

Abstract
The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that plays a critical role in the pathogenesis of many cancers. EGFR is unique in that its ligand-induced dimerization is established solely by contacts between regions of the receptor that are occluded within the monomeric, unliganded state. Activation of EGFR depends on the formation of an asymmetric dimer of the intracellular module of two receptor molecules, a configuration observed in crystal structures of the EGFR kinase domain in the active state. Coupling between the extracellular and intracellular modules is achieved by a switch between alternative geometries of the transmembrane and juxtamembrane segments within the receptor dimer. As the structure of the full-length receptor is yet to be determined, here we review recent structural studies on isolated modules of EGFR and molecular dynamics simulations that have provided much of our current understanding of its signaling mechanism, including how its regulation is compromised by oncogenic mutations.
Keywords: receptor tyrosine kinase, ligand-induced dimerization, asymmetric dimer, oncogenic mutations, transmembrane coupling
Introduction
The transition from unicellular life forms to multicellular ones occurred independently several times in evolution (1). A very early and defining step in the evolutionary branch that led to animal life was the emergence of signaling systems that are built on top of core signaling components that arose earlier in life. These signaling systems include pathways that control intercellular signaling, cell-cell adhesion and the development of the organism. Among the most important components of these systems are the tyrosine kinases, which signal through the generation of phosphotyrosine residues, molecular beacons that recruit proteins containing Src homology (SH2) domains and phosphotyrosine binding (PTB) domains (2).
Receptor tyrosine kinases are the outermost sentinels of the SH2-based signaling pathways. The extracellular modules of these receptors receive signals in the form of peptide hormones or cell surface proteins, and respond by activating intracellular tyrosine kinase domains (Figure 1A). Two other broad classes of receptors also trigger tyrosine kinase activity, but do not contain kinase domains themselves. These are the cytokine receptors and various receptors involved in immune responses, such as the T-cell and B-cell receptors and the Fc receptors (3).
Figure 1. Model for activation, domain architecture and evolutionary lineage of EGFR family members.
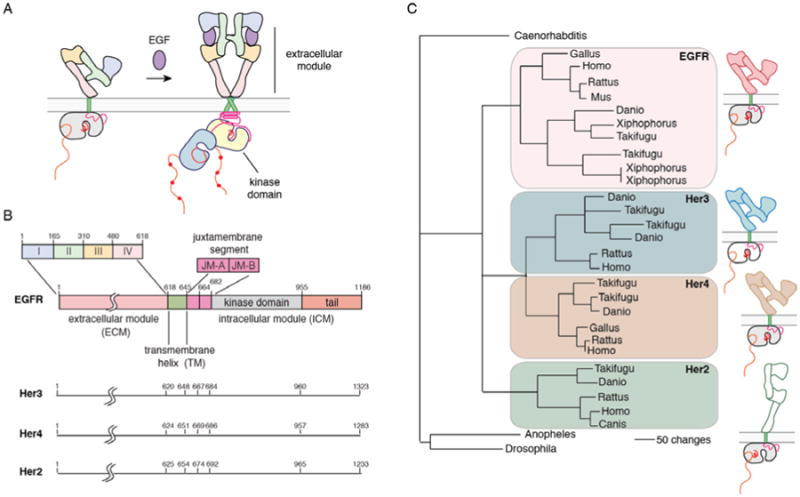
A) Model for activation of EGFR family members. B) Domain boundaries in EGFR family members. C) Dendrogram of the evolution of EGFR family members (modified from (140)). Her2 does not have a known ligand (denoted by an open box), and the kinase domain of Her3 is defunctional (denoted by an open box).
The receptor tyrosine kinases, the cytokine receptors and the immunological receptors have one architectural feature in common, which is that the protein subunits comprising the receptor complexes have only one transmembrane helix. This fact has caused the intact forms of these receptors to evade high-resolution structural analysis, perhaps because the plasma membrane plays a much more critical role in maintaining their structure than is the case for G-protein coupled receptors (GPCRs) or ion channels, which are now understood at an impressive level of molecular detail. Despite this limitation, structural, biochemical and biophysical probing of the mechanisms of receptor tyrosine kinases has yielded an increasingly deep understanding of how these proteins work. In this review, we discuss what is known about the structure and function of one family of receptor tyrosine kinases, of which the epidermal growth factor receptor (EGFR) is the prototypical member.
Human EGFR (also known as Her1/ErbB1, after the viral erythroblastoma gene) and its three close relatives, human epidermal growth factor receptors 2, 3 and 4 (Her2/ErbB2, also known as the neu oncogene, Her3/ErbB3 and Her4/ErbB3) control cell growth and differentiation (4-6) (Figure 1B). These receptors elicit potent mitogenic responses, and genetic abnormalities in these receptors represent one of the most prevalent defects in cancer cells (7). In fact, EGFR was the first cell surface receptor to be recognized as an oncogene (8, 9). Over 30% of breast cancers, 60% of non-small-cell lung cancers, and 40% of glioblastomas either overexpress or contain activating mutations in EGFR family members (10-12). The link between this family of receptors and cancer progression led to the first antibody-based therapy for cancer, Herceptin, which targets the extracellular module of Her2 (13).
The four EGFR family members are among the ∼60 receptor tyrosine kinases in the human genome (14). All receptor tyrosine kinases have an N-terminal extracellular ligand-binding module and a cytoplasmic tyrosine kinase catalytic domain linked by the single transmembrane helix. Despite this general similarity in their architecture, the extracellular modules of the principal subfamilies of these receptors are quite different (14). The transmembrane helices are also very divergent in sequence. Although the kinase domains of the receptor tyrosine kinases are grouped together in one major branch of the kinome (15), they differ in their mechanisms of activation.
The analysis of the genomes of organisms that have diverged very early along the evolutionary branch leading to the metazoan lineage shows that the choanoflagellates, unicellular organisms that are not true metazoans, contain receptor tyrosine kinases, but not EGFR (16, 17). EGFR appears in organisms that are on the true metazoan lineage, such as the sponges (16, 17), and its appearance may be the result of a fusion of an extracellular module with a cytoplasmic tryosine kinase that occurred independently of the fusion events that generated other receptor tyrosine kinases. That EGFR may have arisen independently of other receptor tyrosine kinases is also suggested by the fact that the sequence of the EGFR kinase domain is closer to that of Ack1, a nonreceptor tyrosine kinase (18), than to other receptor tyrosine kinases. The creation of receptors by independent fusion events has also led to the family of receptors for transforming growth factor beta (TGFβ) and bone morphogen protein (BMP) (19, 20). Like receptor tyrosine kinases, these receptors have extracellular ligand binding domains that are connected to a cytoplasmic kinase domain by a single transmembrane helix, but their kinase domains have specificity for serine and threonine residues rather than for tyrosine (21).
Two gene duplication events led to the four EGFR family members present in vertebrates (22) (Figure 1C). Gene duplication often results in one of the two resulting proteins becoming degenerate, leading to functional differentiation among members of the same family (23), and this is seen in the EGFR family. Her2 has lost the capacity to bind ligands, and functions primarily by forming heterodimers with other family members (24). Her3 has lost robust kinase activity (25), and so also signals through heterodimerization (26).
The extracellular module that is characteristic of the EGFR family is a tandem duplication of two kinds of domains. The first and third domains (domains I and III) are compact, and have a β-helical fold. The second and fourth domains (domains II and IV) are elongated and contain several cysteine-rich elements. The extracellular module is followed by a transmembrane helix and an intracellular module with a juxtamembrane segment, a kinase domain, and a C-terminal tail (4-6) (Figure 1A). Ligand binding to the extracellular module promotes dimerization, resulting in auto-phosphorylation of tyrosine residues in the intracellular C-terminal tail. This leads to the recruitment of SH2 and PTB domain containing effector proteins to the phosphotyrosine residues and the triggering of downstream signaling cascades. Besides being a recruitment site for these effectors, the C-terminal tail has also been implicated in kinase regulation (27).
The formation of dimers or higher-order oligomers is an essential step for the activation of EGFR family members (28-30). Although ligand binding promotes dimerization, enhanced expression of the receptors can also drive dimerization through mass action, which is important in certain cancers. Heterodimerization between EGFR family members is also an important aspect of their function and, as mentioned earlier, is especially important for Her3 activation since the kinase domain of that receptor is impaired (25). Although Her2 has an active kinase domain, it does not readily form homodimers under normal conditions, and it is activated by heterodimerization with ligand-bound partners, particularly Her3. Her4 forms homodimers, and can signal from the plasma membrane, but it is also cleaved by membrane-associated proteases upon activation, which causes translocation of the kinase domain from the plasma membrane to intracellular compartments (31, 32).
Here, we review the current state of knowledge of the structure of EGFR family members, and the insights that structural information provides about the molecular details of the regulation and activation of these receptors. We cover a rather narrow subset of the extensive literature on EGFR, focusing only on a subset of topics that have a direct bearing on the three-dimensional structure of the receptor.
Structures of the extracellular module: the tethered monomer and the back-to-back dimer
The extracellular modules of all four EGFR family members have been crystallized and their structures determined with either ligand bound (for EGFR and Her4) or without ligand (for all four receptors), as well as in complex with antibodies or antibody mimics. Daniel Leahy has written a particularly insightful review on the EGFR family (33), which can hardly be bettered in terms of the clarity with which it describes the structural principles underlying the construction and function of the extracellular modules.
There are three key points that emerge from these structural studies on the extracellular modules (33). First, dimerization of EGFR has an unexpected feature, which is that the dimer interface is formed entirely by the receptor itself, with the ligand bound on the outside. Second, there are just two principal classes of conformations seen in all the crystal structures. One corresponds to an “extended” form that, whether in a monomeric or a dimeric state, resembles the conformation of one protomer in the active dimer. The other conformation is a folded over or “tethered” form, in which the dimerization element is buried within a monomer. Third, the restriction to two principal conformations of the extracellular modules arises because domains I and III form a relatively rigid unit, as do domains II and IV. As a consequence, the extracellular module appears to “click” into either the compact, tethered form or the extended form that is primed for dimerization (Figure 2).
