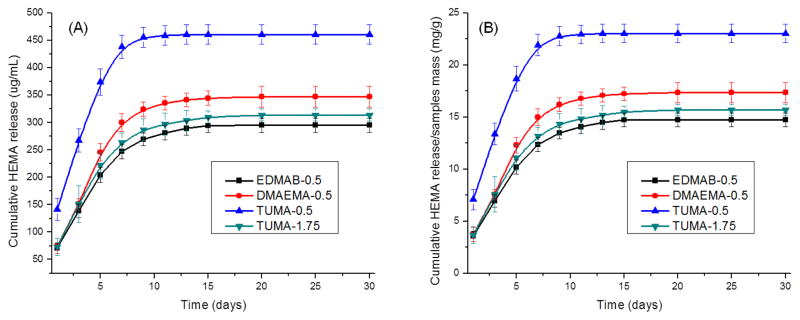
(A) Cumulative HEMA release from dentin adhesives with different co-initiator as a function of incubation time in ethanol. (B) Calculation was done by dividing the mass values of released compounds by the mass values of polymer samples. N= 3 ± SD. The values of 0.5 and 1.75 shown in the symbols mean there are 0.5 or 1.75 wt% of co-initiator in the formulations.
Fig. 8 shows the cumulative BisGMA release as a function of time (The retention time for BisGMA is 18.0 min). The cumulative BisGMA release at 60 days with 0.5 wt % of TUMA (TUMA-0.5: 176.7±9.2 μg/mL) is significantly greater (p<0.05) than EDMAB, DMAEMA, and 1.75 wt% TUMA. EDMAB at 0.5 wt% (90.5±9.1 μg/mL) showed similar results with TUMA at 1.75 wt% (91.4±6.8 μg/mL). DMAEMA at 0.5 wt% showed significantly lower (p<0.05) BisGMA release (63.2±4.2 μg/mL). There would be 8.84±0.46, 4.57±0.34, 4.52±0.53 and 3.16±0.21 mg of BisGMA released from 1 gram polymer samples after 60 days (Fig. 8B) with 0.5 wt% TUMA, 1.75 wt% TUMA, 0.5 wt% EDMAB and 0.5 wt% DMAEMA as co-initiators, respectively.
Fig 8.
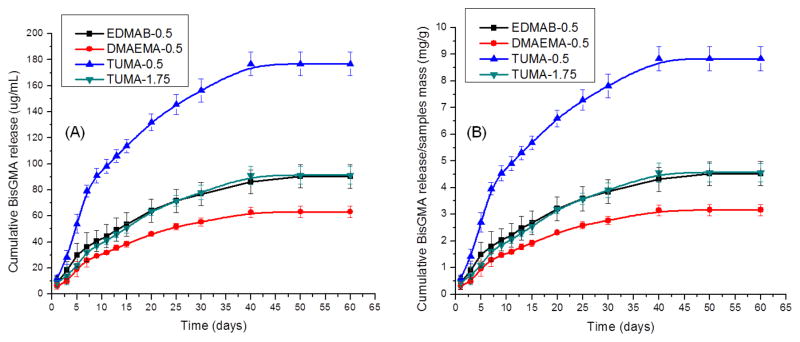
(A) Cumulative BisGMA release from dentin adhesives with different co-initiator as a function of incubation time in ethanol. (B) Calculation was done by dividing the mass values of released compounds by the mass values of polymer samples. N= 3 ± SD. The values of 0.5 and 1.75 shown in the symbols mean there are 0.5 or 1.75 wt% of co-initiator in the formulations.
4. Discussion
This work compared three different amine co-initiators used in dental restorative materials. The chemical stability results indicate EDMAB and TUMA are more stable than DMAEMA. Park and colleagues [2] reported DMAEMA degradation up to 82% during 24 h in water. In comparison, in this study using deuterated dimethyl sufoxide (DMSO-d6) /deionized water (80/20 wt/wt), only 37.6 % degradation of DMAEMA was recorded after 100 days. The mixed (DMSO/water) solvent was used because of its ability to dissolve EDMAB and TUMA, which are hydrophobic compounds.
