

Abstract
Background
Ginsenoside Rd (GSRd), one of the most abundant ingredients of Panax ginseng, protects the heart via multiple mechanisms including the inhibition of Ca2+ influx. We intended to explore the effects of GSRd on L-type Ca2+ current (ICa,L) and define the mechanism of the suppression of ICa,L by GSRd.
Methods
Perforated-patch recording and whole-cell voltage clamp techniques were applied in isolated rat ventricular myocytes.
Results
(1) GSRd reduced ICa,L peak amplitude in a concentration-dependent manner [half-maximal inhibitory concentration (IC50) = 32.4 ± 7.1 μmol/L] and up-shifted the current–voltage (I–V) curve. (2) GSRd (30 μmol/L) significantly changed the steady-state activation curve of ICa,L (V0.5: −19.12 ± 0.68 vs. −16.26 ± 0.38 mV; n = 5, p < 0.05) and slowed down the recovery of ICa,L from inactivation [the time content (ζ) from 91 ms to 136 ms, n = 5, p < 0.01]. (3) A more significant inhibitive effect of GSRd (100 μmol/L) was identified in perforated-patch recording when compared with whole-cell recording [65.7 ± 3.2% (n = 10) vs. 31.4 ± 5.2% (n = 5), p < 0.01]. (4) Pertussis toxin (Giprotein inhibitor) completely abolished the ICa,L inhibition induced by GSRd. There was a significant difference in inhibition potency between the two cyclic adenosine monophosphate elevating agents (isoprenaline and forskolin) prestimulation [55 ± 7.8% (n = 5) vs. 17.2 ± 3.5% (n = 5), p < 0.01]. (5) 1H-[1,2,4]Oxadiazolo[4,3-a]-quinoxalin-1-one (a guanylate cyclase inhibitor) and N-acetyl-l-cysteine (a nitric oxide scavenger) partly recovered the ICa,L inhibition induced by GSRd. (6) Phorbol-12-myristate-13-acetate (a protein kinase C activator) and GF109203X (a protein kinase C inhibitor) did not contribute to the inhibition of GSRd.
Conclusion
These findings suggest that GSRd could inhibit ICa,L through pertussis toxin-sensitive G protein (Gi) and a nitric oxide–cyclic guanosine monophosphate-dependent mechanism.
Keywords: ginsenoside Rd, L-type calcium channels, Panax ginseng, patch-clamp techniques
1. Introduction
Panax ginseng, a traditional herbal medicine, has been used to prevent or treat cardiovascular diseases for at least 2000 years. Ginsenosides, a special group of triterpenoid saponins, are found nearly exclusively in ginseng and have been reported to show potential cardiovascular benefits through diverse mechanisms: antioxidation, modifying vasomotor function, reducing platelet adhesion, influencing ion channels, altering autonomic neurotransmitters release, and improving lipid profiles [1–3]. Among the various ginsenosides, such as Rb, Rc, Rd, Re, Rf, and Rg, ginsenoside Rd (GSRd) is one of the most abundant ingredients in the ginseng root and consequently has been accepted as one of the marker compounds of ginseng quality. GSRd can be produced through the hydrolysis of sugar moieties from the major ginsenosides, making it inexpensive for pharmaceutical use [4]. GSRd has exhibited an encouraging neuroprotective effect in both laboratory and clinical studies [5]. Recently, it has become important to show that GSRd attenuates myocardial ischemia/reperfusion injury in a rat model and in cultured neonatal rat cardiomyocyte model, which is related to the beneficial effects of ginseng in the treatment of heart diseases directly [6]. Multiple mechanisms were elucidated to be involved in the cardioprotective effects of GSRd, which should act synergically in ischemic myocardium. Therefore, in addition to being highly lipophilic and the fact that it is capable of easy diffusion across biological membranes, GSRd may have a potential clinical benefit on heart diseases.
In the mechanism research, GSRd-mediated cardioprotective effects against myocardial ischemia/reperfusion were found by both reducing intracellular reactive oxygen species and inhibiting mitochondria-mediated apoptosis. The activation of Akt/GSK-3β signaling is involved in the cardioprotective effect of GSRd [6]. Moreover, some data have shown that GSRd blocked Ca2+ influx through receptor- and store-operated Ca2+ channels in vascular smooth muscle cells [7]. Because Ca2+ antagonists effectively protect the myocardium from ischemic injury, we speculated that GSRd might exert its protective effects via blocking of Ca2+ channel in cardiomyocytes. In this study, we focused on the effects of GSRd on L-type calcium channel current (Ca2+, the antagonists' target) in isolated rat ventricular myocytes and its potential mechanism.
2. Materials and methods
2.1. Care and use of experimental animals
The experiment was performed in accordance with the institutional guidelines on the care and use of experimental animals set by the Chinese Academy of Sciences.
2.2. Cell preparation and solutions
The ventricular myocytes were enzymatically isolated from the hearts of male Sprague–Dawley rats (Vital River Laboratory Animal Technology Co. Ltd., Beijing, China) and stored in Kraft–Bruthe (KB) medium as previously described [8]. Briefly, the hearts were quickly removed and retrogradely perfused through the aorta with a Ca2+-free Tyrode's solution for 5 min at 37°C, followed by a low Ca2+ concentration Tyrode's solution containing type II collagenase (0.4 mg/mL) (Worthington, USA), Protease XIV (0.03 mg/mL), and bovine serum albumin (1 mg/mL) for 18–25 min. Tyrode's solution contained NaCl (135 mmol/L), KCl (5.4 mmol/L), MgCl2 (1 mmol/L), NaH2PO4 (0.33 mmol/L), 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (10 mmol/L), and glucose (10 mmol/L) (pH 7.4 NaOH), and was oxygenated with 100% oxygen. The ventricles were then minced and gently triturated in Kraft-Bruthe (KB) medium containing KOH (70 mmol/L), KCl (40 mmol/L), KH2PO4 (20 mmol/L), glutamic acid (50 mmol/L), MgCl2 (3 mmol/L), taurine (20 mmol/L), EGTA (0.5 mmol/L), HEPES (10 mmol/L), and glucose (10 mmol/L) (pH 7.4 KOH). Myocytes were harvested using a 200-mm nylon mesh and stored in KB medium at 4°C.