Figure 2. Extracellular module structures for the EGFR family members.
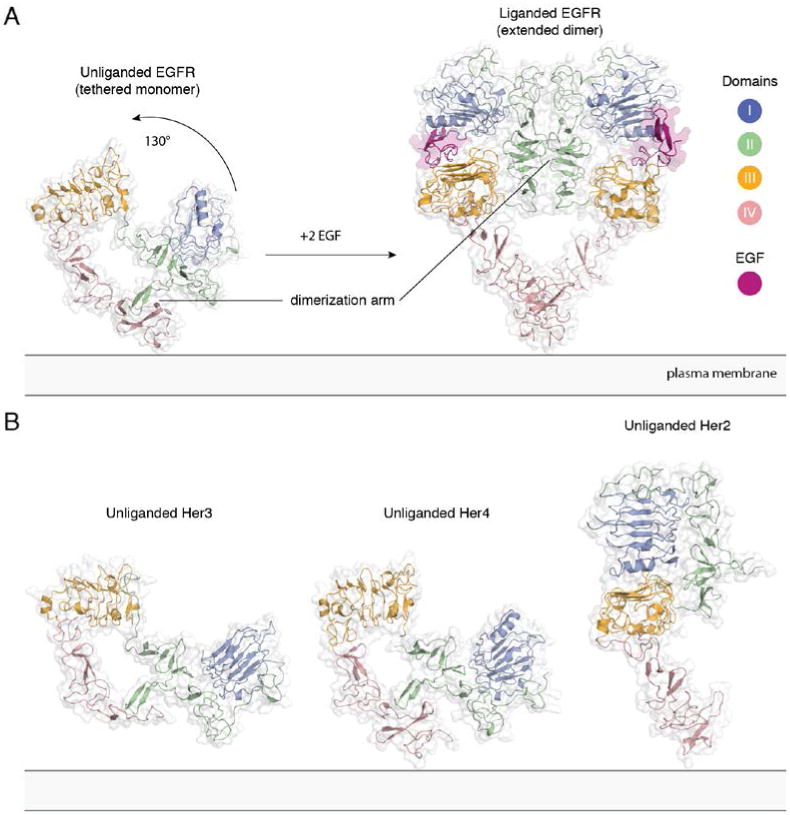
A) The conformational change induced by ligand binding. The tethered conformation of EGFR (left, PDB ID 1NQL, EGF bound at low pH was removed for clarity) rearranges to the extended conformation of EGFR (right, PDB ID 3NJP) upon ligand binding. B) Unliganded Her3 (PDB ID 1M6B) and Her4 (PDB ID 2AHX) can adopt a tethered conformation similar to EGFR, while Her2 (PDB ID 1N8H) is in an extended conformation, even in the absence of ligand.
Knowledge of the ligand-bound structure is based on the crystal structure of a construct of the extracellular module of human EGFR lacking almost the entire domain IV, in complex with TGFα (34), the structure of the entire extracellular module of human EGFR in complex with EGF (35, 36), and also on a crystal structure of the human HER4 extracellular region complexed with its ligand Neuregulin-1β (37). Dimerization of the extracellular module is mediated principally by domain II. A rigid loop is inserted into the cysteine-rich repeats of this domain. This “dimerization arm” interacts with the corresponding element in the dimer partner. The domain II dimerization arm is completely occluded by intramolecular interactions with domain IV in the monomeric tethered conformation. This conformation has been seen in the crystal structure of the isolated extracellular module of human EGFR, which includes an EGF molecule bound with very low affinity as well (38), in a crystal structure containing the entire extracellular region of human HER3 (39), and also in a crystal structure containing the entire extracellular region of human HER4 (40). Ligand binding results in a huge conformational change, a ∼130° rotation of domains I and II with respect to domains III and IV, converting the extracellular module from a bent-over conformation to an extended one, generating a heart-shaped “back-to-back” dimer configuration, in which the ligand is nestled between domains I and III of each subunit of the dimer (Figure 2A).
The structure of the extracellular module of Her2 has also been determined, and it is unique compared to other family members because it adopts the extended conformation without ligand binding. This is seen in the crystal structure of the entire extracellular region of rat Her2 (13), and a truncated construct of human Her2 (41) (Figure 2B). Two key residues in the autoinhibitory domain IV contact region are not conserved (Gly 563 and His 565 of EGFR are replaced with Pro and Phe, respectively, in Her2), which could explain the absence of the autoinhibitory domain II–IV contact in Her2. The extracellular module of Her2 does not homodimerize in solution, perhaps because of subtle conformational differences between the extended extracellular module of Her2 and the extended conformation seen in dimeric EGFR.
The structure of the extracellular module of Her2 resembles that of Drosophila melanogaster EGFR (dEGFR). dEGFR is regulated by growth factor ligands, but a crystal structure shows that it, too, lacks the intramolecular tether seen in human EGFR (42). Instead, a distinct set of autoinhibitory interactions between domains I and III hold unliganded dEGFR in an inactive state, which could provide an alternative means of autoinhibition in the human Her2 extracellular module as well. A structure of a heterodimer including Her2 is not available at present.
The extracellular modules of EGFR family members are heavily glycosylated, containing nearly 40 kDa of sugar moieties (9). Twelve N-glycosylation sites have been identified in EGFR (43-45), and early studies using inhibitors of N-glycosylation have shown that the glycosylation of EGFR is important for its translocation to the cell surface and maturation (46). Several recent studies have investigated specific glycosylation sites in EGFR (47-49). For example, the N420D mutant of EGFR was found to be constitutively phosphorylated, and this ligand-independent activation was due to spontaneous oligomer formation (47). (We use a numbering system that does not count the 24 residue long signal peptide. An alternative numbering scheme has residue numbers increased by 24.) Removal of N-glycosylation at Asn 579 was shown to weaken the tethering interaction between domains II and IV, leading to a more relaxed extracellular module conformation and increased ligand binding affinity (48).
Structures of the extracellular module in complex with therapeutic antibodies
EGFR and HER2 were among the very first receptors to be identified and associated with human tumors, and ever since then, these receptors have been targets for therapeutic intervention (7). Inhibitors of the EGFR family fall into two major classes: monoclonal antibodies that target the extracellular module of the receptor, and small molecules that target the intracellular tyrosine kinase domain. Among the FDA-approved antibodies, Cetuximab and Panitumumab target EGFR, while Pertuzumab and Trastuzumab target Her2.
Several crystal structures of therapeutic antibodies binding to EGFR and HER2 are now available (13, 50, 51) (Figure 3). Cetuximab binds to and blocks the ligand binding site on EGFR domain III, and also sterically prevents EGFR from extending into its active conformation (51). Pertuzumab binds directly to the HER2 dimerization arm and thereby blocks receptor dimerization and activation (50). Trastuzumab binds to domain IV of HER2, proximal to the membrane, but this binding does not block either dimerization or activation of HER2 (13). Instead, Trastuzumab inhibits the proteolytic cleavage of the HER2 extracellular module (52). In the plasma membrane, metalloproteases are known to cleave the extracellular domains of many proteins, including EGFR family members (53, 54). This cleavage leaves behind the transmembrane helix and the tyrosine kinase domain, and such a construct is constitutively active (as discussed later).
Figure 3. Therapeutic antibodies target EGFR and Her2 in versatile ways.
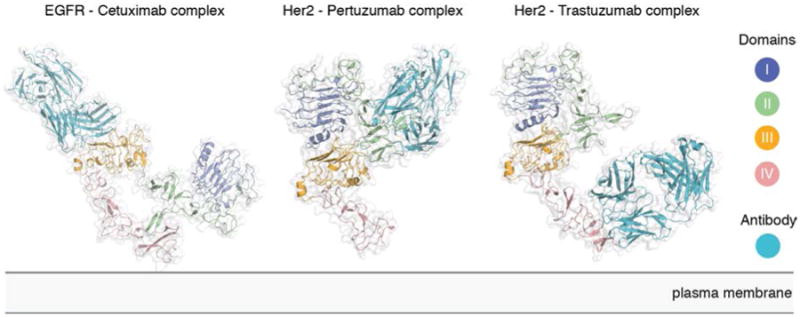
Structure of the EGFR-Cetuximab (PDB ID 1YY9), Her2-Pertuzumab (PDB ID 1S78), and Her2-Trastuzumab (PDB ID 1N8Z) complexes.
An important consideration in antibody therapeutics is whether an immune response will be elicited through recognition of the Fc region of the IgG molecule by the Fc receptor of effector cells to induce cytotoxicity, termed antibody-dependent cell-mediated cytotoxicity (55). Variation in the Fc region of antibodies influences the extent of cytotoxic effector responses, and has been shown to be an important component of trastuzumab action (55).
Nanobodies are small antigen-binding elements from the variable regions of camelid antibodies, which contain only a heavy chain. Nanobodies have been successfully developed to target a smaller concave EGFR epitope, which is inaccessible to the flatter and bigger monoclonal antibodies (56). In addition to a large amount of antibody engineering work on EGFR, alternative methods to block the extracellular modules have also been explored. These include the development of synthetic binding proteins (57) and RNA aptamers (58) that target the extracellular modules. These are valuable tools for investigations into the activation mechanism of these receptors (59).
Kinase activation through the formation of an asymmetric dimer of kinase domains
The first view of the structure of the EGFR kinase domain came from crystallization of the unphosphorylated form with the cancer drug erlotinib, by scientists at Genentech (60). This structure revealed the canonical kinase fold in an active conformation (Figure 4A). Protein kinases contain a conserved Asp-Phe-Gly (DFG) motif at the base of the so-called activation loop or segment, a key regulatory element. The aspartate sidechain is flipped out of the catalytic center in the inactive conformations of many protein kinases (the “DFG out” conformation), preventing it from coordinating ATP, as it must for catalytic activity. In the erlotinib-EGFR complex, the DFG motif is in the “in” conformation, and the activation loop is open and properly configured to bind peptide substrate. Another important structural element of the kinase active site is helix αC, in the N-lobe of the kinase, which packs closely against the rest of the kinase in the active conformation. In the erlotinib complex, helix αC is in the canonical active conformation, rotated inwards towards the ATP binding site. This conformation of the helix presents a conserved glutamate sidechain (Glu 738) for an ion pairing interaction with a conserved lysine sidechain (Lys 721).
