

Abstract
Multiple sclerosis (MS) is a demyelinating disease of the CNS characterized by inflammation and neurodegeneration. Animal models that enable the study of remyelination in the context of ongoing inflammation are greatly needed for the development of novel therapies that target the pathological inhibitory cues inherent to the MS plaque microenvironment. We report the development of an innovative animal model combining cuprizone-mediated demyelination with transfer of myelin-reactive CD4+ T cells. Characterization of this model reveals both Th1 and Th17 CD4+ T cells infiltrate the CNS of cuprizone-fed mice, with infiltration of Th17 cells being more efficient. Infiltration correlates with impaired spontaneous remyelination as evidenced by myelin protein expression, immunostaining, and ultrastructural analysis. Electron microscopic analysis further reveals that demyelinated axons are preserved but reduced in caliber. Examination of the immune response contributing to impaired remyelination highlights a role for peripheral monocytes with an M1 phenotype. This study demonstrates the development of a novel animal model that recapitulates elements of the microenvironment of the MS plaque and reveals an important role for T cells and peripheral monocytes in impairing endogenous remyelination in vivo. This model could be useful for testing putative MS therapies designed to enhance remyelination in the setting of active inflammation, and may also facilitate modeling the pathophysiology of denuded axons, which has been a challenge in rodents because they typically remyelinate very quickly.
Keywords: axons, cuprizone, demyelination, inflammation, multiple sclerosis, oligodendrocyte precursor cells
Introduction
Multiple sclerosis (MS) is a demyelinating disease of the CNS characterized by inflammation and subsequent destruction of oligodendrocytes and axons (Noseworthy et al., 2000). A defining characteristic of disease progression is the dampening or failure of endogenous remyelination, likely due to a variety of inhibitory cues within the plaque (Sloane et al., 2010; Hagemeier et al., 2012). To facilitate remyelination, the microenvironment surrounding a lesion must support oligodendrocyte progenitor cell (OPC) proliferation, recruitment, and differentiation into myelinating oligodendrocytes. A number of recent studies have increased our understanding of how the factors necessary to drive or inhibit these processes can be influenced by inflammation (Arnett et al., 2001; Mason et al., 2001; Chew et al., 2005; Tanner et al., 2011; Tirotta et al., 2011; Kang et al., 2012; Kang et al., 2013; Miron et al., 2013; Miron and Franklin, 2014). However, much remains to be elucidated. Current therapies for MS target the immune response and have demonstrated little efficacy for promoting repair. One potential reason for this lack of effective neuroprotective therapies is the limited availability of animal models that facilitate the study of remyelination in the presence of inflammation.
Experimental autoimmune encephalomyelitis (EAE) has been used for many years to study MS. While studies in the EAE model have led to the development of several effective new therapies for MS, the utility of the model for understanding the effects of inflammation on resident glial cells in an attempt to identify strategies for remyelination and repair is limited in several aspects. Lesion localization in EAE is variable from animal to animal (Gold et al., 2006), and the infiltrating immune cells initiate a cascade of events culminating in the early destruction of axons limiting the ability to test strategies that may enhance remyelination (Wujek et al., 2002; Wang et al., 2005; Jones et al., 2008; Soulika et al., 2009).
In contrast, toxin-based models that are commonly used to study remyelination in the CNS, such as lysolecithin and cuprizone, although useful for identifying factors contributing to the repair process, largely lack the chronic immune response that is known to exist in the MS plaque and have robust endogenous remyelination leaving a smaller window to show the beneficial effects of a putative remyelinating therapy (Denic et al., 2011). For these reasons, a model that combines a myelin-specific immune response with primary demyelination is greatly needed both to more fully understand factors that may contribute to the molecular pathogenesis of MS and to develop novel therapies that will be effective at limiting inflammation while promoting repair of myelin and damaged axons to halt disease progression.
Herein we report the development of an innovative animal model, which combines cuprizone-mediated demyelination with adoptive transfer (AT) of myelin reactive CD4+ T cells. We demonstrate that the presence of myelin-reactive T cells in the CNS of cuprizone-fed animals significantly delays spontaneous remyelination highlighting the important role inflammation likely plays in impairing the reparative processes in MS.
Materials and Methods
Western blot analysis.
Mice were deeply anesthetized with sodium pentobarbital (100 mg/kg BW) and perfused transcardially with chilled HBSS. Using a microdissection scope, the corpus callosum was carefully dissected from each hemisphere. Tissue samples were then homogenized in RIPA buffer (Boston BioProducts) using a hand-held homogenizer. Protein concentration was determined using Bio-Rad protein assay. For MBP and 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP) immunoblots, protein samples were denatured in Laemli's SDS sample buffer containing β-mercaptoethanol. A total of 20 μg of each sample was separated on a 12% Tris HCl gel (Bio-Rad) and transferred to a nitrocellulose membrane. Membranes were blocked in 0.1% Tween 20 TBS containing 5% nonfat milk for 1 h before incubation with primary antibodies. Primary antibodies were diluted in the same solution as follows: anti-CNP (1:750, clone 11–5B; Millipore), anti-MBP (1:1000, clone SMI99; Covance), and anti-β-actin (1:1000, clone 20–33; Sigma). Anti-mouse or anti-rabbit secondary antibodies conjugated to IRDye 680 or 800 were incubated for 1 h before scanning the membrane on a LI-COR Odyssey imager. Images were analyzed using the Odyssey infrared imaging system application software version 3.0. Protein expression was normalized to β-actin.
Transmission electron microscopy.
Mice were anesthetized with pentobarbital (100 mg/kg BW) and perfused transcardially with 2% PFA (freshly prepared from EM grade prill form), 2% glutaraldehyde, 3 mm MgCl2, in 0.1 M sodium cacodylate buffer, pH 7.2, for 30 min at a rate of 2 ml/min. After perfusion, mice were placed in the cold for ∼2–3 h, before removing the brain. Brains were removed and placed in fresh fixative overnight at 4°C before dissection of the corpus callosum. After a buffer rinse, samples were postfixed in reduced 2% osmium tetroxide, 1.6% potassium ferrocyanide in buffer (2 h) on ice in the dark. Following a distilled H2O rinse, samples were stained with 2% aqueous uranyl acetate (0.22 μm filtered, 1 h, dark), dehydrated in a graded series of ethanol, propylene oxide, and embedded in Eponate 12 (Ted Pella) resin. Samples were polymerized at 60°C overnight.
Thin sections (60–90 nm) were cut with a diamond knife on the Reichert-Jung Ultracut E ultramicrotome and picked up with copper slot (1 × 2 mm) grids. Grids were stained with 2% uranyl acetate and 1% lead citrate and observed with a Hitachi 7600 TEM at 80 kV. Images were captured with an AMT CCD (5 megapixel) camera. National Institutes of Health ImageJ software was used to measure the number of myelinated and unmyelinated axons per unit area. G-ratios were determined by calculating the diameter of axon/diameter of axon with myelin. Unmyelinated axons were not included in g-ratio determinations. Ten images were quantified per mouse. Images were selected at random from the ROI (mid-caudal corpus callosum above the fornix).
