

Abstract
Numerous cellular studies have indicated that RhoA signaling is required for oncogenic Ras-induced transformation, suggesting that RhoA is a useful target in Ras induced neoplasia. However, to date very limited data exist to genetically attribute RhoA function to Ras-mediated tumorigenesis in mammalian models. In order to assess whether RhoA is required for K-Ras-induced lung cancer initiation, we utilized the K-RasG12D Lox-Stop-Lox murine lung cancer model in combination with a conditional RhoAflox/flox and RhoC-/- knockout mouse models. Deletion of the floxed Rhoa gene and expression of K-RasG12D was achieved by either CCSP-Cre or adenoviral Cre, resulting in simultaneous expression of K-RasG12D and deletion of RhoA from the murine lung. We found that deletion of RhoA, RhoC or both did not adversely affect normal lung development. Moreover, we found that deletion of either RhoA or RhoC alone did not suppress K-RasG12D induced lung adenoma initiation. Rather, deletion of RhoA alone exacerbated lung adenoma formation, whereas dual deletion of RhoA and RhoC together significantly reduced K-RasG12D induced adenoma formation. Deletion of RhoA appears to induce a compensatory mechanism that exacerbates adenoma formation. The compensatory mechanism is at least partly mediated by RhoC. This study suggests that targeting of RhoA alone may allow for compensation and a paradoxical exacerbation of neoplasia, while simultaneous targeting of both RhoA and RhoC is likely to lead to more favorable outcomes.
Introduction
In the United States, lung cancer kills more people each year than breast, prostate and colon cancer combined [1]. Lung adenocarcinoma is the most common subtype of lung cancer and often harbors activating mutations of K-Ras [2]. K-Ras is a founding member of the Ras GTPase superfamily and is a key signal transduction protein that integrates extracellular stimuli and promotes cell proliferation and survival. Activating mutations of K-Ras disrupt the GTPase activity of the protein, increasing levels of GTP-bound K-Ras, which results in continuous signaling. Despite being among the very first oncogenes discovered, direct pharmacological inhibition of K-Ras has remained elusive [3]. An alternative strategy to inhibiting K-Ras directly is to target its downstream signaling pathways. While the RAF-MEK-ERK signaling pathway is the most important transducer of K-Ras signaling, several other downstream signaling axes including the PI3K-AKT-mTOR pathway, Ral GTPases, and the Rho family of GTPases have each been implicated as required elements for Ras induced transformation.
The mammalian Rho GTPase family includes over 20 members, of which RhoA, Rac1 and Cdc42 are among the best characterized. These Rho GTPases regulate the cell cycle and actin cytoskeleton and are thus critical regulators of processes such as cell shape, adhesion, migration, polarity and proliferation [4]. Given these essential functions of Rho GTPases and the availability of pre-clinical and clinical inhibitors of Rho GTPase signaling, they pose an important topic in cancer research [5–8]. Importantly, Rho GTPases have been shown to be critical for Ras-induced transformation of fibroblasts and epithelial cells [9–13]. Over a decade ago, several classic studies in the GTPase field demonstrated that blocking RhoA signaling could suppress Ras-induced transformation, and conversely that constitutively active RhoA could cooperate synergistically with Raf to promote cell transformation [9,12,13]. Additionally, studies have shown that RhoA plays an important permissive role in cell cycle progression through the G1-S phase: namely, increased RhoA activity inhibits p21Waf1/Cip1 and results in increased amounts of cyclin D1 and p27Kip1 [14–16]. Indeed, blockage of RhoA signaling is thought to induce INK4 activity, in turn halting the cell cycle [16]. Thus, the plurality of evidence shows RhoA is a positive regulator of the cell cycle. Subsequently, RhoA has been found to be either overexpressed or hyperactive in a variety of cancers, and RhoA activity is correlated with negative outcomes in gastric, hepatocellular, esophageal squamous cell, breast and lung carcinomas [17–22].
More recently, greater attention has been paid to the role of Rac1 in tumorigenesis due to the availability of murine genetic models. These studies have confirmed a positive role of Rac1 in tumor initiation. Rac1 was found to be required for K-Ras-induced lung adenoma formation in mice and a Rac1 splicing variant with increased activity, Rac1b, appears to promote tumorigenesis in this context [23,24]. Two other recent studies have indicated that pharmacological inhibition of the downstream mediators of RhoA signaling, ROCK and FAK, are promising therapeutic targets [25,26]. However, to date no mammalian genetic studies have directly assessed whether RhoA is required for oncogenic Ras-mediated tumorigenesis.
In the present work, we have sought to investigate the role of RhoA and the closely related Rho GTPase, RhoC, in K-Ras-induced tumorigenesis using a well-established inducible K-RasG12D knock-in mouse model of lung adenoma induction in combination with our RhoA conditional knockout, RhoAflox/flox, and RhoC-/- mouse models. Our investigation of these genetic studies produced surprising results that individually RhoA and RhoC are dispensable for the oncogenic K-Ras induced lung tumorigenesis and loss of RhoA alone exacerbates, rather than suppresses, tumor initiating activity. Our study implies that pharmacological targeting of multiple Rho family members, e.g. RhoA and RhoC in this context, is required to prevent tumorigenesis.
Results
We first sought to determine whether mice would be viable without RhoA expression in their bronchiolar epithelium. We crossed a RhoA conditional mouse (RhoAflox/flox) with mice harboring the Club Cell Secretory Promoter Cre (CCSP-Cre), as outlined in Table 1. This promoter is expressed mainly in Club cells, eponymously referred to as Clara cells, which are a major component of the bronchial epithelium [27].
Table 1. Breeding Schematic for Transgenic Mice.
| Transgenes | ||||
|---|---|---|---|---|
| Shorthand | RhoWT | RhoAcKO | RhoC-/- | Double Knockout (DKO) |
| RhoA genotype | RhoA+/+ | RhoAflox/flox | RhoA+/+ | RhoAflox/flox |
| RhoC genotype | RhoC+/+ | RhoC+/+ | RhoCΔ2–3 | RhoCΔ2–3 |
| Additional Transgenes: | ||||
| K-Ras knock-in | LSL-K-RasG12D | |||
| Cre-recombinase | Adeno-Cre or CCSP-Cre | |||
Mice harboring CCSP-Cre RhoAflox/flox transgenes were born at the expected Mendelian ratio, did not differ in weight during weaning or adulthood and lived as long as Cre-negative littermates. These mice did not display any signs of overt respiratory distress. Their lungs were normal appearing both grossly and microscopically (Fig 1A and 1B). We wondered whether the Cre expression would be altered by RhoA deletion so we crossed these mice to the double-reporter “mTmG” mouse. These mice constitutively express tdTomato, which switches to eGFP with the expression of Cre-recombinase [28]. We found Cre expression in a patchy distribution within bronchiolar epithelium, which was similar between RhoAcKO and RhoWT backgrounds (Fig 1C).