Figure 4. Kinase domain structures of the EGFR family members.
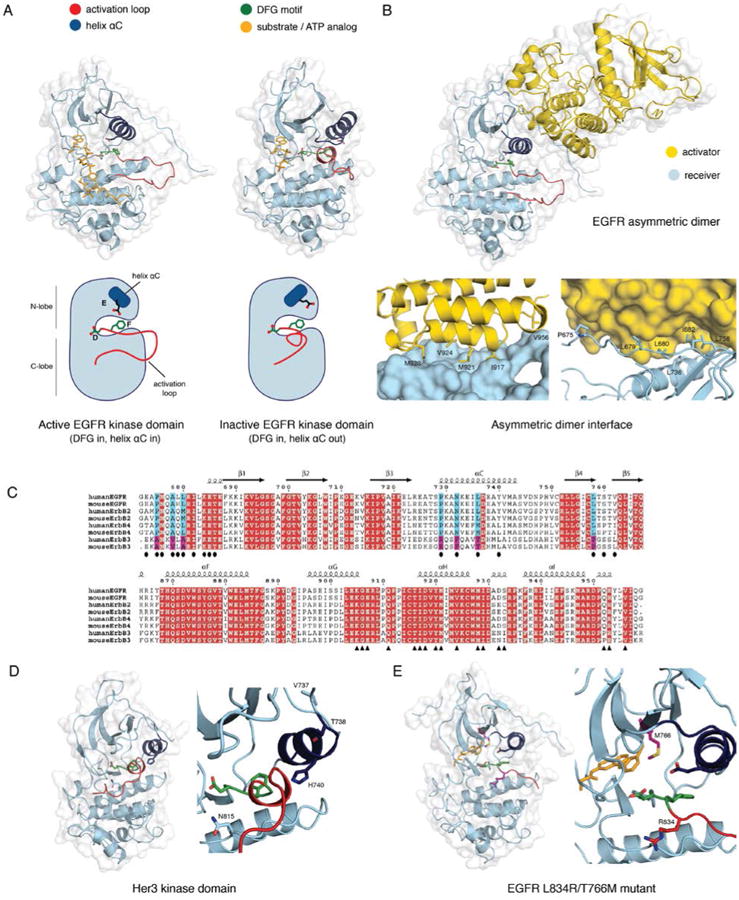
A) The active (PDB ID 2GS6) and the inactive (PDB ID 2GS7) conformation of the EGFR kinase domain. Helix αC is colored blue, the DFG motif green, and the activation loop red. The active structure has an ATP analog - peptide conjugate bound, and the inactive structure has AMP-PNP bound (colored yellow). B) The asymmetric dimer of the EGFR kinase domain (PDB ID 2GS6). The activator kinase is colored yellow, and the receiver (enzymatically active) kinase is colored blue. Residue contacts important on the activator and the receiver are highlighted. C) A sequence alignment of the EGFR family members from human and mouse. Two regions containing the residues involved in the N- and C-lobe faces of the dimer interface are shown in the upper and lower panels, respectively. Identical residues are colored in red. Residues in the N- and C-lobe faces of the dimer interface are denoted by ovals and triangles, respectively. Blue and magenta highlight residues in the dimer interface that are conserved among EGFR, Her2, and Her4 but not in Her3. D) Structure of the Her3 kinase domain (PDB ID 3KEX) with a zoom-in of the active site, and residues resulting in a catalytically impaired kinase are highlighted. The catalytic Asp 813 in EGFR is replaced by Asn 815 in Her3, the critical Glu 738 in helix αC of EGFR is replaced with His 740 in Her3, and Val 737 and Thr 738 in Her3 stabilize the inactive conformation of helix αC. E) A structure of the L834R/T766M double mutant EGFR kinase domain (PDB ID 4LL0) with a zoom-in of the active site. The bound inhibitor, PD168393, is colored yellow, and the mutations are highlighted.
The active conformation of the unphosphorylated EGFR kinase domain was unexpected (60). Studies on the insulin receptor kinase domain had demonstrated a crucial connection between activation loop phosphorylation and the adoption of the active conformation in that kinase (61, 62). In addition, a large body of work on other kinases, particularly Ser/Thr kinases, had led to the assumption that activation loop phosphorylation is required for adoption of the active conformation.
The puzzle regarding the conformation of the EGFR kinase domain deepened when scientists at GlaxoSmithKline determined the structure of the unphosphorylated EGFR kinase domain bound to the cancer drug lapatinib, which revealed a conformation very similar to that of inactive Src-family kinases and Cdk (63). In this conformation helix αC is swung outward from the N-lobe and the activation loop forms a short helix (Figure 4A). Although it is possible that the particular conformation of the kinase domain seen in crystal structures is induced by the drugs, this seems unlikely because of the high affinity of binding. Indeed, erlotinib has been shown to also bind to the inactive conformation of EGFR (64, 65). Mutational studies demonstrated that activation loop phosphorylation is not an absolute requirement for EGFR activation (66). What, then, triggers the switch from the inactive to the active conformation of the EGFR kinase domain?
A breakthrough in our understanding of how EGFR activates came with the discovery that two EGFR kinase domains can interact in an asymmetric fashion so that one, termed the ‘activator’, switches on the other, termed the ‘receiver’, in an allosteric mechanism that does not rely on transphosphorylation (67) (Figure 4B). The clue to the activation mechanism was actually hidden in the original structure of EGFR bound to erlotinib (60), which showed an extensive interface between the N-lobe of one kinase domain and the C-lobe of the other in the crystal lattice. The mechanism by which the kinase domain of EGFR is activated resembles the activation of cyclin-dependent kinase by cyclin. The C-lobe of the activator kinase domain plays the role of the cyclin, even though it is not related structurally to cyclins (68).
A key experiment showed that although the EGFR kinase domain has low activity in solution, its specific activity increases substantially when concentrated on lipid vesicles (67). This activation does not require the tyrosine in the activation loop. Instead, it requires the intact asymmetric dimerization interface, as revealed by mutagenesis of both the full-length EGFR in cells and the kinase domain in vitro. EGFR variants containing single mutations at the asymmetric dimer interface - such as the activator-impaired V924R mutant and the receiver-impaired I682Q mutant - do not respond to EGF binding. However, a combination of the activator-impaired and the receiver-impaired allows the formation of an intact activating interface between kinase domains.
The discovery of the asymmetric dimer provided a conceptual framework for understanding how the degenerate kinase domain of Her3 activates other members of the EGFR family, particularly Her2. Sequence alignments show that residues in Her3 that correspond to the C-lobe (activator) interface are very similar to the residues found at the corresponding location in other members in the family, while residues that correspond to the N-lobe (receiver) interface are divergent in Her3 (Figure 4C). These, and other, features of Her3 suggest that its kinase domain functions as an activator for other EGFR family members. Experimental evidence for an allosteric activation mechanism that is driven by protein-protein interaction rather than by phosphorylation came from a study on Her2/Her3 heterodimers that showed, using mass spectrometry, that conformational changes correlated with activation do not require tyrosine phosphorylation in the activation loop (69).
The structure of the Her4 kinase domain in an active conformation confirmed the importance of the asymmetric dimer (70). The structures of the kinase domains in the activator and receiver positions in the Her4 asymmetric dimer are nearly superimposable on the structure of the asymmetric dimer formed by the kinase domains of EGFR. The structure of the Her2 kinase domain in a homodimeric configuration also revealed the formation of a very similar asymmetric dimer (71).
The potential significance of the asymmetric dimer was anticipated by Groenen et al., who modeled the structure of the EGFR kinase domain based on that of the insulin receptor, and noticed that there are exposed hydrophobic patches in the distal surfaces of both the N- and the C-lobes of the modeled EGFR kinase domain (72). These authors speculated that the two lobes might interact to stabilize the active conformation, which is a remarkable insight considering that the structure of the EGFR kinase domain was determined only later on.
The hydrophobic surface on the N-lobe of the EGFR kinase dimain is a feature that is specific to the active conformation, as the residues that form this surface are sequestered by conformational change to the inactive Src/Cdk conformation. The hydrophobic surface in the C-lobe of the activator, in contrast, is not in a region where conformational changes are observed in kinase domains, and it is likely to remain available as an interaction surface even in the inactive state. This is exploited by the feedback inhibitor Mig6. The crystal structure of a complex between the EGFR kinase domain and a fragment of Mig6 shows that Mig6 binds to the distal surface of the C-lobe of the kinase domain, and inhibition is achieved by blocking the activating dimer interface (73).
The formation of asymmetric dimers by the kinase domains are important for substrate presentation in case of FGF receptors (74), and heterodimerization of kinase domains is a critical step in the activation of RAF kinases (75). These dimers involve completely different interfaces than that seen in EGFR, and sequence comparisons suggest that the activation mechanism described here for the EGFR family is likely to be unique for this family.
Long-timescale molecular dynamics simulations have shown in atomic detail how the kinase domain of EGFR transitions between the active and inactive states (76). The simulations suggest that local unfolding, or “cracking,” at the hinge region between the N- and C-lobe of the kinase, is a necessary step in the transition, leading to a set of conformations in which there is room for a rearrangement of the activation loop. The intermediate conformations revealed by the simulations differ significantly from both the inactive and active compact structures determined crystallographically.