Immunohistochemistry.
Mice were deeply anesthetized with sodium pentobarbital (100 mg/kg BW) and perfused transcardially with chilled PBS followed by 4% PFA. Brains were collected, postfixed overnight, and cryoprotected in 30% sucrose for ∼48 h before freezing. Tissue was sectioned on a cryostat (20 μm) and mounted onto glass slides (Superfrost Plus; Fisher). Sections were blocked and permeabilized in PBS containing 5% normal goat serum and 0.4% Triton X-100 for 1 h at room temperature and then incubated overnight at 4°C in PBS containing 3% normal goat serum, 0.1% Triton X-100, and primary antibody as follows: mouse anti-SMI32 (clone SMI32, 1:1000, Covance), mouse anti-SMI31 (clone SMI31, 1:1000, Covance), mouse anti-APP (clone 22C11, 1:80, Millipore), and rabbit anti-CD3 (1: 200, Dako). Species-specific secondary antibodies directly conjugated to Alexa fluorophores (1:1000, Invitrogen) were used to visualize immunostaining. Sections were incubated in secondary antibodies for 1 h at room temperature before mounting in anti-fade reagent with DAPI.
Mounted slides were imaged using a Zeiss Axio Observer Z1 epifluorescence microscope and Axiovision software with the appropriate excitation and emission filters. A total of 3 or 4 sections were examined per mouse, and 3–6 mice were analyzed per group. Areas were chosen randomly within the regions of interest. National Institutes of Health ImageJ software was used to create a binary image of the staining and to subsequently quantify staining intensity.
Black Gold staining (Black Gold II myelin staining kit, Millipore) was performed according to the manufacturer's instructions. In short, slides were incubated in 0.3% Black Gold at 60°C for 12–20 min until the thinnest fibers were stained. The slides were then fixed in 1% sodium thiosulfate for 3 min, counterstained with 0.1% Cresyl Violet (Millipore) for 3 min, dehydrated using a series of gradated alcohols, cleared in xylene, and coverslipped with VectaMount Permanent Mounting Media (Vector Laboratories). Slides were imaged on a BX41 Olympus microscope using ImageProPlus5.1 software (Media Cybernetics).
Mice.
C57BL/6 (CD45.2+) and CD45.1+ congenic mice were purchased from the National Cancer Institute (Frederick, MD). Mice with a transgenic T-cell receptor specific for MOG 35–55 (2D2 mice) were purchased from The Jackson Laboratory and were bred and maintained in our animal facility. All mice were maintained in a federally approved animal facility at the Johns Hopkins University (Baltimore) in accordance with the Institutional Animal Care and Use Committee. Male mice 8–12 weeks of age were used in all of the experiments. Age-, sex-, and genotype-matched mice were used in all experiments as controls.
Analysis of CNS-infiltrating mononuclear cells.
At 3, 7, or 14 d after transfer, mice were killed for analysis of CNS-infiltrating cells. Mice were anesthetized with sodium pentobarbital (100 mg/kg BW) (Oak Pharmaceuticals) and perfused through the left ventricle with 20 ml of cold 1× HBSS (Cellgro). The whole brain was collected. Cell suspensions were made by passing the tissue through a 100 μm nylon cell strainer (BD Biosciences) and digested for 20 min at room temperature with Liberase TM Research Grade (Roche). The tissue was pelleted and washed with 1× HBSS before resuspending in 4 ml of 37% isotonic Percoll (GE Healthcare), diluted in 10× HBSS, then underlaid with 2 ml of 70% isotonic Percoll and overlaid with 2 ml 1× HBSS. Tissue was centrifuged, and mononuclear cells were isolated from the 37/70 interphase. Cells were then washed and resuspended in 2% FBS in PBS for flow cytometric analysis.
Immunostaining and flow cytometry.
To stain intracellular cytokines, cells were stimulated with Cell Stimulation Mixture plus protein transport inhibitors (eBioscience) for 4–6 h. Surface antigens were stained with the following antibodies: anti-CD4 (RM4–5, BD Biosciences), anti-CD8 (53–6.7, BD Biosciences), anti-CD11b (M1/70, BioLegend), anti-CD45.1 (A20, BD Biosciences), anti-CD45.2 (104, eBioscience), anti-INOS (R&D Systems), and anti-Ly6C (AL-21, BD Biosciences). For intracellular staining, cells were fixed and permeabilized with the Foxp3 staining buffer kit (eBioscience) and stained for intracellular cytokines and proteins with anti-IFN-γ (XMG1.2, BD Biosciences), anti-IL-17 (eBio17B7, eBioscience), and anti-iNOS (6, BD Biosciences) antibodies. Flow cytometric analyses were performed on a FACSCalibur instrument (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Cell culture and polarization.
Spleens and lymph nodes were isolated from naive mice, and single-cell suspensions were made by passing through a 70 μm nylon cell strainer (BD Biosciences). T helper cells were isolated from splenocytes by negative selection using EasySep Mouse CD4+ T cell Enrichment Kit (Stem Cell Technologies), following the manufacturer's protocol. Cells were cultured in RPMI 1640 (Invitrogen) supplemented with 10% v/v FBS (Invitrogen), 100 μg/ml penicillin and streptomycin (Quality Biological), 0.5 μm 2-mercaptoethanol (Invitrogen), 10 mm HEPES buffer (Quality Biological), 1 mm sodium pyruvate (Sigma-Aldrich), and MEM NEAA (Sigma-Aldrich). For Th17 polarization, cells were cultured in complete RPMI and activated with irradiated WT splenocytes and 20 μg/ml myelin oligodendrocyte glycoprotein (MOG 35–55) peptide (Johns Hopkins Peptide Synthesis Core Facility, Baltimore) in the presence of 10 ng/ml IL-1β, 30 ng/ml IL-6 (PeproTech), 3 ng/ml TGF-β (Invitrogen), 20 μg/ml anti-IFN-γ (XMG1.2, BioLegend), and anti-IL-4 (11B11, BioLegend) for 72 h. Cells were then split in half with the addition of new media and 20 ng/ml IL-23 (R&D Systems) and further cultured for 96 h. Live cells were separated by ficoll gradient (Lymphocyte Separation Medium, MP Biomedicals) and restimulated with anti-CD3 (145-2C11, BD Biosciences) and anti-CD28 (37.51, BD Biosciences) for 72 h.
Cuprizone.
C57BL/6 mice were fed 0.2% w/w cuprizone (bis(cyclohexanone) oxaldihydrazone (Sigma-Aldrich) mixed with powdered, irradiated 18% protein rodent diet (Teklad Global) for a total duration of 4 weeks.
AT.
C57BL/6 mice were immunized subcutaneously over two sites on the lateral abdomen with 100 μg of MOG 35–55 peptide in Complete Freund's Adjuvant (CFA) containing 8 μg/ml Mycobacterium tuberculosis H37Ra (Difco Laboratories), followed by intraperitoneal injections of 250 ng of Bordetella pertussis toxin (List Biological Laboratories) on day 0 and 2 after immunization. At day 9 after immunization, mice were killed and draining lymph nodes were collected, a single cell suspension was made by passing them through a 70 μm nylon cell strainer (BD Biosciences) and cultured in the presence of IL-23 for 72 h. Cells were then transferred into cuprizone-fed animals and controls; alternatively, Th17 cells polarized as described above were transferred into the animals.
