

CASE PRESENTATION
History of Present Illness
This patient was a former 32 week premature female infant, born by elective caesarian section due to concern for hydrops. After delivery she was found to have ascites and ultimately conjugated hyperbilirubinemia, but was without other congenital abnormalities. She had a normal newborn screen and no family history of congenital or metabolic disorders. An extensive evaluation for biliary atresia, perinatal iron storage disorders, Wilson disease, alpha-1 antitrypsin, and metabolic diseases was unrevealing. Liver biopsy on day of life 25 showed mild chronic hepatitis with portal and periportal fibrosis and mild persistence of extramedullary hematopoiesis. She had a prolonged neonatal intensive care unit stay secondary to respiratory insufficiency, and remained on supplemental oxygen at time of discharge. Throughout her hospitalization she had persistently elevated C-reactive protein as well as aminotransferases and direct bilirubin, although these had stabilized prior to discharge.
Starting at two months of age, she developed recurrent episodes of fever, respiratory distress, abdominal distension and feeding intolerance, lasting 5–7 days and occurring every 3–5 weeks, and beginning shortly after her first round of immunizations. During episodes she developed transient hepatosplenomegaly and ascites, elevated aminotransferases and CRP, anemia and thrombocytopenia. Empiric antibiotics were typically started, but all cultures were negative. With her third episode she required mechanical ventilation for 5 weeks. During this prolonged hospitalization an extensive diagnostic evaluation was pursued, summarized in Table 1. Repeat liver biopsy was performed showing chronic hepatitis with moderate periportal and pericellular fibrosis. Bone marrow biopsy showed mild granulocytic hyperplasia, mild dyserythropoiesis, and increased interstitial histiocytes without hemophagocytic activity. Ultimately, due to suspected autoimmune process, methylprednisolone 2mg/kg/day was administered, after which she was weaned off all respiratory support. However, as steroids were weaned she continued to have febrile episodes lasting several days, which were managed with increasing steroids and empiric antibiotics. Due to a finding of reduced transitional B cells and concern for B cell immunodeficiency, at age 9 months she was started on monthly intravenous immunoglobulin therapy; however this did not alter the frequency of her febrile episodes.
Table 1.
| Test | Result |
|---|---|
| MPV 17-related disorder | No mutations found |
| Glycogen storage disease type IV GBE1 sequencing | No mutations found |
| CFTR mutation analysis | No mutations found |
| Transferrin isoelectric focusing for congenital disorders of glycosylation | Normal |
| Lysosomal enzyme screen | Normal |
| Lactate and pyruvate | Normal |
| Very long chain fatty acids | Normal |
| Anti-enterocyte antibody | Weak positive (1:20) |
| TTG IgA | Negative |
| Endomysial IgA | Negative |
| Gliadin IgA and IgG | Negative |
| Anti-nuclear antibody | Negative |
| Anti-neutrophil cytoplasmic antibody | Negative |
| Anti-Smooth muscle antibody | Negative |
| Anti-mitochondrial M2 antibody | Negative |
| Anti-liver cytosol or soluble antibodies | Negative |
| Anti LKM antibody | Negative |
| Anti-F-actin antibody | Negative |
| Quantitative immunoglobulins | IgG 1833 (286-1680), IgA 55 (10-131), IgM 295 (21-192) |
| IgG subclasses | IgG1 1520 (143-394), IgG2 100 (23-147), IgG3 154 (4-70), IgG4 1 (1-14) |
| Lymphocyte subpopulations | Low B-lineage lymphocyte percentages, increased CD8 T cell percentages |
| B cell populations | Decreased percentage of transitional B cells and CD5+ B cells |
| Mitogen proliferation assay | Normal |
| Parvovirus IgM and IgG | Negative |
| CMV quantitative PCR | Initial positive log 3.7, repeat negative x3 |
| EBV quantitative PCR | Negative |
| Hepatitis C virus PCR | Negative |
| Mycoplasma IgM and IgG | Negative |
| Legionella urine antigen | Negative |
| Respiratory viral panel | Negative |
Social and Family History
The patient lives with her parents and two healthy siblings. No travel outside the United States. No daycare exposure. Family history was unremarkable without autoimmune diseases, congenital abnormalities, or developmental delay.