2.3. Perforated and whole-cell patch clamp recording
A small aliquot of ventricular myocytes were placed into a 2-mL chamber mounted on the stage of a microscope (DMIRB, LAICA, GER) and superfused with an external solution via a pump (BT100-2J, LONGER, CHN) at a rate of 3 mL/min. GSRd and some drug solutions were directly applied to the myocyte body using a pressurized bath perfusion system (BPS-8, ALA, USA). Patch pipettes were pulled using a puller (PULL100, WPI, USA) and had a tip resistance of 2–4 MΩ when filled with a pipette solution. Perforated patch clamp recording used an amphotericin B-perforated patch, which minimized the dialysis of intracellular contents with the internal pipette solution [9]. Amphotericin B was dissolved in dimethyl sulfoxide at a concentration of 60 mg/mL, and then added to the internal pipette solution at a final amphotericin B concentration of 0.2–0.3 mg/mL. The pipette solution containing amphotericin B was sonicated prior to use. The internal pipette solution contained CsCl (140 mmol/L), MgCl2 (2 mmol/L), CaCl2 (1 mmol/L), EGTA (11 mmol/L), MgATP (5 mmol/L), and HEPES (10 mmol/L) (pH 7.2 CsOH). Tyrode's solution was used to record ICa, L. Immediately after gigaseal formation (seal resistance >1 GΩ), the access resistance was monitored for 3–5 min. The internal pipette solution and the external solution for whole-cell patch recording were in accordance with perforated patch recording. The membrane was ruptured with a gentle suction after gigaseal formation to obtain the whole-cell patch clamp configuration. Voltage clamp recording was performed in single ventricular myocyte using an amplifier (EPC-10, HEKA, GER). Voltage command protocols were provided by a software package (PULSE10.0, HEKA, GER). ICa,L was activated by clamping the cells from a holding potential of -40-0 mV for 250 ms every 10 s. The current can be virtually inhibited by the calcium blocker (diltiazem, 100μmol/L) and is proven to be ICa,L. Peak ICa,L was measured with respect to steady-state current and was not compensated for leak currents.
2.4. Chemical drug
GSRd (a white powder with purity ≥98%) was obtained from the National Institute of Control of Pharmaceutical and Biological Products of China. Other chemical drugs were purchased from Sigma-Aldrich China Inc.
2.5. Data analysis
Software Origin8.0 (OriginLab Corp., USA) and Excel 2007 (Microsoft, Redmond, WA, USA) were used for data analysis. Data are presented as mean ± standard deviation. Student paired or unpaired t test was applied to determine the difference between the two groups, and p < 0.05 was considered statistically significant.
3. Results
3.1. Effects of GSRd on ICa,L in rat ventricular myocytes
GSRd inhibited the peak amplitude of ICa,L in a concentration-dependent manner (Fig. 1). The time course of block of ICa,L by GSRd (10 μmol/L, 30 μmol/L, and 100 μmol/L) is illustrated in Fig. 1A. The recovery was incomplete during the washout period. In superimposed traces, there was a virtual change in peak amplitude and no change in inactivation course and peak time (Fig. 1B). The dose–response curve for GSRd-induced inhibition of ICa,L was constructed from the mean peak ICa,L at each concentration of GSRd (Fig. 1C). On the basis of cell-by-cell fits, the half-maximal inhibitory concentration (IC50) of GSRd was 32.4 ± 7.1 μmol/L with a Hill coefficient of 1.71 (n = 21 cells).
Fig. 1.
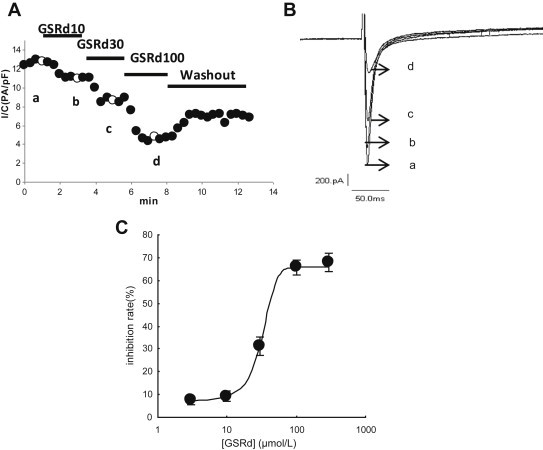
Effect of ginsenoside Rd (GSRd) on peak ICa,L in adult rat ventricular myocytes. Myocytes were voltage-clamped at −40 mV, and ICa,L was repetitively elicited by a single voltage pulse to 0 mV. (A) Consecutive exposure to different concentration GSRd (10 μmol/L, 30 μmol/L, and 100 μmol/L) resulted in consecutive further suppression of peak ICa,L. Upon washout of GSRd, ICa,L partially recovered. (B) Initial superimposed current traces of control and GSRd at times (a–d) indicated in A. (C) The concentration–response curve. Data fitted cell by cell to the following equation: IR = IRmax/[1 + ([C]/IC50)b], where IR is the inhibitory rate [(ICa-Cont − ICa-GSRd)/ICa-Cont × 100%]. [C], concentration of GSRd; IC50, half-maximum inhibition; b = Hill index. IC50 of GSRd was 32.4 ± 7.1 μmol/L, with a Hill coefficient of 1.71 (n = 21 cells).