ELISA.
To quantify cytokine production, supernatants from cultured cells were collected after 72 h, and the levels of IFN-γ and IL-17 were quantified using ELISA kits according to the manufacturer's instructions (BioLegend).
Statistical analysis.
Statistical analysis was conducted using the GraphPad Prism software (GraphPad). Two-tailed Student's t test was used to analyze normally distributed data. Results were considered significant if the p value was <0.05. Error bars indicate SEM in all the figures.
Results
Adoptively transferred CD4+ T cells secreting IL-17 migrate more efficiently to the corpus callosum of cuprizone-fed mice than IFN-γ /IL-17 dual-secreting cells
CD4+ T cells of the Th17 lineage have been demonstrated to migrate more efficiently to the brains of EAE animals than cells of the Th1 lineage (Muller et al., 2000; Wensky et al., 2005; Lees et al., 2008; Pierson et al., 2012). These differences are revealed by characterization of the cellular infiltrate as well as behavioral changes in mice receiving Th17 cells. To effectively mimic the chronic inflammation present during demyelination that is seen in MS patients, it is essential for inflammatory T cells to be present at the site of primary demyelination (the corpus callosum) in the cuprizone-fed recipients. To this end, two different strategies of myelin-specific T-cell generation were compared for their ability to traffic into the brain and subsequently delay remyelination in cuprizone-fed animals. The first method involved traditional AT EAE, induced via immunization of WT C57BL/6 donor animals with MOG 35–55 peptide emulsified in CFA with two injections of pertussis toxin. Draining lymph nodes were isolated from donor animals at 9–10 d after immunization and restimulated in vitro with MOG 35–55 peptide + recombinant (r) IL-23. For the purposes of our study, rIL-23 was used to maintain a mixed population of IL-17/IFN-γ-producing T cells. Following 72 h of culture, the MOG-specific cells were transferred into mice that had received a cuprizone diet for 4 weeks immediately beforehand (Fig. 1A). Alternatively, 2D2 TCR transgenic mice, which express the Vα3Vβ11 cell receptor specific for MOG 35–55 peptide, were used as a source of MOG specific T cells. Spleens and lymph nodes were isolated from 2D2 mice, CD4+ T cells were isolated and polarized to a Th17 phenotype with polarizing cytokines, MOG 35–55 peptide, and irradiated antigen-presenting cells as described previously, and transferred into cuprizone-fed recipients (Fig. 1A). Immediately before transfer, supernatants from cell cultures were collected and ELISAs were performed to establish differences in IFN-γ and IL-17 production from AT T cells versus Th17 cells, respectively. MOG-specific CD4+ T cells generated via traditional AT secreted significantly more IFN-γ than cells generated via Th17 polarization of 2D2 cells (Fig. 1B,C). Importantly, the ratio of IFN-γ:IL-17 was higher in this population of cells compared with 2D2 Th17 cells where IL-17 production was greater than that of IFN-γ. These results were confirmed via intracellular cytokine staining, demonstrating a mixed population of IL-17/IFN-γ + T cells in the AT-generated cells (Fig. 1D) compared with single-positive IL-17-secreting CD4+ T cells from the Th17-polarized cells (Fig. 1E).
Figure 1.
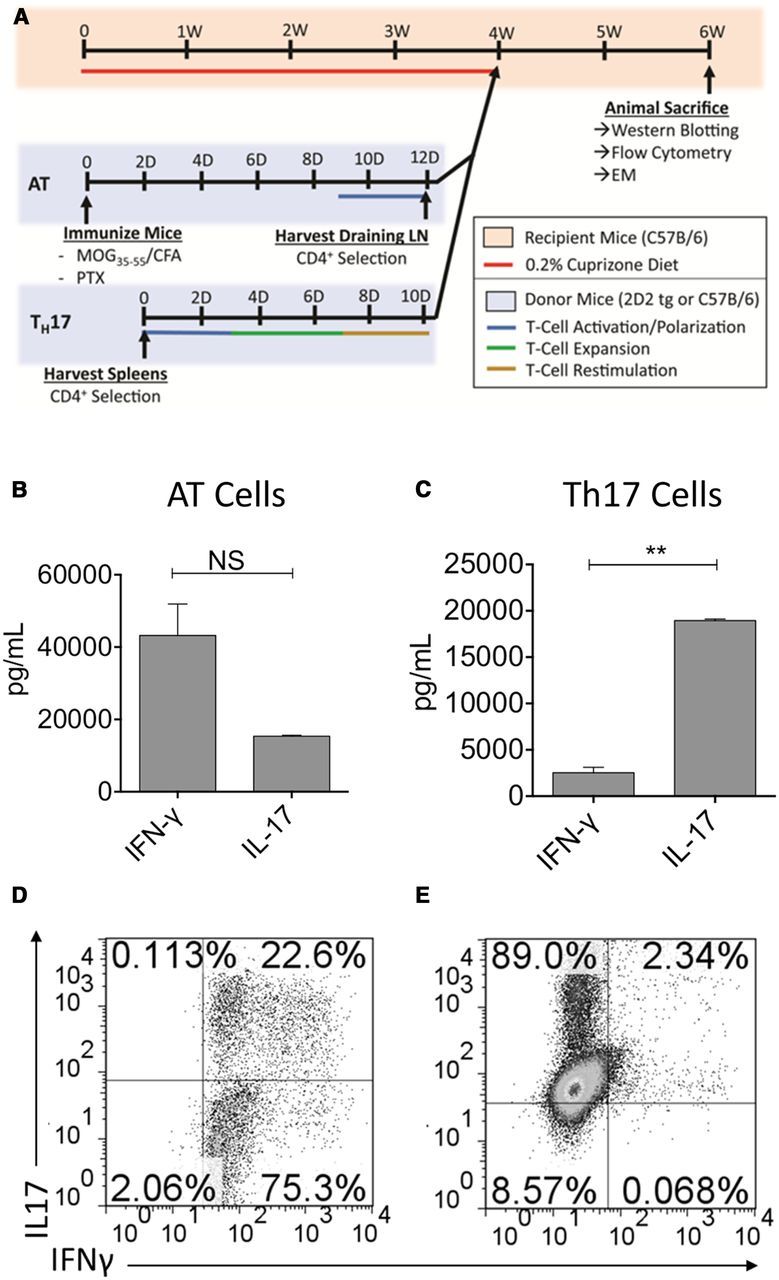
CD4+ T cells restimulated from MOG35–55-immunized donors produce both IL-17 and IFN-γ, whereas CD4+ T cells isolated from unimmunized 2D2 TCR transgenic mice and cultured under Th17-polarizing conditions express mainly IL-17 before transfer. A, Schematic timeline for cuprizone AT model. C57BL/6 male mice were fed a 0.2% cuprizone diet for 4 weeks to induce demyelination. Mice were subsequently switched to normal chow, and CD4+ T cells generated either via traditional AT (highlighted in blue) or Th17 polarization of 2D2 cells (labeled Th17 and highlighted in blue) were transferred by intraperitoneal injection. Mice were monitored for an additional 2 weeks before death and analysis. B, C, ELISA analysis for IFN-γ and IL-17 from supernatants taken from AT (A) or Th17 (C) cultures before transfer into recipient animals. D, E, Intracellular cytokine staining and flow cytometry were performed on AT or Th17-polarized CD4+ T cells before transfer, and IFN-γ and IL-17 production was evaluated. Data shown are representative of 4 replicate wells per experiment and three independent experiments. **p < 0.005 (two-tailed Student's t test). ns, No significant difference.