Among the EGFR family members, the kinase domain of Her3 is unique in that it lacks several conserved residues that are critical for kinase activity. The structure of the Her3 kinase domain, reported by Jura et al. (25) and also by Shi et al. (77), shows how sequence changes have resulted in the disruption of the canonical active configuration of kinases (Figure 4D). Key catalytic residues are missing, helix αC is shortened and distorted, and the activation loop is unable to take on the canonical active configuration. Nevertheless, the activator surface is intact, and Jura et al. showed that Her3 can serve as an activator for EGFR (25). They also showed that the Her3 kinase domain has essentially no detectable kinase activity towards peptide substrates.
The ability of the Her3 kinase domain to activate that of Her2 allosterically has been demonstrated by using carboxyl group footprinting mass spectrometry to analyze HER2/HER3 kinase domain heterodimers (69). These experiments demonstrate that HER2 and HER3 kinase domains preferentially form asymmetric heterodimers, with HER3 and HER2 monomers occupying the activator and receiver kinase positions, respectively.
Shi et al. report the remarkable finding that Her3 has a very low, but detectable autophosphorylation rate when concentrated on vesicles (77). The authors used quantum mechanical calculations to delineate a reaction pathway for Her3-catalyzed phosphoryl transfer that does not require the conserved catalytic base and that can be catalyzed by the “inactive-like” conformation observed crystallographically. A modified model for Her3 signaling was proposed based on this low level of Her3 autophosphorylation activity. In this model, Her2 “preactivates” Her3 through transphosphorylation, and as phosphorylated Her3 demonstrates increased kinase activity, the authors argue that activated Her3 monomers can then form functional homodimers (78). Nevertheless, the specific activity of Her3 autophosphorylation is extremely low, and has been estimated to be ∼1000-fold lower than that of EGFR (25). The functional role for Her3 autophosphorylation remains to be established.
Oncogenic mutations in the kinase domain
The clinical importance of EGFR and Her2 in cancer development has resulted in substantial interest in oncogenic mutations in the kinase domains (7). One of the most common of these mutations, the L834R substitution, is located in the activation loop of the kinase domain. The crystal structure of the L834R mutant (79) shows that it is in the active conformation, and its oncogenic property has been attributed to its ability to lock the enzyme in the active conformation. This explains the increased sensitivity of this mutant to tyrosine kinase inhibitors (80) that may prefer the active conformation (60, 79), such as gefitinib and erlotinib.
A more recent study suggests that the oncogenity of L834R is the result of enhancing dimerization, not necessarily just stabilizing an active conformation (81). Using long-timescale molecular dynamics simulations, Shan et al. show that the N-lobe dimerization interface of the kinase domain is intrinsically disordered and only becomes ordered upon dimerization. They demonstrate that the L834R mutation facilitates EGFR dimerization by suppressing this local disorder. Static light scattering, native gel analysis and enzyme assays indicated that this mutation causes abnormally high activity by promoting EGFR dimerization, rather than by just allowing activation without dimerization. Another molecular dynamics study suggests a reconciliation of both views, and proposes that both the change in the relative stability of the active versus the inactive state and the suppression of disorder seem to play a role in the activation of EGFR variants with the L834R mutation (82).
Unfortunately, resistance to EGFR inhibitors often arises due to a second mutation, T766M. Thr 766 is the “gatekeeper” residue in the kinase domain, so called because the nature of the residue at this position is a key determinant of inhibitor specificity. Structural analysis of the T766M variant of EGFR suggests that it can accommodate inhibitors that target the active state (83). This is in agreement with molecular dynamics simulations, which suggest that the methionine interacts with hydrophobic residues surrounding the active site to stabilize a compact, active conformation (82). An unexpected finding is that the T766M mutation in EGFR increases the affinity of the oncogenic L834R variant for ATP by more than an order of magnitude (83). The increased affinity for ATP relative to EGFR inhibitors is likely the primary mechanism by which the T766M mutant confers drug resistance.
Red Brewer et al. demonstrated a so-called “super-acceptor” activity of these mutant kinases, especially the L834R/T766M double mutant (84). EGFR kinase domains are thought to assume the activator or receiver roles in a random manner, but the EGFR mutants found in lung cancer preferentially assume the receiver position in the presence of wild-type EGFR or Her2, leading to hyperphosphorylation of the wild-type activator. The crystal structure of the T766M/L834R double mutant shows that the kinase domain adopts an active conformation, with an intact asymmetric dimer interface (84) (Figure 4E). Red Brewer et al. argue that the double mutant adopts the receiver position because the energetic cost of inducing the active conformation in the L834R/T766M mutant is lower relative to wild-type receptors. Consequently, in a mixed population of wild-type and oncogenic EGFR, the mutants preferentially assume the active, receiver position. Thus, if wild-type receptors are expressed together with mutant EGFR, this will increase the net activity of the mutant, implying a critical role for wild-type EGFRs in tumorigenesis alongside their mutated counterparts.
The juxtamembrane region as an activating segment
The intracellular juxtamembrane segment is known to play crucial regulatory roles in several receptor kinases. Structures of the type I TGFβ receptor kinase domain, a Ser/Thr kinase, showed that the unphosphorylated juxtamembrane segment interacts with helix αC, holding it in an inactive conformation that is further buttressed by FKBP12 (85). This autoinhibition is released by phosphorylation of serine residues within the juxtamembrane segment. This mechanism bears superficial resemblance to that seen in Ephrin receptors, where the juxtamembrane segment also holds the kinase in an inactive conformation by interacting with helix αC (86). This theme is repeated in kinases such as FLT3 (87) and c-Kit (88). Given these findings, it came as a surprise that deletion of the juxtamembrane region results in loss of phosphorylation in the EGFR tail (89), suggesting a crucial role for this segment in activation of the kinase.
The EGFR juxtamembrane segment consists of two major portions: JM-A (residues 645–663) and JM-B (residues 664–682). The role of the JM-B segment was worked out independently and simultaneously by two groups. Red Brewer et al. (90) determined the structure of a construct of EGFR that included the kinase domain and the juxtamembrane segment. Jura et al. (91) reinterpreted crystal lattice contacts in a structure of the Her4 kinase domain that included the JM-B segment (92). In both structures, the JM-B segment forms a clamp or a latch that reaches across from the N-lobe of the receiver kinase domain in an asymmetric dimer to engage the C-lobe of the activator kinase domain (Figure 5A).
Figure 5. Structures of the juxtamembrane latch and the transmembrane helices of EGFR.
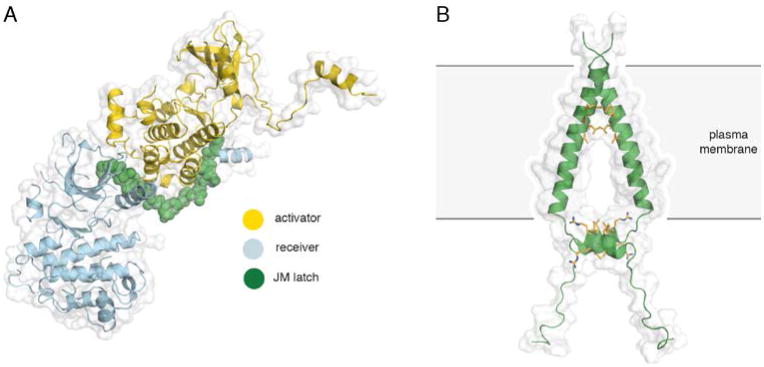
A) The asymmetric dimer of EGFR with the juxtamembrane latch highlighted in green (PDB ID 3GOP). B) An NMR structure of the transmembrane and JM-A helices of EGFR in lipid bicelles (PDB ID 2M20). Interactions between the N-terminal GxxxG-like motifs on the transmembrane segments and the LRRLL motifs of the juxtamembrane segments are highlighted in yellow.
Red Brewer et al. (90) performed alanine-scanning mutagenesis and showed that mutations in the C-terminal 19 residues of the juxtamembrane segment abolish EGFR activation. Phosphorylation of T654 and T669 destabilizes, while the V665M lung cancer mutation stabilizes, the active dimer conformation by reducing and increasing the strength of the juxtamembrane latch interaction, respectively. By using in vitro kinase assays, Jura et al. (91) demonstrated that the juxtamembrane region dimerizes and activates the EGFR kinase, and that it is required on both the activator and the receiver. By designing several mutations in the juxtamembrane segment, based on the Her4 structure, they provide evidence that the JM-B segment latches the activated kinase domain to the activator. They also present the structure of an alternative, symmetric and inactive EGFR kinase dimer, in which the formation of the activating juxtamembrane latch is prevented by the C-terminal tail, suggesting a means to prevent ligand-independent activation.
In the structure of the EGFR kinase domain determined by Red Brewer et al. (90) the JM-A segment packs against another kinase domain, but this crystal contact is unlikely to be maintained in the intact receptor at the plasma membrane. Based on results obtained by NMR, Jura et al. (91) speculated that the JM-A segments of the two kinases instead form an antiparallel helical dimer in the active configuration of the receptor.
Replacement of the juxtamembrane segment in the context of the full-length receptor with an unstructured linker abolishes phosphorylation of EGFR, without measurable effects on receptor dimerization or ligand binding, which supports the critical role of the juxtamembrane region in the formation of the asymmetric dimer (93). Scheck et al. demonstrate the formation of an antiparallel coiled-coil within JM-A by fluorescence spectroscopy (94). They propose that this conformational transition is functionally coupled to receptor activation by EGF, while TGFα binding is communicated to the intracellular domains through formation of an alternative helical interface, suggesting that the juxtamembrane segment may differentially relay the signal initiated by binding of different ligands.
Apart from serving as a latch between asymmetric kinase dimers, it has been suggested that the juxtamembrane interacts with negatively charged lipids in the membrane and also with calmodulin (95), that it mediates feedback signals through threonine phosphorylation (96) and also that it plays a role in sorting and recycling (97). The juxtamembrane segment has also been implicated in negative cooperativity in ligand binding by EGFR, which we discuss later.