3.2. Effects of GSRd on current–voltage relationship of ICa,L
Fig. 2 shows the I–V relationship of ICa,L in the absence or presence of GSRd. Family members of ICa,L were elicited from −40 to between −40 and +70 mV within 250 ms (Fig. 2A). The effects of GSRd on the current–voltage relationship of ICa,L was shown by the mean peak current density from each depolarizing step pulses. ICa,L was activated at −30 mV, and the peak amplitude occurred at the potential of 0 mV. GSRd (10 μmol/L and 30 μmol/L) up-shifted the I–V curve, and the current density at potential of 0 mV was decreased from −12.98 ± 1.39 pA/pF to −10.74 ± 1.32 and −8.81 ± 0.66 pA/pF (n = 5, p < 0.05) (Fig. 2B). GSRd did not alter the characteristics of the I–V relationship (the maximal activation voltage and reversal potential).
Fig. 2.
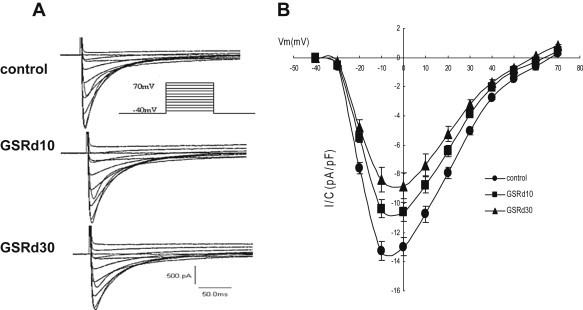
Effect of ginsenoside Rd (GSRd) on current–voltage relationship curves. (A) Initial current–voltage superimposed traces of control and GSRd (10 μmol/L and 30 μmol/L); 250 ms steps from −40 mV to between −40 mV and +70 mV at 1 Hz. (B) Current–voltage relationship curves prior to and after GSRd (10 μmol/L and 30 μmol/L).
3.3. Effects of GSRd on activation and inactivation of ICa,L
Steady-state activation and inactivation curves were obtained before and after GSRd application (Fig. 3). The activation curves were derived from the current–voltage relationship and fitted with the Boltzmann equation:
Fig. 3.
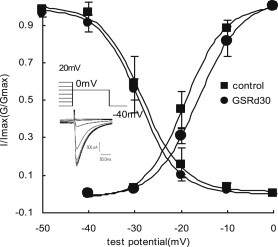
Effect of ginsenoside Rd (GSRd) on steady-state activation and inactivation. For the activation curves, the voltage dependence of the conductance activation variable were fitted to Boltzmann distribution: G/Gmax = 1/{1 + exp[(V − V0.5)/κ]}, where G/Gmax is the ratio of conductances [G = I/(V − Vrev), where Vrev is the reversal potential from each I–V curve)] to maximum conductance (Gmax, measured at 0 mV); V is the membrane potential, V0.5 is the midpoint, and κ is the slope. V0.5 and κ were −19.12 ± 0.68 mV and 4.26 ± 0.78 mV in the control, and −16.26 ± 0.38 mV and 4.61 ± 0.32 mV in GSRd (30 μmol/L). For the inactivation curves, protocol and representative recordings used to assess availability (I/Imax) are shown at lower left. I/Imax were also fitted to Boltzmann distribution. Currents (I) at 0 mV after 1-s conditioning pulses between −50 mV and 20 mV to 0 mV with 250 ms square wave were normalized by maximum current (Imax). V0.5 and κ were −28.34 ± 0.45 mV and 4.41 ± 0.42 mV in the control, and −28.99 ± 0.28 mV and 4.21 ± 0.28 mV after GSRd application.
GSRd (30 μmol/L) shifted V0.5 from −19.12 ± 0.68 to −16.26 ± 0.38 mV (n = 5, p < 0.05) and shifted κ from 4.26 ± 0.78 to 4.61 ± 0.32 (n = 5, p > 0.05). The steady-state inactivation of ICa,L was obtained using a double-pulse protocol. The inactivation curves before and after GSRd application were well described by a Boltzmann equation:
In this investigation, V0.5 and κ were −28.34 ± 0.45 mV and 4.41 ± 0.42 mV in the controls and −28.99 ± 0.28 mV and 4.21 ± 0.28 mV after 30 μmol/L GSRd was applied (n = 5, p > 0.05).
3.4. Effects of GSRd on recovery from inactivation of ICa,L
The effect of GSRd (30 μmol/L) on recovery from inactivation of ICa,L was examined using a double-pulse protocol separated by various intervals (Fig. 4). The ICa,L recovery was not complete (87.2 ± 2.4%, n = 5). The mean recovery time constant (ζ) of ICa,L from inactivation significantly increased from 91 ms to 136 ms after 30 μmol/L GSRd was added (n = 5, p < 0.01).
Fig. 4.
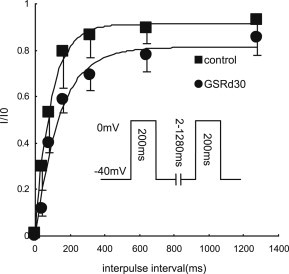
Effect of ginsenoside Rd (GSRd) on the recovery from inactivation. Kinetics of recovery assessed with two 200 ms test pulses separated by various intervals from 2 ms to 1280 ms at a holding potential of −40 mV. The time course of recovery from inactivation was estimated by plotting the ratio of peak ICa,L of the second pulse to that of the first pulse versus interpulse interval. Mean values were fitted by a single exponential function. Recovery was only 87.2 ± 2.4% (n = 5), and the time constants were 90.62 ± 7.05 ms for the control and 135.98 ± 6.39 ms for 30 μmol/L GSRd (n = 5).