Relative to 0.5 wt% EDMAB and 0.5 wt% DMAEMA, DC is lower when 0.5 wt% TUMA is used as the co-initiator. Since the molecular weight of TUMA is higher than EDMAB and DMAEMA, the low mole concentration of TUMA may limit the DC of monomers. Thus, TUMA was increased to 1.75 wt %, so that it had the same mole concentration as EDMAB (0.5 wt %). The DC was subsequently increased to 70.1% (TUMA-1.75). The aromatic amine (EDMAB) had the fastest polymerization rate. The polymerization rate of TUMA is the lowest, even with the increase to 1.75 wt %. The reduced polymerization rate of TUMA may be attributed to the larger size of this co-initiator. Even with a lower polymerization rate, DC with TUMA was comparable with EDMAB and DMAEMA. The lower polymerization rate may offer benefit since lower polymerization rates have been associated with reduced polymerization-induced shrinkage stress [10, 32–37].
Ethanol was used in the leachables study to promote the solubility of the hydrophobic compounds, i.e. EDMAB, TUMA and BisGMA. Based on the HPLC results, more than half of the EDMAB could be released from the polymer, although the chemical stability of EDMAB is comparable to TUMA. Since DMAEMA could be polymerized into the polymer network, there is less release for DMAEMA (11.2%) than EDMAB (53.6%). Furthermore, there was no TUMA release or it was below the detectable limit. TUMA with three methacrylate-urethane groups can be co-polymerized into the polymer network.
The monomer release studies indicate more HEMA and BisGMA released from polymers with low DC. Monomer release is very dependent on DC. With 0.5 wt % TUMA as the co-initiator, more monomer was released and this was related with the lower DC. By increasing TUMA to 1.75 wt %, the monomer release was comparable with the samples formulated with EDMAB and DMAEMA. The release of CQ and DPIHP could not be detected. This might be attributed to the instability of CQ and the ionization of DPIHP in the mobile phase. In regard to the release rate of different compounds, HEMA release (~15 days) is more rapid than BisGMA (~50 days); DMAEMA release (~20 days) is more rapid than EDMAB (~40 days). The results indicate that more hydrophobic (or larger sized) compounds are released slowly from polymers aged in ethanol.
5. Conclusion
A new tertiary amine co-initiator was synthesized and evaluated by comparing with commercial co-initiators, EDMAB and DMAEMA. The chemical stability of EDMAB and TUMA is better than DMAEMA. Degree of conversion of the methacrylate monomers was comparable with EDMAB and DMAEMA when TUMA was used as the co-initiator at 1.75 wt%. The maximum polymerization rate with TUMA was lower than that with EDMAB and DMAEMA. The leachables study indicates amine co-initiator release could be prevented when TUMA is used as the co-initiator. With the increase in the weight content of TUMA, the release of HEMA and BisGMA are comparable with the polymers formulated with EDMAB and DMAEMA. These results provide important information for future development of biocompatible dentin adhesives/composites. TUMA is a promising co-initiator that offers good stability, comparable degree of conversion and prevents amine leaching even with extended aging.
Table 1.
Chemical structures used in the adhesive formulations
| 3-component photoinitiator system (Photosensitizer /amine co-initiator/ iodonium salt) | |||
|---|---|---|---|
| Photosensitizer |
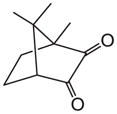 CQ |
Co-initiator (Electron donor) |
EDMAB |
| Iodonium salt |
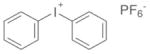 DPIHP |
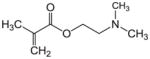 DMAEMA |
|
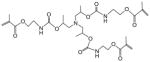 TUMA | |||
| Monomer system | |||
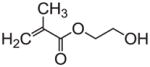 HEMA |
BisGMA |
||
Acknowledgments
This investigation was supported by Research Grant: R01 DE022054 from the National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, MD 20892.
