

Abstract
Chronic pain attenuates midbrain dopamine (DA) transmission, as evidenced by a decrease in opioid-evoked DA release in the ventral striatum, suggesting that the occurrence of chronic pain impairs reward-related behaviors. However, mechanisms by which pain modifies DA transmission remain elusive. Using in vivo microdialysis and microinjection of drugs into the mesolimbic DA system, we demonstrate in mice and rats that microglial activation in the VTA compromises not only opioid-evoked release of DA, but also other DA-stimulating drugs, such as cocaine. Our data show that loss of stimulated extracellular DA is due to impaired chloride homeostasis in midbrain GABAergic interneurons. Treatment with minocycline or interfering with BDNF signaling restored chloride transport within these neurons and recovered DA-dependent reward behavior. Our findings demonstrate that a peripheral nerve injury causes activated microglia within reward circuitry that result in disruption of dopaminergic signaling and reward behavior. These results have broad implications that are not restricted to the problem of pain, but are also relevant to affective disorders associated with disruption of reward circuitry. Because chronic pain causes glial activation in areas of the CNS important for mood and affect, our findings may translate to other disorders, including anxiety and depression, that demonstrate high comorbidity with chronic pain.
Keywords: addiction, affective disorder, chronic pain, dopamine, emotion, opioids
Introduction
The pain experience involves activation of multiple brain regions, including those encoding salience and affect (Legrain, 2011; Mouraux et al., 2011). The fact that negative emotional experiences can produce the sensation of pain in the absence of tissue damage and that cognitive processes such as expectation can modulate the effectiveness of analgesia highlights the importance of salience and affect in the experience of pain. Therefore, changes that modulate the activity of motivational, salience, and affective systems will affect how the brain evaluates pain and analgesia.
There is extensive interaction between systems involved in pain processing and other affective and motivational systems, and it has been proposed that aberrant functioning of the affect and salience circuits may contribute to chronic pain (Borsook et al., 2013). It is common for chronic pain to be comorbid with mood disorders (e.g., depression) and it has been posited that dysfunction of mesolimbic circuitry, including dopamine (DA) neurotransmission, precipitates mood disorders and impairs motivated behaviors (Leknes and Tracey, 2008; Cahill et al., 2013). In fact, chronic pain is second only to bipolar disorder as the major cause of suicide among all medical illnesses and the high comorbidity of depression in patients with chronic pain syndromes further highlights the significance of the negative affect associated with chronic pain (Asmundson and Katz, 2009; Elman et al., 2013).
The mesolimbic DA system, comprising dopaminergic neurons in the VTA that project to the NAc, is an integral circuit encoding salience, motivation, and reward (Berridge, 2007). Diseases linked to deficits in the midbrain DA system, including disorders of mood and affect, substance abuse, and Parkinson's disease, are often comorbid with chronic pain (Jarcho et al., 2012) and can lead to impaired reward behaviors (Flagel et al., 2011; Grob et al., 2012). Imaging studies of chronic pain patients indicate that perturbation of the mesolimbic DA system is linked with chronic pain syndromes (Hagelberg et al., 2003a; Hagelberg et al., 2003b; Wood et al., 2007). Further, total striatal DA levels and opioid-stimulated DA release are markedly reduced in animal models of chronic pain (Ozaki et al., 2002; Taylor et al., 2014). Alterations in DA signaling as a result of chronic pain are associated with motivational deficits and reduced rewarding effects of natural and opioid rewards (Martin et al., 2007; Russo and Nestler, 2013; Schwartz et al., 2014).
How chronic pain affects the mesolimbic DA system is relatively unexplored, but there are sufficient reports to validate a role of altered DA signaling, not only in the chronicity of pain, but also in its response to opioid drugs. Here, we describe a mechanism by which chronic pain activates microglia in the VTA to inhibit DA transmission and disrupt reward behaviors. Inhibiting microglial activation restored normal DA signaling and recovered reward behavior. This work identifies microglial activation as being a critical component modulating reward behavior in chronic pain and suggests inhibiting microglial activation as a potential strategy for restoring disrupted DA transmission that is linked to disorders such as depression and addiction.
Materials and Methods
Animals
Male C57BL/6J and GAD65-GFP on C57BL/6J background mice (The Jackson Laboratory), 8–9 weeks old, were housed in groups of four unless otherwise indicated, and kept on a 12 h light/dark cycle with food and water available ad libitum. Male Sprague Dawley rats (Charles River Laboratories), 8–9 weeks old, were housed in groups of two and kept on a 12 h light/dark cycle with food and water available ad libitum. All behavioral tests were performed during the light phase. All procedures were conducted in accordance with the National Institutes of Health's Guide for the Care of Use of Laboratory Animals and were approved by the University of California–Los Angeles, University of California–Irvine, and Queen's University Institutional Animal Care and Use Committees.
Peripheral nerve injury
Animals were randomly assigned into two surgery groups, sham or peripheral nerve injury (PNI). Surgery was performed as described previously (Mosconi and Kruger, 1996). All animals received acetaminophen (32 mg/ml, p.o) preoperatively and were anesthetized with gaseous isoflurane (induction at 5% and maintenance at 2.0–2.5% in oxygen). An incision was made in the lateral left thigh followed by blunt dissection to expose the sciatic nerve. For PNI, the nerve was encased with a polyethylene tubing (PE50, 2 mm). The control group (sham) received a similar surgery but without dissection of the nerve.
Drugs
Morphine sulfate (1–10 mg/kg, i.p.; Sigma-Aldrich), cocaine hydrochloride (10 mg/kg, i.p.; Sigma-Aldrich), amphetamine hydrochloride (1 mg/kg, i.p.; Sigma-Aldrich), and d-Ala2, N-MePhe4, Gly-OH]-enkephalin (DAMGO, 1–25 ng; Tocris Biosciences) were dissolved in 0.9% saline. These drugs were selected for their ability to stimulate DA release via distinct targets either in the region of the DA terminal (cocaine, amphetamine) or VTA (morphine, DAMGO). Minocycline hydrochloride (30 mg/kg, i.p.; Sigma-Aldrich) was selected for its ability to inhibit microglial activation (Raghavendra et al., 2003; Nutile-McMenemy et al., 2007; Latrémolière et al., 2008; Guasti et al., 2009). Minocycline was dissolved in 0.1 m PBS, pH 7.4, and administered at nerve ligation surgery and every 12 h until the end of experimentation. During the conditioned place preference assay (CPP), minocycline was administered at least 6 h before drug conditioning sessions.
CPP
Systemic drugs.