Fig 1. CCSP-promoter driven deletion of RhoA does not affect normal lung development.
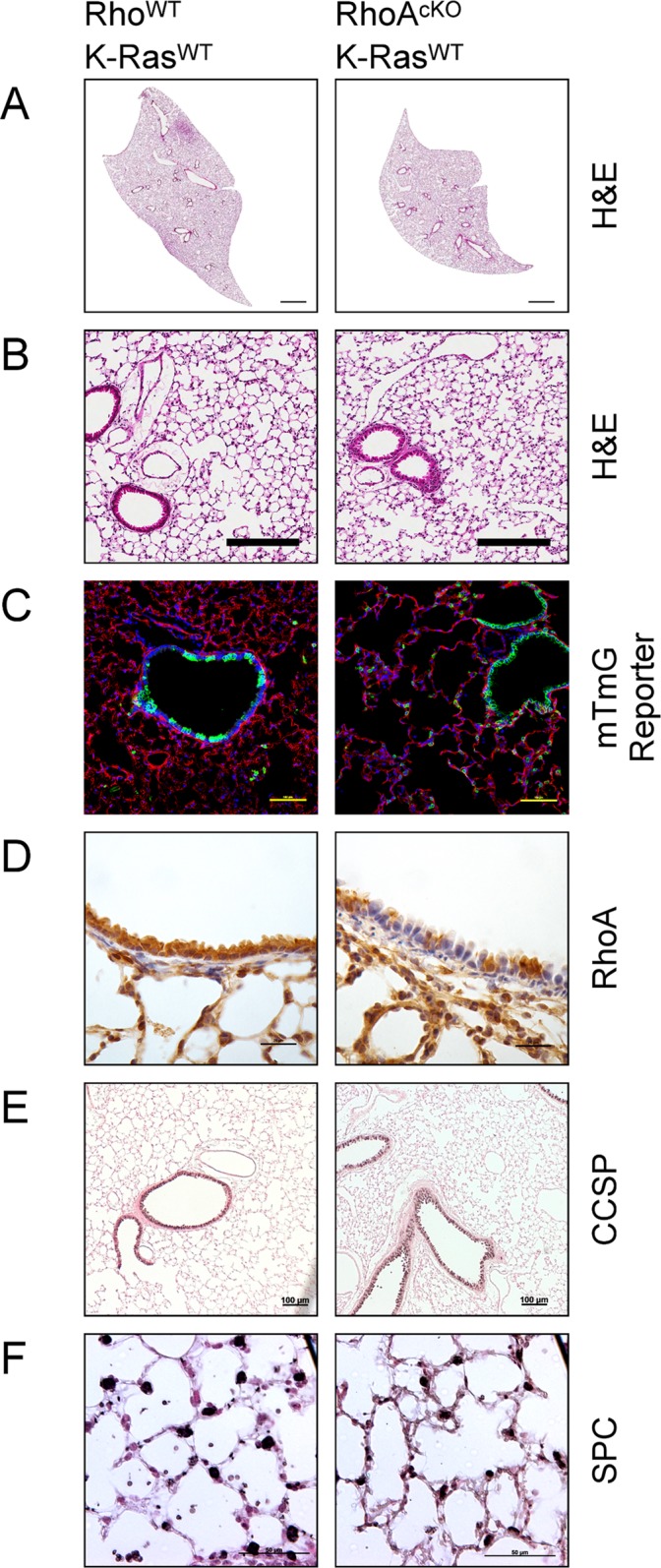
RhoWT and RhoAflox/flox mice were mated with CCSP-Cre mice on a K-RasWT background. (A & B) Lungs by H&E (bars represent 1mm and 200μm for panels A & B respectively). (C) Mice were crossed to a tdTomato to eGFP reporter line to assess for difference is Cre-expression pattern (bars represent 100μm). Green represents eGFP and Cre activity. Red presents tdTomato and the absence of Cre activity. Blue represents DAPI staining. (D) Immunohistochemistry for RhoA (brown) with hematoxylin counterstain (bars represent 25μm). (E) Immunohistochemistry for CCSP (black), counterstained with nuclear fast red (bars represent 100μm). (F) Immunohistochemistry for SPC (black), counterstained with nuclear fast red (bars represent 50μm).
We next assessed for the penetrance of RhoA deletion. Using immunohistochemistry, we found RhoA was deleted from the bronchioles in a patchy pattern similar to the distribution of eGFP in the reporter mice (Fig 1D). Recombination of the RhoAflox/flox allele was also evident in RhoAcKO via PCR with RhoA deletion specific primers (S1 Fig). Moreover, we did not find any qualitative differences in the expression pattern of Club cells or Type II alveolar cells as assessed by CCSP and SPC staining, respectively (Fig 1E and 1F). Thus, deletion of RhoA from Club cells by the CCSP-Cre resulted in viable mice with no signs of respiratory dysfunction. There were no overt differences between RhoAcKO mice and control mice with regards to lung architecture or the amount and distribution of major lung cell types.
Next we sought to determine whether RhoA is required for oncogenic K-Ras-induced tumor initiation in vivo. We bred the Lox-STOP-Lox K-RasG12D (LSL-K-RasG12D) transgene into the CCSP-Cre;RhoAflox/flox mice. At least two different constructs of CCSP-Cre (also referred to as CC10-Cre) have been successfully used to induce lung tumors in mice [29–32]. It was also previously shown that CCSP-Cre LSL-K-RasG12D mice develop a strong inflammatory component to their disease with abundant infiltration of alveolar macrophages and neutrophils [29,30]. Consistent with previous works, we found the CCSP-Cre;LSL-K-RasG12D;RhoAflox/flox mice to have grossly abnormal lungs with abundant infiltration and adenoma formation (Fig 2A and 2B). These mice also displayed prominent hyperplasia of the bronchiolar epithelium and atypical adenomatous hyperplasia (AAH) [33]. We did not find any differences in the disease pathology of RhoAcKO mice in comparison to RhoWT mice and both groups formed adenomas. Only a proportion of the adenomas in either group stained positively for pERKThr202/Tyr204, in agreement with previous reports (Fig 2C) [26,34]. Immunohistochemical staining of the lungs of these mice demonstrated positive RhoA staining for all adenomas in the RhoWT group, whereas a fraction of adenomas stained positively in the RhoAcKO group (Fig 2D and 2E). Although only ~15% of the adenomas observed were RhoA-null in the RhoAcKO group, we found numerous RhoA-null hyperplastic growths, including AAH (S2 Fig).
Fig 2. RhoA is not essential for CCSP-promoter driven, K-RasG12D-induced, lung adenoma formation in mice.