3.5. Different potency by whole-cell and perforated patch recording
In order to examine whether the mechanism of GSRd-induced inhibition was attributable to intracellular signaling cascades or banding to the calcium channel directly, we undertook an additional experiment by using the conventional whole-cell patch technique. In whole-cell recording, the cutting off of the peak currents turned weaker as a result of the application of 100 μmol/L GSRd (Fig. 5) [63.4 ± 9.1% in perforated patch recording (n = 6) vs. 31.4 ± 5.2% in whole-cell recording reduction (n = 5), p < 0.01].
Fig. 5.
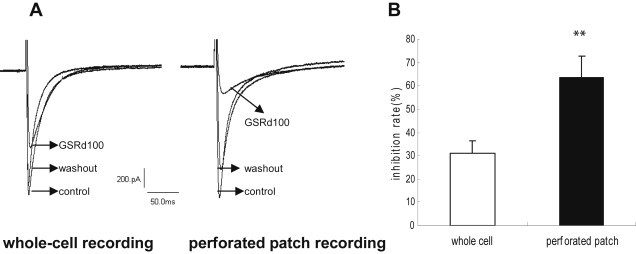
Different inhibition potency by whole-cell recording and perforated patch recording. (A) Example of the effect of ginsenoside Rd (GSRd; 100 μmol/L) on basal ICa,L using whole-cell recording and perforated patch recording, respectively. (B) Percent inhibition of ICa,L induced by GSRd (100 μmol/L) though whole-cell recording or perforated patch recording.
3.6. Effects of pertussis toxin on GSRd-induced inhibition
We hypothesized that Gi protein activation might be responsible for the GSRd-induced inhibition on ICa,L. Myocytes were pretreated with pertussis toxin (PTX) for 6 h at 37°C to decouple Gi from associated stimulation. In PTX-treated cells, under the control condition, ICa,L was similar to that of untreated cells [10.24 ± 2.13 pA/pf (n = 5) vs. 9.88 ± 1.72 pA/pf (n = 5), p > 0.05]. After the application of 30 μmol/L GSRd, ICa,L did not show any change when compared with controls (n = 5; Fig. 6A, 6B).
Fig. 6.
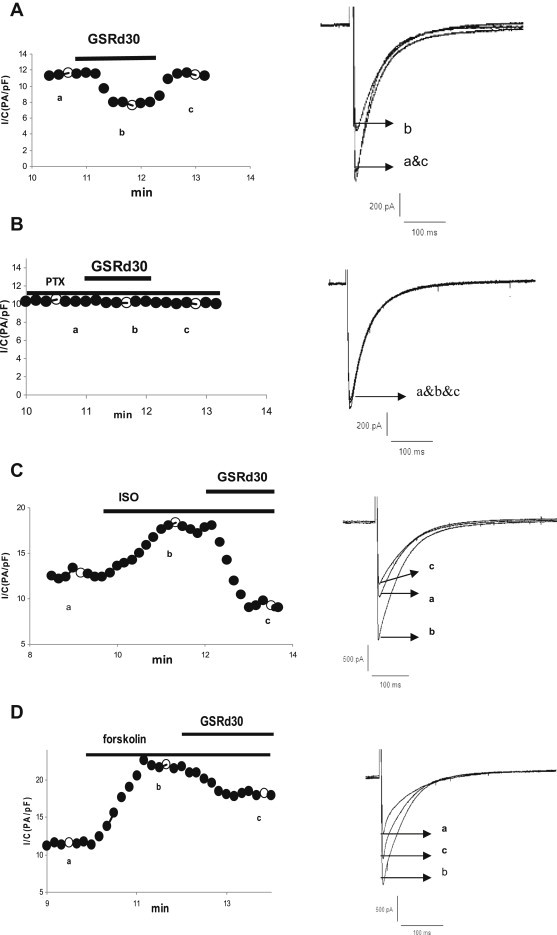
Effect of ginsenoside Rd (GSRd) on ICa,L of myocyte pretreated with pertussis toxin, ISO, and forskolin. The left panels in this and the following figures represent peak amplitude of ICa,L in a representative myocyte against the time. The timing of the drug applications is shown on the top. The right panel depicts superimposed current traces recorded at the timing indicated by lower-case letters in the left panel. (A) Effects of GSRd (30 μmol/L) on ICa,L. (B) Effects of GSRd (30 μmol/L) on ICa,L of myocyte pretreated with pertussis toxin. (C) Effects of GSRd (30 μmol/L) on ICa,L that had been stimulated by ISO (1μmol/L). (D) Effects of GSRd (30 μmol/L) on ICa,L stimulated by forskolin (10 μmol/L).