Transmembrane coupling
The transmembrane segment of EGFR is a 24 residue long single α-helix. The structure of the transmembrane segment of the Her2 homodimer (98), and later that of the EGFR-Her2 heterodimer (99) in lipid bicelles has been studied by NMR. In both cases, the transmembrane segments associate through N-terminal GxxxG-like motifs. These motifs have been identified as general dimerization motifs for transmembrane helices based on the glycophorin A transmembrane segment (100, 101), and they have been implicated in the self-association of transmembrane helices in EGFR family members (102). The observed dimeric structures of the transmembrane helices in lipid bicelles provide an explanation for the effect of some oncogenic mutations, such as the I655V and the V659E substitutions in the transmembrane helix of Her2. Both residues participate in stabilizing dimerization through the N-terminal GxxxG-like motifs (98, 99). The I655V mutation may stabilize homodimerization through the replacement of a large side chain by a smaller one, and the V659E substitution may do so because of intermolecular hydrogen bonding of the glutamate sidechain.
The structure of the transmembrane and the cytoplasmic juxtamembrane segment of EGFR in lipid bicelles has been determined by NMR, aided by molecular dynamics simulations (103, 104) (Figure 5B). This structure explained how the configuration of the transmembrane helices in the EGFR dimer can couple to the conformation of the juxtamembrane segments, and how these conformations are compatible with the asymmetric kinase dimer. The structure of the transmembrane segment reveals a helical dimer consistent with the Her2 structures of Bocharov et al. (98) and Mineev et al. (99). The C-terminal ends of the transmembrane helices are separated by ∼20 Å, which provides the appropriate spacing for an antiparallel interaction between the JM-A helices. This interface is formed by LRRLL motifs in the juxtamembrane segment. Mutating all four of the small residues in the N-terminal dimerization interface in the transmembrane helix to isoleucine (4I, T624I/G625I/G628I/A629I) results in significant inhibition of EGFR in cellular assays, providing experimental evidence for the importance of the N-terminal association between transmembrane helices in receptor activation (103).
The transmembrane helices of EGFR and Her2 contain a second dimerization motif towards their C-terminal ends. Fleishman et al. have speculated that the presence of both N-terminal and C-terminal dimerization motifs generates a transmembrane switch, in which the dimer of transmembrane helices toggles between two configurations, with the N-terminal (extracellular) ends close together in one, and the C-terminal (intracellular) ends close together in the other (105). The authors propose that these configurations correspond to the active and inactive states of the receptor, respectively. Using NMR and molecular dynamics simulations, Endres et al. and Arkhipov et al. show that stabilizing interactions through the C-terminal dimerization motif, by an I640E mutation, leads to the disruption of the N- terminal interface between the transmembrane helices and also disruption of the antiparallel JM-A interaction (103, 104). Using cell-based assays, they demonstrate that the I640E mutation in the intact receptor results in impairment of EGF-dependent activation at low surface densities. The two conserved GxxxG-like motifs have also been suggested, by others, to play a role in mediating and/or stabilizing homo- and heterodimers (106, 107).
According to the model proposed by Endres et al. and Arkhipov et al. (103, 104), the extracellular modules in ligand-bound dimers assume a configuration favoring dimerization of the transmembrane helices near their N terminal ends, dimerization of the juxtamembrane segments, and formation of asymmetric kinase dimers. In ligand-free dimers, by holding apart the N terminal ends of the transmembrane helices, the extracellular modules instead favor C-terminal dimerization of the transmembrane helices, juxtamembrane segment dissociation and membrane burial, and formation of symmetric, inactive kinase dimers. Our current understanding of what the active state of the full-length receptor might look like is depicted in Figure 7A.
Figure 7. Full-length EGFR and its oligomerization states.
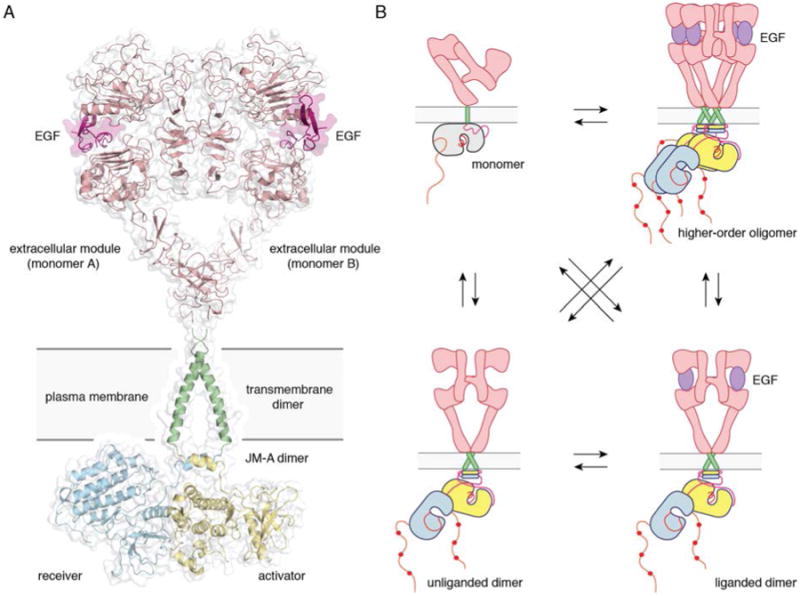
A) A proposed composite model of full-length EGFR based on the structures of individual modules (PDB ID 3NJP for the extracellular module, PDB ID 2M20 for the transmembrane - JM-A helices, and PDB ID 2GS6 for the kinase domains). B) Schematics for possible oligomerization states of EGFR in cells.
Analysis of the monomer and dimer forms of EGFR by chemical and disulfide cross-linking suggested that EGFR has a preformed dimeric structure without bound ligand, and that ligand binding induces rotation of the extracellular juxtamembrane region, hence the transmembrane segment, which reorients the cytoplasmic module and results in activation of the kinase domains (108). Using an elegant protein engineering approach Bell et al. demonstrate similar rotational coupling in Her2 (109). They designed a series of transmembrane helix mutants that sequentially move two Glu residues, within a simplified transmembrane segment, across the entire transmembrane region. The movement of this dimerization motif is expected to rotate the kinase domains ∼103° per residue. Rotation of this interface does not affect dimerization, but leads to a periodic oscillation in kinase activation.
Lu et al. have argued for a more passive role played by the transmembrane helix than is indicated by the preceding discussion (36). Analysis of disulfide cross-linking experiments indicate that EGF-induced dimerization of the transmembrane helices involves an interface less extensive than that found in glycophorin A and integrin, two receptors that dimerize in the absence of activation. Systematic mutagenesis of residues in the transmembrane helix of EGFR to leucine and phenylalanine shows that no single mutation disrupts transmembrane signaling. However, as shown by Das et al. (103), multiple mutations are required to disrupt this interface, which supports the model of active transmembrane coupling.
Negative cooperativity in ligand binding to EGFR
A puzzling aspect of the binding of EGF to EGFR in cellular assays is that the Scatchard plot is not linear, as would be expected for independent binding sites on the receptor with no cooperativity. Instead, the Scatchard plot is curved in a concave-up manner. This feature of the binding isotherm was interpreted in terms of two populations of EGFR in cells (110). This interpretation suggests that a small population of the receptors (∼10%) have very high affinity for the ligand (Kd ∼50 pM), while the majority of receptors have much lower affinity (Kd ∼3 nM). It was proposed that EGF binds with high affinity to the dimeric form of the receptor, but with low affinity to the monomeric form (28). Another interpretation is that spatial segregation of populations of the receptor underlies the heterogeneity of binding (111, 112).
The thinking in the field has now shifted to considering the concave-up Scatchard plot as being due instead to negative cooperativity in dimeric receptors. Pike and colleagues have made extensive measurements of EGF binding to EGFR-bearing cells. They generated stable cell lines expressing EGFR under an inducible promoter, which allowed control of the level of EGFR expression. Using this system, they measured the binding of radiolabeled EGF to these cells as a function of EGFR expression level. For the wild-type receptor, the binding isotherms shift from left to right with increasing EGFR density, that is, the apparent binding becomes weaker at higher EGFR densities on the cell surface (113). Modeling of the data suggests that the affinity of EGF for the second site on the EGFR dimer (2.9 nM) is substantially less than the affinity for the first site (190 pM), i.e. the dimer exhibits negative cooperativity.
Pike and colleagues also obtained information on the structural requirements of negative cooperativity by performing binding analyses on cells expressing increasing levels of various mutant forms of EGFR. These findings link the extracellular juxtamembrane region, and particularly the region responsible for the tethering interaction in domain IV (residues 561-585), to negative cooperativity (114). The intracellular juxtamembrane portion of the receptor was also shown to be involved in the generation of negative cooperativity, since deletion of the kinase domain as well as the C-terminal tail yielded a receptor with negative cooperativity, whereas deletion of the entire intracellular module led to a receptor that showed no cooperativity (115).
By performing a more detailed analysis of various intracellular juxtamembrane mutants, Pike and colleagues have linked the stepwise binding of two ligands to kinase activation (116). In this model, the binding of EGF to the first site on the dimer induces the formation of one asymmetric kinase dimer, then the binding of EGF to the second site is required to disrupt the initial asymmetric dimer and allow the formation of the reciprocal asymmetric dimer. Thus, some of the energy of binding to the second site is used to reorient the first asymmetric dimer, leading to a lower binding affinity and the observed negative cooperativity. Implications of their model for negative cooperativity have been studied using various assays (117-120).
One of the difficulties of explaining negative cooperativity in human EGFR is that the isolated extracellular module does not show negative cooperativity in vitro. In contrast, the isolated extracellular module of the Drosophila receptor (dEGFR) exhibits negative cooperativity in binding to its ligand, Spitz, when the binding is analyzed in vitro using purified proteins. A breakthrough in understanding occurred with the structural and biophysical analysis of dEGFR by Lemmon and co-workers. The crystal structure of a singly-liganded asymmetric extracellular dimer of dEGFR reveals the structural basis for negative cooperativity in that receptor (121) (Figure 6).
