

Abstract
Vitamin A and its derivatives, the retinoids, are micronutrient necessary for the human diet in order to maintain several cellular functions from human development to adulthood and also through aging. Furthermore, vitamin A and retinoids are utilized pharmacologically in the treatment of some diseases, as, for instance, dermatological disturbances and some types of cancer. In spite of being an essential micronutrient with clinical application, vitamin A exerts several toxic effects regarding redox environment and mitochondrial function. Moreover, decreased life quality and increased mortality rates among vitamin A supplements users have been reported. However, the exact mechanism by which vitamin A elicits its deleterious effects is not clear yet. In this review, the role of mitochondrial dysfunction in the mechanism of vitamin A-induced toxicity is discussed.
1. Introduction
Vitamin A (retinol) is a micronutrient present in both vegetal and animal diets [1, 2]. However, humans may be exposed to vitamin A and its derivatives (the retinoids) also pharmacologically, as in the case of therapy for dermatological disturbances, acute promyelocytic leukemia, and immunodeficiency treatment [3–9], to cite a few. During leukemia treatment, vitamin A at doses exceeding 150,000–300,000 IU/day is administrated to children at different ages and young adults [8]. Moreover, vitamin A (as retinol palmitate) is administrated to very-low-weight-preterm infants (which may born weighting 0.8–1.1 kg) at doses exceeding 8500 IU/kg·day−1 during weight gain therapy for undetermined period [10]. Recently, it was reported that vitamin A supplementation at 100,000 to 200,000 IU to children aged 6 to 23 months did not prevent mortality in Guinea-Bissau [11]. It is important to keep in mind that the Recommended Dietary Allowance (RDA) for vitamin A varies from 400 mcg retinol activity equivalents (RAE to conversion, please utilize 1 IU retinol = 0.3 mcg RAE) to 900 mcg RAE in males from 0 to 6 months to +51 years old and from 400 mcg RAE to 700 mcg RAE in females from the same varying age [2, 12]. Then, the levels of circulating vitamin A may be exceeded due to both inadvertent or clinical utilization.
Really, a panoply of side effects has been observed that result from vitamin A intoxication that varies from acute intoxication including, for example, headache, hepatic swelling, vomiting, and diarrhea to chronic intoxication with induction of cognitive decline in subjects at different ages, as observed in the cases of increased irritability, confusion, anxiety disorders, depression, and suicide ideation [8, 9, 13]. The exact mechanism by which vitamin A and retinoids exert such effects is not clear yet. However, it may include cell cycle disarrangement, mitochondrial dysfunction, oxidative and nitrosative stress induction, and activation of cell death signaling in different experimental models.
In this work, the effects of vitamin A and retinoids on some redox and bioenergetics parameters will be discussed focusing on mitochondrial function in different experimental models.
2. Vitamin A Metabolism: A Brief Overview
Vitamin A (or retinol, a diterpene) is originated from isoprene units and is characterized as an isoprenoid with a hydrocarbon chain containing a hydroxyl group at one end. The oxidation of such hydroxyl group yields retinal (an aldehyde, retinaldehyde) or retinoic acid (a carboxylic acid), the biologically active forms of retinol. In a general view, all retinoids are formed by a β-ionone ring and a polyunsaturated side chain and a chemical group varying from alcohol to carboxylic acid or ester as mentioned above. The presence of conjugated double bonds is noteworthy which may be in either trans-configuration or cis-configuration in the molecule of retinoids [14, 15]. Such chemical structure decreases its solubility in aqueous environments.
Vitamin A (retinol) and its derivatives, the retinoids, participate in a myriad of biological processes during animal life from development to adulthood and aging. Control of cell proliferation, differentiation, induction of cell death through apoptosis, formation and shaping of the embryo, organogenesis, and tissue homeostasis depend on physiological concentrations of vitamin A to occur adequately [9, 14–16]. Among retinoids, all-trans retinoic acid is better studied because it is the most biologically potent vitamin A derivative [9, 12, 14, 15]. Vitamin A and retinoids may exert their functions by binding to nuclear receptors (genomic action: induction or repression of the expression of target genes) or though regulation of signaling pathway dependent on phosphorylation of specific targets (nongenomic action: a rapid way to regulate cell events through the action of protein kinases and phosphatases) that culminate in a cellular response to such stimulus [12, 14, 15, 17–19].
Vitamin A may be obtained from both vegetal and animal diets. β-Carotene (an isoprenoid compound) is converted to two molecules of all-trans-retinal by centric oxidative cleavage and all-trans-retinal is reduced to all-trans-retinol, which may be esterified and stored in large amounts in tissues as liver, lung, and fat [12, 20, 21]. In the eyes, retinoids are converted to 11-cis-retinal, which is a visual chromophore that binds to opsin in order to translate light into an electrical signal [22, 23]. Esterified retinol in the form of retinol palmitate is a major source of vitamin A from diet of animal origin, as, for instance, liver, which stores the excess of vitamin A [14, 15, 20, 24].
The absorption of fat-soluble micronutrients occurs very similarly to that observed in lipids in the upper gastrointestinal tract [25, 26] after dissolution via formation of lipid droplets in both stomach and duodenum [27, 28]. The esterified forms of vitamin A (mainly retinol palmitate) are firstly hydrolyzed in the duodenum and the free form is then absorbed by the intestinal mucosa [29]. It is suggested that two pancreatic enzymes perform such hydrolysis, namely, cholesterol ester hydrolase and pancreatic lipase [30–32]. Then, the enterocyte will absorb vitamin A and carotenoids which are incorporated into micelles with other lipids from diet [25, 26]. It was reported that the efficiency of retinol absorption is around 75% [33] and 100% [34–37]. On the other hand, the efficiency of β-carotene absorption was estimated to be from 3% to 90% [36–38]. It was proposed that enterocytes present a specific retinol transporter that functions very efficiently [39, 40]. The absorption of carotenoids occurs mainly through passive diffusion [41].
After enterocyte uptake, retinol is esterified by lecithin retinol acyltransferase (LRAT, which utilizes phosphatidylcholine as acyl group donor) and acyl-CoA acyltransferase (ARAT), leading to the formation of retinol palmitate, retinol oleate, and retinol linoleate, among others [25, 42]. Carotenoids may follow one of these paths inside enterocytes: stay not metabolized (around 40% of provitamin A carotenoids), be cleavage generating retinal via the reaction mediated by β-carotene-15,15′-monooxygenase, or be cleavage by mitochondrial β-carotene-9′,10′-dioxygenase, which is responsible for the formation of apocarotenoids [43].
In the cytosol of the enterocyte, retinol and its derivatives (mainly retinal and retinoic acids) bind to specific proteins called cellular retinol-binding protein II (CRBP II) [25]. In other cells, as, for instance, the hepatocytes, CRBP I is responsible for free retinol transport. Additionally, the binding of retinol to CRBP is necessary to its esterification by LRAT or ARAT. In the hepatocytes, esterified retinol and retinal are also transported by CRBP I. In the plasma, retinol is transported by retinol binding protein (RBP) to general distribution to tissues [14, 15]. Retinol is converted to retinal by either microsomal or cytosolic retinol dehydrogenase (RoDH) isozymes. In turn, retinal is converted in retinoic acid by cytosolic retinal dehydrogenase (RalDH) [14, 15, 44]. Retinoic acids bind to cellular retinoic acid binding protein (CRABP) in cytosol and it is suggested that this complex migrates to nucleus to exert its effects through binding to nuclear receptors to retinoic acid (RAR or RXR) [44, 45].
Central nervous system (CNS) cells also possess nuclear receptors, CRBPs and CRABPs, as well as enzymes necessary to the local metabolism of vitamin A and derivatives. Additionally, it has been postulated that retinoids may act through a nongenomic way in different cell types, including neurons [46, 47]. The role of retinoids is not restricted to development of CNS. It has been shown that retinoids are responsible for synaptic plasticity of the hippocampus, for maintenance of dopamine signaling in mesolimbic and mesostriatal neurons, and for survival of nigrostriatal dopaminergic neurons [48–50].
3. The Relationship of Vitamin A and Retinoids with Biological Membranes
The hydrophobicity of vitamin A and retinoids is a chemical limiting its distribution in the aqueous compartments of the body. As mentioned above, it is necessary to bind such molecules to transport protein to increase its solubility. Indeed, vitamin A reacts with hydrophobic environments, as, for example, biological membranes, and may interfere with its physiology by perturbing phospholipid and steroids homeostasis. It was previously demonstrated that retinol induced hemolysis by penetrating rabbit erythrocytes and disrupting physical structure of the membranes [51]. According to the authors, such effect did not depend on oxidation of retinol and formation of free radicals. However, it was observed that cotreatment with vitamin E alleviated hemolysis. In that work, it was suggested that vitamin E did act by decreasing permeability and fluidity and not through its antioxidant capacity. In other works, the authors found that the retinol-induced hemolysis was dependent on hydroxyl radical formation [52]. Goodall et al. demonstrated that retinol and retinoids (retinaldehyde, α-retinoic acid, iso-13-retinol, and retinyl acetate) induced cell fusion, hemolysis, and swelling of mitochondria [53]. Actually, intravascular administration of all-trans retinoic acid to patients under treatment of acute promyelocytic leukaemia induced hemolysis and complicated the continuation of this clinical procedure [54].
Overall, such findings indicate a potential ability of vitamin A and its derivatives to negatively interact with biological membranes, an event that may lead to organelle stress, as, for instance, mitochondrial dysfunction, and to cell apoptosis or necrosis.