3.7. Effects of GSRd on ICa prestimulated with two cyclic adenosine monophosphate elevating agents
It is known that Gi counteracts the Gs-coupled activation of adenylate cyclase, reducing the production of cyclic adenosine monophosphate (cAMP) [10–12]. We examined how GSRd affected the calcium current in the myocytes pretreated with isoproterenol (ISO), which has the Gs-coupled activation of adenylate cyclase via the β-adrenergic pathway that leads to promotion of the production of cAMP, activation of protein kinase A, and phosphorylation of many proteins including the L-type Ca2+ channel [13,14]. Superfusion of myocytes with ISO (1 μmol/L) induced a significant enhancement effect on ICa,L (196 ± 37% of control, n = 10). GSRd (30 μmol/L) injection in the presence of ISO caused a substantial inhibition of ICa,L (Fig. 6A, 6C). Forskolin, a direct activator of adenylate cyclase, was also used to test how GSRd affected ICa,L. Following superfusion with forskolin (10 μmol/L), ICa,L was increased to 250 ± 16% of the controls (n = 8), and GSRd (30 μmol/L) also inhibited the ICa,L of myocytes prestimulated with forskolin (Fig. 6D). However, in contrast to the substantial inhibition of ICa,L by GSRd in ISO-pretreated myocytes, GSRd application in the presence of forskolin had a relatively weaker inhibitory effect on ICa,L [17.2 ± 3.5% (n = 5) vs. 55 ± 7.8% (n = 5), p < 0.01].
3.8. Effect of N-acetyl-l-cysteine and 1H-[1,2,4]oxadiazolo[4,3-a]-quinoxalin-1- one
Because some ginsenosides such as Re,Rg3 involve the actions of nitric oxide (NO) [15], we hypothesized a possible involvement of the NO–cyclic guanosine monophosphate (cGMP) signal pathway in the actions of GSRd. We first examined the effects of N-acetyl-l-cysteine (NAC), an NO scavenger, on the inhibition by GSRd. Cells were incubated with NAC (10 mmol/L) for 15 min. There was no change in peak ICa,L in the absence or presence of NAC. In the presence of NAC, the inhibition depth of GSRd (30 μmol/L) was decreased when compared with the control cell treated with the same concentration of GSRd (Fig. 6A, 7A). The inhibition rate of NAC-incubating myocytes (15.7 ± 3.2%, n = 5) was significantly different from that of control cells (31.4 ± 4%, n = 6; p < 0.01). An inhibitor of NO-sensitive guanylate cyclase, 1H-[1,2,4]oxadiazolo[4,3-a]-quinoxalin-1-one (ODQ), was also tested to observe the inhibitory effect of GSRd. In the presence of ODQ (10 μmol/L), even though the peak ICa,L was slightly lower (95.7 ± 1.4% relative to the control, n = 5), the inhibition rate of 30 μmol/L GSRd declined from 31.4 ± 4% (n = 6) to 21.4 ± 5.2% (n = 5) (p < 0.05; Fig. 6A, 7B).
Fig. 7.
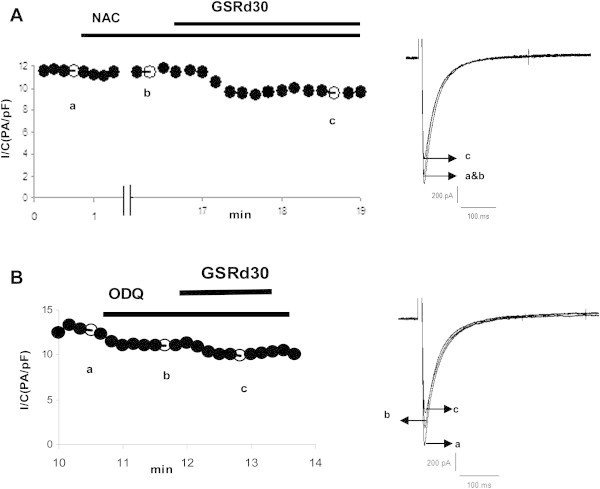
Effect of NAC and ODQ on the inhibition of ICa,L by GSRd. (A) Effect of an NO scavenger, NAC (10 mmol/L), on ICa,L inhibited by GSRd (30 μmol/L). (B) Effects of an inhibitor of guanylate cyclase, ODQ (10 μmol/L), on the inhibition of ICa,L by GSRd (30 μmol/L). GSRd, ginsenoside Rd; NAC, N-acetyl-l-cysteine; NO, nitric oxide; ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]-quinoxalin-1-one.
3.9. Effect of phorbol-12-myristate-13-acetate and GF109203X
We also investigated if the effect of GSRd on ICa,L was mediated through protein kinase C (PKC) signals. Phorbol-12-myristate-13-acetate (PMA), a PKC activator, slightly increased the peak amplitude (∼10% that of controls, n = 5). In the presence of 10 μmol/L PMA, GSRd (30 μmol/L) cut off the peak amplitude. There was no significant difference in GSRd-induced ICa,L inhibition between non-PMA (31.4 ± 4%, n = 6) and PMA (36.4 ± 5.9%, n = 6) (Fig. 6A, 8A). In the presence of a PKC inhibitor, GF109203X (10 μmol/L), no change in peak ICa,L was found. The presence of GF109203X did not affect GSRd-induced ICa,L inhibition [31.4 ± 4% (n = 6) vs. 34.7 ± 5.8% (n = 5), p > 0.05; Fig. 6A, 8B).
Fig. 8.
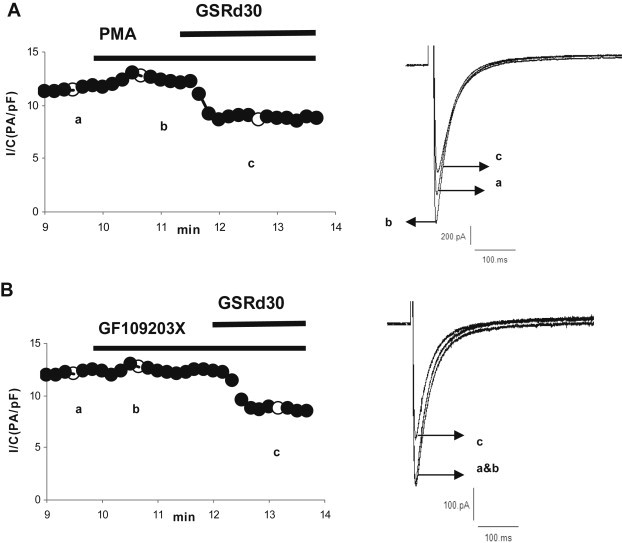
Effect of PMA and GF109203X on inhibition of ICa,L by GSRd. (A) Effect of a PKC activator, PMA (10 μmol/L), on the inhibition of ICa,L by GSRd (30 μmol/L). (B) Effects of a PKC inhibitor, GF109203X (10 μmol/L), on the inhibition of ICa,L by GSRd (30 μmol/L). GSRd, ginsenoside Rd; PKA, protein kinase A; PKC protein kinase C; PMA, phorbol-12-myristate-13-acetate.