CPP is a Pavlovian conditioning test to measure the motivational reward effects of a drug (Cunningham et al., 2006). This test allows for a within-subject analysis of the time spent in the vehicle compared with drug-paired conditioning chamber, which allows for paired analysis to assess the rewarding and/or motivational effects of a drug. CPP was conducted using an unbiased, counterbalanced, two- or three-chamber apparatus. Each square box (28 × 28 × 19 cm for mice; 45 × 45 × 30 cm for rats) was divided into two equal-sized conditioning chambers separated by a guillotine door. The two chambers were distinguished with visual and tactile cues so that the animals could discriminate between chambers. To counterbalance the groups before the conditioning sessions, animals were placed in the CPP apparatus and allowed free access to all chambers. The time spent in each chamber was recorded over 15–30 min using a CCD camera attached to a computer running behavioral tracking software (EthoVision; Noldus). The drug-paired chamber was assigned such that any innate bias for one chamber over the other was balanced between treatment groups. During the conditioning session, animals were confined to only one of the compartments so that they would associate the contextual cues of the compartment with the motivational effects of the stimulus (drug or vehicle, counterbalanced every other day). Conditioning sessions consisted of 8 sessions with 4 trials of morphine sulfate (1 or 10 mg/kg, i.p., n = 5–8) or cocaine hydrochloride (10 mg/kg, i.p., n = 7) and 4 trials of saline on alternating days, and were confined to the chambers for 15 (cocaine) or 30 (morphine) minutes. In a separate experiment, animals received only 1 trial of morphine (10 mg/kg, i.p.) and 1 trial of saline (n = 7–8 per group). The CPP assay began 6 d after peripheral nerve injury at a time previously shown to be during peak pain behavior (Taylor et al., 2014). On the postconditioning day (day 9 after the 8 conditioning trial assay or day 3 for the 2 conditioning trial assay), no treatment was given to the animals so that they could be tested in a drug-free state. Animals were allowed free access to all chambers of the apparatus and the time spent in the drug-paired chamber measured by motion capture Noldus software was determined over 15–30 min.
Intra-VTA DAMGO.
Male Sprague Dawley rats were anesthetized with isoflurane and mounted on a stereotaxic frame to allow implantation of a bilateral intra-VTA cannula (CMA Microdialysis; coordinates from bregma: AP −5.4, ML ± 0.75, DV −8.0). Rats were chosen to ensure accurate targeting of drug to the VTA. We have demonstrated comparable systemic opioid CPP responses between rats and mice in control and chronic pain models (Cahill et al., 2013). Animals were housed individually and allowed to recover for 7 d after intra-VTA cannula insertion, at which point they received a sham or nerve ligation surgery, as described above (n = 6–9 per group). Six days after nerve ligation surgery, animals were subjected to preconditioning and CPP was performed as above. During the conditioning sessions, animals received 4 trials with DAMGO (1 or 25 ng/0.5 μl/side at a rate of 0.25 μl/min) and 4 trials of sterile saline injected directly into the VTA and were confined to the chambers for 30 min. On the postconditioning day (day 9 from the beginning of conditioning trials), animals were allowed free access to both chambers in a drug-free state and the time spent in the drug-paired chamber was measured over 30 min. Animals were killed at the end of the experiment and brains were isolated and sectioned on a cryostat to confirm cannula placement.
Intra-NAc cocaine.
Male C57BL/6J mice (n = 6–8 per group) were anesthetized with isoflurane and mounted on a stereotaxic frame to allow implantation of a bilateral intra-NAc cannula (CMA Microdialysis; coordinates from bregma: AP+1.5, ML +0.8, DV −3.5). Animals also received nerve ligation or sham surgery as described above and were housed singly for 7 d. Cocaine CPP was performed as described above beginning 6 d after surgery. Briefly, after preconditioning, mice received 3 conditioning trials of intra-NAc cocaine (100 μg/0.4 μl at 0.1 μl/min) and 3 trials of PBS over 6 d. Mice were confined to the conditioning chamber during intra-NAc injection for 10 min. On the postconditioning day (day 7), mice were allowed free access to both chambers in a drug-free state and the time spent in the drug-paired chamber was measured over 15 min. Animals were killed at the end of the experiment and brains were isolated and sectioned on a cryostat to confirm cannula placement.
Evoked pain tests
Mechanical allodynia.
Withdrawal thresholds to a mechanical stimulus applied with calibrated von Frey filaments were measured in all groups (n = 6 per group), as described previously (Chaplan et al., 1994). Mice were placed atop a mesh grid floor in a clear Plexiglas enclosure. Von Frey filaments were applied to the plantar surface of the ipsilateral hindpaw in an up–down manner in which filaments with increasing stiffness were applied until a paw retraction was observed. After the first positive reaction, the next less force filament was applied. If no reaction, the next higher force filament was applied; if a reaction was observed, the next less force filament was applied. This was repeated five times per animal and the 50% withdrawal threshold was calculated. Baseline measurements were taken for all animals before surgery and 2 weeks postoperatively.
Thermal tail withdrawal.
Morphine and cocaine antinociception was measured using the tail withdrawal assay at 2 weeks after nerve injury (n = 6–9 per group). Mice were gently restrained by cupping them in a soft plastic conical sleeve and 2.5 cm of the tail was immersed in 49°C water. The time to tail withdrawal was measured. After 3 basal measurements, mice were injected with morphine (10 mg/kg, i.p.) or cocaine (10 mg/kg, i.p.). Tail withdrawal latencies were measured every 10 or 20 min for 1 h. Mice were returned to their home cages between testing. A cutoff of 15 s was imposed to avoid tissue damage based on baseline latencies between 4 and 6 s. Data were expressed as percent maximum possible effect, calculated as follows: (test latency − baseline latency)/(cutoff latency − baseline latency) × 100.
Immunoblot assay
Animals were anesthetized with isofluorane and the brains were isolated (n = 4 per group). Brain punches (1 mm × 1 mm × 1 mm) were taken from the VTA and homogenized in radioimmunoprecipitation assay buffer with protease inhibitors. Equal amounts of protein (25 μg) were loaded onto precast 10% polyacrylamide gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were incubated with an antibody against BDNF (1:150; Millipore) and visualized with a goat anti-rabbit antibody conjugated to HRP (Jackson Immunoresearch). BDNF levels were normalized to the housekeeping protein β-actin (1:5000; Sigma-Aldrich). Results were analyzed with a computer-assisted densitometry analysis (ImageJ).