RhoWT and RhoAflox/flox mice were mated with CCSP-Cre mice on a K-RasG12D background. (A & B) K-RasG12D lungs by H&E (bars represent 1mm and 200μm for panels A & B respectively). (C) Immunohistochemistry for pERKThr202/Tyr204 (brown) with hematoxylin counterstain (bars represent 100μm). (D) Immunohistochemistry for RhoA (brown) with hematoxylin counterstain (bars represent 100μm). (E) Quantification of the RhoA-status of adenomas as assessed by immunohistochemistry. Greater than 30 tumors were counted from four mice per group.
Given that most adenomas evaded RhoA-deletion by CCSP-Cre, our results imply that there is selection pressure for the maintenance of RhoA signaling during KRasG12D-induced transformation. Our results are similar to the finding that Rac1 is required for KRas-induced adenoma formation by Kissil et al. [23]. In their findings, Kissil et al. utilized a similar approach with Rac1flox/flox transgenic mice and found the Rac1 locus was never recombined, and always remained undeleted in adenomas. Our findings differ in that we do find RhoA-null adenomas, thus demonstrating that RhoA signaling is not essential for oncogenic K-Ras-induced adenoma formation.
We wondered if other closely related Rho GTPases may be redundant for RhoA function, and decided to investigate RhoC as a candidate since RhoC shares a very high degree of sequence homology with RhoA and is also thought to be pro-tumorigenic [35]. We did not investigate the third Rho subfamily member, RhoB, because gene targeting and other studies have shown it plays an opposite function to RhoA [36–39]. As previously reported, constitutively RhoC null mice, RhoC-/- mice, are viable and do not demonstrate any overt phenotype [40]. Similar to RhoA deletion alone, CCSP-Cre;RhoAflox/flox;RhoC-/- double-knockout (DKO) mice were born at the expected Mendelian ratio, did not display any signs of overt respiratory distress and their lungs were grossly normal (Fig 3A and 3B). When crossed with the mTmG reporter line we found no differences in Cre expression between RhoWT, RhoC-/- or DKO mice (Fig 3C). We found RhoA expression, as assessed by immunohistochemistry, to be similar between RhoC-/- and RhoWT mice (Fig 3D). RhoA expression in double-knockout mice was similar to that of RhoAcKO mice (Fig 3D). Specifically, we found RhoA deletion from the bronchiolar epithelium in DKO mice to be in a patchy distribution (Fig 3D). Effective RhoA-locus recombination was also evident by PCR (S1 Fig). We did not find any qualitative differences in the expression pattern of Club cells or Type II alveolar cells in any of these mice as assessed by CCSP and SPC staining, respectively (Fig 3E and 3F). Thus, dual RhoA and RhoC deletion from a large proportion of the bronchiolar epithelium does not have any substantial effect on normal lung development or function.
Fig 3. Deletion of RhoA and RhoC together does not impair normal lung development.

RhoWT, RhoC-/- and DKO mice were mated with CCSP-Cre mice on a K-RasWT background. (A & B) Lungs by H&E (bars represent 1mm and 200μm for panels A & B respectively). (C) Mice were crossed to a tdTomato to eGFP reporter line to assess for difference is Cre-expression pattern (bars represent 100μm). Green represents eGFP and Cre activity. Red presents tdTomato and the absence of Cre activity. Blue represents DAPI staining. (D) Immunohistochemistry for RhoA (brown) with hematoxylin counterstain (bars represent 25μm). (E) Immunohistochemistry for CCSP (black), counterstained with nuclear fast red (bars represent 100μm). (F) Immunohistochemistry for SPC (black), counterstained with nuclear fast red (bars represent 50μm).
We further assessed whether double deletion of RhoA and RhoC would affect K-RasG12D induced adenoma formation. We crossed RhoAflox/flox;RhoC-/- mice with the CCSP-Cre;LSL-K-RasG12D line and observed adenoma formation in both RhoC-/- and DKO mice (Fig 4A and 4B). It was previously shown that RhoC-/- mouse embryonic fibroblasts are able to form Ras-induced colonies in agar, and that RhoC-/- mice can form MMTV-PyMT induced breast tumors [40], but to the best of our knowledge this is the first time RhoC has been shown to be dispensable for Ras-induced tumor formation in vivo. We found that these adenomas stained positively for pERKThr202/Tyr204 by immunohistochemistry (Fig 4C). Additionally, immunohistochemistry for RhoA showed that almost all adenomas in the RhoC-/- and DKO stained positively for RhoA (Fig 4D and 4E). These results raise the possibility that there is a strong selective disadvantage for RhoA deletion during adenoma formation in the absence of RhoC. While a RhoA deletion band is present by PCR (S1 Fig), it may be derived from heterozygous RhoA lox recombination or non-adenoma tissue. Deletion of either RhoA or RhoC did not result in differences in Ki67 or cleaved-caspase 3 staining of adenomas suggesting that RhoA and RhoC status do not affect cell proliferation or survival (S2 Fig)
Fig 4. Neither RhoA nor RhoC is required for K-RasG12D-induced adenoma formation in a CCSP-Cre model.

RhoWT, RhoC-/- and DKO mice were mated with CCSP-Cre mice on a K-RasG12D background. (A & B) K-RasG12D lungs by H&E (bars represent 1mm and 200μm for panels A & B respectively). (C) Immunohistochemistry for pERKThr202/Tyr204 (brown) with hematoxylin counterstain (bars represent 100μm). (D) Immunohistochemistry for RhoA (brown) with hematoxylin counterstain (bars represent 100μm). (E) Quantification of the RhoA-status of adenomas as assessed by immunohistochemistry. Greater than 30 tumors were counted from four mice per group.
Importantly, we found that “RhoA-null” tumors in DKO mice maintained pMLCSer20 staining, suggesting compensatory activity for RhoA loss (S3 Fig). Taken together, these results indicate that while RhoA and RhoC are each dispensable for K-RasG12D mediated tumorigenesis, together they contribute to adenoma formation.
To eliminate potential developmental effects of K-RasG12D and RhoA deletion during mouse development, as well as to achieve more robust RhoA deletion, we next used adenoviral mediated induction of lung adenomas to yield a sporadic tumor model [41,42]. Six- to eight-week-old mice were administered intratracheal Cre-expressing adenovirus (Adeno-Cre) in order to simultaneously induce oncogenic K-Ras and delete the floxed Rhoa gene. Twelve weeks post-adenoviral induction mice were euthanized and their lungs harvested. Hematoxylin and eosin (H&E) staining demonstrated adenoma formation in RhoWT, RhoAcKO, RhoC-/- and DKO mice (Fig 5A). As before, only a proportion of the adenomas stained positively for pERKThr202/Tyr204 in each group (Fig 5B). Immunohistochemical staining revealed positive RhoA staining in all adenomas from the RhoWT, RhoC-/- and DKO groups (Fig 5C), whereas only a subset of adenomas from the RhoAcKO group expressed RhoA (Fig 5C and 5D). Therefore, similar to the CCSP-Cre model, RhoA-null adenomas were able to form by Adeno-Cre induction in K-RasG12D;RhoAflox/flox mice, indicating RhoA alone is not essential for K-RasG12D mediated tumorigenesis.