4. Discussion
The present study demonstrates that GSRd causes ICa,L inhibition in rat ventricular myocytes, which is coupled with PTX-sensitive G protein (Gi) and partly mediated through an NO–cGMP-dependent mechanism.
4.1. Effects of GSRd on ICa,L
We found for the first time that GSRd inhibited ICa,L in rat ventricular myocytes in a concentration- and voltage-dependent manner. The results suggest that GSRd can prohibit the influx of excessive Ca2+ through the inhibition of voltage-dependent calcium channel (VDCC) in ventricular myocytes, which may be a new mechanism for protecting the heart against ischemia reperfusion injury. However, these results are not in accordance with previous reports that GSRd had no effect on the VDCC and could inhibit Ca2+ influx via receptor- and store-operated Ca2+ channels in basilar arterial vascular smooth muscle cells. In fact, some calcium channel blockers have different sensitivities to the VDCC in the cardiac or smooth muscle cells, which have been proven to contribute to cardiac and arterial smooth muscle Cav1.2 calcium channels isoforms diversified by alternative splicing [16–18]. More studies are needed to explore whether or which calcium channel isoforms are related to the different impacts of GSRd.
GSRd was assumed to cause the later activation of calcium channels, as well as the slow and incomplete recovery from the inactivation state. These might reflect the slow unbinding rate of the dissociation of the chemical drug, and the fact that about 10% of the channels would not be dissociated.
4.2. Possible mechanism of GSRd on ICa,L
Almost all calcium antagonists, such as dihydropyridines, benzothiazepines, and phenylalkylamines, act by directly binding to the calcium channel protein; however, many extracellular hormones and drugs can also regulate the calcium channel by binding to a variety of transmembrane receptors, initiating multiple signaling cascades [19,20]. When conventional whole-cell patch recording voltage clamp is used, the intracellular environment of the test cell would be altered by the dialysis of the cytoplasmic constituents. This phenomenon results to a change in the effective potency of some drugs that regulate the calcium channel via a signal pathway. The different potency in whole-cell and perforated patch recording led to us focus on the signal pathway as a modulation mechanism of GSRd on ICa,L. In some reports, a PTX-sensitive G protein (Gi) was assumed to participate in N-type and other high-threshold Ca2+ channels inhibition evoked by some ginsenosides such as Rf, Rb1, Rc, Re, and Rg1 in rat sensory neurons [21,22], so we hypothesized that Gi activation might be responsible for the GSRd-induced inhibition of ICa,L. In the present study, GSRd-induced inhibition of ICa,L in rat ventricular myocytes was also virtually eliminated by PTX. Thus, it is reasonable to deduce that the Gi signal pathway is responsible for the GSRd-induced inhibition of ICa,L. It is known that Gi counteracts the Gs-coupled activation of adenylate cyclase, thereby reducing the production of cAMP, so we tested the effects of GSRd on ICa prestimulated with two cAMP elevating agents. In the presence of ISO (β1-AR activator), GSRd caused more significant ICa,L inhibition (ISO vs. non-ISO: 55 ± 7.8% vs. 31.4 ± 4%) and showed an antiadrenergic effect. In contrast to the substantial inhibition in the ISO-pretreated myocytes, 10 μmol/L forskolin caused a relatively weaker ICa,L inhibition (17.2 ± 3.5% vs. 55.0 ± 7.8%). Because forskolin is thought to be a direct activator of adenylate cyclase (AC), it is proposed that Gi-induced inhibition of AC is only relative to the Gs-mediated AC stimulation, and direct activation of AC cannot be fully suppressed by Gi, which may explain the different effects induced by ISO and forskolin. Therefore, the difference inhibition potency between the two elevating cAMP agents suggests GSRd-induced inhibition links to the Gi signal pathway.
The downstream signaling pathways from Gi responsible for L-type calcium channel in cardiac myocytes are not well known. Some evidence shows NO-dependent mechanisms involved in the regulation of L-type calcium channel coupled with Gi: (1) phosphatidylinositol 3-kinase (PI3K), a downstream signaling pathway of Gi, acted on NO synthase-3 [23–26]; (2) NO was shown to inhibit ICa,L [27]; and (3) cGMP, a downstream signal molecule of NO, was involved in the reduced basal and cAMP-elevated L-type calcium current [28,29]. Our data showed that NAC, an NO scavenger, and ODQ, an inhibitor of NO-sensitive guanylate cyclase, partly alleviated GSRd-induced ICa,L inhibition, suggesting that GSRd inhibits ICa,L via an NO action in which the cGMP-dependent pathway is responsible. Thus, GSRd-induced inhibition linked to the NO–cGMP signal pathway may be a Gi protein-dependent mechanism.