Immunocytochemistry
Animals (n = 4–7 per group) were transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB). The brain was dissected from the skull, postfixed for 30 min in the above-described fixative, and cryoprotected in 30% sucrose before cutting 40 μm sections collected in PBS containing 0.2% Triton X-100. For microglial labeling, sections were incubated with anti-IBA-1 (1:2000; Wako) and a goat anti-rabbit IgG conjugated to Alexa Fluor 488 (1:1000; Invitrogen). For K+/Cl− cotransporter (KCC2) staining, sections were incubated with anti-KCC2 (1:500; Millipore) and a donkey anti-rabbit IgG conjugated to Alexa Fluor 594 (1:500; Invitrogen). Images were taken on a Nikon Eclipse 90i fluorescence microscope with a 20× or 40× objective. For KCC2 quantification, images were acquired with a high-resolution digital camera with constant exposure and gain settings. Fluorescent intensity was measured with ImageJ software. For IBA-1 quantification, the degree of microglial activation in the VTA was measured using a semiquantitative method, as described previously (Lynch, 2009). Experimenters were blinded to conditions at all points of quantification. The level of microglial activation was scored on a linear scale of 0–4 ranging from resting microglia (0) to highly activated (4), as described previously (Brettschneider et al., 2012). In addition, the branching points of the filopodia were counted and compared between groups.
Fluorescence lifetime imaging microscopy
For fluorescence lifetime imaging microscopy (FLIM), 300-μm-thick coronal midbrain sections corresponding to the VTA of C57BL/6 and GAD65-GFP knock-in mice (n = 7–12 per group) were incubated in artificial CSF (ACSF) containing the following (in mm); 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, 26 NaHCO3, and 1.25 NaH2PO4, along with a 5 mm concentration of the Cl− indicator N-6-methoxyquinolinium acetoethylester (MQAE; Invitrogen) for 30–40 min at 34°C. Slices were then transferred to a perfusion chamber (2 ml/min) in the presence of 1 μm tetrodotoxin, 10 μm CNQX, 40 μm AP5, 0.5 μm strychnine, and 10 μm gabazine. Furosemide (125 μm; Sigma-Aldrich) was applied to some sections to block KCC2 Cl− transport. Some slices were incubated with anti-trkB (5 μg/ml; Sigma-Aldrich) for 1 h before MQAE. MQAE fluorescence was excited using a Zeiss LSM510 laser-scanning microscope coupled to a femtosecond-pulsed Ti-sapphire laser (Chameleon Ultra) tuned at 750 nm (Gagnon et al., 2013). Fluorescence of MQAE was acquired through a 40× water-immersion objective and fluorescence lifetime was recorded with a Becker & Hickl SPC-830 module through the nondescanned port of the Zeiss LSM510 using a band-pass filter (469/35 nm) coupled to a laser block (short-pass 750 nm). Photons were detected using a PMC-100-1 photosensor (Hamamatsu). Experimental instrument response function of the detection path was acquired using an 80 nm gold nanoparticle suspension to generate a second-harmonic signal.
GABAergic cells were identified visually as part of the VTA and could be distinguished from DA and glutamatergic cells on the basis of their morphology and colabeling in sections taken from GAD65-GFP mice. To confirm brain region specificity, tissue slices were postfixed with PFA and subjected to tyrosine hydroxylase (TH) immunolabeling (1:1000; Calbiochem) to confirm imaging was performed on VTA sections.
Two-photon fluorescence lifetime images were acquired every 10 s for a period of 7 min, followed by perfusion with ACSF containing 15 mm KCl to reverse KCC2-mediated Cl− transport and to force Cl− accumulation inside the cell, leading to a quenching of the measured lifetime (Chorin et al., 2011). The imaging plane was set at least 60 μm under the slice surface to ensure recording of healthy MQAE-loaded cells. For each condition, we recorded cells from at least 7 animals, at least 2 slices per animal (14–20 slices/condition), and 70–100 cells (on average 5/slice). Fluorescence lifetime in each cell was averaged over the whole-cell body area and extracted for each time point using home-made MATLAB software (The MathWorks). Briefly, following the results of previous studies (Digman et al., 2008), we converted the photon timing histograms of each acquired lifetime image to phasor plots. Regions of interest corresponding to cell bodies were selected and added to a new phasor. Each VTA slice had between three and 15 cells, and the lifetimes of these cells were averaged to give one lifetime measurement per time point throughout the recording.
Microdialysis
Mice (n = 4–6 per group) were anesthetized with isoflurane and placed into a stereotaxic frame to allow implantation of a guide cannula (CMA Microdialysis) to an area just dorsal to the right side of the NAc (coordinates from bregma AP +1.5, ML +0.8, DV −3.5). This surgery was performed at the same time as sham and nerve ligation surgery. A separate group received PNI surgery and chronic treatment with minocycline (30 mg/kg, i.p.) every 12 h starting the day of surgery and continuing throughout the experiment. When microdialysis samples were collected, minocycline injection occurred at least 6 h before baseline sampling. Animals were housed individually and allowed to recover for 10–14 d. The day before the experiment, animals were implanted with a concentric microdialysis probe equipped with a 1-mm-long, 300-μm-diameter polyacrylonitril membrane (CMA Microdialysis). Probes were continuously perfused with ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 0.9 NaH2PO4, 5 Na2HPO4, 1.2 CaCl2, and 1 MgCl2, pH 7.4, using syringe pumps at a rate of 1 μl/min. Dialysates were collected at 4°C in 20 min intervals into plastic microvials preloaded with 2 μl of 12.5 mm perchloric acid/250 μm EDTA. Twelve hours after insertion of microdialysis probes, drug (10 mg/kg morphine, 10 mg/kg cocaine, 1 mg/kg amphetamine, or saline; i.p.) was injected every 1–5 h. Each animal was treated with every drug in one day (saline, morphine, cocaine, and amphetamine). The order of drug treatment was randomized and no effect of drug order was observed. For saline, cocaine, and amphetamine, drugs were given 1 h (saline) or 1.5 h (amphetamine and cocaine) apart. Because of the longer effect of morphine on extracellular DA levels, subsequent drug administration was given 5 h after morphine administration. Dialysate samples were analyzed with HPLC analysis (Koizumi et al., 2004). Animals were immediately killed and brains preserved in histological fixative (4% PFA). Brains were coronally sectioned and mounted on glass slides. Sections were counterstained with cresyl violet, dehydrated, and coverslipped to confirm probe placement.