Fig 5. RhoA and RhoC are important in combination, but not individually, for K-RasG12D induced sporadic lung adenoma formation.

Mice from different Rho backgrounds were administered Adeno-Cre virus endotracheally. Lungs were harvested after 12 weeks. (A) H&E of adenomas (bars represent 100μm). (B) Immunohistochemistry for pERKThr202/Tyr204 (brown) with hematoxylin counterstain (bars represent 100μm). (C) Immunohistochemistry for RhoA (brown) with hematoxylin counterstain (bars represent 100μm). (D) Western blots of microdissected tumors (N = normal lung, T = tumor). (E) Quantification of tumor burden. Means represent the quantification of four separate, evenly spaced and similarly sized lung sections, from four mice for each group (n = 4, * = p ≤ 0.001). Data representative of two separate experiments. (F) Quantification of tumor sizes. Data points represent the area of individual tumors expressed in μm2. Red bars represent the mean tumor area. (G) Proportion of RhoA-positive and RhoA-null tumors. Displayed as RhoA staining by the level of staining intensity (levels are as follows: no staining; normal = same intensity staining as adjacent normal alveoli; high = greater than adjacent normal alveoli; very high = much greater intensity staining than adjacent normal alveoli).
We quantified the tumor burden of the Adeno-Cre mice via serial sectioning of their lungs. Surprisingly, we found that there were many more adenomas in the RhoAcKO group as compared to the control, RhoWT group (Fig 5E), and in addition to quantity, many adenomas in the RhoAcKO group are larger in size than those in controls (Fig 5F). Interestingly, analysis of the RhoA status of the larger adenomas within the RhoAcKO group revealed that the relatively larger adenomas (>10,000 μm2) were ones in which RhoA was not fully deleted. Quantification of the number and size of tumors revealed similar numbers and sizes of tumors between RhoWT and RhoC-/- mice (Fig 5E and 5F). However, DKO mice had fewer adenomas and these tumors were smaller than those formed in either RhoWT or RhoC-/- mice (Fig 5E and 5F). Interestingly, we found noticeably increased amounts of hyperplastic lesions in the RhoAcKO group (S4 Fig). Similar to the CCSP-Cre model, increased hyperplasia did not translate to differences in Ki67 or cleaved-caspase 3 staining of adenomas regardless of RhoA status (S4B and S4C Fig).
Due to the heterogeneity of RhoA-positive and RhoA-null adenomas in the RhoAcKO mouse group, we subdivided tumors based on their RhoA deletion status in our analysis. The RhoA expression status of each adenoma was quantified by immunohistochemistry. We found that all adenomas in the RhoWT, RhoC-/- and DKO groups expressed RhoA in the Adeno-Cre induction model (Fig 5G). In particular, every adenoma found in the DKO group retained RhoA and escaped recombination of the RhoAflox/flox allele (Fig 5G) whereas a portion of the tumors in the RhoAcKO group was completely depleted of RhoA, similar to that of the CCSP-Cre model (Fig 5D and 5G). However, the sum of the RhoA-null tumors in the RhoAcKO group did not account for the overall increased numbers of tumors in this group (Fig 5G). This data suggests that there is a strong selection advantage to RhoA expression in the absence of RhoC during K-RasG12D-driven adenoma formation.
Finally, we microdissected Adeno-Cre induced adenomas from mice with different Rho backgrounds and assessed downstream signaling via Western blot (Fig 5D). Both RhoA and RhoC are known to bind to and signal via ROCK and regulate phosphorylation of MLC [43,44]. We found that RhoA-null adenomas maintained or upregulated pMLCSer19 signaling, suggesting likely compensation by RhoC or other related Rho GTPases. The upregulated levels of pMLCSer19 in RhoAcKO mice correlate with the increased tumorigenesis in these mice. Moreover, we found that pMLCSer19 levels are not increased in DKO mice, correlating with the observation that these mice have both smaller and fewer tumors than RhoWT, RhoAcKO or RhoC-/- background mice. These observations are consistent with the interpretation that there is a selection pressure to maintain downstream pMLC signaling when RhoA signaling is lost.
Discussion
Several previous studies of the relationship between RhoA and Ras signaling in cell transformation prompted our current investigation of whether RhoA or RhoC are required for K-Ras-induced tumor formation [9–11]. The main readouts of the earlier studies were anchorage-independent growth in soft agar, cell proliferation, decreased requirement of serum for growth, and focus formation in the Petri dish. These studies addressed two important questions: whether increased RhoA signaling could lead to transformation of cells and whether abrogation of RhoA signaling could prevent Ras-induced transformation. To answer the first question, several studies reported that over expression of wild type RhoA, or constitutively active RhoA or Rac1, can weakly transform cells, suggesting an oncogenic role of active RhoA [9,10,12,13,45,46]. Transformation of cells expressing active Raf were synergistically enhanced with constitutively active RhoA [9]. Answering the second question, it was found that dominant negative forms of RhoA can block Ras-induced transformation [9,10]. However, these classical cell biology studies were limited by the nature of the approaches—dominant-negative or constitutively active mutant over expression—by the models—mostly fibroblasts and tumor cell lines—and by the in vitro nature of the readouts. Nevertheless, these works laid out the conventional rationale that RhoA is a proto-oncogene product and RhoA targeting may be of therapeutic value [35].
Our investigation focuses on addressing the role of RhoA in K-Ras-driven tumorigenesis in vivo in established mouse models of lung cancer. First, it appears that neither RhoA nor RhoC is required for normal lung function in mice. Second, our data shows K-RasG12D-induced adenomas can form in the absence of RhoA, indicating that RhoA is not required for K-RasG12D-induced tumorigenesis. Third, the closely related RhoA homolog, RhoC, which has been implicated as an important pro-tumor metastasis molecule, is also dispensable for K-RasG12D-induced tumorigenesis. Fourth, we found an increase in the numbers of adenomas in the RhoAcKO sporadic lung tumor model in which RhoA is deleted by adenoviral expression of Cre. Fifth, an attempt at combined deletion of RhoA and RhoC resulted in reduced adenoma formation and retention of RhoA in all adenomas. These results indicate that there is a selective advantage to retain RhoA in the absence of RhoC, and there is likely a redundant and/or compensatory role of RhoC in RhoA signaling downstream of oncogenic K-Ras in adenoma formation. Consistent with this interpretation, the phosphorylation status of MLC, downstream of RhoA and RhoC signaling, was not decreased in either RhoA or RhoC-null adenomas, nor in adenomas from DKO mice, in which RhoA is retained.