Physical Exam
On examination at 11 months of age, patient appeared small for age but alert, interactive and without dysmorphic features. She had splenomegaly and hepatomegaly with estimated liver span of 6 cm. There were no signs of rash or arthritis. She had normal muscle tone and bulk. Developmental assessment demonstrated typical social, verbal and fine motor development but gross motor delay, with infant able to sit only with support and unable to roll over. The remainder of her physical exam was normal.
Case Summary
This is a now 11 month old former premature female infant with recurrent episodes of fever, elevated inflammatory markers, anemia, thrombocytopenia and cholestatic liver dysfunction.
Differential diagnosis
The differential diagnosis for this infant is broad, and includes infectious, inflammatory, metabolic and neoplastic processes, as well as both congenital and acquired conditions. Her extensive prior evaluation has been negative for infectious causes. Additionally, she has had negative testing for a large number of autoantibodies associated with known autoimmune disorders. Finally, her course does not clearly fit a primary immunodeficiency. Although the etiology of her mild decrease in transitional B cells is unclear her normal immunoglobulin levels, lack of sinopulmonary infections and negligible response to IVIG argue against a functional B cell deficiency.
Autoimmune hepatitis (AIH)
AIH is an idiopathic autoimmune disease characterized by high levels of immunoglobulins and presence of autoantibodies. It typically presents as an acute onset severe hepatitis often progressing quickly to liver failure. Although it typically affects older children, it can occur in infancy. The two most well described subtypes are AIH-1, associated with antinuclear or anti-smooth muscle antibodies, and AIH-2, associated with anti-LKM antibodies (1), all of which were negative in this child. Autoantibody negative AIH has been reported only rarely in children and little is known regarding its etiology and pathogenesis (2).
Metabolic diseases
Metabolic abnormalities are common causes of neonatal onset liver dysfunction; however, investigation of her liver disease including repeat biopsy has not revealed any known congenital or metabolic abnormalities. One further consideration is Niemann-Pick Disease Type C, which is a lysosomal storage disease leading to progressive organomegaly and neurologic dysfunction (3). Recent work has suggested that liver and neurologic dysfunction in Niemann-Pick is an inflammatory process, although signs of severe systemic inflammation as seen in this patient are uncommon (4). Additionally, liver biopsy did not show evidence for a storage disease. Another consideration is mevalonic aciduria (MA), a severe deficiency in mevalonate kinase in the isoprenoid biosynthesis pathway, leading to dysmorphic features, progressive neonatal onset psychomotor retardation, cerebellar ataxia, visual impairment and recurrent febrile crises (5). Additionally there are rare reports of severe liver dysfunction associated with MA (6–9).
Periodic fever syndrome
The autoinflammatory periodic fever syndromes are a family of rare, heritable disorders which share the characteristic of recurrent inflammatory episodes with no or trivial triggers. They have all been linked to uncontrolled activation of the innate immune system, most notably activation of the inflammasome leading to dysregulated production of proinflammatory cytokines including IL-1β(10). They include familial Mediterranean fever, cyropyrin-associated periodic fever syndrome, TNF-receptor associated periodic fever syndrome, and mevalonate kinase deficiency (MKD), also known as hyper immunoglubulinemia D and periodic fever syndrome (HIDS). Periodic fever syndromes typically present in early childhood, but less severely affected patients may not present until late childhood or as adults. The pattern of this patient’s inflammatory episodes, occurring irregularly every 1–2 months and lasting for several days, could be consistent with MKD (11). However, liver dysfunction is reported only rarely in MKD. Moreover, it is usually reported in more severely affected children who are classified as MA with the characteristic dysmorphic features, growth retardation, developmental delay, ocular, and neurological dysfunction (see above).
Familial hemophagocytic lymphohistiocytosis (HLH)
Primary or familial HLH is a rare, autosomal recessive condition characterized by episodes of overwhelming inflammation associated with multiorgan system dysfunction including cytopenias, coagulopathy, liver dysfunction, CNS abnormalities and hyperferritinemia (12). Its pathological hallmark is natural killer (NK) cell dysfuction, expansion of CD8+ T lymphocytes and hemophagocytic macrophages. It can be fatal, although can be cured with hematopoietic stem cell transplant. More than 75% of patients have identified mutations in the gene encoding perforin, or those encoding proteins involved in granule exocytosis including Munc13-4, syntaxin 11 or syntaxin-binding protein 2. This child’s episodes of hyperinflammation, cytopenias and liver dysfunction are consistent with HLH. Although no evidence of hemophagocytosis was found on initial bone marrow examination, this does not rule out HLH due to sampling error and since visible hemophagocytosis may take time to develop.