Statistics
All data are expressed as mean ± SEM. For analysis of behavior, Western blotting, and immunohistochemical labeling, data were analyzed using either a one-way or two-way ANOVA followed by post hoc analysis. Fluorescence lifetime Cl− measurements were analyzed by programmed MATLAB software. The average slope (change in Cl− over time) per slice over the first 40 s after high K+ perfusion was averaged across the group. The effects of treatment were analyzed by a one-way ANOVA. Differences were considered statistically different with p < 0.05.
Results
To test whether chronic pain disrupts DA transmission, we measured evoked extracellular DA release using in vivo microdialysis. In the sham surgery group (control), systemic morphine (10 mg/kg) evoked a robust increase in extracellular DA in the NAc (Fig. 1). However, morphine failed to elicit an increase in extracellular DA in the chronic pain group (Fig. 1; Fpain state(2,138) = 19.73, p = 0.0001, Ftime(13,138) = 2.74, p = 0.002, Finteraction(26,138) = 1.01, p = 0.45). Cocaine also showed deficits in its ability to evoke DA release in the chronic pain group (Fpain state(2,52) = 5.95, p = 0.004, Ftime(6,52) = 4.28, p = 0.001, Finteraction(12,52) = 1.44, p = 0.18) compared with the control group. Interestingly, amphetamine, which causes an increase in extracellular DA by promoting vesicular release and transporter blockade, stimulated extracellular DA levels in both control and chronic pain groups, with no difference observed between groups (Fpain state(2,38) = 1.32, p = 0.23, Ftime(6,38) = 9.1, p = 0.0001, Finteraction(12,38) = 1.01, p = 0.46).
Figure 1.

Loss of opioid- and cocaine-evoked DA release in chronic pain. Morphine (10 mg/kg) failed to stimulate extracellular DA in the PNI group, but treatment with minocycline (PNI+MC; 30 mg/kg) restored morphine-evoked extracellular DA levels. Similarly, cocaine-evoked (10 mg/kg) DA levels were lower in the PNI group compared with the sham group and cocaine-evoked extracellular DA could be recovered by pretreatment with minocycline (PNI+MC). Amphetamine-evoked (1 mg/kg) extracellular DA was not significantly different between PNI and sham groups and was not altered by MC treatment. ***p < 0.001. Error bars indicate SEM. Insets depict area under the curve (AUC) for the entire measurement period of DA dialysis for each group. *p < 0.05. Error bars indicate SEM.
To determine whether the loss of evoked release of DA in the mesolimbic system of chronic pain animals was associated with a loss of reward-like behavior, we determined the effects of morphine and cocaine-induced CPP in control and chronic pain animals. Systemic morphine (10 mg/kg) produced a robust place preference in both control and chronic pain groups after conditioning for 8 d (Fig. 2a; Fdrug side(1,15) = 9.45, p = 0.008; Fpain state(1,15) = 0.51, p = 0.49; Finteraction(1,15) = 0.0005, p = 0.94). However, when animals were conditioned for only 2 d (once with morphine and once with saline), the chronic pain group failed to show a place preference to morphine, whereas in the control group, morphine place preference was observed (Fig. 2a; Fdrug side(1,26) = 10.61, p = 0.003, Fpain state(1,26) = 0.02, p = 0.90, Finteraction(1,26) = 2.80, p = 0.1). To determine whether the chronic pain animals exhibited state-dependent learning to systemic morphine after only two conditioning sessions, mice were injected with morphine (10 mg/kg, i.p.) 72 h after conditioning. A state-dependent effect was evident with a difference score between morphine and saline compartments of 540 ± 198 s (t = 2.732, p < 0.05). A lower dose of morphine (1 mg/kg) produced a trend toward a place preference, but was not significant in either the control or the chronic pain group (Fdrug side(1,8) = 3.56, p = 0.09, Fpain state(1,8) = 2.94, p = 0.12, Finteraction(1,8) = 0.002, p = 0.96). Contrary to expectations, systemic administration of cocaine (10 mg/kg) also produced a place preference in both the control and chronic pain groups (Fig. 2b; Fdrug side(1,12) = 37.78, p = 0.0001, Fpain state(1,12) = 0.006, p = 0.94, Finteraction(1,12) = 0.45, p = 0.62). To determine whether systemic administration of cocaine or morphine had the potential to produce negative reinforcement in chronic pain animals (via eliciting analgesic effects), we assessed whether cocaine and morphine increased tail withdrawal latencies in both control and chronic pain animals. The analgesic effects of cocaine and morphine on nerve injury-induced pain hypersensitivities of the hindpaw could not be assessed because of drug-induced hyperlocomotion; therefore, the analgesic effects were measured via the tail immersion assay. Systemic morphine (10 mg/kg, i.p.) produced a large antinociceptive response that was not significantly different in chronic pain animals compared with controls (Fig. 2c). Similarly, systemic injection of cocaine (10 mg/kg, i.p.) produced a small but robust antinociceptive effect in both control and chronic pain groups, and this effect was not significantly different between groups.
Figure 2.

Systemic opioid- and cocaine-induced place preference in chronic pain. a, Systemic morphine (10 mg/kg) produced a place preference in control and chronic pain (PNI) groups. When animals received limited conditioning trials (one exposure to 10 mg/kg morphine and one to saline), only the sham group displayed a robust place preference. Systemic low dose of morphine (1 mg/kg) produced a trend toward a place preference, but was not significant in either the sham or the PNI group. *p < 0.05, **p < 0.01. Error bars indicate SEM. b, Systemic cocaine (10 mg/kg) produced a place preference in both sham and PNI groups. **p < 0.01, ***p < 0.001. Error bars indicate SEM. c, Morphine (10 mg/kg) and cocaine (10 mg/kg) antinociception was measured using the tail withdrawal assay. Neither morphine nor cocaine antinociception was significantly different between sham or PNI groups. Further, inhibition of microglia in chronic pain animals (PNI+MC) did not affect morphine antinociception. Error bars indicate SEM. n.s., Not significant; %MPE, percent maximum possible effect.
Several prior studies have indicated that systemically administered opioids can produce a place preference independently of DA by engaging reward circuits outside of the mesolimbic DA system (Hnasko et al., 2005; Vargas-Perez et al., 2009). To isolate the contribution of DA signaling to reward behavior, we assessed the ability of opioids or cocaine microinjected into the VTA or NAc to produce a place preference in control and chronic pain animals. DAMGO (1 ng) injected directly into the VTA produced a place preference in the control group; however, it failed to produce a place preference in the chronic pain group (Fig. 3a; Fdrug side(1,15) = 10.49, p = 0.006, Fpain state(1,15) = 1.72, p = 0.21, Finteraction(1,15) = 15.18, p = 0.0014). Injections of a higher dose of DAMGO (25 ng) into the VTA produced a place preference in both the control and chronic pain groups (Fdrug side(1,11) = 58.59, p = 0.0001, Fpain state(1,11) = 2.46, p = 0.14, Finteraction(1,11) = 3.72, p = 0.08). Intra-NAc injection of cocaine (100 μg), which stimulates increased extracellular DA in the NAc by transporter blockade at the axonal terminals, produced a place preference in the control group, but not in the chronic pain group (Fig. 3b; Fdrug side(1,22) = 1.35, p = 0.26, Fpain state(1,22) = 0.33, p = 0.57, Finteraction(1,22) = 6.69, p = 0.02).
