

Abstract
We investigated clozapine (CLZ) tissue pharmacokinetics in vivo by using carbon-11-labeled CLZ (11C-CLZ) and positron emission tomography (PET). Eight healthy volunteers underwent 11C-CLZ studies wherein computed tomography image acquisition was followed by PET scans (whole-body, four; brain, four). After bolus intravenous 11C-CLZ injection, PET images were acquired at various timepoints for 2–3 hours. Tissue 11C-CLZ signals were plotted over time, and pharmacokinetic parameters were determined. High 11C-CLZ radioactivity was detected in the liver and brain, implying CLZ hepatic metabolism and efficient blood–brain barrier penetration. The urinary and hepatobiliary tracts were involved in 11C-CLZ excretion. Moderate to high radioactivity was observed in the dopaminergic and serotonergic receptor-rich brain regions, indicating CLZ binding to multiple receptor types. To our knowledge, this is the first study to report the determination of 11C-CLZ tissue pharmacokinetics in humans. PET using radiolabeled drugs can provide valuable information that could complement plasma pharmacokinetic data.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ The pharmacological profile of CLZ has been derived from conventional blood PK data and there is a lack of direct tissue PK data in human. PET is uniquely suited to in vivo tissue pharmacology studies. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ In an attempt to illustrate in vivo tissue PKs of CLZ, we performed microdosing studies using PET and 11C-CLZ for whole-body organs and brain regions. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ High 11C-CLZ radioactivity was detected in the liver and brain, implying CLZ hepatic metabolism and efficient BBB penetration. The urinary and hepatobiliary tracts were involved in 11C-CLZ excretion. Moderate to high radioactivity was observed in the dopaminergic and serotonergic receptor-rich brain regions, indicating CLZ binding to multiple receptor types. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ The results provide valuable information that could complement plasma pharmacokinetic data of CLZ. PET under a microdosing concept has a very important role in drug research and development because it enables direct measurement of tissue drug concentrations.
The pharmacokinetic (PK) profile of clozapine (CLZ), a widely used atypical antipsychotic drug, has been characterized through half a century of studies and therapeutic application since its invention in the 1960s. Many researchers have investigated the pharmacological profile of CLZ and revealed the practical and biological aspects required to assure its safety and efficacy.1 Peak plasma concentration occurs 0.5 to 4 hours after oral intake and the plasma elimination half-life (T1/2) after single oral administration is about 12 hours.2 CLZ is moderately metabolized by the hepatic P450 microsomal enzyme system, mainly to N-oxide and N-desmethyl compounds, and at least 80% of administered CLZ appears in the urine or feces as metabolites.3 CLZ is excreted via the biliary (30%) and renal (50%) excretion routes.4 Agranulocytosis is a major adverse effect associated with CLZ therapy.5,6
Although the pharmacological profile of clozapine has been well characterized, most of this information has been derived from conventional blood PK data and there is a lack of direct tissue PK data in human. The lack of information regarding tissue distribution of drug often hampers interpretation of PK and pharmacodynamic properties of a drug.7 This occurs since most drugs exert their effects in tissue rather than in blood, and multiple factors, such as differences in perfusion, regional pH, and permeability of cell membrane govern drug exchange between blood and tissue. Furthermore, a drug's effect can be influenced by several interactive factors in tissues as well and those are not predictable from a single blood PK assessment.8
Positron emission tomography (PET) has gained widespread clinical acceptance in recent decades and is now firmly established as a key 3D medical imaging technique. In addition, PET is uniquely suited to in vivo pharmacology studies and is recognized as a pioneering system that supplies essential information, such as tissue PKs, for developing and refining pharmacology.9–13 Recently, PET studies performed with radiolabeled drugs for PK assessment have provided advantages over conventional blood PK studies using the microdosing concept.14,15 This is achieved by visualizing and quantifying in vivo tissue PK properties of drugs throughout the body, as well as in specific target organs and lesions, with exceptional sensitivity and good temporal and spatial resolution.9,11,16–18
In an attempt to illustrate in vivo tissue PKs of CLZ, we performed microdosing studies using PET and carbon-11-labeled CLZ (11C-CLZ) for whole-body organs and brain regions, respectively.