Systemic juvenile idiopathic arthritis (SJIA)
SJIA is a severe inflammatory disorder of childhood of unknown etiology (13). It is classified as a subtype of JIA, although it has several distinctive clinical and epidemiologic features, including a lack of sex bias and no peak age of onset. It is also unique in having prominent extra-articular features, such as prolonged spiking fevers, rash, lymphadenopathy and serositis. It is a clinical diagnosis, although it is notable for markedly elevated inflammatory markers including ferritin (14). Children with SJIA are also at significant risk for development of macrophage activation syndrome (MAS), an episode of overwhelming inflammation that is considered a form of secondary reactive HLH (15). While this patient’s clinical symptoms resemble SJIA, the fever in SJIA is typically persistent rather than periodic. In addition, there was no report of arthritis, although this can be a late feature in some patients. Finally, while liver dysfunction can be associated with MAS, it is not typically a feature of SJIA.
Clinical Course
Ultimately this patient was transferred to our institution during an episode of fever, respiratory distress and abdominal distention. Labs were notable for white blood cells 40×103/μl, hemoglobin 6.4 g/dL, platelets 159×103/μl, CRP 13 mg/dL (normal <1mg/dl), aspartate transaminase 839 U/L and alanine transaminase 1198 U/L. An evaluation for hemophagocytic syndromes was initiated. The serum ferritin was mildly elevated at 790 ng/ml (normal 15–450), soluble IL-2 receptor was elevated at 4977 U/ml (normal <3000), and fibrinogen and triglycerides were normal. NK cell functional testing revealed a decreased proportion of cells expressing CD107A upon stimulation (8%, normal 11–35%), as well as decreased intensity of CD107A surface expression (mean fluorescence intensity 178, normal 207–678), indicative of abnormal degranulation. There was also a mildly decreased proportion of NK cells expressing perforin (83%, normal 87–95%) as well as reduced perforin intensity (MFI 92, normal 98–181). Further examination of both prior liver biopsies showed unusually abundant intersinusoidal histiocytes, highlighted by CD163 with occasional cells appearing to have engulfed nucleated white blood cells including mature neutrophils and mononucleated cells likely to be nucleated red blood cell precursors (Figure 1). Genetic testing for familial HLH revealed a R28C substitution in one allele of the perforin gene PRF1. There were no polymorphisms found in genes encoding Munc 13–4, Rab27a, Syntaxin 11, or STXBP2.
Figure 1.
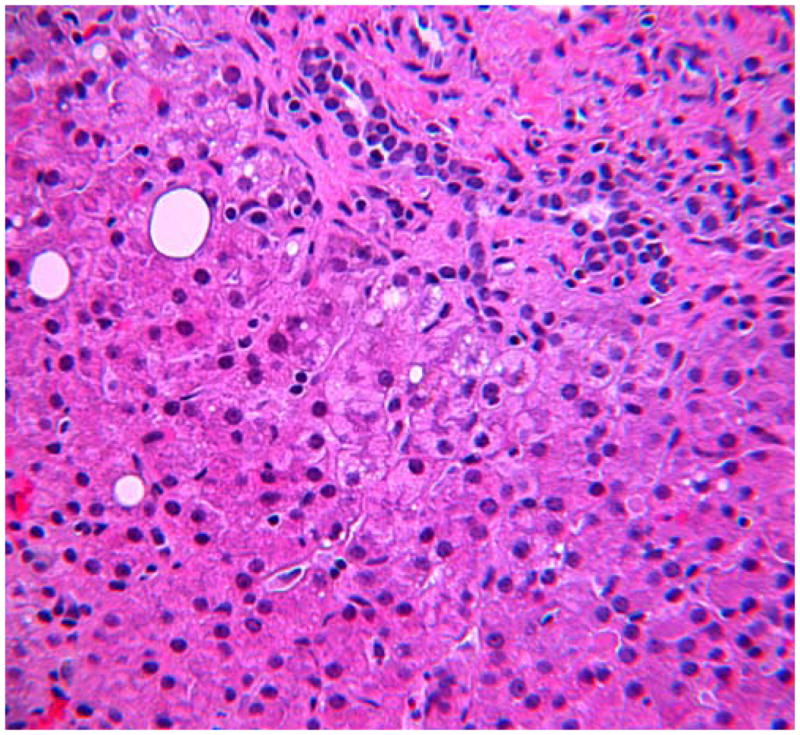
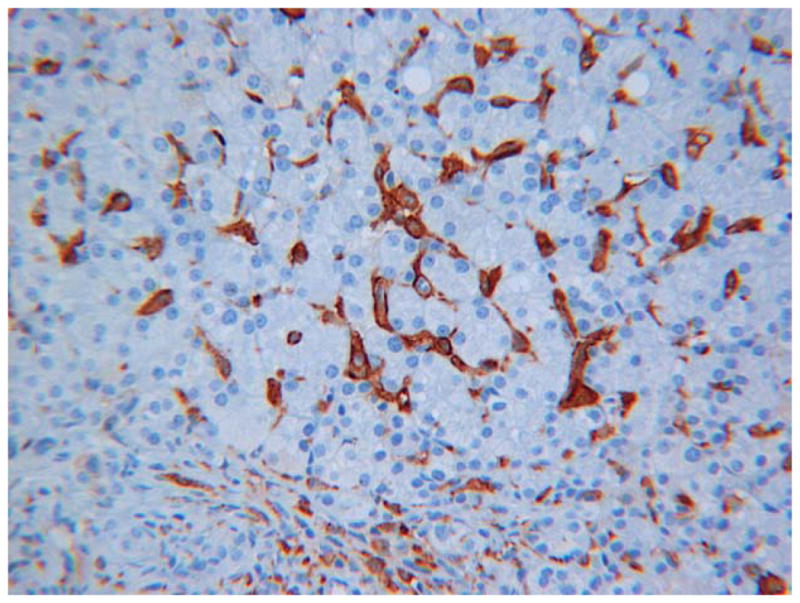

(A) H&E stained section of patients liver biopsy showing mild portal area inflammation. Zone 1 hepatocytes show non-specific reactive changes and rare lipid droplets. Lobular inflammation is deceptively minor. (B) CD163 immunostaining reveals marked increase in activated macrophages within the sinusoids of the liver. (C) Transmission electron microscopy shows erythrophagocytosis of a nucleated red blood cell (RBC) in one of three visualized reactive histiocytes (RH) obstructing the sinusoids of the liver. H=hepatocyte.