Our counterintuitive findings are in line with several recent studies that have begun to challenge the conventional paradigm that RhoA is a simple proto-oncogene. Two whole-exome sequencing studies of human lymphomas found recurrent Gly17Val mutations in RhoA which confer a dominant negative or loss of function phenotype [47,48]. Another very recent study using two different mouse models of colon cancer found that mice carrying a dominant negative RhoA transgene (RhoAT19N) had increased tumor burden [49]. Furthermore, a study of K-Ras-driven liver tumorigenesis in Zebrafish has found that an active form of RhoA (RhoAG14V) hindered tumor formation whereas dominant-negative RhoA (RhoAT19N) accelerated tumorigenesis [50]. While the pro-oncogenic mechanism of RhoA function loss remains unclear, our work suggests that other Rho family members such as RhoC may compensate for RhoA loss, or possibly even “overcompensate”, resulting in a paradoxical increase in tumorigenesis.
A limitation of our mouse model in this study is that RhoA is deleted at the same time as K-RasG12D is expressed, and thus our work does not address whether RhoA plays a role in tumor maintenance or progression. It is possible that once oncogenic K-Ras induced tumors have developed, they become addicted to permissive signaling from wild-type RhoA. In this case, RhoA signaling would be required for tumor maintenance. Indeed, an examination of human cancer cell lines treated with RhoA-specific siRNA or the Rho inhibitor Rhosin shows that they are dependent on RhoA function for proliferation and progression (data not shown). These findings pose an intriguing possibility that targeted inhibition of RhoA activity alone is useful for suppressing advanced tumors in certain instances, but may exacerbate preneoplastic lesions. Another caution interpreting our results is that both mouse cancer models are accompanied by significant inflammatory responses which could potentially be modulated by RhoA status, which in turn may be promoting the inflammatory component (i.e. effects through tumor microenvironment).
The interplay between RhoA and RhoC signaling is in some ways similar to that of B-Raf and c-Raf in oncogenic K-Ras-driven adenoma initiation. Recent studies have found that c-Raf, but not B-Raf, is required for K-Ras-driven adenoma formation, despite historical evidence in mouse embryonic tissues and fibroblasts suggesting the opposite, that B-Raf but not c-Raf is required for ERK phosphorylation [51–55]. In fact, similar to our findings, pharmacologic targeting of B-Raf results in increased activity of c-Raf and a paradoxical increase in ERK phosphorylation and tumorigenesis in KRas-mutant cancers [53,56–58]; though this is not the case with genetic deletion of B-Raf. Interestingly, Blasco et al. also found that whole organism deletion of both B-Raf and c-Raf resulted in no obvious phenotype in mice, also similar to our finding of overtly healthy mice with dual deletion of RhoA and RhoC from the bronchiolar epithelium [51].
Our study of the relationship between RhoA, RhoC and K-Ras in murine lung adenoma formation sheds light on the broader subject of cellular signal transduction networks, and how signal redundancy and compensation in these networks can result in counterintuitive results when the network is modified. These insights are important considering the increased interest and emphasis on targeted therapeutics against individual pathways. The robustness of signal transduction networks paired with the evolutionary power of clonal selection in cancer suggests the best antineoplastic strategies may need to incorporate multiple drug targets with an anticipation of how cancer cells will adapt to and evade targeted therapies. Our study suggests that simultaneous targeting of both RhoA and RhoC signaling is critical, as targeting of RhoA alone may worsen clinical outcomes. Further study confirming our proposed compensatory mechanism is warranted, as is a deeper understanding of the overlapping and differential functions of RhoA, RhoC and other related Rho GTPases in cancer biology. Last, our study suggests that targeting ROCK signaling, which is a shared downstream mediator of both RhoA and RhoC, may be sufficient to block K-Ras-driven cancer as has been previously suggested [25,59].
Materials and Methods
Animal work
RhoAflox/flox mice generated as previously described [60]. RhoCΔ2–3, LSL-K-Ras, CCSP-Cre and mTmG mice have been previously characterized [28,29,40,61]. Mice were bred and housed in a specific pathogen-free vivarium at Cincinnati Children’s Hospital Medical Center and protocols were approved by the Institutional Animal Use and Care Committee of the Cincinnati Children’s Hospital Research Foundation (IACUC2013-0069), in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care and the Office of Laboratory Animal Welfare. All in vivo experiments were performed using age-matched mice. Genotyping primers can be found in the supplementary information (S1 Table). Adeno-Cre virus (Ad5-CMV-Cre) was obtained from the University of Iowa Gene Transfer Vector Core. Administration of Adeno-Cre virus was performed as previously described using a dose of 108 PFU per mouse [41]. Mice were anesthetized with isoflurane and pain control was achieved with buprenorphine.
Western blotting
Western blotting was performed using standard protocols, with important changes or steps described herein. Briefly, lysis was performed using radioimmunoprecipitation assay buffer (RIPA buffer) supplemented with protease inhibitors cocktail (cOmplete Protease Inhibitor Cocktail Tablets, Roche) and phosphatase inhibitor cocktail consisting of sodium fluoride, sodium orthovanadate, sodium pyrophosphate, ß-glycerophosphate and sodium molybdate (Simple Stop 1 Phosphatase Inhibitor Cocktail, Gold Biotechnology). Transfer was performed using the Trans-Blot Turbo Transfer System (Bio-Rad). Visualization was performed using fluorescently conjugated secondary antibodies and an infrared imaging System (LiCor Odyssey CLx).
Immunohistochemistry
Briefly, tissues were fixed using 3.7% paraformaldehyde in PBS overnight and embedding to paraffin. Secondary antibodies and horse-radish peroxidase (HRP) used consisted of either Signal Stain Boost (Cell Signaling Technology) or anti-rabbit donkey secondary (GE Healthcare) and Vectastain Elite ABC Kit (Vector Labs) avidin-biotin complex. The developing reagent was DAB (3, 3’-diaminobenzidine) HRP substrate (Vector Labs). Counterstaining was performed using Gill’s hematoxylin or nuclear fast red. Quantification of stainings were made by serially sectioning lungs in 250μm step increments and counting a sample of evenly spaced sections for the number of tumors. Tumor size was expressed as tumor area as measured in ImageJ.
Primary antibodies
Primary antibodies for immunoblotting or immunohistochemistry are as follows: RhoA (67B9) Rabbit mAb (1:400 dilution, Cell Signaling Technology [CST] #2117), Myosin Light Chain 2 (D18E2) Rabbit mAb (CST #8505), Phospho-Myosin Light Chain 2 (Ser19) Mouse mAb (1:150 dilution, CST #3675), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) Rabbit mAb (CST #4370), Phospho-MEK1/2 (Ser221) (166F8) Rabbit mAb (CST #2338), GAPDH (D16H11) Rabbit mAb (CST #5174), Anti-Myosin Light Chain (phospho S20) antibody (Abcam ab2480), Ki67 Rabbit mAb, (790–4286 Ventana), cleaved-caspase 3 Rabbit Ab (orb137850 Biorbyt), N-Terminal Pro SPC Rabbit Ab (Seven Hills Bioreagents WRAB-9337), and CCSP Rabbit Ab (Seven Hills Bioreagents WRAB-3950). All antibodies were used at the manufacturers recommended dilutions unless otherwise stated.