Figure 3.
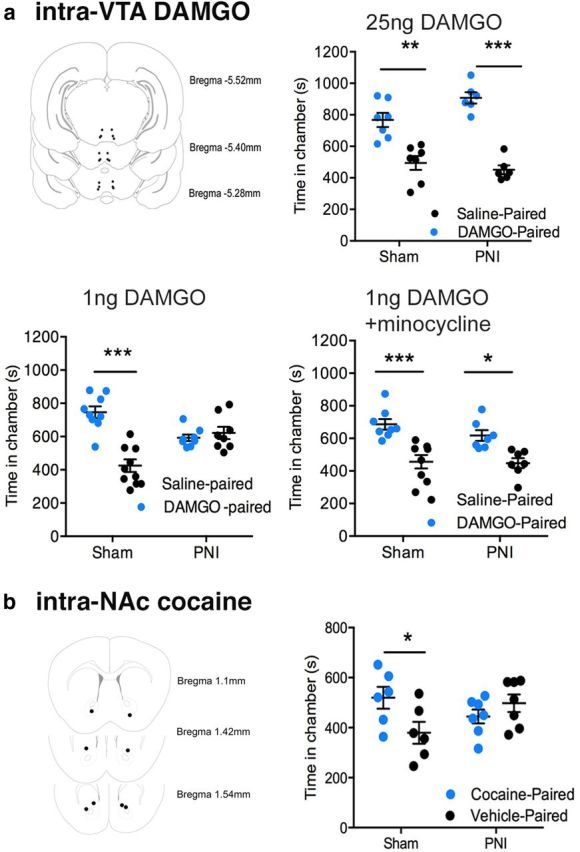
Loss of mesolimbic-specific opioid- and cocaine-induced place preference in chronic pain. a, Repeated conditioning to intra-VTA DAMGO (1 ng) failed to produce a robust place preference in the PNI groups, whereas a high dose of DAMGO (25 ng) produced a robust place preference in both the sham and PNI groups. Pretreatment with the microglial inhibitor minocycline (30 mg/kg) recovered intra-VTA DAMGO (1 ng) place preference in the PNI group. Top left pictograph depicts intra-VTA injection sites. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars indicate SEM. b, Cocaine (100 μg) administered directly into the NAc produced a place preference in the sham group, but not the PNI group. Left pictograph depicts intra-NAc injection sites. *p < 0.05. Error bars indicate SEM.
Microglia have been implicated in the development of chronic pain at the spinal level and play a pivotal role in central sensitization and synaptic plasticity (Taves et al., 2013). We explored the contribution of microglia to the loss of evoked increase in extracellular DA in chronic pain. Microglial activation, as measured by morphological changes in IBA-1 immunolabeling, was evident in the VTA of chronic pain animals compared with controls and chronic treatment with the microglial inhibitor minocycline (30 mg/kg) significantly blocked this activation (Fig. 4a; F(2,35) = 23.56, p = 0.0001). In the chronic pain group, minocycline was found to restore both the morphine- and cocaine-stimulated increase in extracellular DA to comparable levels in the control group (Fig. 1). Treatment with minocycline did not affect drug-evoked extracellular DA in the control group, nor did it alter the amphetamine-stimulated rise in extracellular DA in either control or chronic pain group (Fig. 1). Furthermore, pretreatment with minocycline recovered intra-VTA DAMGO place preference in the chronic pain group (Fig. 3a: Fdrug side(1,26) = 32.29, p < 0.0001, Fpain state(1,26) = 1.224, p = 0.28, Finteraction(1,26) = 0.4, p = 0.7). Inhibiting microglial activation did not prevent the development of mechanical allodynia measured 6 h after minocycline treatment, corresponding to the regimen schedule used in the CPP assay (Fig. 4b; Ftime(1,15) = 35.44, p = 0.0001, Fpain state(2,15) = 3.69, p = 0.05, Finteraction(2,15) = 9.41, p = 0.01). Further, minocycline treatment did not affect morphine or cocaine analgesia in either the control or chronic pain group (Fig. 2c).
Figure 4.

Chronic pain activates microglia in the VTA. a, Microglia were significantly activated in the VTA of the chronic pain (PNI) group and displayed a robust activated phenotypes. Pretreatment with the microglial inhibitor minocycline (MC; 30 mg/kg) blocked microglial activation in the PNI+MC group. **p < 0.01, ***p < 0.001. Error bars indicate SEM. Scale bar, 75 μm. b, Mechanical withdrawal thresholds of the ipsilateral hindpaw were measured with von Frey filaments. The PNI group showed significantly lowered mechanical thresholds 2 weeks after nerve injury. Systemic treatment with the microglial inhibitor MC did not affect evoked pain thresholds. **p < 0.01, ***p < 0.001. Error bars indicate SEM. n.s., Not significant.
Previous research showed that microglial activation contributes to chronic pain by disrupting Cl− homeostasis via a TRkB-KCC2 pathway in GABAA-expressing spinal cord lamina I nociceptive neurons (Coull et al., 2005). We thus investigated whether this pathway also contributes to the loss of mesolimbic DA activity in chronic pain. BDNF protein expression measured by Western blot analysis was significantly upregulated in the VTA of chronic pain animals and this upregulation was blocked by systemic minocycline treatment (Fig. 5f; Ftreatment(1,11) = 8.10, p = 0.02). Quantitative analysis of KCC2 immunolabeling in the VTA showed significantly lower KCC2 protein expression in the chronic pain group, which was also prevented by treatment with minocycline (Fig. 5b; F(2,14) = 8.73, p = 0.004). To determine whether KCC2 function was disrupted within the VTA of chronic pain animals, we prepared VTA coronal slices and measured K+-driven Cl− influx via fluorescent lifetime measurements of the Cl− probe MQAE. This method has been used previously to measure the Cl− transport rate within spinal cord neurons (Doyon et al., 2011; Ferrini et al., 2013; Gagnon et al., 2013). MQAE dye loaded exclusively into GABAergic (GAD65-GFP+) neurons of the VTA (Fig. 5c). The rate of Cl− accumulation induced by elevated K+ was significantly slower in slices from chronic pain animals (Fig. 5d,e; F(3,35) = 4.26, p = 0.01). This decrease in Cl− transport was attenuated by in vivo pretreatment with minocycline or in vitro incubation of the slices with a functional blocking antibody to TrkB.