Finally, we also tested if the effects of GSRd on ICa,L are mediated through the PKC signal pathway. Although some hormones such as angiotensin II and acetylcholine enhance ICa,L via PKC involvement, the effect of PKC on L-type channels is contradictory—enhancement, inhibition, and biphasic effects have been described about PKC activators on ICa,L [30–32]. Discrepant effects on ICa,L can arise from diverse CaV1.2 splice variants and from the intricacies of Gq signaling, including differential Gβγ subunits following receptor activation [33]. Our data suggest that GSRd inhibits ICa,L through a pathway distinct from that of PKC, because a PKC activator and a PKC inhibitor had no effect on the inhibition of GSRd.
Although the present study demonstrates that GSRd causes ICa,L inhibition through the Gi protein and the NO–cGMP signal pathway, we did not address the issue on whether the effect of the NO–cGMP signal pathway is via a pathway dependent on Gi protein action induced by GSRd. Because GSRd had been demonstrated to be antiapoptotic by activating PI3K/Akt and Akt/GSK-3 signaling in rat intestinal epithelial cells and neonatal rat cardiomyocytes [4,34], we hypothesized that NO production induced by GSRd may stem from the action of NO synthase (NOS) carried out by PI3K, a downstream signaling pathway of the Gi protein. The remaining issues to be addressed include finding out whether or not NOS is activated by GSRd and which pathway links GSRd to NOS activation.
Conflicts of interest
All authors have no conflicts of interest to declare.
Acknowledgments
This research would not have been possible without the technical assistance of Dr Hong-Xu Meng from Xiyuan Hospital, China Academy of Chinese Medical Sciences.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
- 1.Attele A.S., Wu J.A., Yuan C.S. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol. 1999;58:1685–1693. doi: 10.1016/s0006-2952(99)00212-9. [DOI] [PubMed] [Google Scholar]
- 2.Chen X., Gillis C.N., Moalli R. Vascular effects of ginsenosides in vitro. Br J Pharmacol. 1984;82:485–491. doi: 10.1111/j.1476-5381.1984.tb10784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X. Cardiovascular protection by ginsenosides and their nitric oxide releasing action. Clin Exp Pharmacol Physiol. 1996;23:728–732. doi: 10.1111/j.1440-1681.1996.tb01767.x. [DOI] [PubMed] [Google Scholar]
- 4.Kim M.K., Lee J.W., Lee K.Y., Yang D.C. Microbial conversion of major ginsenoside Rb(1) to pharmaceutically active minor ginsenoside Rd. J Microbiol. 2005;43:456–462. [PubMed] [Google Scholar]
- 5.Ye R., Zhao G., Liu X. Ginsenoside Rd for acute ischemic stroke: translating from bench to bedside. Expert Rev Neurother. 2013;13:603–613. doi: 10.1586/ern.13.51. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y., Li X., Wang X., Lau W., Wang Y., Xing Y., Zhang X., Ma X., Gao F. Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3β signaling and inhibition of the mitochondria-dependent apoptotic pathway. PLoS One. 2013;8:e70956. doi: 10.1371/journal.pone.0070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan Y.Y., Zhou J.G., Zhang Z., Wang G.L., Cai B.X., Hong L., Qiu Q.Y., He H. Ginsenoside-Rd from Panax notoginseng blocks Ca2+ influx through receptor- and store-operated Ca2+ channels in vascular smooth muscle cells. Eur J Pharmacol. 2006;548:129–136. doi: 10.1016/j.ejphar.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Meng H.X., Wang B., Liu J.X. Effect of salvianolic acid B and tetrahydropalmatine on the L-type calcium channel of rat ventricular myocytes. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2011;31:1514–1517. [PubMed] [Google Scholar]
- 9.Meng H.X., Wang B., Liu J.X. Recording L-type calcium channel current by perforated patch clamp and effect of dehydrocorydaline on L-type calcium channel. Chin Pharmacol Bull. 2011;27:1051–1054. [Google Scholar]
- 10.Zhang Z.S., Cheng H.J., Onishi K., Ohte N., Wannenburg T., Cheng C.P. Enhanced inhibition of L-type Ca2+ current by beta3-adrenergic stimulation in failing rat heart. J Pharmacol Exp Ther. 2005;315:1203–1211. doi: 10.1124/jpet.105.089672. [DOI] [PubMed] [Google Scholar]
- 11.Kashihara T., Nakada T., Shimojo H., Horiuchi-Hirose M., Gomi S., Shibazaki T., Sheng X., Hirose M., Hongo M., Yamada M. Chronic receptor-mediated activation of Gi/o proteins alters basal t-tubular and sarcolemmal L-type Ca2+ channel activity through phosphatases in heart failure. Am J Physiol Heart Circ Physiol. 2012;302:1645–1654. doi: 10.1152/ajpheart.00589.2011. [DOI] [PubMed] [Google Scholar]
- 12.Zhu W., Zeng X., Zheng M., Xiao R.P. The enigma of beta2-adrenergic receptor Gi signaling in the heart: the good, the bad, and the ugly. Circ Res. 2005;97:507–509. doi: 10.1161/01.RES.0000184615.56822.bd. [DOI] [PubMed] [Google Scholar]
- 13.Li H.Y., Bian J.S., Kwan Y.W., Wong T.M. Enhanced responses to 17beta-estradiol in rat hearts treated with isoproterenol: involvement of a cyclic AMP-dependent pathway. J Pharmacol Exp Ther. 2000;293:592–598. [PubMed] [Google Scholar]