4. The Effects of Vitamin A and Its Derivatives on Mitochondrial Membranes and Organelle Physiology
4.1. In Vitro Effects of Vitamin A on Mitochondria
As previously mentioned, retinol induced mitochondrial swelling and disrupted membrane organization in in vitro assays [51–54]. Rigobello et al. [55] did demonstrate that different retinoic acids (namely, all-trans, 9-cis, and 13-cis retinoic acids) were able to induce swelling of the organelle isolated from rat liver. All the retinoids tested induced membrane permeability transition (which was observed as swelling) and decreased membrane potential. Interestingly, neither EGTA (Ca2+ ion chelating agent) nor cyclosporin A (CsA) inhibited the effects elicited by 13-cis retinoic acid. Additionally, 13-cis retinoic acid induced cytochrome c release from the organelle, an event that is necessary to trigger the intrinsic apoptotic pathway [56]. Later, it was reported that retinol also altered mitochondrial structure by inducing swelling and lipid peroxidation in mitochondrial membranes in vitro [57]. In addition, retinol induced cytochrome c release and increased superoxide anion radical (O2 −•) production in a dose-dependent pattern. When analyzed together, such results indicate part of the mechanism by which retinol and retinoids may trigger cell death through the mitochondrial/intrinsic pathway. Also, it demonstrates that vitamin A may exert prooxidant effects by altering mitochondrial function and favoring electron leakage from mitochondria, leading to increased free radical generation.
Really, it was demonstrated that retinol induced apoptosis in cultured Sertoli cells by a mitochondria-dependent pathway [58]. In such work, the researchers found that retinol induced a decrease in cell viability and ATP content and increased O2 −• formation. Additionally, increased cytochrome c release to the cytosol and, consequently, increased caspase-3/7 enzyme activity were observed. Then, from isolated mitochondria assays to cultured cells, the deleterious effects of vitamin A on mitochondria may be observed. Such negative action of this vitamin and its derivatives on mitochondrial function and/or dynamics may result in cell death. The release of cytochrome c to cytosol may lead to two important processes: increased O2 −• production and apoptosis through formation of the apoptosome. However, apoptosis is dependent on sufficient ATP levels because the apoptosome consumes ATP (or dATP) to cleave and activate caspases. Then, deregulated cytochrome c release may lead the cells to die by necrosis, which induces inflammation, an even more deleterious process to cell viability (for a review, please see [56]).
Silva et al. reported that acitretin (a synthetic retinoid that is used in the treatment of severe extensive psoriasis) at 5–20 μM altered the function of rat liver mitochondria in vitro [59]. The authors found impaired phosphorylation capacity, decreased ATP levels and adenine nucleotide translocase (ANT) content, and Ca2+-induced mPTP (mitochondrial permeability transition pore). On the other hand, decreased membrane potential was not observed. Surprisingly, such effects were not reverted by the cotreatment with thiol group reductants or other antioxidant agents, showing, at least in part, that a redox mechanism did not take part in the events observed. On the other hand, mPTP was blocked by ANT ligands, as, for instance, ATP and ADP.
Recently, Sawada et al. reported that all-trans-retinal (10–30 μM), a byproduct of the visual cycle (originated from the chromophore 11-cis-retinal), decreased viability of ARPE-19 cell line and induced oxidative stress-dependent Bax activation through PLC/IP3/Ca2+ signals and by activation of p53 following DNA damage [60]. The authors conclude that all-trans-retinal affected cell viability by a mechanism that increased the concentration of cytosolic Ca2+ ions, which lead to oxidative stress and DNA damage. In turn, it activates p53 through a mechanism dependent on phosphorylation of ser46 residue and translocation to the cytosol, where it activates Bax, triggering apoptosis. This work demonstrates the importance of maintaining the levels of all-trans-retinal under control in retina, since disrupted all-trans-retinal clearance may lead to retinopathy, as previously reported [61]. However, the authors did not investigate the role of mitochondria in the induction of apoptosis in that work.
A synthetic retinoid (ST1926) was recently tested for treating adult T-cell leukemia/lymphoma and demonstrated ability to induce growth arrest and apoptosis of T malignant cells [62]. ST1926 at 1 μM for 48 hours induced apoptosis in HuT-102, MT-2, Jurkat, and Molt-4 cell lines. Even though the apoptotic mechanism depends on caspase-3, any parameter related to mitochondrial dynamics was not investigated in that work.
4.2. In Vivo Effects of Vitamin A on Mitochondria
The effects of vitamin A and retinoids on mitochondrial function were well investigated in vitro. However, recently it was demonstrated that intragastric (gavage) vitamin A supplementation at pharmacological doses (from 1,000 to 9,000 IU/kg·day−1) for 3, 7, or 28 days induced redox (Table 1) and bioenergetics (Table 2) impairments in rat brain regions and other tissues of adult male Wistar rats, as discussed below. Additionally, some abnormalities in behavioral tasks were observed, as, for example, in the open field and light-dark box [63–66].
Table 1.
Summary of the in vivo effects of subacute vitamin A supplementation on mitochondrial membranes parameters.
| Sample | Lipid peroxidation | Protein carbonylation | Protein nitration | Protein thiol content | Reference |
|---|---|---|---|---|---|
| Cerebral cortex | ↑ | ↑ | Not measured | ↓ | [65] |
| Cerebellum | ↑ | ↑ | Not measured | ↓ | [65] |
| Substantia nigra | ↑ | Not measured | Not measured | Not measured | [67] |
| Striatum | ↑ | Not measured | Not measured | Not measured | [67] |
| Hypothalamus | ↑ | Not measured | Not measured | Not measured | [68] |
| Frontal cortex | ↑ | ↑ | ↑ | Unaltered | [69, 70] |
| Hippocampus | ↑ | ↑ | ↑ | Unaltered | [70, 71] |
| Liver | ↑ | ↑ | Not measured | Unaltered | [72, 73] |
| Heart | Not measured | Not measured | ↑ | Not measured | [74] |
| Lung | ↑ | ↑ | ↑ | Unaltered | [75] |
Adult male rats were treated with vitamin A supplementation (1,000–9,000 IU/kg/day) subacutely (see text for details).
Table 2.
Summary of in vivo effects of subacute vitamin A supplementation on mitochondrial function parameters.
| Sample | Complexes I–III | Complexes II-III | Complexes II + SDH | Complex IV | Reference |
|---|---|---|---|---|---|
| Cerebral cortex | Not measured | Not measured | Not measured | Not measured | — |
| Cerebellum | ↑ | Unaltered | Unaltered | ↓ | [76] |
| Substantia nigra | ↑ | ↑ | ↑ | Unaltered | [67] |
| Striatum | ↑ | Unaltered | Unaltered | ↓ | [67] |
| Hypothalamus | ↑ | Unaltered | Unaltered | ↓ | [68] |
| Frontal cortex | ↑ | Unaltered | Unaltered | Unaltered | [69, 70] |
| Hippocampus | ↑ | Unaltered | Unaltered | ↓ | [70, 71] |
| Liver | ↑ | ↑ | ↑ | ↑ | [72, 73] |
| Heart | ↓ | ↓ | ↓ | Not measured | [74] |
| Lung | ↑ | ↑ | ↑ | Not measured | [75] |
Adult male rats were treated with vitamin A supplementation (1,000–9,000 IU/kg/day) subacutely (see text for details).
Vitamin A supplementation increased mitochondrial superoxide anion radical (O2 −•) production (Table 3) and induced lipid peroxidation, protein carbonylation and nitration, and oxidation of protein thiol groups in mitochondrial membranes isolated from rat cerebral cortex, cerebellum, substantia nigra, striatum, frontal cortex, and hypothalamus [67–69, 76, 78]. In the same rat brain areas, increased complex I–III enzyme activity was observed [67–69, 76, 78]. However, a proportional increase in the following complexes of the mitochondrial electron transfer chain (METC) was not found as expected. For example, vitamin A supplementation induced a decrease in complex IV enzyme activity in rat cerebellum, striatum, and hypothalamus [67, 68, 76]. On the other hand, any change in some complexes activities was not observed as follows: complexes II, II-III and succinate dehydrogenase (SDH) (cerebellum) [76]; complex IV (substantia nigra) [67]; complexes II-III and SDH (striatum, hypothalamus) ([67], [68], resp.); complexes II-III, SDH, and complex IV (frontal cortex) [69] (Table 2). Such impairment in electron flux between mitochondrial complexes may favor electron leakage from the electron transfer chain, since the electron flux is higher between some complexes, but the reduction of O2 to water is not occurring at the same rate due to unaltered or even decreased complex IV enzyme activity (Figure 1). Also, more O2 is available to react with electron donors and becomes O2 −• [79, 80]. Furthermore, increased complexes I–III, II-III, and II and SDH and complex IV enzyme activities were also reported in the liver of the animals that receive vitamin A supplementation at clinical doses for 28 days [72]. These findings are different from that observed in brain regions of the animals that received vitamin A for the same period, as described above, since it was demonstrated that complex IV enzyme activity was increased at a very similar rate when compared to complexes I–III in rat liver. However, such increment in the electron flux between the electron transfer chain (ETC) complexes was accompanied by a proportional increase in O2 −• production (Table 3). This result may suggest that O2 −• is being produced by mitochondria isolated from vitamin A-treated rats by a mechanism that is not obligatorily associated with uncoupling of the ETC activity.
Table 3.
Summary of in vivo effects of subacute vitamin A supplementation on mitochondrial redox parameters.
| Sample | Superoxide anion radical | Mn-SOD enzyme activity | MAO enzyme activity | Reference |
|---|---|---|---|---|
| Cerebral cortex | ↑ | Not measured | Not measured | [65] |
| Cerebellum | ↑ | Not measured | Not measured | [65] |
| Substantia nigra | ↑ | ↑ | Unaltered | [67] |
| Striatum | ↑ | ↑ | ↑ | [67] |
| Hypothalamus | ↑ | Not measured | Not measured | [68] |
| Frontal cortex | ↑ | ↑ | ↑ | [77] |
| Hippocampus | ↑ | ↑ | ↑ | [77] |
| Liver | ↑ | Not measured | Not measured | [73] |
| Heart | Not measured | Not measured | Not measured | — |
| Lung | ↑ | Not measured | Not measured | [75] |
Adult male rats were treated with vitamin A supplementation subacutely (see text for details).