Figure 5.

Chronic pain disrupts Cl− transport in VTA GABAergic through a microglia-BDNF signaling pathway. a, KCC2 labeling in the VTA was primarily localized in processes not labeled for TH. Immunolabeling of KCC2 (green) was identified in non-TH (red)-labeled neurons and the labeling was punctate, which is indicative of terminal arborization. Scale bar, 75 μm. The right micrographs represent a single image of a TH+ neuron (middle, arrowhead) surrounded by KCC2-immunoreactive arbors (bottom). The top image is a merge of the bottom two images. b, The chronic pain (PNI) group exhibited decreased KCC2 expression in the VTA that could be recovered by systemic treatment with minocycline (PNI+MC; 30 mg/kg). Left black and white micrographs are representative images of KCC2 labeling in the VTA of sham and PNI groups. The right histogram shows KCC2 quantification. Error bars indicate SEM. *p < 0.05, **p < 0.01. AU, Arbitrary units. c, Top left, KCC2 function was assayed in VTA slices using MQAE FLIM. The VTA was localized with brain atlas coordinates and confirmed with TH labeling (red) after Cl− imaging assay. Bottom left, Representative micrographs showing MQAE loading in GAD65-GFP cells (white arrows) in the VTA (2-photon optical section ∼1 μm). Top right, Pseudocolor images showing lifetime maps from control VTA slices in the presence of 2.5 or 15 mm KCl. d, Representative lifetime plots from VTA slices. e, Graph plotted by averaging the slope in the lifetime plots immediately after administration of 15 mm KCl. The PNI group had significantly slower fluorescence lifetime change rate indicative of a slower transport of Cl− into the cell. Normal transport rate was restored in animals pretreated with minocycline (PNI+MC) or in slices treated with a TrkB antagonist (PNI+anti-trkB). (*p < 0.05; n = 7 animals with at least 2 slices per animal, 14–20 slices/condition, and 70–100 cells, on average 5/slice). Error bars indicate SEM. f, VTA punches from the PNI group showed significantly elevated BDNF protein, which was blocked with pretreatment with MC. *p < 0.05 compared with sham, #p < 0.05 compared with PNI+saline. Error bars indicate SEM.
Discussion
In the present study, we show that chronic pain results in decreased accumbal morphine- and cocaine-stimulated extracellular DA levels that contribute to impaired reward-related behaviors. We have identified activated microglia and BDNF as key mediators in this dysfunction of the mesolimbic DA system by disrupting the Cl− gradient in GABAergic interneurons of the VTA.
The loss of morphine- and cocaine-stimulated extracellular DA in the NAc of the chronic pain group was striking (Fig. 1). A prior study conducted 3 d after nerve injury described a partially attenuated morphine-stimulated DA response (Ozaki et al., 2002). This supports our current findings and suggests that dysfunction in mesolimbic DA circuitry becomes increasingly apparent as animals transition from acute to chronic pain. An impaired mesolimbic DA response in chronic pain is also supported by a recent human fMRI study that found dramatically reduced reward-stimulated activity in the VTA of fibromyalgia patients (Loggia et al., 2014).
Despite the lack of morphine-evoked striatal DA release in chronic pain animals, systemic morphine continued to produce a place preference. One hypothesis is that the retained opioid place preference in chronic pain animals despite the absence of DA activation is due to the negative reinforcement (pain relief) and, indeed, morphine antinociception was found to be intact in chronic pain animals (Fig. 2c; Navratilova et al., 2012). It should be recognized that the role of DA signaling in opioid reward is complex. Several prior studies have indicated that mesolimbic DA activity is not required for opioid place preference (Hnasko et al., 2005; Vargas-Perez et al., 2009) but, rather, that DA release may be involved in the salience or memory processing of rewarding stimuli (Berridge, 2007). Indeed, when only one morphine conditioning trial was run, chronic pain animals failed to display a place preference (Fig. 2a). We suggest that this may be a reflection of a blunted DA release impairing reward-learning behavior in this shortened conditioning trial paradigm. When an opioid agonist (1 ng of DAMGO) was injected directly into the VTA, it failed to produce a place preference in the chronic pain group (Fig. 3a). This further supports the premise that chronic pain leads to a loss of mesolimbic DA-driven reward. Systemic cocaine, like morphine, exhibited a place preference in both control and chronic pain groups. However, cocaine has a well documented analgesic effect (Fig. 2c; Lin et al., 1989) and it is possible that cocaine place preference could be a result of negative reinforcement as a result of pain relief. The presence of morphine and cocaine place preference after systemic drug administration despite the absence of drug-evoked striatal DA release suggests that circuitry outside of the mesolimbic DA system, possibly related to relief of pain and negative reinforcement, drives place preference in chronic pain states.
One parsimonious hypothesis for the loss of opioid-evoked DA release is a loss of function or expression of the mu opioid (MOP) receptor in the VTA (Niikura et al., 2008). Injection of a higher dose of DAMGO (25 ng) into the VTA produced a full CPP in chronic pain animals, suggesting that MOP receptors are still functional, but higher doses of opioids are needed to evoke increases in extracellular DA in chronic pain animals. This result would be consistent with a desensitized opioid system. To address this potential confound, we have probed the mesolimbic system with nonopioid, DA-stimulating drugs. Loss of DA-mediated reward was not restricted to opioids and a similar phenomenon was observed with cocaine. Injection of cocaine into the NAc produced a place preference in the control group, but not in the chronic pain group. These data, along with microdialysis experiments, serve to reinforce the notion of a hyporeactive mesolimbic DA system to drug reward stimuli in chronic pain.
Our prior studies have determined that chronic pain leads to a major reduction in total tissue levels of striatal DA; however, this was not attributed to a loss of DA cell bodies in the VTA (Taylor et al., 2014). Given our results with cocaine, a drug that blocks the DA reuptake transporter, we challenged the system with amphetamine, a drug that evokes increased extracellular DA via a reverse efflux mechanism. Interestingly, amphetamine-evoked DA levels were equivalent in the control and chronic pain groups. Our data are consistent with increased inhibitory tone on DA neurons in chronic pain animals given the lack of cocaine- or morphine-stimulated extracellular DA levels. However, the unaltered amphetamine-stimulated extracellular DA levels suggest that the free intracellular DA pool is maintained in chronic pain animals.