METHODS
Subjects
Subjects were recruited via Internet advertisements and a hospital bulletin board. Eight healthy male volunteers participated in the present study. Four subjects (mean age [± SD], 28.3 ± 3.9 years) were allocated for the whole-body PET study and the remaining four subjects (mean age, 28.3 ± 4.9 years) for the brain PET study. Subjects had no clinically significant medical or neurologic conditions, and no clinically significant abnormalities on physical, neurologic, or laboratory examinations. The study was approved by the Institutional Review Board of the Medical Science Research Institute of Seoul National University Bundang Hospital. Written informed consent was obtained from all subjects prior to participation.
Synthesis of 11C-CLZ
11C-CLZ was synthesized from norclozapine by 11C-methylation using 11C-methyl triflate in the TRACERlab FX C-pro module (GE Healthcare, Milwaukee, WI) with minor modification of previously published methods.19 Briefly, 11C-carbon dioxide was produced via the 14N(p,α)11C nuclear reaction by bombardment of nitrogen gas (99.9999%) with a 13-MeV proton beam produced by the KOTRON-13 cyclotron (Samyoung Unitech, Seoul, Korea). 11C-methyl iodide was converted from 11C-carbon dioxide on-line by gas-phase conversion in an automated module, and then converted into 11C-methyl triflate using a silver triflate column. 11C-methyl triflate carried in a flow of helium gas was then bubbled into a solution of acetone (0.45 mL) containing 1 mg of norclozapine at −20 °C. When radioactivity had peaked in the reactor, the solution was heated to 70 °C and maintained for 4 minutes. After cooling, the reaction mixture was quenched by addition of a high-performance liquid chromatography (HPLC) solvent (1.2 mL) and subsequently injected into a reverse-phase HPLC system. The HPLC purification was performed in 50% CH3CN:0.1 NH4OAc at a flow rate of 3 mL/min by using an ultraviolet detector at 254 nm and a gamma-ray detector. The collected solution was exchanged with 7% ethanol–saline solution using a C18 plus Sep-Pak cartridge to remove the HPLC solvent. The radiochemical yield was 37.9 ± 6.2% with over 99% radiochemical purity.
PET data acquisition
Subjects were directed not to ingest anything, except for water, for at least 6 hours prior to the PET study. Subjects arrived at the PET center of Seoul National University Bundang Hospital 2 hours before the scan and rested in a quiet room to stabilize their physical condition. Subjects were then positioned supine on the bed of the PET scanner with their arms positioned besides their torso and resting on the bed. Fabric bands were fastened around the head and limbs to prevent movement during the scan. PET images were acquired in 3D mode using a Discovery VCT PET/Computed Tomography (CT) scanner (GE Healthcare) with a 70-cm transaxial field of view, 15.7-cm axial field of view, and 3.27-mm axial slice thickness. The scanner provided a 4.8-mm axial spatial resolution at the center of the field of view at full-width at half-maximum. The scanner was calibrated on a quarterly basis with the experimental phantom filled with a known concentration of 11C to guarantee its performance reliability for radioactivity quantification. A low-dose helical CT scan was conducted prior to the PET scan. The CT acquisition protocol employed a peak kilovoltage of 140 kV, X-ray tube current of 95 mA, exposure time of 912 ms, pitch 1.75:1, and a rotation cycle of 0.8 seconds.