Figure 1.
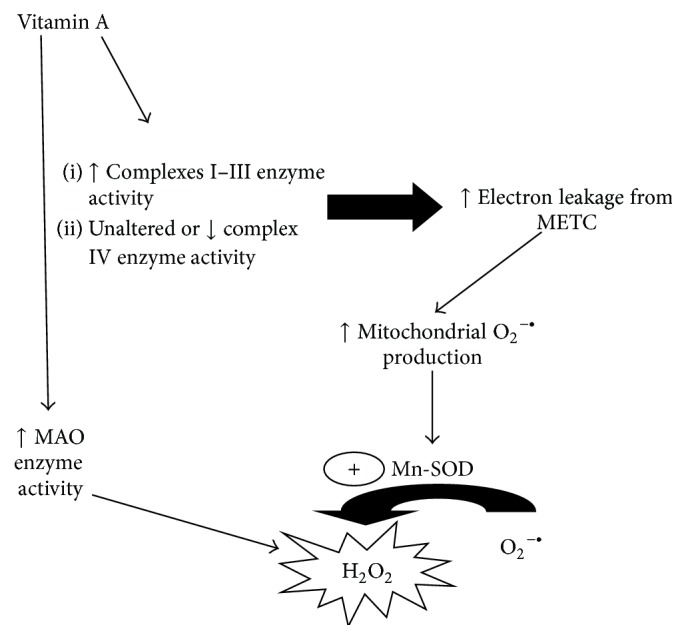
A schematic diagram summarizing the effects of in vivo vitamin A supplementation on mitochondrial function regarding the mitochondrial electron transfer chain (METC) enzyme activity. Mitochondrial dysfunction may lead to increased O2 −• production through electron leakage and partial reduction of O2. Mn-SOD converts O2 −• to H2O2 and, together with MAO, favors an increase in the levels of H2O2 in different cell types (please see text for details). H2O2 is able to react with iron ions generating •OH (the most powerful ROS) through Fenton chemistry reaction (not shown), for example, leading to widespread redox disturbances.
More recently, it was published that vitamin A supplementation induced an increase in total 3-nitrotyrosine content in rat cerebral cortex, hippocampus, substantia nigra, striatum, hypothalamus, heart, and lung [67, 68, 71, 74, 75, 81]. In addition, increased 3-nitrotyrosine content in proteins located in the mitochondrial membranes isolated from frontal cortex, hippocampus, heart, and lung of vitamin A-treated rats was reported [69, 71, 74, 75] (Table 1). The formation of 3-nitrotyrosine is a consequence of increased levels of O2 −• and NO•, which give rise to peroxynitrite (ONOO−), that may react with tyrosine residues in proteins leading to the formation of 3-nitrotyrosine. Additionally, ONOO− may give rise to peroxynitrous acid (ONOOH), which produces nitryl cation (NO2+), nitrogen dioxide radical (•NO2), and hydroxyl radical (•OH) through homolytic fission reaction [82, 83]. At least in part, the increase in 3-nitrotyrosine content may be explained by the increase in mitochondrial O2 −• production elicited directly or indirectly by vitamin A supplementation. In order to investigate whether NO• production (as indirectly assessed through 3-nitrotyrosine formation) participates in mitochondrial dysfunction and behavioral disturbances observed in the experimental model of vitamin A supplementation, the role of a cotreatment with L-NG-nitroarginine methyl ester was tested (L-NAME; 30 mg/kg four times a week), a nonspecific nitric oxide synthase (NOS) inhibitor, on such parameters. Interestingly, L-NAME cotreatment did not exert any effect on the redox unbalance elicited by vitamin A on rat frontal cortex, hippocampus, substantia nigra, and striatum [77].
It was previously described that increased formation rates of 3-nitrotyrosine favor protein aggregation, which may lead to serious consequences regarding mitochondrial function, such as import of molecules (from metabolic substrates to proteins necessary to the ETC function, among others) from cytosol and other complex processes as mitochondrial fusion and fission. Both α-synuclein and α-tubulin may be nitrated and form protein aggregates that accumulate in cytoplasm [84–86]. α-Synuclein has been implicated in the mechanism behind the pathogenesis of neurodegenerative synucleinopathies [84, 87]. Recently, it was shown that α-synuclein may interact negatively with mitochondria causing it to lose transmembrane potential and decrease phosphorylation capacity [88]. In fact, α-synuclein may bind to the inner mitochondrial membrane in α-helical conformation [89]. Interestingly, increased levels of α-synuclein, but unaltered levels of β-synuclein, in brain regions of vitamin A-treated rats were demonstrated [67, 71, 77]. However, neither alterations in α-synuclein structure nor interactions of such protein with mitochondria in the experimental model of vitamin A supplementation were investigated.
On the other hand, it was shown that vitamin A supplementation for 28 days increased monoamine oxidase (MAO) enzyme activity in both areas of the nigrostriatal axis and hippocampus [71, 77, 90] (Table 3). MAO is responsible for the chemical inactivation of dopamine and serotonin and produces H2O2 in such reaction [87, 91]. MAO is located in the outer mitochondrial membrane facing the cytosol, but H2O2 is a membrane soluble ROS and may enter mitochondria or other organelles [91]. Taken together, such data indicate mitochondria as an important source of H2O2, since manganese-superoxide dismutase (Mn-SOD; mitochondrial) and MAO enzyme activities were found increased in the hippocampus and nigrostriatal axis of vitamin A-treated rats [71, 90] (Table 3). H2O2, which is also water soluble, may diffuse to places far away from its origin, disseminating the redox impairment from one cellular environment to another [92–97] (Figure 1). Interestingly, CAT enzyme activity was found either unaltered or decreased in brain areas of vitamin A-exposed rats [63, 64, 66]. Such finding suggests that an impairment exists also on the ratio between SOD and CAT enzyme activities, which may favor an increase in H2O2 production. Furthermore, accumulated O2 −• is able to inhibit CAT enzyme activity, as well as other enzymes [98] (Figure 2). Then, it may be suggested that, in the experimental model of vitamin A supplementation, mitochondria is a biological source of H2O2 in some rat brain regions and such effect may be linked to the oxidative stress observed in some reports (Figure 3).
Figure 2.
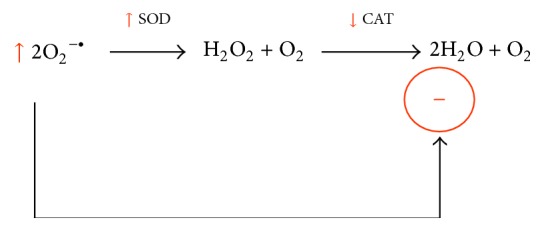
Unbalanced SOD/CAT ratio resulting in increased H2O2 production. Additionally, increased O2 −• levels inhibit CAT enzyme activity allosterically leading to even more high H2O2 concentration due to accumulation of this ROS.
Figure 3.

A general view of the effects of in vivo vitamin A supplementation in an animal experimental model. It has been hypothesized that vitamin A may induce mitochondrial dysfunction by different ways as follows: (1) by decreasing BDNF levels, (2) by inducing ER stress and calcium ion metabolism deregulation, and/or (3) by increasing α-synuclein levels. The increased O2 −• levels may induce redox unbalance in the organelle that, in turn, may generate more O2 −• in a vicious cycle. Increased H2O2 production (by Mn-SOD and MAO enzymes) may disseminate redox impairment from one region to another.
In addition to a possible H2O2 generation increase, increased glutathione S-transferase (GST; an enzyme that is responsible for phase II detoxification reactions of conjugation in several cell types) enzyme activity in the vitamin A supplementation experimental model was detected [67, 76]. Such enzyme consumes reduced glutathione (GSH) to produce more polar xenobiotics that are easily excreted from cells [99]. By consuming GSH at increased rates, it may facilitate the perpetuation of H2O2 prooxidant signal, since GSH is utilized by GPx in the conversion of H2O2 to water [87, 100, 101]. In the nigrostriatal axis, there is a high Fe2+ content that may react with H2O2 through Fenton chemistry reaction in cases of hypervitaminosis A, for example, leading to increased production of •OH, the most powerful free radical in biological systems [87, 102, 103]. Indeed, it may facilitate dopaminergic neuronal death by either apoptosis or necrosis, leading detrimental effects on movement control, as observed in patients suffering from Parkinson's disease [104, 105]. Although redox impairment was found in such rat brain areas, any alteration regarding cellular markers of cell death was not observed, such as caspase-3 or caspase-8 enzyme activity [67–69, 78, 90].
4.3. Ex Vivo Effects of Vitamin A on Mitochondria
Vitamin A supplementation at clinical doses for 3 or 7 days induced several prooxidant effects also on rat liver, which is the main site of vitamin A storage in mammals [14, 15, 47]. It was observed that vitamin A supplementation (1,000 to 9,000 IU/kg·day−1) for 3 or 7 days induced oxidative stress in mitochondrial membranes and increased O2 −• production [73]. Also, increased complexes I–III enzyme activity was demonstrated without any effect on complexes II-III and IV. However, the more surprising in that work is the fact that intact mitochondria isolated from the liver of the animals that received vitamin A supplementation were found to be more sensitive to an incubation of 10 minutes with CaCl2 at low concentration (75 μM; ex vivo assay). Calcium ions mediate mitochondrial dysfunction by increasing reactive oxygen species (ROS) production and triggering mPTP, resulting in apoptosis as reviewed elsewhere [106–108]. A 2.5- to 2.9-fold increase in lipid peroxidation levels in the mitochondria isolated from vitamin A-treated rats when exposed to CaCl2 was detected. Similar effects were seen when protein carbonylation and thiol oxidation markers were quantified in such experimental model. Cotreatment with DTT, GSH, superoxide dismutase (SOD), or catalase (CAT) did decrease the prooxidant effect induced by CaCl2. Neither CsA nor bongkrekic acid (BKA) (mPTP inhibitors) did alter the effect induced by CaCl2 [73]. Then, such data suggest that the prooxidant effects that appeared after exposure to CaCl2 are not related to mPTP formation. Additionally, CaCl2 amplified O2 −• production in intact mitochondria isolated from vitamin A-treated animals. However, only cotreatment with GSH or SOD did decrease CaCl2-induced O2 −• production [73]. Then, it may be concluded that in vivo vitamin A supplementation increased the ex vivo mitochondrial susceptibility to a challenge that indirectly induces a prooxidant state in the organelle. However, it was not associated with mPTP formation, as indicated through the utilization of mPTP inhibitors. At least in part, some of the findings presented above are similar to the effects elicited by the treatment with a synthetic retinoid (acitretin) on mitochondrial function in vitro [59].