To evaluate whether these aforementioned differences in cytokine production and corresponding changes in integrin and chemokine receptor expression resulted in differential trafficking to the corpus callosum of cuprizone-fed recipients, animals were killed at 3, 7, and 14 d after transfer, and the frequencies of CD4+ T cells in the brains were evaluated by flow cytometry. The infiltration of CD4+ T cells was found to peak at 14 d after transfer, and Th17 cells trafficked more efficiently than AT cells (10% vs 30%, respectively) (Fig. 2C,D,G,H). The frequencies of Th17 cells in the whole brain also correlated with the number of CD4+ T cells in the corpus callosum of the animals at 14 d after transfer of 2D2 Th17 cells (Fig. 2I), confirming that Th17 cells traffic efficiently to the site of primary demyelination. In further confirmation of this result, CD3+ T cells were localized to the corpus callosum of mice that received Th17 cells following cuprizone in much greater abundance than in mice receiving either cuprizone or Th17 cells alone (Fig. 3).
Figure 2.

Adoptively transferred CD4+ T cells secreting IL-17 migrate more efficiently to the corpus callosum of cuprizone-fed mice than IFN-γ/IL-17-secreting CD4+ T cells. A–D, Myelin-reactive CD4+ T cells were generated via traditional AT as outlined in Figure 1. A, C, The frequency of CD4+ T cells that were detected by flow cytometry in the brains of recipient mice at 7 d (A) and 14 d (C) after cell transfer, respectively. B, D, Bar graphs represent quantitative data from 4 animals per group. E–H, Myelin-reactive CD4+ T cells were generated via Th17 polarization of 2D2 T cells. E, G, The frequency of CD4+ T cells that were detected in the brains of recipient mice by flow cytometry at 7 d (E) and 14 d (G) after transfer, respectively. F, H, Bar graphs represent quantitative data collected from 4 animals per experimental group. I, Flow cytometric analysis demonstrating the frequency of CD4+ T cells infiltrating the corpus callosum of recipient animals 14 d after transfer of Th17 cells. All data are representative of three independent experiments. *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test).
Figure 3.

CD3+ T cells infiltrate the corpus callosum of cuprizone-fed mice following Th17 cell transfer. Mice were killed 14 d after cessation of the cuprizone diet and T-cell transfer. CD3+ cells are occasionally observed in the corpus callosum of mice receiving either Th17 cells or cuprizone alone but are robustly present in those receiving both treatments. Representative images from two independent experiments. N = 3 mice per group. Scale bar, 50 μm.
Transfer of MOG-specific T cells significantly reduces the amount of myelin protein in the corpus callosum
Because we hypothesize that chronic inflammation in the CNS of MS patients contributes to failure to effectively remyelinate, we examined myelin protein expression in the corpora callosa of cuprizone-fed mice in the presence or absence of transferred MOG-specific CD4+ T cells. As predicted, because cuprizone-fed mice have been demonstrated to spontaneously remyelinate beginning from the third week of toxin ingestion (Doan et al., 2013), we observed an ∼60% recovery of MBP expression compared with no cuprizone controls 14 d after transfer (4 weeks on cuprizone and 2 weeks off; Fig. 4A,B). Interestingly, in mice receiving MOG-specific Th17 cells, this recovery was significantly decreased compared with cuprizone alone, indicating a delay or impediment in spontaneous remyelination (Fig. 4A,B). CNPase expression was also significantly decreased in the corpora callosa of mice receiving either AT T cells or MOG-specific Th17 cells at 14 d after transfer, supporting the notion of delayed remyelination in the presence of inflammation (Fig. 4A,B). This delay in remyelination was also observed via myelin staining with Black Gold (Fig. 5A,C).
Figure 4.

The transfer of myelin-reactive CD4+ T cells significantly reduces the amount of myelin protein in the corpus callosum. A, Protein was isolated from the corpus callosum 14 d after cessation of cuprizone diet and T-cell transfer. MBP and CNPase levels were determined by Western blot. B, Densitometric analysis of Western blots. The transfer of AT T cells decreased the abundance of CNPase below that attained with cuprizone alone while Th17 cells significantly reduced both MBP and CNPase. Total protein levels were normalized to β-actin. Data are mean ± SEM; N = 8 mice per group; *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test). ****p < 0.0001 (two-tailed Student's t test). Blots are representative of three independent experiments.
Figure 5.

Th17-transferred cells suppress endogenous remyelination after acute cuprizone exposure. Mice were fed 0.2% cuprizone for 4 weeks before transfer of Th17 cells and return to a regular diet. Remyelination and axonal integrity were evaluated 14 d after Th17 transfer. A, Black Gold staining demonstrates reduced myelin levels following transfer of Th17 cells to cuprizone-demyelinated mice. Scale bar, 100 μm. B, Electron microscopy of corpus callosum under indicated conditions. Left, original magnification ×10,000. Scale bar, 5 μm. Right, original magnification ×50,000. Scale bar, 1 μm. C, The percentage of the callosum that was stained by Black Gold was determined by dividing the area stained with Black Gold by the total area of the callosum. N = 5 or 6 animals per group. D, The percentage of myelinated axons was determined by counting myelinated axons and total axons in 10 images at ×10,000 magnification from 4 to 6 animals per experimental group. E, G-ratios are indicative of remyelination following cuprizone treatment. F, Small-caliber axons were counted at ×50,000. Th17 cells increase the abundance of small-caliber axons after cuprizone treatment. Data are mean ± SEM. *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test). Data are representative of two independent experiments.
Together with the T-cell infiltration data shown in Figure 2, these data suggest that, although differences likely exist in the homogeneity of the myelin-reactive T cells generated via traditional AT compared with 2D2 TCR transgenic T cells, MOG reactive T cells generated via Th17 polarization of 2D2 CD4+ T cells are more ideally suited for the creation of chronic inflammation in this model and provide a novel way to study the effects of potential new or existing drugs on remyelination in MS as well as expand our knowledge of MS pathogenesis. Therefore, Th17 cells were used for the remainder of the study. Of note, mice receiving Th17 cells following cuprizone treatment were subject to a similar incidence (∼70% for both groups) and severity of EAE compared with controls (average disease score was 1.2 ± 1.2 for the no cuprizone group and 1.5 ± 1.2 for the cup + Th17 group).