Figure 6. Structural basis for negative cooperativity in ligand binding to Drosophila. EGFR.
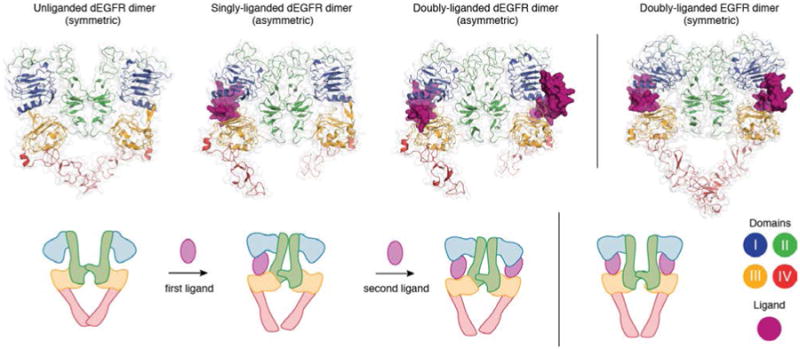
The unliganded (PDB ID 3I2T), singly-liganded (PDB ID 3LTG) and doubly-liganded (PDB ID 3LTF) dEGFR dimer. The structure of the doubly liganded human EGFR dimer (PDB ID 3NJP) is shown on the right.
Before interacting with Spitz, the ligand-binding sites are identical in pre-formed dimers of dEGFR. The first (highest-affinity) binding event yields the singly ligated, asymmetric dimer, because Spitz “wedges” itself between domains I and III in one subunit of the dimer, and pushes them apart. This distorts domain II and forces a substantial reorientation of the dimerization arm, and allows formation of a more extensive, asymmetric dimer interface. In this asymmetric dEGFR dimer, domain II in the unoccupied receptor is structurally restrained, and can no longer bend to allow Spitz to wedge itself fully into the unoccupied ligand-binding site without disrupting the extensive asymmetric interface. This, binding of Spitz to the second site will be weaker, leading to negative cooperativity.
Although structures of human EGFR with only one ligand bound have not been determined, Tynan et al. suggest that in humans, such asymmetry is brought forth by interactions with the plasma membrane (122). FRET imaging reveals that a high-affinity ligand-binding human EGFR conformation is consistent with the extracellular region aligned flat on the plasma membrane, in such a way that the second binding site is occluded. Molecular dynamics simulations also suggest that asymmetrical interactions with the membrane may be responsible for this negative cooperativity (123). Such simulations further indicate that heterodimers formed by extracellular modules of Her2 and other EGFR family members assume an asymmetric conformation similar to that of Drosophila EGFR dimers (124).
Studies on the full-length receptor
Two groups have succeeded in purifying nearly full-length recombinant EGFR (lacking most of the C-terminal tail) in detergent micelles (125, 126). This approach allows the biochemical characterization of ligand-induced activation of the intact receptor, previously only feasible by indirect methods in cells. Wang et al. studied the two most common non-small cell lung cancer mutations in EGFR, L834R and a deletion mutant, Δ722–726, and they found both to be at least as active as the EGF-bound receptor, and that activity was still strongly coupled to asymmetric kinase dimer formation (127). Previous studies of the isolated kinase domains had indicated that the L834R mutation was sufficient to promote full constitutive activity, and the authors claim that such discrepancies underscore the limitations of attempting to understand the regulation of this complex receptor by studying isolated fragments.
Negative-stain electron microscopy has been used to visualize the coupling between the extracellular and the intracellular modules of EGFR at low resolution (128, 129). The unliganded receptor adopts a monomeric and tethered extracellular module conformation. Intriguingly, the EGF-bound receptor adopts two distinct dimer conformations: a rod-like kinase domain structure consistent with the asymmetric dimer and a more globular intracellular region consistent with a more symmetric association. These two types of dimers coexist with a monomeric form. Inhibitors that stabilize the active or inactive conformation of the kinase active site, as well as various mutations, have been shown to shift the equilibrium among the three observed forms. The coupling of one conformation of an activated receptor extracellular module to multiple kinase-domain arrangements suggests that the linkage between the extra- and intracellular regions of EGFR is unexpectedly flexible. It is important to note that these experiments have been carried out on detergent solubilized EGFR, which could substantially alter the behavior of the receptor when compared to that in lipid bilayers.
The oligomeric state of the receptor: ligand-independent dimers and higher order oligomers
A large number of studies suggest that the classical model of unliganded monomer to EGF-bound dimer transition might not be so simple (Figure 7B). Several groups have been addressing the question of what the resting state of the receptor is, whether it is predominantly a monomer or rather an unliganded dimer (108, 130-136). Very briefly, the main conclusion arising from these reports is that in the basal state EGFR exists predominantly as monomer, while it is in an equilibrium with ligand-independent dimers. Clustering before ligand binding is rare for EGFR, but might be a characteristic of Her2. The fraction of EGFR that dimerizes before ligand binding depends primarily on receptor surface densities and the cell types used.
In a related question regarding the oligomeric state of EGFR, Burgess and colleagues argue that a dimer-tetramer transition is required for receptor activation (137-139). Their data are consistent with a significant fraction of liganded EGFR tetramers, and they propose that such tetramers comprise two dimers juxtaposed in a side-by-side (or slightly staggered) arrangement (137), and suggest that tetrameric EGFR is the main signaling unit (138, 139).
Concluding remarks
Structural analysis of the extracellular and the intracellular modules of EGFR have provided invaluable insight into how this receptor, central in cancer development, functions. Crystallography combined with biochemical, biophysical and cell biology experiments have revealed the detailed mechanism of how ligand binding leads to conformational change, and how the extracellular module dimerizes through an interface completely mediated by the receptor itself. These approaches demonstrated how the EGFR kinase domains activate through an allosteric mechanism involving asymmetric kinase dimers. NMR provided insight into transmembrane-juxtamembrane coupling, and we now understand the structural basis for negative cooperativity in ligand binding in Drosophila EGFR.
Despite all these advances, we are still lacking a complete understanding of how the full-length receptor functions. This is mainly due to the technical difficulty of crystallizing single-pass transmembrane proteins, of which few, if any, structures are currently available. New avenues in membrane protein crystallography and electron microscopy, as well as long-timescale molecular dynamics simulations are promising directions that might provide a breakthrough in the field. There are also several mechanistic questions that cannot be tackled from a purely structural perspective, and for which the integration of sophisticated cell biology approaches will be necessary. These include understanding the oligomerization state of the receptor, both in the resting state and as the active signaling unit, understanding the precise mechanism of tail phosphorylation, and whether the asymmetry present at the kinase level is translated into differential phosphorylation of the activator and the receiver tail. We believe that these efforts will ultimately enable us to fully understand the complex regulation of this receptor family, knowledge that is essential for improving cancer therapeutics directed against this class of receptors.
Contributor Information
Erika Kovacs, Email: kovacs@berkeley.edu.
Julie Anne Zorn, Email: jazorn@berkeley.edu.
Yongjian Huang, Email: coomean@gmail.com.
Tiago Barros, Email: tiago@berkeley.edu.