In four subjects, whole-body PET was used to assess the time-course of 11C-CLZ distribution within various organs. Subjects underwent a 3-hour dynamic series of 15 whole-body PET scans starting simultaneously with a 20-second intravenous injection of 11C-CLZ: 2 scans of 105 seconds, 2 scans of 210 seconds, 2 scans of 315 seconds, 2 scans of 420 seconds, and 7 scans of 1,260 seconds. Voiding of urinary bladder was scheduled 2 hours after injection. The injected 11C-CLZ dose was 840.5 ± 243.7 MBq and the specific activity at the time of injection was 65.5 ± 13.5 GBq/μmol. The injected mass of CLZ was 4.3 ± 1.1 μg. In a further four subjects, a 120-minute dynamic brain PET scan was obtained after a 20-second intravenous injection of 11C-CLZ to investigate the regional PK of 11C-CLZ in the brain. The scan protocol consisted of 12 frames of 10 seconds, 16 frames of 30 seconds, 8 frames of 60 seconds, 18 frames of 240 seconds, and 3 frames of 600 seconds. Higher amounts of radioactivity (injected dose = 1,035.4 ± 118.1 MBq) were administered for brain scans than for whole-body scans in order to obtain high-quality brain images of 11C-CLZ distribution during early scan frames that have short scanning times (10–60 seconds). The specific activity at the time of injection was 99.6 ± 64.2 GBq/μmol. The injected mass of CLZ was 13.4 ± 21.3 μg.
The decay- and CT-based attenuation-corrected recordings of annihilation photons were reconstructed by Fourier rebinning filtered back projection by using a 6-mm Hanning filter. The reconstruction process resulted in whole-body PET images with 3.91 × 3.91 × 3.27 mm voxel spacing (x, y, z) and 128 × 128 × 323 voxel dimension (x, y, z), while brain PET images had 0.98 × 0.98 × 3.27 mm voxel spacing (x, y, z) and 256 × 256 × 47 voxel dimension (x, y, z).
PET data analysis
Volume-of-interest (VOI) analysis of whole-body organs was conducted using MRIcron software (http://www.mccauslandcenter.sc.edu/mricro/mricron/index.html). Canvas images of VOI drawings were chosen between CT and PET, depending on organ visibility and mobility. VOIs for the liver, lung, spleen, brain, stomach, heart, and kidney were drawn on individual subject's CT images in a slice-by-slice manner, whereas those for the gallbladder, intestine, and urinary bladder were drawn on averaged PET images by adjusting iso-contour VOIs. PMOD software (PMOD v. 3.1, PMOD Technologies, Zurich, Switzerland) was used to perform VOI analysis of the brain regions. Realignment and spatial normalization of the individual dynamic brain PET data were carried out to apply the automated anatomical labeling method embedded in the PMOD software.20 The following VOIs were selected: the amygdala, anterior cingulate cortex, frontal cortex, hippocampus, insula, occipital cortex, parietal cortex, posterior cingulate cortex, striatum, temporal cortex, and cerebellum.
The radioactivity concentration of 11C-CLZ in the whole-body organ was expressed as the percentage injected dose (ID) per organ (%ID/organ) calculated as voxel mean concentration in organ VOI (Bq/ml)/injected dose (Bq) × organ volume (ml) × 100. VOI sizes are shown in Table 1. Radioactivity concentration in brain regions was expressed as the percentage injected dose per gram of tissue (%ID/g) calculated as voxel mean concentration in each brain region VOI (Bq/ml)/injected dose (Bq) × 100. The concentration was plotted over time to generate the time–concentration profiles and allow calculation of PK parameters in the organs or brain regions, such as time to maximal plasma concentration (Tmax), peak plasma concentration (Cmax), T1/2, and area under the curve (AUC). T1/2 was estimated by nonlinear least square curve fitting using the multiple exponential function in Prism (v. 5, GraphPad Software, La Jolla, CA).
Table 1.