Transfer of MOG-specific Th17 cells to cuprizone-fed mice reduces the level of endogenous remyelination through a mechanism independent of axonal damage or loss
Demyelination is a hallmark feature of the MS plaque (Noseworthy et al., 2000). For this reason, animal models developed to study MS have attempted to accurately recapitulate this feature. However, although loss of myelin is a prominent feature of the EAE model, this demyelination is also accompanied by axonal degeneration as evidenced by axonal vacuolization and fragmentation as well as atrophy (Wujek et al., 2002; Wang et al., 2005; Herrero-Herranz et al., 2008; Jones et al., 2008; Soulika et al., 2009). With decreased numbers of intact demyelinated axons present, as well as the variable localization of demyelinated axons, studies focused on remyelination become difficult. For these reasons, most remyelination studies thus far have focused on the cuprizone or lysolecithin models of primary demyelination. These models, although preserving axons, spontaneously remyelinate at a rapid rate and lack the ongoing inflammation that exists in MS (Denic et al., 2011). Therefore, it was essential to demonstrate that transfer of myelin-specific T cells to cuprizone-fed animals could delay spontaneous remyelination without tremendous axonal loss. To explore this, we examined the corpora callosa of the animals at 14 d after transfer via electron microscopy. Remarkably, the percentage of demyelinated axons was significantly increased in animals receiving cuprizone and Th17 cells compared with cuprizone alone (Fig. 5B,D), suggesting that, although the cuprizone-fed animals have initiated spontaneous remyelination, this process is inhibited when myelin-reactive T cells are transferred. There was no difference in the percentage of myelinated axons in mice receiving Th17 cells without cuprizone, and this number was comparable to the no cuprizone-no cells controls. The number of total axons with a diameter <0.3 μm in a fixed area was found to be increased in the cuprizone + Th17 group compared with cuprizone alone (Fig. 5F). Consistent with remyelination, we noted a change in the average g-ratio of cuprizone-treated mice, which was unaffected by the transfer of Th17 cells. This result indicates that Th17 cells do not influence myelin thickness when remyelination does occur (Fig. 5E). Axonal integrity was also evaluated by immunostaining for SMI31, SMI32, and APP. SMI31 staining was preserved or slightly increased in the cuprizone and cuprizone + Th17 conditions consistent with the EM findings showing intact axons. Moreover, increased SMI31 staining has been reported to be seen in situations of increased axonal stress, including demyelination (Petzold et al., 2008). As expected, significantly increased APP and SMI32 staining was also observed in cuprizone-treated animals that received Th17 cells compared with the cuprizone alone group showing that demyelinated axons have impaired fast transport of βAPP and nonphosphorylated neurofilament and are consistent with findings in acute lesions of MS patients (Watson et al., 1989; Xie et al., 2010) (Fig. 6A–D). Importantly, these data indicate that demyelinated axons, although stressed, are present in increased numbers in cuprizone + Th17 animals, supporting the finding that transfer of myelin-reactive T cells to cuprizone-fed recipients delays the spontaneous remyelination but does not result in axonal loss, as is commonly observed in the EAE model. In addition, as loss of phosphorylation of neurofilaments causes closer spacing and has been shown to result in more compact axons, this could explain the finding of an increased number of small-caliber axons in the cuprizone-fed mice receiving Th17 cells (Yin et al., 1998).
Figure 6.

Transfer of Th17 cells induces axonal pathology consistent with demyelination. Mice were fed 0.2% cuprizone for 4 weeks before transfer of Th17 cells and return to a regular diet. Axonal integrity was evaluated 14 d after Th17 transfer. A, Immunostaining for APP, SMI32, and SMI31 demonstrates that axons are stressed following transfer of Th17 cells, consistent with previously reported pathology of demyelinated axons. Scale bars, 100 μm. B–D, The percentage of the callosum that was stained by APP, SMI32, or SMI31 was determined by dividing the area stained by the total area of the callosum; N = 4–6 animals per group. Mice that received Th17 cells exhibit significantly increased accumulation of APP (B). An increase in both nonphosphorylated NF-H (SMI-32) (C) and phosphorylated NF-H (SMI-31) (D) also suggests axonal stress secondary to sustained demyelination in mice receiving Th17 cells following cuprizone exposure. Data are mean ± SEM. *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test). Data are representative of two independent experiments.
The phenotype of transferred MOG-reactive T cells is altered following migration to the CNS in cuprizone-fed mice
In an attempt to dissect the immunological mechanisms that contribute to delayed remyelination in cuprizone-fed mice receiving Th17 cells, mononuclear cells were isolated from the brains of the animals at 14 d after transfer and the phenotype of CD4+ T cells present was examined by flow cytometry. As expected, more CD4+ T cells were present in the brains of animals receiving AT of 2D2 Th17 cells compared with animals that did not receive cells. Interestingly, when the phenotype of the T cells was examined, we observed a relatively even distribution of IL-17 only and IL-17/IFN-γ double-producing CD4+ T cells in mice receiving cells but no cuprizone. However, the CD4+ T cells in mice receiving cells and cuprizone were comprised of significantly more double-positive IFN-γ/IL-17-producing cells (Fig. 7A,B). It has been hypothesized that double-positive IFN-γ/IL-17 cells are the major pathogenic T cells both in MS and EAE (Hirota et al., 2011). Therefore, these finding suggest that the phenotypic shift could be contributing to the delayed remyelination in the cuprizone + Th17 animals. To determine whether the host immune response is contributing significantly to the delayed remyelination, we transferred 2D2 Th17 cells into cuprizone-fed CD45.1 congenic recipients and examined the proportion of transferred cells versus host cells in the CNS of the animals. Interestingly, at 14 d after transfer, the majority of CD4+ T cells found in the brains of both cuprizone-fed and non–cuprizone-fed mice were CD45.2-transferred T cells, suggesting that it is the transferred myelin-reactive CD4+ T cells that are driving the immune response in the CNS and impairing remyelination (Fig. 7C,D).
Figure 7.

The phenotype of transferred MOG-reactive T cells is altered following migration to the CNS in cuprizone-fed mice and is comprised more of donor than host T cells. A, The phenotype of CD4+ T cells infiltrating the brains of cuprizone-fed mice was characterized 14 d after transfer using flow cytometric analysis. Intracellular cytokine staining was performed, and IFN-γ and IL-17 production was analyzed. Frequencies of IFN-γ positive, IL-17-positive, or double-positive cells were determined following gating of live CD4+ cells and indicate an increased frequency of double-positive cells in the brains of cuprizone-fed mice compared with no cuprizone controls. B, Bar graph represents quantitative data representing 4 animals per group. C, CD4+ Th17 cells were generated from CD45.2 + 2D2 TCR transgenic mice and transferred into cuprizone-fed CD45.1 congenic recipients to allow for distinction between transferred and host CD4+ T cells. T-cell infiltrate was characterized from the brains of recipient mice at 14 d after transfer by flow cytometric analysis. Frequencies of CD4+CD45.1 host T cells were examined compared with CD45.2 transferred T cells. D, Bar graphs represent quantitative data representing 6 animals per group. All data are representative of three independent experiments. *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test).