The effects of vitamin A supplementation on a mitochondrial challenging with CaCl2 in the case of rat liver analyses were discussed above. However, it was also investigated whether in vivo vitamin A supplementation altered brain mitochondria response to an ex vivo challenge with H2O2 or β-amyloid peptide1–40 and peptide1–42 [70, 90]. As expected, vitamin A supplementation increased the susceptibility of mitochondria (isolated from the nigrostriatal axis and from frontal cortex and hippocampus) to H2O2 (a ROS) and to β-amyloid peptide1–40 and peptide1–42 (which accumulate at both extra- and intraneuronal environments in the case of Alzheimer's disease) [87]. β-Amyloid peptide1–40 and peptide1–42, which may accumulate in the extracellular environment, also are able to enter neurons and interact with organelles, such as mitochondria, leading to membrane rupture, among other effects, and general dysfunction [109–113]. It is an important finding demonstrating that even recommended doses of vitamin A (which have been considered to be secure to humans) facilitate mitochondrial damage when such organelles are exposed to reactive molecules (with or without radical nature) (Figure 4).
Figure 4.
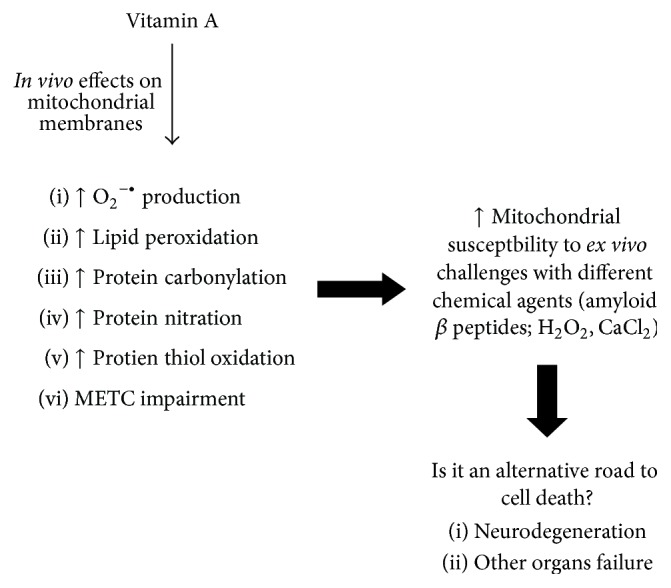
A general view of the consequences of in vivo vitamin A supplementation on the susceptibility of mitochondria to ex vivo challenges with different chemical agents. Mitochondria isolated from vitamin A-treated rats are more sensitive to different chemical insults including amyloid β, H2O2, and CaCl2, as discussed in the text.
4.4. Other Evidences of Vitamin A-Induced Toxicity on Mammalian Mitochondria
It was also observed that vitamin A supplementation (1,000–9,000 IU/kg·day−1 for 28 days) induced a decrease in the levels of brain-derived neurotrophic factor (BDNF) in rat hippocampus [71]. BDNF is a major neurotrophin in the mammalian brain and is involved in the induction of neuronal proliferation and maintenance of neuron survival [114–116]. Furthermore, BDNF may signal mitochondrial biogenesis in different cell types including neurons [117, 118]. Then, BDNF is also responsible, at least in part, for maintaining ATP homeostasis in mammalian cells. However, a causal link between mitochondrial dysfunction and deregulated BDNF levels was not established yet.
Some evidences point to vitamin A as an inducer of endoplasmic reticulum (ER) stress since increased BiP/Grp78 levels in the hippocampus of vitamin A-treated rats was reported [71]. BiP (a protein chaperone) is a major regulator of ER function and participates, for example, in protein folding and assembly, binding to Ca2+ ions, and controlling ER stress sensors activation [130, 131]. Whether vitamin A or one of its derivatives alter ER function was not demonstrated yet; but by inducing ER stress, vitamin A may deregulate Ca2+ ions homeostasis, which may lead to mitochondrial dysfunction and cell death [132] (Figure 3).
5. Clinical Hypothesis of the Impact of Hypervitaminosis A on Human Health
Mitochondrial dysfunction gives rise to a myriad of consequences. It includes bioenergetics deficits, increased production of reactive oxygen or nitrogen species (ROS and RNS, resp.), and apoptosis or necrosis. Then, it is very important to maintain mitochondrial homeostasis to avoid loss of cellular quality and death by mechanisms that may culminate in inflammation, for example.
It has been shown that retinoids possess an ability to alter cell cycle and to induce apoptosis in some experimental models. It was published that the treatment of adult mice with 13-cis-retinoic acid at 1 mg/kg·day−1 (a clinical dose commonly applied in the treatment of nodular acne) for 1–6 weeks suppressed hippocampal cell division (neurogenesis) and, consequently, decreased capacity to learn in behavioral task [133]. Accordingly, Sakai et al. demonstrated increased cell loss in the hippocampus of mice treated for 3 weeks with 13-cis-retinoic acid at 1 mg/kg·day−1 [134]. The mechanism by which 13-cis-retinoic acid altered neurogenesis and induced cell death in mice hippocampus is not clear, but it has been reported that this retinoid may trigger apoptosis through activation of caspase-3 and by modulating bcl2 and p53 gene expression in melanoma cells [135]. Reinforcing the finding that a retinoid may induce negative consequences to hippocampal function, it was reported that vitamin A supplementation with retinol palmitate induced anxiety-like behavior in adult rats [63]. Anxiety is a behavior closely related to alterations in the function of hippocampus and significantly decreases human life quality [136–138]. Furthermore, studies in humans demonstrated that the use of 13-cis-retinoic acid (as treatment to nodular acne) decreased metabolism in orbitofrontal cortex, a region associated with depression [119]. Indeed, there is a strong body of evidence showing that 13-cis-retinoic acid (isotretinoin) induced depression and increased both suicide ideation and suicide rates among some patients under such treatment [120–124]. However, it remains to be elucidated whether there is a causal link between bioenergetics impairment and neuronal dysfunction that leads to detrimental alteration in human behavior.
In fact, the capacity of retinoids to induce mitochondrial dysfunction and cell death has been utilized pharmacologically as a strategy to treat several human diseases from dermatological disturbances to some types of cancer (Table 4). On the other hand, it is not clear whether a vitamin A overload would be beneficial to cells under constant stress and low antioxidant defenses, as, for instance, neurons [87, 139, 140]. Increased cell death rates are observed in the case of Parkinson's disease and Alzheimer's disease [87], and increased ingestion or other forms of exposure to such vitamin may favor a more drastic situation with accelerated neuronal loss and increased neuroinflammation. Really, it has been reported that vitamin supplements utilization (including vitamin A and carotenoids) by well-nourished subjects may increase risk of mortality among them [125–127]. Indeed, the ingestion of antioxidant supplements in the primary prevention of chronic diseases or mortality in agreement with recent dietary guidelines is not suggested [127]. Additionally, it is alarming that the combination of β-carotene (30 mg; vitamin A precursor from vegetal diet) and retinol palmitate (25,000 IU) supplementation increased lung cancer incidence among men and women in a clinical trial that has to be stopped due to increased lung cancer and death among the volunteers [141]. However, the mechanisms by which vitamin A and retinoids, among other lipophilic vitamins, alter cell function leading to death remain to be elucidated.
Table 4.
Clinical utilization of vitamin A and retinoids.
| Retinoid | Utilization | Reference |
|---|---|---|
| Various | Prevention of infectious diseases | [4] |
| Retinol palmitate | Treatment of acute promyelocytic leukemia | [5, 7] |
| Retinol palmitate | Treatment of acute nonlymphocytic leukemia | [6] |
| Various | Weight gain therapy in preterm infants | [10] |
| Retinol palmitate/acetate | Immunotherapy (with vaccination) | [11] |
| Isotretinoin | Acne therapy | [119–124] |
| Various | Antioxidant therapy, increased longevity (supplements) | [125–127] |
| Retinyl esters | Treatment of infants born from HIV-positive women (immunodeficiency therapy) | [128] |
| Various | Antioxidant therapy in heart disease | [16] |
| Various | Utilization in general dermatology | [129] |
6. Conclusion
Vitamin A and its derivatives, the retinoids, disrupt mitochondrial function by a mechanism that is not completely understood. However, it accounts with impaired electron flux between the complexes of the METC, increased ROS production, and induction of oxidative and nitrosative stress to mitochondrial membranes. Additionally, vitamin A and retinoids alter the mitochondrial structure by causing swelling of the organelle. More investigations are needed to elucidate how vitamin A and retinoids affect mitochondria and whether there is a causative link between such event and the clinical manifestations observed both experimentally and in humans.