A metabolic genetics evaluation was also initiated. Urine organic acid, obtained during an inflammatory attack but not during a fever spike, confirmed a prominent peak of mevalonic acid. Genetic testing identified compound heterozygous mutations in mevalonate kinase (V310M and G336S). Given a presumed diagnosis of MKD she was started on anakinra 2mg/kg/day, and after which was without fever throughout her hospitalization. Her markers of inflammation including CRP, ESR, ferritin and sIL2R normalized, and her serum aminotransferase levels decreased. At last follow-up three months later the patient was doing well without any further episodes of fever.
Discussion
Here, we report an infant with MKD and recurrent inflammatory episodes with liver dysfunction, successfully treated with anakinra. MKD is a metabolic autoinflammatory syndrome caused by mutations in mevalonate kinase, a key enzyme in the non-sterol isoprenoid biosynthesis pathway (11). How metabolic abnormalities in MKD lead to hyperinflammation is not fully known, although defective prenylation may potentiate activation of the inflammasome (16–18). Indeed, treatment with anti-IL-1 therapy appears to be highly efficacious in these patients (19). The diagnosis of MKD can be challenging, as IgD levels may be normal, mevalonic acid excretion may be intermittent in more mild forms of the disease, and the clinical phenotype can be difficult to recognize in infancy (20, 21). Prior to establishing the molecular basis of MKD, two distinct clinical phenotypes were mapped to mutations in mevalonate kinase: hyper immunoglubulinemia D and periodic fever syndrome (HIDS) and mevalonic aciduria (MA), though these likely represent the extremes of a phenotypic spectrum (21). Children with HIDS develop recurrent episodes of fever, abdominal pain, adenopathy, hepatosplenomegaly, rash and arthralgias. In contrast, though children with MA also have recurrent inflammatory episodes, they also exhibit dysmorphic features, growth retardation, profound developmental delay, ocular, and neurological dysfunction. While the patient presented here is on the severe end of MKD spectrum, she lacks the dysmorphic features and global developmental delay classically seen in MA (11). Similarly, significant liver dysfunction as reported here has only been reported in severely affected MA patients (6–9). Although there is some genotype-phenotype correlation in MKD, these associations are imprecise (22). This child was found to be a compound heterozygote, with one allele (V310M) previously described in severely affected patients with MA (7, 22), while the other allele (G336S) has been described in a patient with HIDS (23). Taken together, this case enhances our understanding of the clinical spectrum seen in MKD, and underscores that patients can exhibit a combination of features seen in classic HIDS and MA.