Remyelination remains impaired 21 d after the transfer of Th17 cells
We next sought to determine whether the reduction in remyelination we observed following Th17 cell transfer persisted beyond 14 d. Notably, the number of CD4+ T cells present in the brain 21 d after transfer was found to have declined from ∼35% at 14 d after transfer, to ∼25% (Fig. 8A,B). However, we still observed a significant decrease in the abundance of both MBP and CNPase protein in mice receiving Th17 cells following cuprizone (Fig. 8C–E). To confirm these data at the ultrastructural level, we again compared the number of myelinated axons across groups. Although the percentage of myelinated axons in the group receiving Th17 cells after cuprizone treatment increased from ∼5% at 14 d after transfer to ∼11% at day 21, this was still significantly less than the cuprizone only group (∼23%; Fig. 9A,B). The effect of the Th17 cells on remyelination therefore persists to 21 d after transfer. The number of small-caliber axons (<0.3 μm) was also determined to be greater in the cuprizone + Th17 group compared with cuprizone only. However, the magnitude of this effect was diminished from the 2 week time-point (∼55% at 3 weeks vs ∼64% at 2 weeks; Fig. 9C). We did not find a significant difference in the total number of axons between experimental groups (Fig. 9D).
Figure 8.

CD4+ T cells persist in the brain 21 d after Th17 cell transfer and significantly reduce the amount of myelin protein in the corpus callosum. A, The frequency of CD4+ T cells that were detected in the brains of recipient mice by flow cytometry at 21 d after transfer. B, Bar graphs represent quantitative data collected from 4 animals per experimental group. C, Protein was isolated from the corpus callosum 21 d after cessation of cuprizone diet and T-cell transfer. MBP and CNPase levels were determined by Western blot. D, E, Densitometric analysis of Western blots. The transfer of Th17 cells decreased the abundance of MBP and CNPase below that attained with cuprizone alone. Total protein levels were normalized to β-actin. Data are mean ± SEM; N = 9 mice per group; *p < 0.05 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test). Blots are representative of two independent experiments.
Figure 9.

Remyelination remains impaired 21 d after transfer of Th17 cells. A, Electron microscopy of corpus callosum under indicated conditions. Left, original magnification ×10,000. Scale bar, 2 μm. Right, original magnification ×50,000. Scale bar, 0.5 μm. B, The percentage of myelinated axons was determined by counting myelinated axons and total axons in 10 images ×10,000 from 4 to 6 animals per experimental group. C, Small-caliber axons were counted at ×50,000. Transfer of Th17 cells increases the abundance of small-caliber axons following cuprizone treatment. D, Total axon counts were not significantly changed across experimental groups. Data are mean ± SEM. *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). Data are representative of two independent experiments.
Peripheral monocytes are recruited to the CNS of cuprizone-fed animals receiving Th17 cells compared with cuprizone-fed controls and display an M1 phenotype
When myelin reactive CD4+ T cells are transferred into recipient animals, these cells traffic to the perivascular space via the glia limitans or the choroid plexus where they must get reactivated by perivascular macrophages (Bartholomäus et al., 2009). It is this reactivation of the T cells that results in the recruitment of peripheral monocytes and lymphocytes to the CNS. These recruited peripheral monocytes are thought to play a major role in promoting chronic inflammation in the MS lesion and in impairing the endogenous remyelination process (Yamasaki et al., 2014). OPC migratory pathways depend, at least in part, on signals sent via microglia and/or astrocytes from the damaged axon (Miron and Franklin, 2014). The presence of inflammatory monocytes is essential for clearance of myelin debris, but excessive production of reactive oxygen species can result in remyelination failure (Miron and Franklin, 2014). Therefore, we investigated whether the AT of myelin-reactive T cells to cuprizone-fed recipients resulted in the recruitment of peripheral monocytes of the M1 phenotype that could contribute to the delayed remyelination observed. To this end, mononuclear cells were isolated from the brains of the animals at 7 and 14 d after transfer, and peripheral monocyte recruitment was examined via flow cytometry. There was a significant increase in CD45 high/CD11b high peripheral monocytes in mice receiving Th17 cells, beginning at 7 d after transfer and peaking at 14 d after transfer with cuprizone + Th17 mice having the highest frequencies of cells (Fig. 10A–C). Further examination revealed that this population of cells was also positive for the inflammatory monocyte marker Ly6C and the M1 marker iNOS (Fig. 10D–G). These data suggest that the mechanism by which transfer of myelin-reactive Th17 cells to cuprizone-fed mice results in impaired remyelination is likely due, at least in part, to the recruitment of M1-like macrophages, which may be producing ROS that can impair clearance of myelin debris and effect OPC maturation.
Figure 10.

Peripheral inflammatory monocyte infiltration is increased in the CNS of cuprizone-fed animals at 14 d after transfer of myelin-reactive Th17 cells. Mononuclear cells were isolated from the brains of recipient animals at 7 d (A) and 14 d (B) after transfer of Th17 cells. The frequencies of CD11b+CD45hi monocytes and CD11b+CD45intermediate microglia were determined by flow cytometric analysis. Representative FACS plots are shown. Bar graphs represent quantitative analysis from 4 animals per group at 7 d (C) and 14 d (D) after transfer. The phenotype of CD11b+CD45hi monocytes was examined for the inflammatory markers Ly6C and iNOS (D, F). Representative FACS plots are shown following gating on CD11b+ cells. Bar graphs represent quantitative analysis from 6 animals per group at 14 d after transfer for CD45+Ly6C+ cells (E) and CD45+iNOS+ cells (G). All data are representative of three independent experiments. *p < 0.05 (two-tailed Student's t test). **p < 0.005 (two-tailed Student's t test). ***p < 0.001 (two-tailed Student's t test). ns, Not significant.
Discussion
Chronic inflammation plays an important role in MS pathogenesis, and the ability to remyelinate is likely dictated by a number of different factors, including age, disease severity, disease duration, and environmental cues. An increased understanding of how inflammation alters cell intrinsic and extrinsic factors to, in turn, influence OPC proliferation, recruitment, differentiation, and ultimately, remyelination is crucial for the development of novel therapies that target disease progression. Moreover, a need exists to elucidate the factors that contribute to successful remyelination as well as those that result in its failure. The lack of available animal models to study these processes has limited the advancement of such therapies. Herein we present an innovative animal model that demonstrates impaired endogenous remyelination in cuprizone-fed animals when myelin-reactive CD4+ T cells are transferred. Specifically, we have shown that CD4+ T-cell infiltration peaks at 14 d after transfer and Th17 cells migrate more efficiently to the corpus callosum than CD4+ T cells producing more IFN-γ than IL-17. This increased infiltration of T cells correlates with decreased MBP and CNPase protein expression and reduced myelin staining. Importantly, the number of demyelinated axons is increased in mice receiving CD4+ T cells; and, although distressed, as indicated by APP and SMI32 staining, the total number of axons is preserved. Notably, we find that mice receiving Th17s after cuprizone treatment have significantly more small-caliber axons (<0.3 μm) that may fall below the threshold required to elicit myelination. Moreover, the effects of Th17 cell transfer were still apparent 21 d after transfer as evidenced by a persistent increase in the number of demyelinated axons compared with cuprizone treatment alone, increased numbers of small-diameter axons, and a reduction in the abundance of the myelin proteins CNPase and MBP. An examination of the immunologic mechanisms contributing to this delayed repair reveals a role for CD4+ T cells secreting IFN-y and IL-17 as well as infiltrating peripheral monocytes.