Data analysis and presentation
Data was gathered and analyzed in either ImageJ 1.47 (NIH), GraphPad Prism (GraphPad) or the GNU Image Manipulation Program 2.8 (GIMP). Analysis in GraphPad Prism comprised of unpaired two-way student T-tests or ANOVA.
Supporting Information
DNA was isolated from mouse lungs of different Rho background mice mated to either CCSP-Cre K-RasWT (K-WT) or CCSP-Cre LSL-K-RasG12D mice (KRas). Pairs of K-RasWT and LSL-K-RasG12D mice for each Rho background are shown. Deletion bands for LSL-K-RasG12D mice demonstrate expression of K-RasG12D (deletion band is the larger band at 315bp). Deletion bands for RhoA demonstrate recombination of the RhoAflox/flox locus (deletion band at 667bp). Yellow arrows point to the 500bp band of a 100bp ladder.
(TIFF)
(A) RhoA immunohistochemistry (brown) of hyperplastic lesions with hematoxylin counterstain of CCSP-Cre LSL-K-RasG12D mice from different Rho backgrounds (bars represents 100μm). (B) Percentage of Ki67 positive cells in CCSP-Cre LSL-K-RasG12D driven adenomas (n = 4, p >0.05). (C) Representative image of cleaved-caspase 3 (CC3) staining of CCSP-Cre LSL-K-RasG12D driven adenomas. Positive control is injured renal tubules (20X). Bars represent 100μm.
(TIFF)
Immunohistochemistry of serially sectioned adenomas from CCSP-Cre LSL-K-RasG12D mice. Rows represent serial sections of the same adenoma from their respective Rho backgrounds. Columns are of RhoA, pMLCSer20, pMEKSer221 and pERKThr202/Tyr204 stainings respectively with hematoxylin counterstain (bars represents 100μm).
(TIFF)
(A) RhoA immunohistochemistry (brown) with hematoxylin counterstain of Adeno-Cre induced LSL-K-RasG12D mice from different Rho backgrounds (bars represents 100μm). (B) Percentage of Ki67 positive cells in Adeno-Cre LSL-K-RasG12D driven adenomas (n = 4, p >0.05). (C) Representative image of cleaved-caspase 3 (CC3) staining of Adeno-Cre LSL-K-RasG12D driven adenomas. Positive control is injured renal tubules (20X). Bars represent 100μm.
(TIFF)
(TIFF)
Table of genotyping and deletion primers used for PCR. Specific PCR protocols can be found in the references for transgenic mice under Materials and Methods.
(DOCX)
Acknowledgments
Thanks to Drs Jeffery A Whitsett, Anna Perl and Lee Grimes for the transgenic mice they provided us. Many thanks to Drs Timothy D. LeCras, Vladimir V. Kalinichenko, John C. Morris, James C. Mulloy, Leesa L. Sampson and Kathryn A. Wikenheiser-Brokamp for their insightful comments and guidance. Additionally, we would like to acknowledge support from the University of Cincinnati Medical Scientist Training Program and the Molecular and Developmental Biology Graduate Program at Cincinnati Children’s Hospital Medical Center.
Data Availability
All relevant data are within the research article and supporting information files.
Funding Statement
Funding was provided by the National Institutes of Health through the following grants: R01CA150547 (YZ), R01CA141341 (YZ) and 5T32GM063483 (IZ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64: 9–29. Available: http://www.ncbi.nlm.nih.gov/pubmed/24399786. 10.3322/caac.21208 [DOI] [PubMed] [Google Scholar]
- 2. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, et al. (2012) Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120. Available: http://www.ncbi.nlm.nih.gov/pubmed/22980975. 10.1016/j.cell.2012.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gysin S, Salt M, Young A, McCormick F (2011) Therapeutic strategies for targeting ras proteins. Genes Cancer 2: 359–372. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3128641&tool=pmcentrez&rendertype=abstract. 10.1177/1947601911412376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269. Available: http://www.ncbi.nlm.nih.gov/pubmed/16212495. [DOI] [PubMed] [Google Scholar]
- 5. Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101: 7618–7623. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=419655&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shang X, Marchioni F, Evelyn CR, Sipes N, Zhou X, et al. (2013) Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc Natl Acad Sci U S A 110: 3155–3160. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3581902&tool=pmcentrez&rendertype=abstract. 10.1073/pnas.1212324110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, et al. (2012) Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem Biol 19: 699–710. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3383629&tool=pmcentrez&rendertype=abstract. 10.1016/j.chembiol.2012.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, et al. (1997) Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389: 990–994. Available: http://www.ncbi.nlm.nih.gov/pubmed/9353125. [DOI] [PubMed] [Google Scholar]
- 9. Khosravi-Far R, Solski P a, Clark GJ, Kinch MS, Der CJ (1995) Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol 15: 6443–6453. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=230895&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qiu RG, Chen J, McCormick F, Symons M (1995) A role for Rho in Ras transformation. Proc Natl Acad Sci U S A 92: 11781–11785. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=40486&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du J, Jiang B, Coffey RJ, Barnard J (2004) Raf and RhoA cooperate to transform intestinal epithelial cells and induce growth resistance to transforming growth factor beta. Mol Cancer Res 2: 233–241. Available: http://www.ncbi.nlm.nih.gov/pubmed/15140945. [PubMed] [Google Scholar]
- 12. Qiu RG, Chen J, Kirn D, McCormick F, Symons M (1995) An essential role for Rac in Ras transformation. Nature 374: 457–459. Available: http://www.ncbi.nlm.nih.gov/pubmed/7700355. [DOI] [PubMed] [Google Scholar]
- 13. Prendergast GC, Khosravi-Far R, Solski PA, Kurzawa H, Lebowitz PF, et al. (1995) Critical role of Rho in cell transformation by oncogenic Ras. Oncogene 10: 2289–2296. Available: http://www.ncbi.nlm.nih.gov/pubmed/7784077. [PubMed] [Google Scholar]
- 14. Croft DR, Olson MF (2006) The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol Cell Biol 26: 4612–4627. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1489131&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olson MF, Paterson HF, Marshall CJ (1998) Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 394: 295–299. Available: http://cosmos.asu.edu/publications/papers/Davies_83NATURE.pdf. [DOI] [PubMed] [Google Scholar]
- 16. Zhang S, Tang Q, Xu F, Xue Y, Zhen Z, et al. (2009) RhoA regulates G1-S progression of gastric cancer cells by modulation of multiple INK4 family tumor suppressors. Mol Cancer Res 7: 570–580. Available: http://www.ncbi.nlm.nih.gov/pubmed/19372585. 10.1158/1541-7786.MCR-08-0248 [DOI] [PubMed] [Google Scholar]