There is also emerging evidence that severe febrile attacks in MKD may resemble MAS, an episode of overwhelming inflammation leading to multisystem organ dysfunction (24–26). Indeed, multiple features support this child manifesting a hemophagocytic process such as MAS. First, her episodes were associated with elevated ferritin and sIL2r, cytopenias, decreased NK cell function, and liver dysfunction; indeed, she would satisfy diagnostic criteria for HLH (27). Second, both liver biopsies showed abundant sinusoidal histiocytes with hemophagocytosis. Although there is one previous report of a child with MKD and significant liver infiltration by CD68+ monocytes, there was no evidence of hemophagocytosis (28). Finally, she had mildly reduced perforin expression along with a heterozygous variant in the perforin gene (R28C). Perforin mutations are found in 20–50% of children with familial HLH, as well as some children with systemic juvenile idiopathic arthritis (SJIA) who develop MAS (29–31). Although there are no previous reports of MAS-like syndrome occurring in association with this sequence variant, it is a rare variant found in only 5 of approximately 2400 PRF1 alleles sequenced through our Clinical Genetics Laboratory, and predicted to have radical effects on protein structure. It has also been identified in a child with SJIA without history of MAS (30). Interestingly, along with decreased perforin expression our patient was found to have abnormal NK cell degranulation, suggesting a second, unidentified, defect in NK cell function. Although the etiology of MAS remains unclear, a growing body of evidence supports cytolytic dysfunction as central to the pathogenesis of primary familial and secondary reactive HLH (30–34). One emerging theory is a two-hit hypothesis, in which MAS develops when a genetically susceptible individual, with defects in immunoregulatory mechanisms including NK cell function such as perforin, experiences a hyperinflammatory state due to infection or rheumatic disease (35). Indeed, studies of multiple independent cohorts of children with SJIA have found that approximately one third of those who develop MAS have polymorphisms in genes implicated in familial HLH, most commonly perforin (30, 36, 37).
Likewise, MAS is increasingly recognized as a complication of the periodic fever syndromes, including MKD (25, 38, 39). However, to our knowledge this is the first report of MAS associated with perforin variant in a child with an autoinflammatory periodic fever syndrome. Severe episodes of MAS are typically managed with intravenous corticosteroids and, if refractory, other immunosuppressive therapies (40). Here, while our patient did improve clinically with high-dose steroids, she continued to have inflammatory episodes with features of MAS until she achieved clinical remission with IL-1 blockade. This observation further supports the need to treat the underlying triggers for inflammation which trigger MAS, and highlights the emerging role of IL-1 antagonists in the management of MAS.
Hepatosplenomegaly is commonly reported in MKD; however, there are only rare reports of hepatitis (28, 41) or more severe cholestatic liver disease in some children with severe MA (6–9) (Table 2). These children are all on the severe end of the disease spectrum, with most progressing to death or end-stage liver disease. The pathophysiology of this liver disease is unknown, and it is unclear to what extent liver damage in these cases could be due to an MAS-like process, or whether specific immunosuppressive therapy would have led to improvement in liver function. Interestingly, there is one report of a child with MKD and severe liver dysfunction and a markedly elevated ferritin level; no further investigation for MAS/HLH was reported (7). This child underwent orthotopic liver transplant leading to significant functional improvement but continued febrile episodes, including biopsy-proven liver inflammation. Ultimately she underwent bone marrow transplant, leading to full clinical remission. That report as well as the present case is highly suggestive that in some MKD patients MAS-like episodes may underlie significant liver disease.