- 14.Skeberdis V.A., Jurevicius J., Fischmeister A.R. Beta-2 adrenergic activation of L-type Ca2+ current in cardiac myocytes. J Pharmacol Exp Ther. 1997;283:452–461. [PubMed] [Google Scholar]
- 15.Bai C.X., Sunami A., Namiki T., Sawanobori T., Furukawa T. Electrophysiological effects of ginseng and ginsenoside Re in guinea pig ventricular myocytes. Eur J Pharmacol. 2003;476:35–44. doi: 10.1016/s0014-2999(03)02174-5. [DOI] [PubMed] [Google Scholar]
- 16.Zhang H.Y., Liao P., Wang J.J., Yu de J., Soong T.W. Alternative splicing modulates diltiazem sensitivity of cardiac and vascular smooth muscle Ca(v)1.2 calcium channels. Br J Pharmacol. 2010;160:1631–1640. doi: 10.1111/j.1476-5381.2010.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao P., Zhang H.Y., Soong T.W. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch. 2009;458:481–487. doi: 10.1007/s00424-009-0635-5. [DOI] [PubMed] [Google Scholar]
- 18.Liao P., Yong T.F., Liang M.C., Yue D.T., Soong T.W. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc Res. 2005;68:197–203. doi: 10.1016/j.cardiores.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 19.Zahradník I., Minarovic I., Zahradníková A. Inhibition of the cardiac L-type calcium channel current by antidepressant drugs. J Pharmacol Exp Ther. 2008;324:977–984. doi: 10.1124/jpet.107.132456. [DOI] [PubMed] [Google Scholar]
- 20.Treinys R., Jurevicius J. L-type Ca2+ channels in the heart: structure and regulation. Medicina (Kaunas) 2008;44:491–499. [PubMed] [Google Scholar]
- 21.Nah S.Y., McCleskey E.W. Ginseng root extract inhibits calcium channels in rat sensory neurons through a similar path, but different receptor, as mu-type opioids. J Ethnopharmacol. 1994;42:45–51. doi: 10.1016/0378-8741(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 22.Nah S.Y., Park H.J., McCleskey E.W. A trace component of ginseng that inhibits Ca2+ channels through a pertussis toxin-sensitive G protein. Proc Natl Acad Sci U S A. 1995;92:8739–8743. doi: 10.1073/pnas.92.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo S.H., Leblais V., Wang P.H., Crow M.T., Xiao R.P. Phosphatidylinositol 3-kinase functionally compartmentalizes the concurrent G(s) signaling during beta2-adrenergic stimulation. Circ Res. 2002;91:46–53. doi: 10.1161/01.res.0000024115.67561.54. [DOI] [PubMed] [Google Scholar]
- 24.Dedkova E.N., Wang Y.G., Blatter L.A., Lipsius S.L. Nitric oxide signaling by selective beta(2)-adrenoceptor stimulation prevents ACh-induced inhibition of beta(2)-stimulated Ca(2+) current in cat atrial myocytes. Circ Res. 2005;97:566–573. doi: 10.1113/jphysiol.2002.023341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Z., Jiang Y.P., Xu X.H., Ballou L.M., Cohen I.S., Lin R.Z. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes. 2007;56:2780–2789. doi: 10.2337/db06-1629. [DOI] [PubMed] [Google Scholar]
- 26.He J.Q., Balijepalli R.C., Haworth R.A., Kamp T.J. Crosstalk of beta-adrenergic receptor subtypes through Gi blunts beta-adrenergic stimulation of L-type Ca2+ channels in canine heart failure. J Physiol. 2002;542:711–723. doi: 10.1161/01.RES.0000181160.31851.05. [DOI] [PubMed] [Google Scholar]
- 27.Gallo M.P., Malan D., Bedendi I., Biasin C., Alloatti G., Levi R.C. Regulation of cardiac calcium current by NO and cGMP-modulating agents. Pflugers Arch. 2001;441:621–628. doi: 10.1007/s004240000475. [DOI] [PubMed] [Google Scholar]
- 28.Ziolo M.T., Lewandowski S.J., Smith J.M., Romano F.D., Wahler G.M. Inhibition of cyclic GMP hydrolysis with zaprinast reduces basal and cyclic AMP-elevated L-type calcium current in guinea-pig ventricular myocytes. Br J Pharmacol. 2003;138:986–994. doi: 10.1038/sj.bjp.0705112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartzell H.C., Fischmeister R. Opposite effects of cyclic GMP and cyclic AMP on Ca current in single heart cells. Nature. 1986;323:273–275. doi: 10.1038/323273a0. [DOI] [PubMed] [Google Scholar]
- 30.Rose R.A., Giles W.R. Natriuretic peptide C receptor signalling in the heart and vasculature. J Physiol. 2008;586:353–366. doi: 10.1113/jphysiol.2007.144253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurokawa H., Murray P.A., Damron D.S. Propofol attenuates beta- adrenoreceptor-mediated signal transduction via a protein kinase C-dependent pathway in cardiomyocytes. Anesthesiology. 2002;96:688–698. doi: 10.1097/00000542-200203000-00027. [DOI] [PubMed] [Google Scholar]
- 32.Weiss S., Doan T., Bernstein K.E., Dascal N. Modulation of cardiac Ca2+ channel by Gq-activating neurotransmitters reconstituted in Xenopus oocytes. J Biol Chem. 2004;279:12503–12510. doi: 10.1074/jbc.M310196200. [DOI] [PubMed] [Google Scholar]
- 33.Satin J. The long and short of PKC modulation of the L-type calcium channel. Channels (Austin) 2013;7:57–58. doi: 10.4161/chan.24147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura T., Cui X., Sakaguchi N., Akashi M. Ginsenoside Rd prevents and rescues rat intestinal epithelial cells from irradiation-induced apoptosis. Food Chem Toxicol. 2008;46:3080–3090. doi: 10.1016/j.fct.2008.06.011. [DOI] [PubMed] [Google Scholar]