Then, even though more investigations in this field are necessary, it is more secure to take some caution when vitamin A has been ingested at higher than recommended levels by individuals with familial history of neurodegenerative diseases, for instance, Alzheimer's disease and Parkinson's disease, or are already affected by such irreversible disorders. Really, the fact that vitamin A increased susceptibility of mitochondria to some common cellular stress inducer agents (CaCl2 and H2O2 and not only β-amyloid peptide1–40 and peptide1–42) must be considered in the case of utilization of such micronutrient as supplement or fortified food in any case of disease, not only those from neuronal origin.
Overall, caution must be taken when utilizing vitamin A or its derivatives in some specific conditions, since such molecules regulate cell cycle and cell fate (survival or death) by different ways and its toxic effects may also lead to irreversible damage.
Acknowledgments
Thanks are due to Fernanda Rafaela Jardim, M.S., for English grammar revision. Some of the data discussed here were obtained from research that was funded by CNPq.
Abbreviations
- ANT:
Adenine nucleotide translocase
- ARAT:
Acyl-CoA acyltransferase
- BDNF:
Brain-derived neurotrophic factor
- BKA:
Bongkrekic acid
- CNS:
Central nervous system
- CRABP:
Cellular retinoic acid binding protein
- CRBP I:
Cellular retinol-binding protein I
- CRBP II:
Cellular retinol-binding protein II
- CAT:
Catalase
- CsA:
Cyclosporin A
- ER:
Endoplasmic reticulum
- ETC:
Electron transfer chain
- GSH:
Glutathione
- GST:
Glutathione S-transferase
- L-NAME:
L-NG-nitroarginine methyl ester
- LRAT:
Lecithin retinol acyltransferase
- MAO:
Monoamine oxidase
- METC:
Mitochondrial electron transfer chain
- Mn-SOD:
Manganese-superoxide dismutase
- mPTP:
Mitochondrial permeability transition pore
- NOS:
Nitric oxide synthase
- RAE:
Retinol activity equivalents
- RalDH:
Retinal dehydrogenase
- RAR:
Retinoic acid receptor
- RBP:
Retinol binding protein
- RDA:
Recommended Dietary Allowance
- RNS:
Reactive nitrogen species
- RoDH:
Retinol dehydrogenase
- ROS:
Reactive oxygen species
- SDH:
Succinate dehydrogenase
- SOD:
Superoxide dismutase.
Conflict of Interests
The author declares that there is no conflict of interests regarding the publication of this paper.
References
- 1.Ross D. A. Recommendations for vitamin A supplementation. Journal of Nutrition. 2002;131:2902S–2906S. doi: 10.1093/jn/132.9.2902S. [DOI] [PubMed] [Google Scholar]
- 2.Tanumihardjo S. A. Assessing vitamin A status: past, present and future. The Journal of Nutrition. 2004;134(1):290S–293S. doi: 10.1093/jn/134.1.290S. [DOI] [PubMed] [Google Scholar]
- 3.Allen L. H., Haskell M. Estimating the potential for vitamin A toxicity in women and young children. Journal of Nutrition. 2002;132(9):2907S–2919S. doi: 10.1093/jn/132.9.2907S. [DOI] [PubMed] [Google Scholar]
- 4.Glasziou P. P., Mackerras D. E. M. Vitamin A supplementation in infectious diseases: a meta-analysis. British Medical Journal. 1993;306(6874):366–370. doi: 10.1136/bmj.306.6874.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsutani H., Iwasaki H., Kawai Y., et al. Reduction of leukemia cell growth in a patient with acute promyelocytic leukemia treated by retinol palmitate. Leukemia Research. 1990;14(7):595–600. doi: 10.1016/0145-2126(90)90013-y. [DOI] [PubMed] [Google Scholar]
- 6.Tsutani H., Ueda T., Uchida M., Nakamura T. Pharmacological studies of retinol palmitate and its clinical effect in patients with acute non-lymphocytic leukemia. Leukemia Research. 1991;15(6):463–471. doi: 10.1016/0145-2126(91)90057-Z. [DOI] [PubMed] [Google Scholar]
- 7.Fenaux P., Chomienne C., Degos L. Treatment of acute promyelocytic leukaemia. Best Practice and Research: Clinical Haematology. 2001;14(1):153–174. doi: 10.1053/beha.2000.0121. [DOI] [PubMed] [Google Scholar]
- 8.Myhre A. M., Carlsen M. H., Bøhn S. K., Wold H. L., Laake P., Blomhoff R. Water-miscible, emulsified, and solid forms of retinol supplements are more toxic than oil-based preparations. American Journal of Clinical Nutrition. 2003;78(6):1152–1159. doi: 10.1093/ajcn/78.6.1152. [DOI] [PubMed] [Google Scholar]
- 9.O'Reilly K., Bailey S. J., Lane M. A. Retinoid-mediated regulation of mood: possible cellular mechanisms. Experimental Biology and Medicine. 2008;233(3):251–258. doi: 10.3181/0706-mr-158. [DOI] [PubMed] [Google Scholar]
- 10.Mactier H., Weaver L. T. Vitamin A and preterm infants: what we know, what we don't know, and what we need to know. Archives of Disease in Childhood: Fetal and Neonatal Edition. 2005;90(2):F103–F108. doi: 10.1136/adc.2004.057547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisker A. B., Bale C., Rodrigues A., et al. High-dose vitamin A with vaccination after 6 months of age: a randomized trial. Pediatrics. 2014;134(3):e739–e748. doi: 10.1542/peds.2014-0550. [DOI] [PubMed] [Google Scholar]
- 12.van Loo-Bouwman C. A., Naber T. H. J., Schaafsma G. A review of vitamin A equivalency of β-carotene in various food matrices for human consumption. The British Journal of Nutrition. 2014;111(12):2153–2166. doi: 10.1017/s0007114514000166. [DOI] [PubMed] [Google Scholar]
- 13.Snodgrass S. R. Vitamin neurotoxicity. Molecular Neurobiology. 1992;6(1):41–73. doi: 10.1007/BF02935566. [DOI] [PubMed] [Google Scholar]
- 14.Napoli J. L. Retinoic acid: its biosynthesis and metabolism. Progress in Nucleic Acid Research and Molecular Biology. 1999;63:139–188. doi: 10.1016/s0079-6603(08)60722-9. [DOI] [PubMed] [Google Scholar]
- 15.Napoli J. L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids. 2012;1821(1):152–167. doi: 10.1016/j.bbalip.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palace V. P., Khaper N., Qin Q., Singal P. K. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radical Biology and Medicine. 1999;26(5-6):746–761. doi: 10.1016/s0891-5849(98)00266-4. [DOI] [PubMed] [Google Scholar]
- 17.Li Y., Wongsiriroj N., Blaner W. S. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surgery and Nutrition. 2014;3(3):126–139. doi: 10.3978/j.issn.2304-3881.2014.05.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piskunov A., Al Tanoury Z., Rochette-Egly C. The Biochemistry of Retinoic Acid Receptors I: Structure, Activation, and Function at the Molecular Level. Vol. 70. Dordrecht, The Netherlands: Springer; 2014. Nuclear and extra-nuclear effects of retinoid acid receptors: how they are interconnected; pp. 103–127. (Subcellular Biochemistry). [DOI] [PubMed] [Google Scholar]
- 19.Evans R. M., Mangelsdorf D. J. Nuclear receptors, RXR, and the big bang. Cell. 2014;157(1):255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Lintig J. Provitamin A metabolism and functions in mammalian biology. American Journal of Clinical Nutrition. 2012;96(5):1234S–1244S. doi: 10.3945/ajcn.112.034629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore T. Vitamin A and carotene: VI. The conversion of carotene to vitamin A in vivo . Biochemical Journal. 1930;24(3):692–702. doi: 10.1042/bj0240692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palczewski K. G protein-coupled receptor rhodopsin. Annual Review of Biochemistry. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Lintig J., Kiser P. D., Golczak M., Palczewski K. The biochemical and structural basis for trans-to-cis isomerization of retinoids in the chemistry of vision. Trends in Biochemical Sciences. 2010;35(7):400–410. doi: 10.1016/j.tibs.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olson J. A., Gunning D. The storage form of vitamin A in rat liver cells. Journal of Nutrition. 1983;113(11):2184–2191. doi: 10.1093/jn/113.11.2184. [DOI] [PubMed] [Google Scholar]
- 25.Reboul E. Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins. Nutrients. 2013;5(9):3563–3581. doi: 10.3390/nu5093563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borel P. Factors affecting intestinal absorption of highly lipophilic food microconstituents (fat-soluble vitamins, carotenoids and phytosterols) Clinical Chemistry and Laboratory Medicine. 2003;41(8):979–994. doi: 10.1515/CCLM.2003.151. [DOI] [PubMed] [Google Scholar]
- 27.Tyssandier V., Reboul E., Dumas J.-F., et al. Processing of vegetable-borne carotenoids in the human stomach and duodenum. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2003;284(6):G913–G923. doi: 10.1152/ajpgi.00410.2002. [DOI] [PubMed] [Google Scholar]
- 28.Borel P., Pasquier B., Armand M., et al. Processing of vitamin A and E in the human gastrointestinal tract. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2001;280(1):G95–G103. doi: 10.1152/ajpgi.2001.280.1.G95. [DOI] [PubMed] [Google Scholar]
- 29.Carrière F., Barrowman J. A., Verger R., Laugier R. Secretion and contribution to lipolysis of gastric and pancreatic lipases during a test meal in humans. Gastroenterology. 1993;105(3):876–888. doi: 10.1016/0016-5085(93)90908-u. [DOI] [PubMed] [Google Scholar]
- 30.Lombardo D., Guy O. Studies on the substrate specificity of a carboxyl ester hydrolase from human pancreatic juice. II. Action on cholesterol esters and lipid-soluble vitamin esters. Biochimica et Biophysica Acta. 1980;611(1):147–155. doi: 10.1016/0005-2744(80)90050-9. [DOI] [PubMed] [Google Scholar]
- 31.Zahalka H. A., Cheng S. C., Burton G. W., Ingold K. U. Hydrolysis of stereoisomeric alpha-tocopheryl acetates catalyzed by bovine cholesterol esterase. Biochimica et Biophysica Acta—Lipids and Lipid Metabolism. 1987;921(3):481–485. doi: 10.1016/0005-2760(87)90075-0. [DOI] [PubMed] [Google Scholar]