References
- 1.Olabisi O. Interpretations of Polymer-Polymer Miscibility. Abstr Pap Am Chem S. 1981;181:83. -Ched. [Google Scholar]
- 2.Schroeder WF, Vallo CI. Effect of different photoinitiator systems on conversion profiles of a model unfilled light-cured resin. Dent Mater. 2007;23:1313–1321. doi: 10.1016/j.dental.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Anagnostou M, Chatzigianni E, Doucoudakis S, Potamianou A, Tesseromatis C. Biocompatibility of resin composites subcutaneously implanted in rats with experimentally induced arthritis. Dent Mater. 2009;25:863–867. doi: 10.1016/j.dental.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Bouillaguet S. Biological risks of resin-based materials to the dentin-pulp complex. Crit Rev Oral Biol M. 2004;15:47–60. doi: 10.1177/154411130401500105. [DOI] [PubMed] [Google Scholar]
- 5.Geurtsen W. Biocompatibility of resin-modified filling materials. Crit Rev Oral Biol M. 2000;11:333–355. doi: 10.1177/10454411000110030401. [DOI] [PubMed] [Google Scholar]
- 6.Moharamzadeh K, Brook IM, Van Noort R. Biocompatibility of Resin-based Dental Materials. Materials. 2009;2:514–548. [Google Scholar]
- 7.Schedle A, Ortengren U, Eidler N, Gabauer M, Hensten A. Do adverse effects of dental materials exist? What are the consequences, and how can they be diagnosed and treated? Clin Oral Implan Res. 2007;18:232–256. doi: 10.1111/j.1600-0501.2007.01481.x. [DOI] [PubMed] [Google Scholar]
- 8.Schweikl H, Spagnuolo G, Schmalz G. Genetic and cellular toxicology of dental resin monomers. J Dent Res. 2006;85:870–877. doi: 10.1177/154405910608501001. [DOI] [PubMed] [Google Scholar]
- 9.Oxman JD, Jacobs DW, Trom MC, Sipani V, Ficek B, Scranton AB. Evaluation of initiator systems for controlled and sequentially curable free-radical/cationic hybrid photopolymerizations. J Polym Sci Pol Chem. 2005;43:1747–1756. [Google Scholar]
- 10.Furuse AY, Mondelli J, Watts DC. Network structures of Bis-GMA/TEGDMA resins differ in DC, shrinkage-strain, hardness and optical properties as a function of reducing agent. Dent Mater. 2011;27:497–506. doi: 10.1016/j.dental.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Bakopoulou A, Papadopoulos T, Garefis P. Molecular Toxicology of Substances Released from Resin-Based Dental Restorative Materials. Int J Mol Sci. 2009;10:3861–3899. doi: 10.3390/ijms10093861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo X, Wang Y, Spencer P, Ye Q, Yao X. Effects of water content and initiator composition on photopolymerization of a model BisGMA/HEMA resin. Dent Mater. 2008;24:824–831. doi: 10.1016/j.dental.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park J, Ye Q, Topp EM, Misra A, Kieweg SL, Spencer P. Effect of photoinitiator system and water content on dynamic mechanical properties of a light-cured bisGMA/HEMA dental resin. J Biomed Mater Res A. 2010;93A:1245–1251. doi: 10.1002/jbm.a.32617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Spencer P, Yao X, Ye Q. Effect of coinitiator and water on the photoreactivity and photopolymerization of HEMA/camphoquinone-based reactant mixtures. J Biomed Mater Res A. 2006;78A:721–728. doi: 10.1002/jbm.a.30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye Q, Park J, Topp E, Spencer P. Effect of photoinitiators on the in vitro performance of a dentin adhesive exposed to simulated oral environment. Dent Mater. 2009;25:452–458. doi: 10.1016/j.dental.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knaus S, Gruber HF. Photoinitiators with Functional-Groups .3. Water-Soluble Photoinitiators Containing Carbohydrate Residues. J Polym Sci Pol Chem. 1995;33:929–939. [Google Scholar]
- 17.Kolar A, Gruber HF, Greber G. Photoinitiators with Functional-Groups .2. Silicon-Containing Photoinitiators. J Macromol Sci Pure. 1994;A31:305–318. [Google Scholar]
- 18.Knaus S, Gruber HF. Photoinitiators with functional groups .4. Water-soluble photoinitiators containing quaternary ammonium groups. J Macromol Sci Pure. 1996;A33:869–881. [Google Scholar]
- 19.Klos R, Gruber H, Greber G. Photoinitiators with Functional-Groups .1. Polymer Photoinitiators. J Macromol Sci Chem. 1991;A28:925–947. [Google Scholar]