References
- 1.Ruiz-Trillo I, Burger G, Holland PW, King N, Lang BF, et al. The origins of multicellularity: a multi-taxon genome initiative. Trends in genetics : TIG. 2007;23:113–8. doi: 10.1016/j.tig.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Lim WA, Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010;142:661–7. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annual review of biochemistry. 2000;69:373–98. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 4.Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–17. doi: 10.1038/nrm3048. [DOI] [PubMed] [Google Scholar]
- 5.Endres NF, Engel K, Das R, Kovacs E, Kuriyan J. Regulation of the catalytic activity of the EGF receptor. Current opinion in structural biology. 2011;21:777–84. doi: 10.1016/j.sbi.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor perspectives in biology. 2014;6:a020768. doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer cell. 2014;25:282–303. doi: 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Downward J, Yarden Y, Mayes E, Scrace G, Totty N, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521–7. doi: 10.1038/307521a0. [DOI] [PubMed] [Google Scholar]
- 9.Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature. 1984;309:418–25. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 10.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nature reviews Cancer. 2007;7:169–81. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 11.Westphal M, Meima L, Szonyi E, Lofgren J, Meissner H, et al. Heregulins and the ErbB-2/3/4 receptors in gliomas. Journal of neuroGoncology. 1997;35:335–46. doi: 10.1023/a:1005837122181. [DOI] [PubMed] [Google Scholar]
- 12.Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19:6102–14. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 13.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–60. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 14.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 16.Richter DJ, King N. The genomic and cellular foundations of animal origins. Annual review of genetics. 2013;47:509–37. doi: 10.1146/annurev-genet-111212-133456. [DOI] [PubMed] [Google Scholar]
- 17.Nichols SA, Dirks W, Pearse JS, King N. Early evolution of animal cell signaling and adhesion genes. Proc Natl Acad Sci U S A. 2006;103:12451–6. doi: 10.1073/pnas.0604065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokoyama N, Miller WT. Biochemical properties of the Cdc42-associated tyrosine kinase ACK1. Substrate specificity, authphosphorylation, and interaction with Hck. J Biol Chem. 2003;278:47713–23. doi: 10.1074/jbc.M306716200. [DOI] [PubMed] [Google Scholar]
- 19.Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-beta receptor function. Trends in cell biology. 2009;19:385–94. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–96. [PubMed] [Google Scholar]
- 22.Stein RA, Staros JV. Insights into the evolution of the ErbB receptor family and their ligands from sequence analysis. BMC evolutionary biology. 2006;6:79. doi: 10.1186/1471-2148-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–5. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 24.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 25.Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A. 2009;106:21608–13. doi: 10.1073/pnas.0912101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger MB, Mendrola JM, Lemmon MA. ErbB3/HER3 does not homodimerize upon neuregulin binding at the cell surface. FEBS letters. 2004;569:332–6. doi: 10.1016/j.febslet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 27.Pines G, Huang PH, Zwang Y, White FM, Yarden Y. EGFRvIV: a previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene. 2010;29:5850–60. doi: 10.1038/onc.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yarden Y, Schlessinger J. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987;26:1434–42. doi: 10.1021/bi00379a034. [DOI] [PubMed] [Google Scholar]
- 29.Yarden Y, Schlessinger J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry. 1987;26:1443–51. doi: 10.1021/bi00379a035. [DOI] [PubMed] [Google Scholar]
- 30.Honegger AM, Kris RM, Ullrich A, Schlessinger J. Evidence that autophosphorylation of solubilized receptors for epidermal growth factor is mediated by intermolecular cross-phosphorylation. Proc Natl Acad Sci U S A. 1989;86:925–9. doi: 10.1073/pnas.86.3.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vecchi M, Baulida J, Carpenter G. Selective cleavage of the heregulin receptor ErbB-4 by protein kinase C activation. J Biol Chem. 1996;271:18989–95. doi: 10.1074/jbc.271.31.18989. [DOI] [PubMed] [Google Scholar]
- 32.Cheng QC, Tikhomirov O, Zhou W, Carpenter G. Ectodomain cleavage of ErbB-4: characterization of the cleavage site and m80 fragment. J Biol Chem. 2003;278:38421–7. doi: 10.1074/jbc.M302111200. [DOI] [PubMed] [Google Scholar]
- 33.Leahy DJ. Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Advances in protein chemistry. 2004;68:1–27. doi: 10.1016/S0065-3233(04)68001-6. [DOI] [PubMed] [Google Scholar]
- 34.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell. 2002;110:763–73. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 35.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–87. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 36.Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, et al. Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol Cell Biol. 2010;30:5432–43. doi: 10.1128/MCB.00742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu P, Cleveland TEt, Bouyain S, Byrne PO, Longo PA, Leahy DJ. A single ligand is sufficient to activate EGFR dimers. Proc Natl Acad Sci U S A. 2012;109:10861–6. doi: 10.1073/pnas.1201114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–17. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 39.Cho HS, Leahy DJ. Structure of the extracellular region of HER3 reveals an interdomain tether. Science. 2002;297:1330–3. doi: 10.1126/science.1074611. [DOI] [PubMed] [Google Scholar]
- 40.Bouyain S, Longo PA, Li S, Ferguson KM, Leahy DJ. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci U S A. 2005;102:15024–9. doi: 10.1073/pnas.0507591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 42.Alvarado D, Klein DE, Lemmon MA. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature. 2009;461:287–91. doi: 10.1038/nature08297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith KD, Davies MJ, Bailey D, Renouf DV, Hounsell EF. Analysis of the glycosylation patterns of the extracellular domain of the epidermal growth factor receptor expressed in Chinese hamster ovary fibroblasts. Growth Factors. 1996;13:121–32. doi: 10.3109/08977199609034572. [DOI] [PubMed] [Google Scholar]
- 44.Sato C, Kim JH, Abe Y, Saito K, Yokoyama S, Kohda D. Characterization of the N-oligosaccharides attached to the atypical Asn-X-Cys sequence of recombinant human epidermal growth factor receptor. Journal of biochemistry. 2000;127:65–72. doi: 10.1093/oxfordjournals.jbchem.a022585. [DOI] [PubMed] [Google Scholar]
- 45.Zhen Y, Caprioli RM, Staros JV. Characterization of glycosylation sites of the epidermal growth factor receptor. Biochemistry. 2003;42:5478–92. doi: 10.1021/bi027101p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gamou S, Shimizu N. Glycosylation of the epidermal growth factor receptor and its relationship to membrane transport and ligand binding. Journal of biochemistry. 1988;104:388–96. doi: 10.1093/oxfordjournals.jbchem.a122478. [DOI] [PubMed] [Google Scholar]
- 47.Tsuda T, Ikeda Y, Taniguchi N. The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J Biol Chem. 2000;275:21988–94. doi: 10.1074/jbc.M003400200. [DOI] [PubMed] [Google Scholar]
- 48.Whitson KB, Whitson SR, Red-Brewer ML, McCoy AJ, Vitali AA, et al. Functional effects of glycosylation at Asn-579 of the epidermal growth factor receptor. Biochemistry. 2005;44:14920–31. doi: 10.1021/bi050751j. [DOI] [PubMed] [Google Scholar]
- 49.Fernandes H, Cohen S, Bishayee S. Glycosylation-induced conformational modification positively regulates receptor-receptor association: a study with an aberrant epidermal growth factor receptor (EGFRvIII/DeltaEGFR) expressed in cancer cells. J Biol Chem. 2001;276:5375–83. doi: 10.1074/jbc.M005599200. [DOI] [PubMed] [Google Scholar]
- 50.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer cell. 2004;5:317–28. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 51.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Baselga J, Albanell J, Molina MA, Arribas J. Mechanism of action of trastuzumab and scientific update. Seminars in oncology. 2001;28:4–11. doi: 10.1016/s0093-7754(01)90276-3. [DOI] [PubMed] [Google Scholar]
- 53.Vecchi M, Carpenter G. Constitutive proteolysis of the ErbB-4 receptor tyrosine kinase by a unique, sequential mechanism. J Cell Biol. 1997;139:995–1003. doi: 10.1083/jcb.139.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Codony-Servat J, Albanell J, Lopez-Talavera JC, Arribas J, Baselga J. Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Res. 1999;59:1196–201. [PubMed] [Google Scholar]
- 55.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature medicine. 2000;6:443–6. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 56.Schmitz KR, Bagchi A, Roovers RC, van Bergen en Henegouwen PM, Ferguson KM. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013;21:1214–24. doi: 10.1016/j.str.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hackel BJ, Neil JR, White FM, Wittrup KD. Epidermal growth factor receptor downregulation by small heterodimeric binding proteins. Protein engineering, design & selection : PEDS. 2012;25:47–57. doi: 10.1093/protein/gzr056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen CH, Chernis GA, Hoang VQ, Landgraf R. Inhibition of heregulin signaling by an aptamer that preferentially binds to the oligomeric form of human epidermal growth factor receptor-3. Proc Natl Acad Sci U S A. 2003;100:9226–31. doi: 10.1073/pnas.1332660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Q, Park E, Kani K, Landgraf R. Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc Natl Acad Sci U S A. 2012;109:13237–42. doi: 10.1073/pnas.1200105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–72. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 61.Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. The EMBO journal. 1997;16:5572–81. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hubbard SR, Wei L, Ellis L, Hendrickson WA. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–54. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 63.Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–9. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- 64.Gajiwala KS, Feng J, Ferre R, Ryan K, Brodsky O, et al. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure. 2013;21:209–19. doi: 10.1016/j.str.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 65.Park JH, Liu Y, Lemmon MA, Radhakrishnan R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. The Biochemical journal. 2012;448:417–23. doi: 10.1042/BJ20121513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gotoh N, Tojo A, Hino M, Yazaki Y, Shibuya M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem Biophys Res Commun. 1992;186:768–74. doi: 10.1016/0006-291x(92)90812-y. [DOI] [PubMed] [Google Scholar]
- 67.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–49. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 68.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Collier TS, Diraviyam K, Monsey J, Shen W, Sept D, Bose R. Carboxyl group footprinting mass spectrometry and molecular dynamics identify key interactions in the HER2-HER3 receptor tyrosine kinase interface. J Biol Chem. 2013;288:25254–64. doi: 10.1074/jbc.M113.474882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16:460–7. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286:18756–65. doi: 10.1074/jbc.M110.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Groenen LC, Walker F, Burgess AW, Treutlein HR. A model for the activation of the epidermal growth factor receptor kinase involvement of an asymmetric dimer? Biochemistry. 1997;36:3826–36. doi: 10.1021/bi9614141. [DOI] [PubMed] [Google Scholar]