Table 2.
| Report | Age at presentation |
Age at diagnosis |
Genotype | Other abnormalities |
Transaminase elevation |
Liver biopsy findings | Extramedullary hematopoeisis |
Treatments | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Hinson et al (1998) | Perinatal | 2 months | ND | None noted | Yes (moderate) | Chronic active cholestatic hepatitis | Cutaneous | unknown | Hypotonia, FTT |
| Prenatal | Unknown | ND | Hypospadias, frontal bossing, low-set ears, down-slanted palpebral fissues, long eyelashes | Yes (moderate) | Marked cholestatis, portal fibrosis | Hepatic | Unknown | Died DOL 87 | |
| Raupp et al (2004) | Prenatal | Post- mortem | ND | None noted | No | ND | No | Antibiotics | Died DOL 15 |
| Prenatal | DOL 20 | L35S homozygote |
None noted | No | ND | No | Antibiotics | Died 2 months | |
| Steiner et al (2011) | Prenatal | Post- mortem | I268T homozygote |
Ventriculomegaly, short limbs, frontal bossing, cardiomegaly | unknown | Marked fibrosis with bile plugging | Cutaneous, lungs, liver, kidneys, adrenals | Antibiotics | Died DOL 20 |
| Chaudhury et al (2012) | Prenatal | 2 weeks | I268T V310M |
Aortic stenosis | Unknown | Micronodular cirrhosis | No | Anakinra, HSCT | OLT age 4, normal growth and typically developing age 8 |
Clinical characteristics, liver findings and outcome of reported patient with MKD and cholestatic liver disease
In conclusion, we report an infant with MKD and recurrent inflammatory episodes associated with liver dysfunction and histiocyte infiltration consistent with MAS. Her presentation illustrates the spectrum of disease seen in MKD, and demonstrates that patients can have severe liver disease in the absence of other features associated with MA. Taken together, we suggest that patients with MKD are at risk for MAS-like episodes, and that occurrence of liver dysfunction should lead to careful evaluation for MAS and initiation of aggressive immunosuppressive therapy for both the underlying autoinflammatory disease as well as this potentially fatal complication.
Significance and Innovation.
We present the unusual case of an 11 month old infant we evaluated for recurrent episodes of fever, elevated inflammatory markers, anemia, thrombocytopenia and cholestatic liver dysfunction, ultimately diagnosed with mevalonate deficiency. Interestingly her inflammatory episodes were characterized by extensive liver infiltration by hemophagocytic histiocytes, as well as NK cell dysfunction and perforin gene mutation, which together with other features is consistent with macrophage activation syndrome (MAS).
This case sheds light on the diagnosis of MKD by examining a patient with severe features such as liver dysfunction occurring without other features of mevalonic aciduria.
This case also highlights an emerging concept which is that hereditary periodic fever patients, particularly those with MKD, are at risk for MAS, and represents the first patient with genetically confirmed hereditary periodic fever with MAS in the setting of perforin gene mutation.
Acknowledgments
Support: Dr. Schulert is supported by K12 HL119986 from the National Institutes of Health. Dr. Grom is supported by RO1-AR059049 and PO1-AR048929 from the National Institutes of Health.
We would like to thank Dr. Amber Begtrup, Assistant Director of the Molecular Genetics Laboratory at CCHMC, for helpful discussions on PRF1 sequencing.