In this study, we have identified a mechanism by which the opioid- and cocaine-stimulated DA response is blunted in chronic pain animals. We describe a decrease in Cl− transport in GABAergic interneurons of the VTA due to a loss of KCC2 expression (Fig. 5). A loss of functional KCC2 has been shown previously to result in a shift in the GABA reversal potential, leading to disinhibition of spinal lamina I neurons (Coull et al., 2003). We identified activated microglia and BDNF as being possible components to this mechanism, in that minocycline treatment or interfering with BDNF signaling in the chronic pain group recovered Cl− transport in GABAergic neurons and recovered morphine- and cocaine-stimulated DA release. Activated microglia are implicated in the development of chronic pain (Raghavendra et al., 2002; Ledeboer et al., 2006; Loggia et al., 2015) and BDNF released from activated microglia contributes directly to the hyperexcitability of the pain pathway (Coull et al., 2005). Previous reports implicate BDNF-TrkB signaling as a negative modulator of opioid function in the VTA in models of opioid dependence (Koo et al., 2012; Vargas-Perez et al., 2014). Our findings extend these results by implicating KCC2 function on GABAergic neurons as driving this negative modulation in a chronic pain model.
Changes in the DA system likely contribute to the affective disorders associated with chronic pain and may also affect the effectiveness of many analgesics, such as morphine, the therapeutic potential of which is strongly linked to its rewarding capacity. Moreover, this work affirms the importance of microglial activation in modulating reward behavior and suggests microglial inhibitors as an effective therapeutic for disrupted DA transmission. We acknowledge that minocycline may have off-target effects and further studies are warranted to explore the utility of inhibiting microglia in this paradigm using more specific and/or local strategies of inhibition.
In conclusion, it is well established that neuroplastic changes in reward circuitry, including molecular and cellular changes within the mesolimbic DA system, lead to the genesis of negative affect (Koob and Le Moal, 2005; Koob and Volkow, 2010; Koob et al., 2014). Our present data show that chronic pain causes activation of microglia in the VTA that modifies reward by disrupting Cl− homeostasis in GABAergic neurons. These findings represent a paradigm shift in the field of chronic pain by identifying a signaling and biophysical mechanism by which microglia alter the reward pathway.
Footnotes
This work was supported by the National Institutes of Health (Grant DA005010 to C.J.E.), the Shirley and Stefan Hatos Foundation (A.M.W.T., A.G., N.P.M.; C.J.E. is the holder of the Stefan and Shirley Hatos endowed chair), and the Canadian Institutes of Health (Research Grants MOP 12942 to Y.D.K. and MOP 123298 to C.M.C.). We thank Miss Sarah Pickens and Miss Zenna Evans for their technical assistance.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Asmundson GJ, Katz J. Understanding the co-occurrence of anxiety disorders and chronic pain: state-of-the-art. Depress Anxiety. 2009;26:888–901. doi: 10.1002/da.20600. [DOI] [PubMed] [Google Scholar]
- Berridge KC. The debate over dopamine's role in reward: the case for incentive salience. Psychopharmacology. 2007;191:391–431. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM, Trojanowski JQ. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7:e39216. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsook D, Edwards R, Elman I, Becerra L, Levine J. Pain and analgesia: the value of salience circuits. Prog Neurobiol. 2013;104:93–105. doi: 10.1016/j.pneurobio.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Xue L, Grenier P, Magnussen C, Lecour S, Olmstead MC. Changes in morphine reward in a model of neuropathic pain. Behav Pharmacol. 2013;24:207–213. doi: 10.1097/FBP.0b013e3283618ac8. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chorin E, Vinograd O, Fleidervish I, Gilad D, Herrmann S, Sekler I, Aizenman E, Hershfinkel M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J Neurosci. 2011;31:12916–12926. doi: 10.1523/JNEUROSCI.2205-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Gremel CM, Groblewski PA. Drug-induced conditioned place preference and aversion in mice. Nat Protoc. 2006;1:1662–1670. doi: 10.1038/nprot.2006.279. [DOI] [PubMed] [Google Scholar]
- Digman MA, Dalal R, Horwitz AF, Gratton E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys J. 2008;94:2320–2332. doi: 10.1529/biophysj.107.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon N, Prescott SA, Castonguay A, Godin AG, Kröger H, De Koninck Y. Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Comput Biol. 2011;7:e1002149. doi: 10.1371/journal.pcbi.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1–27. doi: 10.1016/j.pneurobio.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De Koninck Y. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013;16:183–192. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, Akers CA, Clinton SM, Phillips PE, Akil H. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469:53–57. doi: 10.1038/nature09588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, Perez-Sanchez J, Boudreau D, Wang B, Dumas L, Valade I, Bachand K, Jacob-Wagner M, Tardif C, Kianicka I, Isenring P, Attardo G, Coull JA, De Koninck Y. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med. 2013;19:1524–1528. doi: 10.1038/nm.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob S, Pizzagalli DA, Dutra SJ, Stern J, Mörgeli H, Milos G, Schnyder U, Hasler G. Dopamine-related deficit in reward learning after catecholamine depletion in unmedicated, remitted subjects with bulimia nervosa. Neuropsychopharmacology. 2012;37:1945–1952. doi: 10.1038/npp.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasti L, Richardson D, Jhaveri M, Eldeeb K, Barrett D, Elphick MR, Alexander SP, Kendall D, Michael GJ, Chapman V. Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Mol Pain. 2009;5:35. doi: 10.1186/1744-8069-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagelberg N, Forssell H, Rinne JO, Scheinin H, Taiminen T, Aalto S, Luutonen S, Någren K, Jääskeläinen S. Striatal dopamine D1 and D2 receptors in burning mouth syndrome. Pain. 2003a;101:149–154. doi: 10.1016/S0304-3959(02)00323-8. [DOI] [PubMed] [Google Scholar]
- Hagelberg N, Forssell H, Aalto S, Rinne JO, Scheinin H, Taiminen T, Någren K, Eskola O, Jääskeläinen SK. Altered dopamine D2 receptor binding in atypical facial pain. Pain. 2003b;106:43–48. doi: 10.1016/S0304-3959(03)00275-6. [DOI] [PubMed] [Google Scholar]
- Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005;438:854–857. doi: 10.1038/nature04172. [DOI] [PubMed] [Google Scholar]
- Jarcho JM, Mayer EA, Jiang ZK, Feier NA, London ED. Pain, affective symptoms, and cognitive deficits in patients with cerebral dopamine dysfunction. Pain. 2012;153:744–754. doi: 10.1016/j.pain.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi M, Midorikawa N, Takeshima H, Murphy NP. Exogenous, but not endogenous nociceptin modulates mesolimbic dopamine release in mice. J Neurochem. 2004;89:257–263. doi: 10.1111/j.1471-4159.2003.02322.x. [DOI] [PubMed] [Google Scholar]
- Koo JW, Mazei-Robison MS, Chaudhury D, Juarez B, LaPlant Q, Ferguson D, Feng J, Sun H, Scobie KN, Damez-Werno D, Crumiller M, Ohnishi YN, Ohnishi YH, Mouzon E, Dietz DM, Lobo MK, Neve RL, Russo SJ, Han MH, Nestler EJ. BDNF is a negative modulator of morphine action. Science. 2012;338:124–128. doi: 10.1126/science.1222265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the “dark side” of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addition. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, Jr, George O. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76:370–382. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latrémolière A, Mauborgne A, Masson J, Bourgoin S, Kayser V, Hamon M, Pohl M. Differential implication of proinflammatory cytokine interleukin-6 in the development of cephalic versus extracephalic neuropathic pain in rats. J Neurosci. 2008;28:8489–8501. doi: 10.1523/JNEUROSCI.2552-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Liu T, Shumilla JA, Mahoney JH, Vijay S, Gross MI, Vargas JA, Sultzbaugh L, Claypool MD, Sanftner LM, Watkins LR, Johnson KW. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol. 2006;2:279–291. doi: 10.1017/S1740925X0700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrain V. Where is my pain? Pain. 2011;152:467–468. doi: 10.1016/j.pain.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Leknes S, Tracey I. A common neurobiology for pain and pleasure. Nat Rev Neurosci. 2008;9:314–320. doi: 10.1038/nrn2333. [DOI] [PubMed] [Google Scholar]
- Lin Y, Morrow TJ, Kiritsy-Roy JA, Terry LC, Casey KL. Cocaine: evidence for supraspinal, dopamine-mediated, non-opiate analgesia. Brain Res. 1989;479:306–312. doi: 10.1016/0006-8993(89)91633-8. [DOI] [PubMed] [Google Scholar]
- Loggia ML, Berna C, Kim J, Cahalan CM, Gollub RL, Wasan AD, Harris RE, Edwards RR, Napadow V. Disrupted brain circuitry for pain-related reward/punishment in fibromyalgia. Arthritis Rheumatol. 2014;66:203–212. doi: 10.1002/art.38191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, Riley M, Wasan AD, Zürcher NR, Albrecht DS, Vangel MG, Rosen BR, Napadow V, Hooker JM. Evidence for brain glial activation in chronic pain patients. Brain. 2015;138:604–615. doi: 10.1093/brain/awu377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40:139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Kim SA, Buechler NL, Porreca F, Eisenach JC. Opioid self-administration in the nerve-injured rat: relevance of antiallodynic effects to drug consumption and effects of intrathecal analgesics. Anesthesiology. 2007;106:312–322. doi: 10.1097/00000542-200702000-00020. [DOI] [PubMed] [Google Scholar]
- Mosconi T, Kruger L. Fixed-diameter polyethylene cuffs applied to the rat sciatic nerve induce a painful neuropathy: Ultrastructural morphometric analysis of axonal alterations. Pain. 1996;64:37–57. doi: 10.1016/0304-3959(95)00077-1. [DOI] [PubMed] [Google Scholar]
- Mouraux A, Diukova A, Lee MC, Wise RG, Iannetti GD. A multisensory investigation of the functional significance of the “pain matrix.”. Neuroimage. 2011;54:2237–2249. doi: 10.1016/j.neuroimage.2010.09.084. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, Qu C, Eyde N, Ci S, Ossipov MH, King T, Fields HL, Porreca F. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc Natl Acad Sci U S A. 2012;109:20709–20713. doi: 10.1073/pnas.1214605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura K, Narita M, Narita M, Nakamura A, Okutsu D, Ozeki A, Kurahashi K, Kobayashi Y, Suzuki M, Suzuki T. Direct evidence for the involvement of endogenous beta-endorphin in the suppression of the morphine-induced rewarding effect under a neuropathic pain-like state. Neurosci Lett. 2008;435:257–262. doi: 10.1016/j.neulet.2008.02.059. [DOI] [PubMed] [Google Scholar]
- Nutile-McMenemy N, Elfenbein A, Deleo JA. Minocycline decreases in vitro microglial motility, beta1-integrin, and Kv1.3 channel expression. J Neurochem. 2007;103:2035–2046. doi: 10.1111/j.1471-4159.2007.04889.x. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Narita M, Narita M, Iino M, Sugita J, Matsumura Y, Suzuki T. Suppression of the morphine-induced rewarding effect in the rat with neuropathic pain: implication of the reduction in mu-opioid receptor functions in the ventral tegmental area. J Neurochem. 2002;82:1192–1198. doi: 10.1046/j.1471-4159.2002.01071.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, Malenka RC. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science. 2014;345:535–542. doi: 10.1126/science.1253994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taves S, Berta T, Chen G, Ji RR. Microglia and spinal cord synaptic plasticity in persistent pain. Neural Plast. 2013;2013:753656. doi: 10.1155/2013/753656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Murphy NP, Evans CJ, Cahill CM. Correlation between ventral striatal catecholamine content and nociceptive thresholds in neuropathic mice. J Pain. 2014;15:878–885. doi: 10.1016/j.jpain.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Perez H, Ting-A Kee R, Walton CH, Hansen DM, Razavi R, Clarke L, Bufalino MR, Allison DW, Steffensen SC, van der Kooy D. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science. 2009;324:1732–1734. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Perez H, Bahi A, Bufalino MR, Ting-A Kee R, Maal-Bared G, Lam J, Fahmy A, Clarke L, Blanchard JK, Larsen BR, Steffensen S, Dreyer JL, van der Kooy D. BDNF signaling in the VTA links the drug-dependent state to drug withdrawal aversions. J Neurosci. 2014;34:7899–7909. doi: 10.1523/JNEUROSCI.3776-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood PB, Schweinhardt P, Jaeger E, Dagher A, Hakyemez H, Rabiner EA, Bushnell MC, Chizh BA. Fibromyalgia patients show an abnormal dopamine response to pain. Eur J Neurosci. 2007;25:3576–3582. doi: 10.1111/j.1460-9568.2007.05623.x. [DOI] [PubMed] [Google Scholar]