- 32.Lauridsen C., Hedemann M. S., Jensen S. K. Hydrolysis of tocopheryl and retinyl esters by porcine carboxyl ester hydrolase is affected by their carboxylate moiety and bile acids. Journal of Nutritional Biochemistry. 2001;12(4):219–224. doi: 10.1016/s0955-2863(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 33.Sivakumar B., Reddy V. Absorption of labelled vitamin A in children during infection. British Journal of Nutrition. 1972;27(2):299–304. doi: 10.1079/bjn19720094. [DOI] [PubMed] [Google Scholar]
- 34.O'Neill M. E., Thurnham D. I. Intestinal absorption of β-carotene, lycopene and lutein in men and women following a standard meal: response curves in the triacylglycerol-rich lipoprotein fraction. British Journal of Nutrition. 1998;79(2):149–159. doi: 10.1079/bjn19980026. [DOI] [PubMed] [Google Scholar]
- 35.Novotny J. A., Dueker S. R., Zech L. A., Clifford A. J. Compartmental analysis of the dynamics of β-carotene metabolism in an adult volunteer. Journal of Lipid Research. 1995;36(8):1825–1838. [PubMed] [Google Scholar]
- 36.van Vliet T., Schreurs W. H. P., van den Berg H. Intestinal β-carotene absorption and cleavage in men: response of β-carotene and retinyl esters in the triglyceride-rich lipoprotein fraction after a single oral dose of β-carotene. The American Journal of Clinical Nutrition. 1995;62(1):110–116. doi: 10.1093/ajcn/62.1.110. [DOI] [PubMed] [Google Scholar]
- 37.van Lieshout M., West C. E., van Breemen R. B. Isotopic tracer techniques for studying the bioavailability and bioefficacy of dietary carotenoids, particularly β-carotene, in humans: a review. The American Journal of Clinical Nutrition. 2003;77(1):12–28. doi: 10.1093/ajcn/77.1.12. [DOI] [PubMed] [Google Scholar]
- 38.Faulks R. M., Hart D. J., Wilson P. D. G., Scott K. J., Southon S. Absorption of all-trans and 9-cis β-carotene in human ileostomy volunteers. Clinical Science. 1997;93(6):585–591. doi: 10.1042/cs0930585. [DOI] [PubMed] [Google Scholar]
- 39.Quick T. C., Ong D. E. Vitamin A metabolism in the human intestinal Caco-2 cell line. Biochemistry. 1990;29(50):11116–11123. doi: 10.1021/bi00502a015. [DOI] [PubMed] [Google Scholar]
- 40.Kawaguchi R., Yu J., Honda J., et al. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315(5813):820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 41.Hollander D., Ruble P. E., Jr. beta-carotene intestinal absorption: bile, fatty acid, pH, and flow rate effects on transport. The American Journal of Physiology. 1978;235(6):E686–691. doi: 10.1152/ajpendo.1978.235.6.E686. [DOI] [PubMed] [Google Scholar]
- 42.Sauvant P., Mekki N., Charbonnier M., Portugal H., Lairon D., Borel P. Amounts and types of fatty acids in meals affect the pattern of retinoids secreted in human chylomicrons after a high-dose preformed vitamin A intake. Metabolism: Clinical and Experimental. 2003;52(4):514–519. doi: 10.1053/meta.2003.50082. [DOI] [PubMed] [Google Scholar]
- 43.Castenmiller J. J. M., West C. E. Bioavailability and bioconversion of carotenoids. Annual Review of Nutrition. 1998;18:19–38. doi: 10.1146/annurev.nutr.18.1.19. [DOI] [PubMed] [Google Scholar]
- 44.Napoli J. L. Retinoic acid biosynthesis and metabolism. FASEB Journal. 1996;10(9):993–1001. doi: 10.1096/fasebj.10.9.8801182. [DOI] [PubMed] [Google Scholar]
- 45.Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochemical Journal. 2000;348(3):481–495. doi: 10.1042/0264-6021:3480481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zetterstrom R. H. Localization of cellular retinoid-binding proteins suggests specific roles for retinoids in the adult central nervous system. Neuroscience. 1994;62(3):899–918. doi: 10.1016/0306-4522(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 47.Blomhoff R., Blomhoff H. K. Overview of retinoid metabolism and function. Journal of Neurobiology. 2006;66(7):606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 48.Vergara M. N., Arsenijevic Y., del Rio-Tsonis K. CNS regeneration: a morphogen's tale. Journal of Neurobiology. 2005;64(4):491–507. doi: 10.1002/neu.20158. [DOI] [PubMed] [Google Scholar]
- 49.McCaffery P., Drager U. C. High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(16):7772–7776. doi: 10.1073/pnas.91.16.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krȩzel W., Ghyselinck N., Samad T. A., et al. Impaired locomotion and dopamine signaling in retinoid receptor mutant mice. Science. 1998;279(5352):863–867. doi: 10.1126/science.279.5352.863. [DOI] [PubMed] [Google Scholar]
- 51.Urano S., Inomori Y., Sugawara T., et al. Vitamin E: inhibition of retinol-induced hemolysis and membrane-stabilizing behavior. Journal of Biological Chemistry. 1992;267(26):18365–18370. [PubMed] [Google Scholar]
- 52.Krishnamurthy S., George T., Bai N. J. Hydroxy radical involvement in retinol hemolysis of human erythrocytes in vitro . Indian Journal of Biochemistry and Biophysics. 1984;21(6):397–399. [PubMed] [Google Scholar]
- 53.Goodall A. H., Fisher D., Lucy J. A. Cell fusion, haemolysis and mitochondrial swelling induced by retinol and derivatives. Biochimica et Biophysica Acta. 1980;595(1):9–14. doi: 10.1016/0005-2736(80)90242-4. [DOI] [PubMed] [Google Scholar]
- 54.Hogan C. J., Wiley J. S., Billington T. Intravascular haemolysis complicating treatment of acute promyelocytic leukaemia with all-trans retinoic acid (ATRA) Australian and New Zealand Journal of Medicine. 1997;27(4):450–451. doi: 10.1111/j.1445-5994.1997.tb02213.x. [DOI] [PubMed] [Google Scholar]
- 55.Rigobello M. P., Scutari G., Friso A., Barzon E., Artusi S., Bindoli A. Mitochondrial permeability transition and release of cytochrome c induced by retinoic acids. Biochemical Pharmacology. 1999;58(4):665–670. doi: 10.1016/s0006-2952(99)00149-5. [DOI] [PubMed] [Google Scholar]
- 56.Green D. R., Galluzzi L., Kroemer G. Metabolic control of cell death. Science. 2014;345(6203) doi: 10.1126/science.1250256.1250256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klamt F., de Oliveira M. R., Moreira J. C. F. Retinol induces permeability transition and cytochrome c release from rat liver mitochondria. Biochimica et Biophysica Acta: General Subjects. 2005;1726(1):14–20. doi: 10.1016/j.bbagen.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 58.Klamt F., dal-Pizzol F., Gelain D. P., et al. Vitamin A treatment induces apoptosis through an oxidant-dependent activation of the mitochondrial pathway. Cell Biology International. 2008;32(1):100–106. doi: 10.1016/j.cellbi.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 59.Silva F. S. G., Ribeiro M. P. C., Santos M. S., Rocha-Pereira P., Santos-Silva A., Custódio J. B. A. Acitretin affects bioenergetics of liver mitochondria and promotes mitochondrial permeability transition: potential mechanisms of hepatotoxicity. Toxicology. 2013;306:93–100. doi: 10.1016/j.tox.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 60.Sawada O., Perusek L., Kohno H., et al. All-trans-retinal induces Bax activation via DNA damage to mediate retinal cell apoptosis. Experimental Eye Research. 2014;123:27–36. doi: 10.1016/j.exer.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maeda A., Maeda T., Golczak M., Palczewski K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. Journal of Biological Chemistry. 2008;283(39):26684–26693. doi: 10.1074/jbc.M804505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El Hajj H., Khalil B., Ghandour B., et al. Preclinical efficacy of the synthetic retinoid ST1926 for treating adult T-cell leukemia/lymphoma. Blood. 2014;124(13):2072–2080. doi: 10.1182/blood-2014-03-560060. [DOI] [PubMed] [Google Scholar]
- 63.de Oliveira M. R., Silvestrin R. B., Mello E Souza T., Moreira J. C. F. Oxidative stress in the hippocampus, anxiety-like behavior and decreased locomotory and exploratory activity of adult rats: effects of sub acute vitamin A supplementation at therapeutic doses. NeuroToxicology. 2007;28(6):1191–1199. doi: 10.1016/j.neuro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 64.de Oliveira M. R., de Bittencourt Pasquali M. A., Silvestrin R. B., Mello e Souza T., Moreira J. C. F. Vitamin A supplementation induces a prooxidative state in the striatum and impairs locomotory and exploratory activity of adult rats. Brain Research. 2007;1169(1):112–119. doi: 10.1016/j.brainres.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 65.de Oliveira M. R., Moreira J. C. F. Acute and chronic vitamin A supplementation at therapeutic doses induces oxidative stress in submitochondrial particles isolated from cerebral cortex and cerebellum of adult rats. Toxicology Letters. 2007;173(3):145–150. doi: 10.1016/j.toxlet.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 66.de Oliveira M. R., Silvestrin R. B., Mello e Souza T., Moreira J. C. F. Therapeutic vitamin A doses increase the levels of markers of oxidative insult in substantia nigra and decrease locomotory and exploratory activity in rats after acute and chronic supplementation. Neurochemical Research. 2008;33(3):378–383. doi: 10.1007/s11064-007-9438-2. [DOI] [PubMed] [Google Scholar]