References
- 1.Mieli-Vergani G, Vergani D. Paediatric autoimmune liver disease. Archives of disease in childhood. 2013;98(12):1012–7. doi: 10.1136/archdischild-2013-303848. [DOI] [PubMed] [Google Scholar]
- 2.Bogdanos DP, Mieli-Vergani G, Vergani D. Autoantibodies and their antigens in autoimmune hepatitis. Seminars in liver disease. 2009;29(3):241–53. doi: 10.1055/s-0029-1233533. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Poyato MS, Pineda M. New agents and approaches to treatment in Niemann-Pick type C disease. Current pharmaceutical biotechnology. 2011;12(6):897–901. doi: 10.2174/138920111795542697. [DOI] [PubMed] [Google Scholar]
- 4.Cologna SM, Cluzeau CV, Yanjanin NM, Blank PS, Dail MK, Siebel S, et al. Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. Journal of inherited metabolic disease. 2014;37(1):83–92. doi: 10.1007/s10545-013-9610-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buhaescu I, Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clinical biochemistry. 2007;40(9–10):575–84. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Steiner LA, Ehrenkranz RA, Peterec SM, Steiner RD, Reyes-Mugica M, Gallagher PG. Perinatal onset mevalonate kinase deficiency. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2011;14(4):301–6. doi: 10.2350/11-02-0985-OA.1. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhury S, Hormaza L, Mohammad S, Lokar J, Ekong U, Alonso EM, et al. Liver transplantation followed by allogeneic hematopoietic stem cell transplantation for atypical mevalonic aciduria. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2012;12(6):1627–31. doi: 10.1111/j.1600-6143.2011.03989.x. [DOI] [PubMed] [Google Scholar]
- 8.Raupp P, Varady E, Duran M, Wanders RJ, Waterham HR, Houten SM. Novel genotype of mevalonic aciduria with fatalities in premature siblings. Archives of disease in childhood Fetal and neonatal edition. 2004;89(1):F90–1. doi: 10.1136/fn.89.1.F90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinson DD, Rogers ZR, Hoffmann GF, Schachtele M, Fingerhut R, Kohlschutter A, et al. Hematological abnormalities and cholestatic liver disease in two patients with mevalonate kinase deficiency. American journal of medical genetics. 1998;78(5):408–12. [PubMed] [Google Scholar]
- 10.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annual review of immunology. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clinical immunology. 2013;147(3):197–206. doi: 10.1016/j.clim.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Current opinion in allergy and clinical immunology. 2011;11(6):512–6. doi: 10.1097/ACI.0b013e32834c22f5. [DOI] [PubMed] [Google Scholar]
- 13.Petty RE. Growing pains: the ILAR classification of juvenile idiopathic arthritis. The Journal of rheumatology. 2001;28(5):927–8. [PubMed] [Google Scholar]
- 14.Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nature reviews Rheumatology. 2011;7(7):416–26. doi: 10.1038/nrrheum.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes and immunity. 2012;13(4):289–98. doi: 10.1038/gene.2012.3. [DOI] [PubMed] [Google Scholar]
- 16.Frenkel J, Rijkers GT, Mandey SH, Buurman SW, Houten SM, Wanders RJ, et al. Lack of isoprenoid products raises ex vivo interleukin-1beta secretion in hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis and rheumatism. 2002;46(10):2794–803. doi: 10.1002/art.10550. [DOI] [PubMed] [Google Scholar]
- 17.Kuijk LM, Mandey SH, Schellens I, Waterham HR, Rijkers GT, Coffer PJ, et al. Statin synergizes with LPS to induce IL-1beta release by THP-1 cells through activation of caspase-1. Molecular immunology. 2008;45(8):2158–65. doi: 10.1016/j.molimm.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis and rheumatism. 2006;54(11):3690–5. doi: 10.1002/art.22194. [DOI] [PubMed] [Google Scholar]
- 19.Ter Haar N, Lachmann H, Ozen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Annals of the rheumatic diseases. 2013;72(5):678–85. doi: 10.1136/annrheumdis-2011-201268. [DOI] [PubMed] [Google Scholar]
- 20.van der Hilst JC, Bodar EJ, Barron KS, Frenkel J, Drenth JP, van der Meer JW, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine. 2008;87(6):301–10. doi: 10.1097/MD.0b013e318190cfb7. [DOI] [PubMed] [Google Scholar]
- 21.Simon A, Kremer HP, Wevers RA, Scheffer H, De Jong JG, Van Der Meer JW, et al. Mevalonate kinase deficiency: Evidence for a phenotypic continuum. Neurology. 2004;62(6):994–7. doi: 10.1212/01.wnl.0000115390.33405.f7. [DOI] [PubMed] [Google Scholar]