- 17. Pan Y, Bi F, Liu N, Xue Y, Yao X, et al. (2004) Expression of seven main Rho family members in gastric carcinoma. Biochem Biophys Res Commun 315: 686–691. Available: http://www.ncbi.nlm.nih.gov/pubmed/14975755. [DOI] [PubMed] [Google Scholar]
- 18. Li XR, Ji F, Ouyang J, Wu W, Qian LY, et al. (2006) Overexpression of RhoA is associated with poor prognosis in hepatocellular carcinoma. Eur J Surg Oncol 32: 1130–1134. Available: http://www.ncbi.nlm.nih.gov/pubmed/16806792. [DOI] [PubMed] [Google Scholar]
- 19. Faried A, Nakajima M, Sohda M, Miyazaki T, Kato H, et al. (2005) Correlation between RhoA overexpression and tumour progression in esophageal squamous cell carcinoma. Eur J Surg Oncol 31: 410–414. Available: http://www.ncbi.nlm.nih.gov/pubmed/15837049. [DOI] [PubMed] [Google Scholar]
- 20. Bellizzi A, Mangia A, Chiriatti A, Petroni S, Quaranta M, et al. (2008) RhoA protein expression in primary breast cancers and matched lymphocytes is associated with progression of the disease. Int J Mol Med 22: 25–31. Available: http://www.ncbi.nlm.nih.gov/pubmed/18575772. [PubMed] [Google Scholar]
- 21. Fritz G, Just I, Kaina B (1999) Rho GTPases are over-expressed in human tumors. Int J Cancer 81: 682–687. Available: http://www.ncbi.nlm.nih.gov/pubmed/10328216. [DOI] [PubMed] [Google Scholar]
- 22. Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B (2002) Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer 87: 635–644. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2364248&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Bender Kim CF, et al. (2007) Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res 67: 8089–8094. Available: http://www.ncbi.nlm.nih.gov/pubmed/17804720. [DOI] [PubMed] [Google Scholar]
- 24. Zhou C, Licciulli S, Avila JL, Cho M, Troutman S, et al. (2012) The Rac1 splice form Rac1b promotes K-ras-induced lung tumorigenesis. Oncogene: 1–7. Available: http://www.ncbi.nlm.nih.gov/pubmed/22430205. 10.1038/onc.2012.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar MSS, Hancock DCC, Molina-Arcas M, Steckel M, East P, et al. (2012) The GATA2 Transcriptional Network Is Requisite for RAS Oncogene-Driven Non-Small Cell Lung Cancer. Cell 149: 642–655. Available: http://linkinghub.elsevier.com/retrieve/pii/S0092867412004084. 10.1016/j.cell.2012.02.059 [DOI] [PubMed] [Google Scholar]
- 26. Konstantinidou G, Ramadori G, Torti F, Kangasniemi K, Ramirez RE, et al. (2013) RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov 3: 444–457. Available: http://www.ncbi.nlm.nih.gov/pubmed/23358651. 10.1158/2159-8290.CD-12-0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rawlins EL, Perl A-K (2012) The a“MAZE”ing world of lung-specific transgenic mice. Am J Respir Cell Mol Biol 46: 269–282. Available: http://www.ncbi.nlm.nih.gov/pubmed/22180870. 10.1165/rcmb.2011-0372PS [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605. Available: http://scholar.google.com/scholar?hl=en&btnG=Search&q=intitle:ARTICLE+A+Global+Double-Fluorescent+Cre+Reporter+Mouse#0. [DOI] [PubMed] [Google Scholar]
- 29. Ji H, Houghton M, Mariani TJ, Perera S, Kim CB, et al. (2006) K-ras activation generates an inflammatory response in lung tumors. Oncogene 25: 2105–2112. Available: http://www.ncbi.nlm.nih.gov/pubmed/16288213. [DOI] [PubMed] [Google Scholar]
- 30. Moghaddam SJ, Li H, Cho S-N, Dishop MK, Wistuba II, et al. (2009) Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol 40: 443–453. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2660561&tool=pmcentrez&rendertype=abstract. 10.1165/rcmb.2008-0198OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iwanaga K, Yang Y, Raso MG, Ma L, Hanna AE, et al. (2008) Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res 68: 1119–1127. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2750029&tool=pmcentrez&rendertype=abstract. 10.1158/0008-5472.CAN-07-3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pacheco-Pinedo EC, Durham AC, Stewart KM, Goss AM, Lu MM, et al. (2011) Wnt/β-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J Clin Invest 121: 1935–1945. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3083778&tool=pmcentrez&rendertype=abstract. 10.1172/JCI44871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, et al. (2004) Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res 64: 2307–2316. Available: http://www.ncbi.nlm.nih.gov/pubmed/15059877. [DOI] [PubMed] [Google Scholar]
- 34. Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, et al. (2010) Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468: 567–571. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3011233&tool=pmcentrez&rendertype=abstract. 10.1038/nature09526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sahai E, Marshall CJ (2002) RHO-GTPases and cancer. Nat Rev Cancer 2: 133–142. Available: http://www.ncbi.nlm.nih.gov/pubmed/12635176. [DOI] [PubMed] [Google Scholar]
- 36. Huang M, Prendergast GC (2006) RhoB in cancer suppression. Histol Histopathol 21: 213–218. Available: http://www.ncbi.nlm.nih.gov/pubmed/16329046. [DOI] [PubMed] [Google Scholar]
- 37. Bousquet E, Mazières J, Privat M, Rizzati V, Casanova A, et al. (2009) Loss of RhoB expression promotes migration and invasion of human bronchial cells via activation of AKT1. Cancer Res 69: 6092–6099. Available: http://www.ncbi.nlm.nih.gov/pubmed/19602596. 10.1158/0008-5472.CAN-08-4147 [DOI] [PubMed] [Google Scholar]
- 38. Mazieres J, Antonia T, Daste G, Muro-Cacho C, Berchery D, et al. (2004) Loss of RhoB expression in human lung cancer progression. Clin Cancer Res 10: 2742–2750. Available: http://www.ncbi.nlm.nih.gov/pubmed/15102679. [DOI] [PubMed] [Google Scholar]
- 39. Sato N, Fukui T, Taniguchi T, Yokoyama T, Kondo M, et al. (2007) RhoB is frequently downregulated in non-small-cell lung cancer and resides in the 2p24 homozygous deletion region of a lung cancer cell line. Int J Cancer 120: 543–551. Available: http://www.ncbi.nlm.nih.gov/pubmed/17096327. [DOI] [PubMed] [Google Scholar]
- 40. Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, et al. (2005) RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev 19: 1974–1979. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1199568&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DuPage M, Dooley AL, Jacks T (2009) Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4: 1064–1072. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2757265&tool=pmcentrez&rendertype=abstract. 10.1038/nprot.2009.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, et al. (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15: 3243–3248. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=312845&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sahai E, Marshall CJ (2002) ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol 4: 408–415. Available: http://www.ncbi.nlm.nih.gov/pubmed/11992112. [DOI] [PubMed] [Google Scholar]