- 67.de Oliveira M. R., Oliveira M. W. S., Behr G. A., Hoff M. L. M., da Rocha R. F., Moreira J. C. F. Evaluation of the effects of vitamin A supplementation on adult rat substantia nigra and striatum redox and bioenergetic states: mitochondrial impairment, increased 3-nitrotyrosine and alpha-synuclein, but decreased D2 receptor contents. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(2):353–362. doi: 10.1016/j.pnpbp.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 68.De Oliveira M. R., Oliveira M. W. S., Da Rocha R. F., Moreira J. C. F. Vitamin A supplementation at pharmacological doses induces nitrosative stress on the hypothalamus of adult Wistar rats. Chemico-Biological Interactions. 2009;180(3):407–413. doi: 10.1016/j.cbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 69.de Oliveira M. R., Oliveira M. W. S., Behr G. A., Moreira J. C. F. Vitamin A supplementation at clinical doses induces a dysfunction in the redox and bioenergetics states, but did change neither caspases activities nor TNF-α levels in the frontal cortex of adult Wistar rats. Journal of Psychiatric Research. 2009;43(8):754–762. doi: 10.1016/j.jpsychires.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 70.de Oliveira M. R., da Rocha R. F., Moreira J. C. F. Increased susceptibility of mitochondria isolated from frontal cortex and hippocampus of vitamin A-treated rats to non-aggregated amyloid-β peptides 1–40 and 1–42. Acta Neuropsychiatrica. 2012;24(2):101–108. doi: 10.1111/j.1601-5215.2011.00588.x. [DOI] [PubMed] [Google Scholar]
- 71.de Oliveira M. R., da Rocha R. F., Stertz L., et al. Total and mitochondrial nitrosative stress, decreased brain-derived neurotrophic factor (BDNF) levels and glutamate uptake, and evidence of endoplasmic reticulum stress in the hippocampus of vitamin A-treated rats. Neurochemical Research. 2011;36(3):506–517. doi: 10.1007/s11064-010-0372-3. [DOI] [PubMed] [Google Scholar]
- 72.de Oliveira M. R., Soares Oliveira M. W., Müller Hoff M. L., Behr G. A., da Rocha R. F., Fonseca Moreira J. C. Evaluation of redox and bioenergetics states in the liver of vitamin A-treated rats. European Journal of Pharmacology. 2009;610(1–3):99–105. doi: 10.1016/j.ejphar.2009.03.046. [DOI] [PubMed] [Google Scholar]
- 73.de Oliveira M. R., Oliveira M. W. S., Lorenzi R., Fagundes da Rocha R., Fonseca Moreira J. C. Short-term vitamin A supplementation at therapeutic doses induces a pro-oxidative state in the hepatic environment and facilitates calcium-ion-induced oxidative stress in rat liver mitochondria independently from permeability transition pore formation: detrimental effects of vitamin A supplementation on rat liver redox and bioenergetic states homeostasis. Cell Biology and Toxicology. 2009;25(6):545–560. doi: 10.1007/s10565-008-9111-9. [DOI] [PubMed] [Google Scholar]
- 74.da Rocha R. F., de Oliveira M. R., Schonhofen P., Schnorr C. E., Dal Pizzol F., Moreira J. C. F. Long-term vitamin A supplementation at therapeutic doses induces mitochondrial electrons transfer chain (METC) impairment and increased mitochondrial membrane-enriched fraction (MMEF) 3-nitrotyrosine on rat heart. Free Radical Research. 2010;44(5):505–512. doi: 10.3109/10715761003636849. [DOI] [PubMed] [Google Scholar]
- 75.de Bittencourt Pasquali M. A., de Oliveira M. R., de Bastiani M. A., et al. L-NAME co-treatment prevent oxidative damage in the lung of adult Wistar rats treated with vitamin A supplementation. Cell Biochemistry and Function. 2012;30(3):256–263. doi: 10.1002/cbf.2791. [DOI] [PubMed] [Google Scholar]
- 76.de Oliveira M. R., Moreira J. C. F. Impaired redox state and respiratory chain enzyme activities in the cerebellum of vitamin A-treated rats. Toxicology. 2008;253(1–3):125–130. doi: 10.1016/j.tox.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 77.de Oliveira M. R., da Rocha R. F., Schnorr C. E., Moreira J. C. F. L-NAME cotreatment did prevent neither mitochondrial impairment nor behavioral abnormalities in adult Wistar rats treated with vitamin A supplementation. Fundamental and Clinical Pharmacology. 2012;26(4):513–529. doi: 10.1111/j.1472-8206.2011.00943.x. [DOI] [PubMed] [Google Scholar]
- 78.de Oliveira M. R., Lorenzi R., Schnorr C. E., Morrone M., Moreira J. C. F. Increased 3-nitrotyrosine levels in mitochondrial membranes and impaired respiratory chain activity in brain regions of adult female rats submitted to daily vitamin A supplementation for 2 months. Brain Research Bulletin. 2011;86(3-4):246–253. doi: 10.1016/j.brainresbull.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 79.Grivennikova V. G., Vinogradov A. D. Generation of superoxide by the mitochondrial complex I. Biochimica et Biophysica Acta. 2006;1757(5-6):553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 80.Andreyev A. Y., Kushnareva Y. E., Starkov A. A. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 81.de Oliveira M. R., Oliveira M. W. S., Moreira J. C. F. Pharmacological doses of vitamin A increase caspase-3 activity selectively in cerebral cortex. Fundamental & Clinical Pharmacology. 2010;24(4):445–450. doi: 10.1111/j.1472-8206.2009.00789.x. [DOI] [PubMed] [Google Scholar]
- 82.Radi R. Peroxynitrite, a stealthy biological oxidant. The Journal of Biological Chemistry. 2013;288(37):26464–26472. doi: 10.1074/jbc.r113.472936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carballal S., Bartesaghi S., Radi R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochimica et Biophysica Acta. 2014;1840(2):768–780. doi: 10.1016/j.bbagen.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giasson B. I., Duda J. E., Murray I. V. J., et al. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 85.Souza J. M., Giasson B. I., Chen Q., Lee V. M.-Y., Ischiropoulos H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. The Journal of Biological Chemistry. 2000;275(24):18344–18349. doi: 10.1074/jbc.m000206200. [DOI] [PubMed] [Google Scholar]
- 86.Eiserich J. P., Estévez A. G., Bamberg T. V., Chumley P. H., Beckman J. S., Freeman B. A. Microtubule dysfunction by posttranslational nitrotyrosination of α- tubulin: a nitric oxide-dependent mechanism of cellular injury. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6365–6370. doi: 10.1073/pnas.96.11.6365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry. 2006;97(6):1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 88.Bir A., Sen O., Anand S., et al. α-synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: implications in the pathogenesis of Parkinson's disease. Journal of Neurochemistry. 2014;131(6):868–877. doi: 10.1111/jnc.12966. [DOI] [PubMed] [Google Scholar]
- 89.Robotta M., Gerding H. R., Vogel A., et al. Alpha-synuclein binds to the inner membrane of mitochondria in an α-helical conformation. ChemBioChem. 2014;15(17):2499–2502. doi: 10.1002/cbic.201402281. [DOI] [PubMed] [Google Scholar]
- 90.de Oliveira M. R., da Rocha R. F., Pasquali M. A. D. B., Moreira J. C. F. The effects of vitamin A supplementation for 3 months on adult rat nigrostriatal axis: increased monoamine oxidase enzyme activity, mitochondrial redox dysfunction, increased β-amyloid1-40 peptide and TNF-α contents, and susceptibility of mitochondria to an in vitro H2O2 challenge. Brain Research Bulletin. 2012;87(4-5):432–444. doi: 10.1016/j.brainresbull.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 91.Edmondson D. E. Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: biological implications. Current Pharmaceutical Design. 2014;20(2):155–160. doi: 10.2174/13816128113190990406. [DOI] [PubMed] [Google Scholar]
- 92.Boveris A., Chance B. The mitochondrial generation of hydrogen peroxide. Biochemical Journal. 1973;134(3):707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veal E. A., Day A. M., Morgan B. A. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26(1):1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 94.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nature Immunology. 2002;3(12):1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 95.Rhee S. G., Kang S. W., Jeong W., Chang T.-S., Yang K.-S., Woo H. A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Current Opinion in Cell Biology. 2005;17(2):183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 96.Stone J. R., Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants and Redox Signaling. 2006;8(3-4):243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 97.Halliwell B. Free radicals and antioxidants: updating a personal view. Nutrition Reviews. 2012;70(5):257–265. doi: 10.1111/j.1753-4887.2012.00476.x. [DOI] [PubMed] [Google Scholar]
- 98.Kono Y., Fridovich I. Superoxide radical inhibits catalase. The Journal of Biological Chemistry. 1982;257(10):5751–5754. [PubMed] [Google Scholar]
- 99.Dourado D. F. A. R., Fernandes P. A., Ramos M. J. Mammalian cytosolic glutathione transferases. Current Protein and Peptide Science. 2008;9(4):325–337. doi: 10.2174/138920308785132677. [DOI] [PubMed] [Google Scholar]
- 100.Rashid K., Sinha K., Sil P. C. An update on oxidative stress-mediated organ pathophysiology. Food and Chemical Toxicology. 2013;62:584–600. doi: 10.1016/j.fct.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 101.Szkudelski T., Okulicz M., Bialik I., Szkudelska K. The influence of fasting on liver sulfhydryl groups, glutathione peroxidase and glutathione-S-transferase activities in the rat. Journal of Physiology and Biochemistry. 2004;60(1):1–6. doi: 10.1007/BF03168215. [DOI] [PubMed] [Google Scholar]
- 102.Sian-Hülsmann J., Mandel S., Youdim M. B. H., Riederer P. The relevance of iron in the pathogenesis of Parkinson's disease. Journal of Neurochemistry. 2011;118(6):939–957. doi: 10.1111/j.1471-4159.2010.07132.x. [DOI] [PubMed] [Google Scholar]