It has been suggested that in MS and EAE different regions of the CNS function as distinct microenvironments and can be characterized by different types of inflammation. For example, specific types of immune cells preferentially migrate to the brain as opposed to the spinal cord/optic nerve and vice versa (Pierson et al., 2012). Indeed, an increased ratio of Th17:Th1 cells has been demonstrated to promote infiltration of CD4+ T cells into the brain parenchyma and is associated with “atypical EAE” as opposed to the Th1 migration to spinal cord, which results in “classic EAE” (Stromnes et al., 2008). The mechanisms that promote this differential migration are not completely understood but are likely dependent upon chemokine receptor and integrin expression (Carlson et al., 2008; Reboldi et al., 2009; Cruz-Orengo et al., 2011; Glatigny et al., 2011). The results presented herein support this notion and demonstrate that CD4+ T cells comprised of an increased Th17:Th1 ratio migrate more effectively to the corpus callosum of cuprizone-fed mice compared with T cells secreting more IFN-γ than IL-17. Moreover, T cells accumulate in an increased fashion in mice that have been demyelinated with cuprizone compared with no cuprizone controls (receiving cells). These data suggest that the microenvironment of the lesion may play a role in inflammatory cell recruitment, either in an attempt to facilitate repair or to perpetuate the destruction.
Upon further examination, the presence of CD4+ T cells in the brain correlated with decreased myelin protein expression at 14 d after transfer. Electron microscopy, APP, SMI32, and SMI31 staining also clearly reveal the presence of intact demyelinated axons that, although stressed due to lack of myelin (Xie et al., 2010), are otherwise healthy and awaiting remyelination by oligodendrocytes. Moreover, we observed an increased number of small-caliber axons following the transfer of Th17 cells. These results are consistent with the increase in SMI32 staining and the fact that demyelinated axons, as a result of loss of phosphorylation, have decreased interneurofilament spacing in the axoplasm and smaller caliber (Yin et al., 1998; Petzold et al., 2008). Because these axons have a diameter below that typically associated with myelination (Waxman and Black, 1988; Lee et al., 2012), they may not yet support remyelination. By 3 weeks after transfer, although there are still more small-caliber axons in mice receiving Th17 cells compared with cuprizone treatment alone, the percentage has fallen from 64.4 ± 6.5% at 2 weeks to 55.2 ± 4%. Because we find the total number of axons to be unchanged, this shift may mark the beginning of axonal recovery.
Together, these findings support a role for our model in studying remyelination therapies as transfer of T cells extends the period of demyelination and opens the window to examine whether current or new therapies can promote remyelination in an expedited manner even in the face of inflammation. Indeed, studies have demonstrated that inflammatory mediators, including IFN-γ and IL-17, can have direct effects on OPC proliferation, differentiation, expression of myelin proteins, and survival (Chew et al., 2005; Kang et al., 2013). Moreover, the presence of inflammatory cytokines in MS lesions has been demonstrated to result in increased expression of chemokine receptors, including CXCR1, CXCR2, and CXCR3, on oligodendrocytes, which in turn affects their migration and recruitment during the remyelination process (Omari et al., 2005). IFN-γ has also been demonstrated to affect the ability of OPCs to exit the cell cycle resulting in increased proliferation and decreased differentiation and myelin protein expression (Chew et al., 2005). Along these same lines, IL-17 induced Act-1 signaling in OPCs has been demonstrated to play an important role during remyelination in the cuprizone model (Kang et al., 2012, 2013).
Our data also demonstrate that the phenotype of myelin-reactive Th17 cells changes following migration to the CNS to a predominantly double-positive IL-17/IFN-γ population supporting the notion that Th17 cells can exhibit context-dependent plasticity (Kurschus et al., 2010; Muranski and Restifo, 2013). These cells were found to be CD45.2 transferred T cells as opposed to host CD4+ T cells, and this phenotypic shift was more pronounced in the cuprizone-fed mice compared with the no cuprizone controls, suggesting that both IFN-γ and IL-17 are contributing to the delayed endogenous remyelination that is observed in our model, and that perhaps a microenvironment featuring injury or loss of myelin could play a role in altering the phenotype of the T cells recruited to the lesion. Further investigation is required to determine the molecular mechanisms by which these T cells and the cytokines that they produce effect OPC maturation, proliferation, and survival. However, this is the first evidence, to our knowledge, that double-positive IFN-γ/IL-17-secreting T cells, which have been implicated in the pathogenesis of MS, can delay the spontaneous remyelination that occurs in the cuprizone model. These data highlight the importance of studying remyelination in the context of inflammation and shed light on potential mechanisms contributing to remyelination failure in MS.
Although we would expect the majority of myelin debris to be cleared by the peak of T-cell infiltration (14 d after transfer and return to a regular diet), we cannot rule out the possibility that the transferred Th17 cells could affect any ongoing clearance and thereby influence the rate of remyelination. The macrophage phenotype has also been shown to affect the success of remyelination (Edwards et al., 2006; Miron et al., 2013). Interestingly, we demonstrate increased infiltration of peripheral monocytes of the M1 phenotype in the brains of animals receiving myelin-reactive CD4+ T cells, which is in contrast to the predominant microglial activation typically associated with the cuprizone model (Matsushima and Morell, 2001). M1 macrophages are associated with cytotoxicity, secretion of proinflammatory cytokines, production of reactive oxygen and nitrogen species and can impair the regenerative processes of remyelination (Edwards et al., 2006; Miron et al., 2013). Although M1 macrophages have been demonstrated to play an important role in OPC recruitment, a prolonged M1 phenotype or delay in the switch from an M1 to M2 phenotype can result in impaired OPC differentiation and reduced clearance of myelin debris (Miron and Franklin, 2014). In addition, in contrast to microglia, peripheral monocytes have been demonstrated to play an important role in induction and progression of EAE (Yamasaki et al., 2014). The data from our model suggest that the presence of IFN-γ/IL-17-secreting T cells in the corpus callosum delays the endogenous repair process, at least in part, by the recruitment of M1 macrophages from the periphery. This persistent inflammation likely impairs the necessary shift from an M1 to an M2 phenotype that has been demonstrated to be so essential for initiating the remyelination process in damaged tissue.
In conclusion, the novel animal model presented herein, which combines primary demyelination induced via cuprizone toxin with the AT of myelin-reactive CD4+ T cells, will provide researchers with additional means to help elucidate why remyelination fails so often in MS patients. Moreover, studies using this model could facilitate the development of therapies that might be more likely to promote remyelination in the setting of inflammation as is typically the case in MS.
Footnotes
This work was supported by the National Institutes of Health Grant R37 NS041435 to P.A.C., an National Multiple Sclerosis Society Collaborative Center Award, and the Kenneth and Claudia Silverman Foundation. We thank Barbara Smith for excellent technical assistance with electron microscopy.