- 44. Riento K, Ridley AJ (2003) Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4: 446–456. Available: http://www.ncbi.nlm.nih.gov/pubmed/12778124. [DOI] [PubMed] [Google Scholar]
- 45. Avraham H, Weinberg RA (1989) Characterization and expression of the human rhoH12 gene product. Mol Cell Biol 9: 2058–2066. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=362999&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perona R, Esteve P, Jiménez B, Ballestero RP, Ramón y Cajal S, et al. (1993) Tumorigenic activity of rho genes from Aplysia californica. Oncogene 8: 1285–1292. Available: http://www.ncbi.nlm.nih.gov/pubmed/8479750. [PubMed] [Google Scholar]
- 47. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, et al. (2014) Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet 46: 171–175. Available: http://www.ncbi.nlm.nih.gov/pubmed/24413737. 10.1038/ng.2872 [DOI] [PubMed] [Google Scholar]
- 48. Palomero T, Couronné L, Khiabanian H, Kim M- Y, Ambesi-Impiombato A, et al. (2014) Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 46: 166–170. Available: http://www.ncbi.nlm.nih.gov/pubmed/24413734. 10.1038/ng.2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rodrigues P, Macaya I, Bazzocco S, Mazzolini R, Andretta E, et al. (2014) RHOA inactivation enhances Wnt signalling and promotes colorectal cancer. Nat Commun 5: 5458 Available: http://www.ncbi.nlm.nih.gov/pubmed/25413277. 10.1038/ncomms6458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chew TW, Liu XJ, Liu L, Spitsbergen JM, Gong Z, et al. (2013) Crosstalk of Ras and Rho: activation of RhoA abates Kras-induced liver tumorigenesis in transgenic zebrafish models. Oncogene: 1–11. Available: http://www.ncbi.nlm.nih.gov/pubmed/23812423. 10.1038/onc.2013.34 [DOI] [PubMed] [Google Scholar]
- 51. Blasco RB, Francoz S, Santamaría D, Cañamero M, Dubus P, et al. (2011) c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 19: 652–663. Available: http://www.ncbi.nlm.nih.gov/pubmed/21514245. 10.1016/j.ccr.2011.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karreth F, Frese KK, DeNicola GM, Baccarini M, Tuveson D (2011) C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov 1: 128–136. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3203527&tool=pmcentrez&rendertype=abstract. 10.1158/2159-8290.CD-10-0044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wimmer R, Baccarini M (2010) Partner exchange: protein-protein interactions in the Raf pathway. Trends Biochem Sci 35: 660–668. Available: http://www.ncbi.nlm.nih.gov/pubmed/20621483. 10.1016/j.tibs.2010.06.001 [DOI] [PubMed] [Google Scholar]
- 54. Galabova-Kovacs G, Matzen D, Piazzolla D, Meissl K, Plyushch T, et al. (2006) Essential role of B-Raf in ERK activation during extraembryonic development. Proc Natl Acad Sci U S A 103: 1325–1330. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1360532&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mikula M, Schreiber M, Husak Z, Kucerova L, Rüth J, et al. (2001) Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J 20: 1952–1962. Available: http://embojnl.embopress.org/content/20/8/1952.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, et al. (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464: 431–435. Available: http://www.ncbi.nlm.nih.gov/pubmed/20130576. 10.1038/nature08833 [DOI] [PubMed] [Google Scholar]
- 57. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464: 427–430. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3178447&tool=pmcentrez&rendertype=abstract. 10.1038/nature08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, et al. (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140: 209–221. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2872605&tool=pmcentrez&rendertype=abstract. 10.1016/j.cell.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vigil D, Kim TY, Plachco A, Garton AJ, Castaldo L, et al. (2012) ROCK1 and ROCK2 are required for non-small cell lung cancer anchorage-independent growth and invasion. Cancer Res 72: 5338–5347. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3810962&tool=pmcentrez&rendertype=abstract. 10.1158/0008-5472.CAN-11-2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Melendez J, Stengel K, Zhou X, Debidda M, Andreassen P, et al. (2011) RhoA GTPase is dispensable for actomyosin regulation but is essential for mitosis in primary mouse embryonic fibroblasts. J Biol Chem 286: 15132–15137. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3083211&tool=pmcentrez&rendertype=abstract. 10.1074/jbc.C111.229336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tuveson D, Shaw AT, Willis N, Silver DP, Jackson EL, et al. (2004) Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387. Available: http://www.ncbi.nlm.nih.gov/pubmed/15093544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DNA was isolated from mouse lungs of different Rho background mice mated to either CCSP-Cre K-RasWT (K-WT) or CCSP-Cre LSL-K-RasG12D mice (KRas). Pairs of K-RasWT and LSL-K-RasG12D mice for each Rho background are shown. Deletion bands for LSL-K-RasG12D mice demonstrate expression of K-RasG12D (deletion band is the larger band at 315bp). Deletion bands for RhoA demonstrate recombination of the RhoAflox/flox locus (deletion band at 667bp). Yellow arrows point to the 500bp band of a 100bp ladder.
(TIFF)
(A) RhoA immunohistochemistry (brown) of hyperplastic lesions with hematoxylin counterstain of CCSP-Cre LSL-K-RasG12D mice from different Rho backgrounds (bars represents 100μm). (B) Percentage of Ki67 positive cells in CCSP-Cre LSL-K-RasG12D driven adenomas (n = 4, p >0.05). (C) Representative image of cleaved-caspase 3 (CC3) staining of CCSP-Cre LSL-K-RasG12D driven adenomas. Positive control is injured renal tubules (20X). Bars represent 100μm.
(TIFF)
Immunohistochemistry of serially sectioned adenomas from CCSP-Cre LSL-K-RasG12D mice. Rows represent serial sections of the same adenoma from their respective Rho backgrounds. Columns are of RhoA, pMLCSer20, pMEKSer221 and pERKThr202/Tyr204 stainings respectively with hematoxylin counterstain (bars represents 100μm).
(TIFF)
(A) RhoA immunohistochemistry (brown) with hematoxylin counterstain of Adeno-Cre induced LSL-K-RasG12D mice from different Rho backgrounds (bars represents 100μm). (B) Percentage of Ki67 positive cells in Adeno-Cre LSL-K-RasG12D driven adenomas (n = 4, p >0.05). (C) Representative image of cleaved-caspase 3 (CC3) staining of Adeno-Cre LSL-K-RasG12D driven adenomas. Positive control is injured renal tubules (20X). Bars represent 100μm.
(TIFF)
(TIFF)
Table of genotyping and deletion primers used for PCR. Specific PCR protocols can be found in the references for transgenic mice under Materials and Methods.
(DOCX)
Data Availability Statement
All relevant data are within the research article and supporting information files.