- 20.Pongprueksa P, Senawongse P, Vongphan N. Effect of dentinal tubule orientation on the modulus of elasticity of resin-infiltrated demineralized dentin. Dent Mater J. 2014;33:54–58. doi: 10.4012/dmj.2013-199. [DOI] [PubMed] [Google Scholar]
- 21.Michelsen VB, Lygre H, Skalevik R, Tveit AB, Solheim E. Identification of organic eluates from four polymer-based dental filling materials. Eur J Oral Sci. 2003;111:263–271. doi: 10.1034/j.1600-0722.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- 22.Park JG, Ye Q, Topp EM, Lee CH, Kostoryz EL, Misra A, Spencer P. Dynamic Mechanical Analysis and Esterase Degradation of Dentin Adhesives Containing a Branched Methacrylate. J Biomed Mater Res B. 2009;91B:61–70. doi: 10.1002/jbm.b.31374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JG, Ye Q, Topp EM, Misra A, Spencer P. Water sorption and dynamic mechanical properties of dentin adhesives with a urethane-based multifunctional methacrylate monomer. Dent Mater. 2009;25:1569–1575. doi: 10.1016/j.dental.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye Q, Spencer P, Wang Y, Misra A. Relationship of solvent to the photopolymerization process, properties, and structure in model dentin adhesives. J Biomed Mater Res A. 2007;80A:342–350. doi: 10.1002/jbm.a.30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye QA, Wang Y, Williams K, Spencer P. Characterization of photopolymerization of dentin adhesives as a function of light source and irradiance. J Biomed Mater Res B. 2007;80B:440–446. doi: 10.1002/jbm.b.30615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song LY, Ye Q, Ge XP, Misra A, Laurence JS, Berry CL, Spencer P. Synthesis and evaluation of novel dental monomer with branched carboxyl acid group. J Biomed Mater Res B. 2014;102:1473–1484. doi: 10.1002/jbm.b.33126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ge XP, Ye Q, Song LY, Misra A, Spencer P. Synthesis and evaluation of novel siloxane-methacrylate monomers used as dentin adhesives. Dent Mater. 2014;30:1073–1087. doi: 10.1016/j.dental.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye Q, Park JG, Topp E, Wang Y, Misra A, Spencer P. In vitro performance of nano-heterogeneous dentin adhesive. J Dent Res. 2008;87:829–833. doi: 10.1177/154405910808700911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J, Ye Q, Singh V, Kieweg SL, Misra A, Spencer P. Synthesis and evaluation of novel dental monomer with branched aromatic carboxylic acid group. J Biomed Mater Res B. 2012;100B:569–576. doi: 10.1002/jbm.b.31987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang M, Crivello JV. Synthesis and cationic photopolymerization of epoxy-functional siloxane monomers and oligomers. J Polym Sci Pol Chem. 2003;41:3056–3073. [Google Scholar]
- 31.Liska R. Photoinitiators with functional groups. v. new water-soluble photoinitiators containing carbohydrate residues and copolymerizable derivatives thereof. J Polym Sci Pol Chem. 2002;40:1504–1518. [Google Scholar]
- 32.Ullrich G, Ganster B, Salz U, Moszner N, Liska R. Photoinitiators with functional groups. IX. Hydrophilic bisacylphosphine oxides for acidic aqueous formulations. J Polym Sci Pol Chem. 2006;44:1686–1700. [Google Scholar]
- 33.Jauk S, Liska R. Photoinitiators with functional groups 9: New derivatives of covalently linked benzophenone-amine based photoinitiators. J Macromol Sci A. 2008;45:804–810. [Google Scholar]
- 34.Jauk S, Liska R. Photoinitiators with functional groups, 8 - Benzophenone with covalently bound phenylglycine. Macromol Rapid Comm. 2005;26:1687–1692. [Google Scholar]
- 35.Ullrich G, Herzog D, Liska R, Burtscher P, Moszner N. Photoinitiators with functional groups. VII. Covalently bonded camphorquinone - Amines. J Polym Sci Pol Chem. 2004;42:4948–4963. [Google Scholar]
- 36.Furuse AY, Cunha LF, Moresca R, Paganeli G, Mondelli RFL, Mondelli J. Enamel Wetness Effects on Bond Strength Using Different Adhesive Systems. Oper Dent. 2011;36:274–280. doi: 10.2341/10-163-L. [DOI] [PubMed] [Google Scholar]
- 37.Pontons-Melo JC, Furuse AY, Mondelli J. A direct composite resin stratification technique for restoration of the smile. Quintessence Int. 2011:42. [PubMed] [Google Scholar]