The authors declare no competing financial interests.
References
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Bartholomäus I, Kawakami N, Odoardi F, Schläger C, Miljkovic D, Ellwart JW, Klinkert WE, Flügel-Koch C, Issekutz TB, Wekerle H, Flügel A. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462:94–98. doi: 10.1038/nature08478. [DOI] [PubMed] [Google Scholar]
- Carlson T, Kroenke M, Rao P, Lane TE, Segal B. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med. 2008;205:811–823. doi: 10.1084/jem.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, King WC, Kennedy A, Gallo V. Interferon-gamma inhibits cell cycle exit in differentiating oligodendrocyte progenitor cells. Glia. 2005;52:127–143. doi: 10.1002/glia.20232. [DOI] [PubMed] [Google Scholar]
- Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, McCandless EE, Patel JR, Luker GD, Littman DR, Russell JH, Klein RS. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011;208:327–339. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic A, Johnson AJ, Bieber AJ, Warrington AE, Rodriguez M, Pirko I. The relevance of animal models in multiple sclerosis research. Pathophysiology. 2011;18:21–29. doi: 10.1016/j.pathophys.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan V, Kleindienst AM, McMahon EJ, Long BR, Matsushima GK, Taylor LC. Abbreviated exposure to cuprizone is sufficient to induce demyelination and oligodendrocyte loss. J Neurosci Res. 2013;91:363–373. doi: 10.1002/jnr.23174. [DOI] [PubMed] [Google Scholar]
- Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatigny S, Duhen R, Oukka M, Bettelli E. Cutting edge: loss of alpha4 integrin expression differentially affects the homing of Th1 and Th17 cells. J Immunol. 2011;187:6176–6179. doi: 10.4049/jimmunol.1102515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Hagemeier K, Brück W, Kuhlmann T. Multiple sclerosis: remyelination failure as a cause of disease progression. Histol Histopathol. 2012;27:277–287. doi: 10.14670/HH-27.277. [DOI] [PubMed] [Google Scholar]
- Herrero-Herranz E, Pardo LA, Gold R, Linker RA. Pattern of axonal injury in murine myelin oligodendrocyte glycoprotein induced experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Neurobiol Dis. 2008;30:162–173. doi: 10.1016/j.nbd.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Nguyen TT, Deboy CA, Griffin JW, Whartenby KA, Kerr DA, Calabresi PA. Behavioral and pathological outcomes in MOG 35–55 experimental autoimmune encephalomyelitis. J Neuroimmunol. 2008;199:83–93. doi: 10.1016/j.jneuroim.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Kang Z, Liu L, Spangler R, Spear C, Wang C, Gulen MF, Veenstra M, Ouyang W, Ransohoff RM, Li X. IL-17-induced Act1-mediated signaling is critical for cuprizone-induced demyelination. J Neurosci. 2012;32:8284–8292. doi: 10.1523/JNEUROSCI.0841-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, Li X. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci. 2013;16:1401–1408. doi: 10.1038/nn.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurschus FC, Croxford AL, Heinen AP, Wörtge S, Ielo D, Waisman A. Genetic proof for the transient nature of the Th17 phenotype. Eur J Immunol. 2010;40:3336–3346. doi: 10.1002/eji.201040755. [DOI] [PubMed] [Google Scholar]
- Lee S, Leach MK, Redmond SA, Chong SY, Mellon SH, Tuck SJ, Feng ZQ, Corey JM, Chan JR. A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat Methods. 2012;9:917–922. doi: 10.1038/nmeth.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JR, Golumbek PT, Sim J, Dorsey D, Russell JH. Regional CNS responses to IFN-gamma determine lesion localization patterns during EAE pathogenesis. J Exp Med. 2008;205:2633–2642. doi: 10.1084/jem.20080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. 2001;21:7046–7052. doi: 10.1523/JNEUROSCI.21-18-07046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Franklin RJ. Macrophages and CNS remyelination. J Neurochem. 2014;130:165–171. doi: 10.1111/jnc.12705. [DOI] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller DM, Pender MP, Greer JM. A neuropathological analysis of experimental autoimmune encephalomyelitis with predominant brain stem and cerebellar involvement and differences between active and passive induction. Acta Neuropathol. 2000;100:174–182. doi: 10.1007/s004019900163. [DOI] [PubMed] [Google Scholar]
- Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. 2013;121:2402–2414. doi: 10.1182/blood-2012-09-378653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Omari KM, John GR, Sealfon SC, Raine CS. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain. 2005;128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- Petzold A, Gveric D, Groves M, Schmierer K, Grant D, Chapman M, Keir G, Cuzner L, Thompson EJ. Phosphorylation and compactness of neurofilaments in multiple sclerosis: indicators of axonal pathology. Exp Neurol. 2008;213:326–335. doi: 10.1016/j.expneurol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev. 2012;248:205–215. doi: 10.1111/j.1600-065X.2012.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:14965–14979. doi: 10.1523/JNEUROSCI.3794-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14:337–342. doi: 10.1038/nm1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner DC, Cherry JD, Mayer-Pröschel M. Oligodendrocyte progenitors reversibly exit the cell cycle and give rise to astrocytes in response to interferon-gamma. J Neurosci. 2011;31:6235–6246. doi: 10.1523/JNEUROSCI.5905-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirotta E, Ransohoff RM, Lane TE. CXCR2 signaling protects oligodendrocyte progenitor cells from IFN-gamma/CXCL10-mediated apoptosis. Glia. 2011;59:1518–1528. doi: 10.1002/glia.21195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Ayers MM, Catmull DV, Hazelwood LJ, Bernard CC, Orian JM. Astrocyte-associated axonal damage in pre-onset stages of experimental autoimmune encephalomyelitis. Glia. 2005;51:235–240. doi: 10.1002/glia.20199. [DOI] [PubMed] [Google Scholar]
- Watson DF, Griffin JW, Fittro KP, Hoffman PN. Phosphorylation-dependent immunoreactivity of neurofilaments increases during axonal maturation and beta,beta'-iminodipropionitrile intoxication. J Neurochem. 1989;53:1818–1829. doi: 10.1111/j.1471-4159.1989.tb09248.x. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Black JA. Unmyelinated and myelinated axon membrane from rat corpus callosum: differences in macromolecular structure. Brain Res. 1988;453:337–343. doi: 10.1016/0006-8993(88)90174-6. [DOI] [PubMed] [Google Scholar]
- Wensky AK, Furtado GC, Marcondes MC, Chen S, Manfra D, Lira SA, Zagzag D, Lafaille JJ. IFN-gamma determines distinct clinical outcomes in autoimmune encephalomyelitis. J Immunol. 2005;174:1416–1423. doi: 10.4049/jimmunol.174.3.1416. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol. 2002;61:23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Xie M, Tobin JE, Budde MD, Chen CI, Trinkaus K, Cross AH, McDaniel DP, Song SK, Armstrong RC. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J Neuropathol Exp Neurol. 2010;69:704–716. doi: 10.1097/NEN.0b013e3181e3de90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu Ph, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]