- 22.Mandey SH, Schneiders MS, Koster J, Waterham HR. Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Human mutation. 2006;27(8):796–802. doi: 10.1002/humu.20361. [DOI] [PubMed] [Google Scholar]
- 23.Touitou I. Infevers: an online database for autoinflammatory mutations. [cited 2014 11/03/2014]; Available from: http://fmf.igh.cnrs.fr/ISSAID/infevers/
- 24.Rigante D, Capoluongo E, Bertoni B, Ansuini V, Chiaretti A, Piastra M, et al. First report of macrophage activation syndrome in hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis and rheumatism. 2007;56(2):658–61. doi: 10.1002/art.22409. [DOI] [PubMed] [Google Scholar]
- 25.Bader-Meunier B, Florkin B, Sibilia J, Acquaviva C, Hachulla E, Grateau G, et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics. 2011;128(1):e152–9. doi: 10.1542/peds.2010-3639. [DOI] [PubMed] [Google Scholar]
- 26.Grom AA. Macrophage activation syndrome and reactive hemophagocytic lymphohistiocytosis: the same entities? Current opinion in rheumatology. 2003;15(5):587–90. doi: 10.1097/00002281-200309000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric blood & cancer. 2007;48(2):124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 28.Tahara M, Sakai H, Nishikomori R, Yasumi T, Heike T, Nagata I, et al. Patient with neonatal-onset chronic hepatitis presenting with mevalonate kinase deficiency with a novel MVK gene mutation. Modern rheumatology / the Japan Rheumatism Association. 2011;21(6):641–5. doi: 10.1007/s10165-011-0442-7. [DOI] [PubMed] [Google Scholar]
- 29.de Saint Basile G, Menasche G, Latour S. Inherited defects causing hemophagocytic lymphohistiocytic syndrome. Annals of the New York Academy of Sciences. 2011;1246:64–76. doi: 10.1111/j.1749-6632.2011.06307.x. [DOI] [PubMed] [Google Scholar]
- 30.Vastert SJ, van Wijk R, D’Urbano LE, de Vooght KM, de Jager W, Ravelli A, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology. 2010;49(3):441–9. doi: 10.1093/rheumatology/kep418. [DOI] [PubMed] [Google Scholar]
- 31.Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794–8. doi: 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich A. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. The Journal of pediatrics. 2003;142(3):292–6. doi: 10.1067/mpd.2003.110. [DOI] [PubMed] [Google Scholar]
- 33.Hazen MM, Woodward AL, Hofmann I, Degar BA, Grom A, Filipovich AH, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis and rheumatism. 2008;58(2):567–70. doi: 10.1002/art.23199. [DOI] [PubMed] [Google Scholar]
- 34.Zhang K, Biroschak J, Glass DN, Thompson SD, Finkel T, Passo MH, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis and rheumatism. 2008;58(9):2892–6. doi: 10.1002/art.23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M, Behrens EM, Atkinson TP, Shakoory B, Grom AA, Cron RQ. Genetic defects in cytolysis in macrophage activation syndrome. Current rheumatology reports. 2014;16(9):439. doi: 10.1007/s11926-014-0439-2. [DOI] [PubMed] [Google Scholar]
- 36.Kaufman KM, Linghu B, Szustakowski JD, Husami A, Yang F, Zhang K, et al. Whole exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis & rheumatology. 2014 doi: 10.1002/art.38793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bracaglia C, Sieni E, Da Ros M, De Fusco C, Micalizzi C, Cetica V, et al. Mutaitons of familial hemophagocytic lymphohistiocytosis (FHL) related genes and abnormalities of cytotoxicity function tests in patients with macrophage activation syndrome (MAS) occuring in systemic juvenile idiopathic arthritis (sJIA) Pediatric rheumatology online journal. 2014;12(Suppl 1):P53. [Google Scholar]
- 38.Horneff G, Rhouma A, Weber C, Lohse P. Macrophage activation syndrome as the initial manifestation of tumour necrosis factor receptor 1-associated periodic syndrome (TRAPS) Clinical and experimental rheumatology. 2013;31(3 Suppl 77):99–102. [PubMed] [Google Scholar]
- 39.Mohr V, Schulz A, Lohse P, Schumann C, Debatin KM, Schuetz C. Urticaria, fever, and hypofibrinogenemia. Arthritis & rheumatology. 2014;66(5):1377. doi: 10.1002/art.38345. [DOI] [PubMed] [Google Scholar]
- 40.Schulert GS, Grom AA. Macrophage activation syndrome and cytokine-directed therapies. Best practice & research Clinical rheumatology. 2014;28(2):277–92. doi: 10.1016/j.berh.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leyva-Vega M, Weiss PF, Ganesh J, Conlin L, Spinner NB, Matthews RP. Significant liver disease in a patient with Y116H mutation in the MVK gene. American journal of medical genetics Part A. 2011;155A(6):1461–4. doi: 10.1002/ajmg.a.33915. [DOI] [PubMed] [Google Scholar]