- 103.Friedman A., Galazka-Friedman J., Koziorowski D. Iron as a cause of Parkinson disease—a myth or a well established hypothesis? Parkinsonism and Related Disorders. 2009;15(supplement 3):S212–S214. doi: 10.1016/s1353-8020(09)70817-x. [DOI] [PubMed] [Google Scholar]
- 104.Politis M. Neuroimaging in Parkinson disease: from research setting to clinical practice. Nature Reviews Neurology. 2014;10(12):708–722. doi: 10.1038/nrneurol.2014.205. [DOI] [PubMed] [Google Scholar]
- 105.Girault J.-A. Signaling in striatal neurons: the phosphoproteins of reward, addiction, and dyskinesia. Progress in Molecular Biology and Translational Science. 2012;106:33–62. doi: 10.1016/b978-0-12-396456-4.00006-7. [DOI] [PubMed] [Google Scholar]
- 106.Duchen M. R. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28(5-6):339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- 107.Galluzzi L., Bravo-San Pedro J. M., Kroemer G. Organelle-specific initiation of cell death. Nature Cell Biology. 2014;16(8):728–736. doi: 10.1038/ncb3005. [DOI] [PubMed] [Google Scholar]
- 108.Suen D.-F., Norris K. L., Youle R. J. Mitochondrial dynamics and apoptosis. Genes & Development. 2008;22(12):1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Manczak M., Anekonda T. S., Henson E., Park B. S., Quinn J., Reddy P. H. Mitochondria are a direct site of Aβ accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Human Molecular Genetics. 2006;15(9):1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 110.Chen X., Yan S. D. Mitochondrial Aβ: a potential cause of metabolic dysfunction in Alzheimer's disease. IUBMB Life. 2006;58(12):686–694. doi: 10.1080/15216540601047767. [DOI] [PubMed] [Google Scholar]
- 111.Pavlov P. F., Petersen C. H., Glaser E., Ankarcrona M. Mitochondrial accumulation of APP and Aβ: significance for Alzheimer disease pathogenesis. Journal of Cellular and Molecular Medicine. 2009;13(10):4137–4145. doi: 10.1111/j.1582-4934.2009.00892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Du H., Guo L., Fang F., et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nature Medicine. 2008;14(10):1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yao J., Irwin R. W., Zhao L., Nilsen J., Hamilton R. T., Brinton R. D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(34):14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boyce V. S., Mendell L. M. Neurotrophins and spinal circuit function. Frontiers in Neural Circuits. 2014;8, article 59 doi: 10.3389/fncir.2014.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lu B., Nagappan G., Lu Y. Neurotrophic Factors. Vol. 220. Springer; 2014. BDNF and synaptic plasticity, cognitive function, and dysfunction; pp. 223–250. (Handbook of Experimental Pharmacology). [DOI] [PubMed] [Google Scholar]
- 116.Poo M. M. Neurotrophins as synaptic modulators. Nature Reviews Neuroscience. 2001;2(1):24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 117.Burkhalter J., Fiumelli H., Allaman I., Chatton J.-Y., Martin J.-L. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. Journal of Neuroscience. 2003;23(23):8212–8220. doi: 10.1523/JNEUROSCI.23-23-08212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Markham A., Cameron I., Franklin P., Spedding M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. European Journal of Neuroscience. 2004;20(5):1189–1196. doi: 10.1111/j.1460-9568.2004.03578.x. [DOI] [PubMed] [Google Scholar]
- 119.Bremner J. D., Fani N., Ashraf A., et al. Functional brain imaging alterations in acne patients treated with isotretinoin. American Journal of Psychiatry. 2005;162(5):983–991. doi: 10.1176/appi.ajp.162.5.983. [DOI] [PubMed] [Google Scholar]
- 120.Wolverton S. E., Harper J. C. Important controversies associated with isotretinoin therapy for acne. American Journal of Clinical Dermatology. 2013;14(2):71–76. doi: 10.1007/s40257-013-0014-z. [DOI] [PubMed] [Google Scholar]
- 121.Ballester Sánchez R., De Unamuno Bustos B., Agustí Mejías A., Febrer Bosch M. I. Increase in creatine phosphokinase and a suicide attempt during isotretinoin treatment. Anales de Pediatría. 2012;76(6):365–366. doi: 10.1016/j.anpedi.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 122.Saitta P., Keehan P., Yousif J., Way B. V., Grekin S., Brancaccio R. An update on the presence of psychiatric comorbidities in acne patients, part 2: depression, anxiety, and suicide. Cutis. 2011;88(2):92–97. [PubMed] [Google Scholar]
- 123.Wysowski D. K., Pitts M., Beitz J. An analysis of reports of depression and suicide in patients treated with isotretinoin. Journal of the American Academy of Dermatology. 2001;45(4):515–519. doi: 10.1067/mjd.2001.117730. [DOI] [PubMed] [Google Scholar]
- 124.Nevoralová Z., Dvořáková D. Mood changes, depression and suicide risk during isotretinoin treatment: a prospective study. International Journal of Dermatology. 2013;52(2):163–168. doi: 10.1111/j.1365-4632.2011.05334.x. [DOI] [PubMed] [Google Scholar]
- 125.Bjelakovic G., Nikolova D., Gluud L. L., Simonetti R. G., Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. Journal of the American Medical Association. 2007;297(8):842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 126.Bjelakovic G., Nikolova D., Gluud C. Meta-regression analyses, meta-analyses, and trial sequential analyses of the effects of supplementation with beta-carotene, vitamin A, and vitamin E singly or in different combinations on all-cause mortality: do we have evidence for lack of harm? PloS one. 2013;8(9) doi: 10.1371/journal.pone.0074558.e74558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bjelakovic G., Nikolova D., Gluud C. Antioxidant supplements and mortality. Current Opinion and Clinical Nutrition and Metabolic Care. 2014;17(1):40–44. doi: 10.1097/MCO.0000000000000009. [DOI] [PubMed] [Google Scholar]
- 128.Humphrey J. H., Iliff P. J., Marinda E. T., et al. Effects of a single large dose of vitamin A, given during the postpartum period to HIV-positive women and their infants, on child HIV infection, HIV-free survival, and mortality. Journal of Infectious Diseases. 2006;193(6):860–871. doi: 10.1086/500366. [DOI] [PubMed] [Google Scholar]
- 129.Orfanos C. E., Zouboulis C. C., Almond-Roesler B., Geilen C. C. Current use and future potential role of retinoids in dermatology. Drugs. 1997;53(3):358–388. doi: 10.2165/00003495-199753030-00003. [DOI] [PubMed] [Google Scholar]
- 130.Li J., Lee A. S. Stress induction of GRP78/BiP and its role in cancer. Current Molecular Medicine. 2006;6(1):45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 131.Gutiérrez T., Simmen T. Endoplasmic reticulum chaperones and oxidoreductases: critical regulators of tumor cell survival and immunorecognition. Frontiers in Oncology. 2014;4, article 291 doi: 10.3389/fonc.2014.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Borutaite V., Morkuniene R., Brown G. C. Release of cytochrome c from heart mitochondria is induced by high Ca2+ and peroxynitrite and is responsible for Ca2+-induced inhibition of substrate oxidation. Biochimica et Biophysica Acta. 1999;1453(1):41–48. doi: 10.1016/s0925-4439(98)00082-9. [DOI] [PubMed] [Google Scholar]
- 133.Crandall J., Sakai Y., Zhang J., et al. 13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):5111–5116. doi: 10.1073/pnas.0306336101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sakai Y., Crandall J. E., Brodsky J., McCaffery P. 13-cis retinoic acid (accutane) suppresses hippocampal cell survival in mice. Annals of the New York Academy of Sciences. 2004;1021:436–440. doi: 10.1196/annals.1308.059. [DOI] [PubMed] [Google Scholar]
- 135.Guruvayoorappan C., Pradeep C. R., Kuttan G. 13-cis-Retinoic acid induces apoptosis by modulating caspase-3, bcl-2, and p53 gene expression and regulates the activation of transcription factors in B16F-10 melanoma cells. Journal of Environmental Pathology, Toxicology and Oncology. 2008;27(3):197–207. doi: 10.1615/jenvironpatholtoxicoloncol.v27.i3.40. [DOI] [PubMed] [Google Scholar]
- 136.Bannerman D. M., Grubb M., Deacon R. M. J., Yee B. K., Feldon J., Rawlins J. N. P. Ventral hippocampal lesions affect anxiety but not spatial learning. Behavioural Brain Research. 2003;139(1-2):197–213. doi: 10.1016/s0166-4328(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 137.Bannerman D. M., Rawlins J. N. P., McHugh S. B., et al. Regional dissociations within the hippocampus—memory and anxiety. Neuroscience and Biobehavioral Reviews. 2004;28(3):273–283. doi: 10.1016/j.neubiorev.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 138.Deacon R. M. J., Rawlins J. N. P. Hippocampal lesions, species-typical behaviours and anxiety in mice. Behavioural Brain Research. 2005;156(2):241–249. doi: 10.1016/j.bbr.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 139.Lotharius J., Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nature reviews. Neuroscience. 2002;3(12):932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 140.Salminen L. E., Paul R. H. Oxidative stress and genetic markers of suboptimal antioxidant defense in the aging brain: a theoretical review. Reviews in the Neurosciences. 2014;25(6):805–819. doi: 10.1515/revneuro-2014-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Omenn G. S., Goodman G. E., Thornquist M. D., et al. Risk factors for lung cancer and for intervention effects in CARET, the beta-carotene and retinol efficacy trial. Journal of the National Cancer Institute. 1996;88(21):1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]