- 73.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–4. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bae JH, Boggon TJ, Tome F, Mandiyan V, Lax I, Schlessinger J. Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc Natl Acad Sci U S A. 2010;107:2866–71. doi: 10.1073/pnas.0914157107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu J, Stites EC, Yu H, Germino EA, Meharena HS, et al. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–46. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shan Y, Arkhipov A, Kim ET, Pan AC, Shaw DE. Transitions to catalytically inactive conformations in EGFR kinase. Proc Natl Acad Sci U S A. 2013;110:7270–5. doi: 10.1073/pnas.1220843110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692–7. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Steinkamp MP, Low-Nam ST, Yang S, Lidke KA, Lidke DS, Wilson BS. erbB3 is an active tyrosine kinase capable of homo- and heterointeractions. Mol Cell Biol. 2014;34:965–77. doi: 10.1128/MCB.01605-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer cell. 2007;11:217–27. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–71. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- 81.Shan Y, Eastwood MP, Zhang X, Kim ET, Arkhipov A, et al. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149:860–70. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 82.Sutto L, Gervasio FL. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc Natl Acad Sci U S A. 2013;110:10616–21. doi: 10.1073/pnas.1221953110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Red Brewer M, Yun CH, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci U S A. 2013;110:E3595–604. doi: 10.1073/pnas.1220050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–36. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 86.Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell. 2001;106:745–57. doi: 10.1016/s0092-8674(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 87.Griffith J, Black J, Faerman C, Swenson L, Wynn M, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–78. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 88.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–63. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]
- 89.Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc Natl Acad Sci U S A. 2007;104:19238–43. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, et al. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell. 2009;34:641–51. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jura N, Endres NF, Engel K, Deindl S, Das R, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wood ER, Shewchuk LM, Ellis B, Brignola P, Brashear RL, et al. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc Natl Acad Sci U S A. 2008;105:2773–8. doi: 10.1073/pnas.0708281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.He L, Hristova K. Consequences of replacing EGFR juxtamembrane domain with an unstructured sequence. Sci Rep. 2012;2:854. doi: 10.1038/srep00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scheck RA, Lowder MA, Appelbaum JS, Schepartz A. Bipartite tetracysteine display reveals allosteric control of ligand-specific EGFR activation. ACS Chem Biol. 2012;7:1367–76. doi: 10.1021/cb300216f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McLaughlin S, Smith SO, Hayman MJ, Murray D. An electrostatic engine model for autoinhibition and activation of the epidermal growth factor receptor (EGFR/ErbB) family. J Gen Physiol. 2005;126:41–53. doi: 10.1085/jgp.200509274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hunter T, Ling N, Cooper JA. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature. 1984;311:480–3. doi: 10.1038/311480a0. [DOI] [PubMed] [Google Scholar]
- 97.Hobert ME, Kil SJ, Medof ME, Carlin CR. The cytoplasmic juxtamembrane domain of the epidermal growth factor receptor contains a novel autonomous basolateral sorting determinant. J Biol Chem. 1997;272:32901–9. doi: 10.1074/jbc.272.52.32901. [DOI] [PubMed] [Google Scholar]
- 98.Bocharov EV, Mineev KS, Volynsky PE, Ermolyuk YS, Tkach EN, et al. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem. 2008;283:6950–6. doi: 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]
- 99.Mineev KS, Bocharov EV, Pustovalova YE, Bocharova OV, Chupin VV, Arseniev AS. Spatial structure of the transmembrane domain heterodimer of ErbB1 and ErbB2 receptor tyrosine kinases. J Mol Biol. 2010;400:231–43. doi: 10.1016/j.jmb.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 100.Lemmon MA, Treutlein HR, Adams PD, Brunger AT, Engelman DM. A dimerization motif for transmembrane alpha-helices. Nat Struct Biol. 1994;1:157–63. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 101.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–9. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 102.Mendrola JM, Berger MB, King MC, Lemmon MA. The single transmembrane domains of ErbB receptors self-associate in cell membranes. J Biol Chem. 2002;277:4704–12. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 103.Endres NF, Das R, Smith AW, Arkhipov A, Kovacs E, et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell. 2013;152:543–56. doi: 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arkhipov A, Shan Y, Das R, Endres NF, Eastwood MP, et al. Architecture and membrane interactions of the EGF receptor. Cell. 2013;152:557–69. doi: 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fleishman SJ, Schlessinger J, Ben-Tal N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc Natl Acad Sci U S A. 2002;99:15937–40. doi: 10.1073/pnas.252640799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gerber D, Sal-Man N, Shai Y. Two motifs within a transmembrane domain, one for homodimerization and the other for heterodimerization. J Biol Chem. 2004;279:21177–82. doi: 10.1074/jbc.M400847200. [DOI] [PubMed] [Google Scholar]
- 107.Escher C, Cymer F, Schneider D. Two GxxxG-like motifs facilitate promiscuous interactions of the human ErbB transmembrane domains. J Mol Biol. 2009;389:10–6. doi: 10.1016/j.jmb.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 108.Moriki T, Maruyama H, Maruyama IN. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J Mol Biol. 2001;311:1011–26. doi: 10.1006/jmbi.2001.4923. [DOI] [PubMed] [Google Scholar]
- 109.Bell CA, Tynan JA, Hart KC, Meyer AN, Robertson SC, Donoghue DJ. Rotational coupling of the transmembrane and kinase domains of the Neu receptor tyrosine kinase. Mol Biol Cell. 2000;11:3589–99. doi: 10.1091/mbc.11.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.King AC, Cuatrecasas P. Resolution of high and low affinity epidermal growth factor receptors. Inhibition of high affinity component by low temperature, cycloheximide, and phorbol esters. J Biol Chem. 1982;257:3053–60. [PubMed] [Google Scholar]
- 111.Mayawala K, Vlachos DG, Edwards JS. Heterogeneities in EGF receptor density at the cell surface can lead to concave up scatchard plot of EGF binding. FEBS Lett. 2005;579:3043–7. doi: 10.1016/j.febslet.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 112.den Hartigh JC, van Bergen en Henegouwen PM, Verkleij AJ, Boonstra J. The EGF receptoris an actin-binding protein. J Cell Biol. 1992;119:349–55. doi: 10.1083/jcb.119.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Macdonald JL, Pike LJ. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc Natl Acad Sci U S A. 2008;105:112–7. doi: 10.1073/pnas.0707080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adak S, DeAndrade D, Pike LJ. The tethering arm of the EGF receptor is required for negative cooperativity and signal transduction. J Biol Chem. 2011;286:1545–55. doi: 10.1074/jbc.M110.182899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Macdonald-Obermann JL, Pike LJ. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J Biol Chem. 2009;284:13570–6. doi: 10.1074/jbc.M109.001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Adak S, Yang KS, Macdonald-Obermann J, Pike LJ. The membrane-proximal intracellular domain of the epidermal growth factor receptor underlies negative cooperativity in ligand binding. J Biol Chem. 2011;286:45146–55. doi: 10.1074/jbc.M111.274175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang KS, Ilagan MX, Piwnica-Worms D, Pike LJ. Luciferase fragment complementation imaging of conformational changes in the epidermal growth factor receptor. J Biol Chem. 2009;284:7474–82. doi: 10.1074/jbc.M808041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Macdonald-Obermann JL, Piwnica-Worms D, Pike LJ. Mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc Natl Acad Sci U S A. 2012;109:137–42. doi: 10.1073/pnas.1111316109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li Y, Macdonald-Obermann J, Westfall C, Piwnica-Worms D, Pike LJ. Quantitation of the effect of ErbB2 on epidermal growth factor receptor binding and dimerization. J Biol Chem. 2012;287:31116–25. doi: 10.1074/jbc.M112.373647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Macdonald-Obermann JL, Pike LJ. Different EGF Receptor Ligands Show Distinct Kinetics and Biased or Partial Agonism for Homodimer and Heterodimer Formation. J Biol Chem. 2014 doi: 10.1074/jbc.M114.586826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alvarado D, Klein DE, Lemmon MA. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell. 2010;142:568–79. doi: 10.1016/j.cell.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tynan CJ, Roberts SK, Rolfe DJ, Clarke DT, Loeffler HH, et al. Human epidermal growth factor receptor (EGFR) aligned on the plasma membrane adopts key features of Drosophila EGFR asymmetry. Mol Cell Biol. 2011;31:2241–52. doi: 10.1128/MCB.01431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Arkhipov A, Shan Y, Kim ET, Shaw DE. Membrane interaction of bound ligands contributes to the negative binding cooperativity of the EGF receptor. PLoS computational biology. 2014;10:e1003742. doi: 10.1371/journal.pcbi.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arkhipov A, Shan Y, Kim ET, Dror RO, Shaw DE. Her2 activation mechanism reflects evolutionary preservation of asymmetric ectodomain dimers in the human EGFR family. Elife. 2013;2:e00708. doi: 10.7554/eLife.00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qiu C, Tarrant MK, Boronina T, Longo PA, Kavran JM, et al. In vitro enzymatic characterization of near full length EGFR in activated and inhibited states. Biochemistry. 2009;48:6624–32. doi: 10.1021/bi900755n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mi LZ, Grey MJ, Nishida N, Walz T, Lu C, Springer TA. Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry. 2008;47:10314–23. doi: 10.1021/bi801006s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang Z, Longo PA, Tarrant MK, Kim K, Head S, et al. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat Struct Mol Biol. 2011;18:1388–93. doi: 10.1038/nsmb.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mi LZ, Lu C, Li Z, Nishida N, Walz T, Springer TA. Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat Struct Mol Biol. 2011;18:984–9. doi: 10.1038/nsmb.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lu C, Mi LZ, Schurpf T, Walz T, Springer TA. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J Biol Chem. 2012;287:38244–53. doi: 10.1074/jbc.M112.414391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gadella TW, Jr, Jovin TM. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J Cell Biol. 1995;129:1543–58. doi: 10.1083/jcb.129.6.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martin-Fernandez M, Clarke DT, Tobin MJ, Jones SV, Jones GR. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys J. 2002;82:2415–27. doi: 10.1016/S0006-3495(02)75585-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sako Y, Minoghchi S, Yanagida T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat Cell Biol. 2000;2:168–72. doi: 10.1038/35004044. [DOI] [PubMed] [Google Scholar]
- 133.Low-Nam ST, Lidke KA, Cutler PJ, Roovers RC, van Bergen en Henegouwen PM, et al. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat Struct Mol Biol. 2011;18:1244–9. doi: 10.1038/nsmb.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nagy P, Claus J, Jovin TM, Arndt-Jovin DJ. Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc Natl Acad Sci U S A. 2010;107:16524–9. doi: 10.1073/pnas.1002642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Saffarian S, Li Y, Elson EL, Pike LJ. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys J. 2007;93:1021–31. doi: 10.1529/biophysj.107.105494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature. 2010;464:783–7. doi: 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- 137.Clayton AH, Walker F, Orchard SG, Henderson C, Fuchs D, et al. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J Biol Chem. 2005;280:30392–9. doi: 10.1074/jbc.M504770200. [DOI] [PubMed] [Google Scholar]
- 138.Clayton AH, Orchard SG, Nice EC, Posner RG, Burgess AW. Predominance of activated EGFR higher-order oligomers on the cell surface. Growth Factors. 2008;26:316–24. doi: 10.1080/08977190802442187. [DOI] [PubMed] [Google Scholar]
- 139.Kozer N, Barua D, Henderson C, Nice EC, Burgess AW, et al. Recruitment of the adaptor protein Grb2 to EGFR tetramers. Biochemistry. 2014 doi: 10.1021/bi500182x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gomez A, Volff JN, Hornung U, Schartl M, Wellbrock C. Identification of a second egfr gene in Xiphophorus uncovers an expansion of the epidermal growth factor receptor family in fish. Molecular biology and evolution. 2004;21:266–75. doi: 10.1093/molbev/msh017. [DOI] [PubMed] [Google Scholar]