PK parameters of whole-body organs
| Parameter | Tmax (min) | Cmax (%ID/organ) | T1/2 (min) | AUC0-t (%ID/organ × min) | VOI size (ml) |
|---|---|---|---|---|---|
| Liver | 37.6 ± 4.8 | 15.0 ± 1.2 | 388.0 ± 176.6 | 2,323.5 ± 172.8 | 1,559.4 ± 389.8 |
| Lung | 0.9 ± 0.0 | 31.0 ± 2.6 | 178.4 ± 30.9 | 1,584.3 ± 103.2 | 2,801.6 ± 324.5 |
| Brain | 18.7 ± 3.1 | 7.6 ± 1.1 | 274.3 ± 97.2 | 1,192.0 ± 153.9 | 1,519.0 ± 96.0 |
| Intestine | 140.8 ± 73.7 | 3.0 ± 1.3 | - | 455.9 ± 176.5 | 643.7 ± 183.1 |
| Stomach | 40.5 ± 73.9 | 1.5 ± 0.7 | 684.5 ± 519.5 | 235.3 ± 126.0 | 427.8 ± 203.2 |
| Heart | 0.9 ± 0.0 | 4.2 ± 1.4 | 276.0 ± 112.7 | 222.4 ± 43.5 | 640.0 ± 65.9 |
| Kidney | 0.9 ± 0.0 | 3.5 ± 0.7 | 383.1 ± 61.5 | 140.5 ± 18.3 | 357.4 ± 68.0 |
| Spleen | 2.6 ± 1.9 | 1.3 ± 0.5 | 301.5 ± 88.5 | 95.8 ± 29.0 | 177.9 ± 45.1 |
| Gallbladder | 105.7 ± 81.0 | 0.7 ± 0.3 | - | 85.4 ± 25.0 | 28.1 ± 10.7 |
| Urinary bladder | 129.9 ± 21.7 | 0.4 ± 0.2 | - | 66.6 ± 23.2 | 172.4 ± 36.3 |
Values are mean ± SD.
An AUC ratio method was used to quantitatively determine the routes of excretion of 11C-CLZ. The amounts of 11C-CLZ excreted via hepatobiliary and urinary tracts were estimated using the sum of AUCs for the gallbladder and intestine (AUChepatobiliary) and the sum of AUCs for the kidneys and urinary bladder (AUCurinary), respectively. The source of the excreted 11C-CLZ was calculated as the sum of AUCs for the remaining organs (AUCsource). Nonlinear exploratory stepwise extrapolation was conducted until the sum of AUChepatobiliary and AUCurinary matched AUCsource and the end time of extrapolation (timepoint at which total activity is excreted from the body) was recognized as t′, when AUC0-t′,hepatobiliary + AUC0-t′,urinary = AUC0-t′,source. At t′, the ratio between AUChepatobiliary and AUCsource, and the ratio between AUCurinary and AUCsource were considered to represent the proportion of injected 11C-CLZ excreted via the hepatobiliary and urinary tracts, respectively.
RESULTS
Tissue PK of 11C-CLZ in whole-body organs
Figure 1 represents whole-body PET images obtained serially after the injection of 11C-CLZ and shows the time-course of 11C-CLZ distribution. Figure 2 shows the concentration profiles in the various organs generated using PET data. Organ PK parameters derived from these profiles are shown in Table 1. In the heart, radioactivity rapidly increased after 11C-CLZ injection, followed by fast clearance consistent with cardiac circulation. A rapid and high peak was also observed in the lung, most likely due to high pulmonary blood flow. The lung retained substantial amounts of 11C-CLZ until the end of scanning (AUC0-t = 1,584.3 ± 103.2% ID/organ × min). The radioactivity of the gallbladder and intestine increased steadily during the scanning time. There was rapid distribution of radioactivity followed by fast clearance in the kidney with increasing urinary bladder activity over time (Figure 2). These data indicated that both hepatobiliary and urinary tracts contributed to drug excretion. The liver exhibited the highest AUC0-t (2,323.5 ± 172.8% ID/organ × min) of the organs of interest, supporting its role in CLZ metabolism. In the brain, a high Cmax (7.6 ± 1.1% ID/organ), early Tmax (18.7 ± 3.1 min), long T1/2 (274.3 ± 97.2 min), and high AUC0-t (1,192.0 ± 153.9% ID/organ × min) were observed, indicating good blood–brain barrier (BBB) penetration of CLZ and slow washout from the brain. Indeed, the brain accommodated 18.6 ± 1.8% (%AUC0-t,brain/AUC0-t,total organs) of the total radioactivity distributed in the body during the scanning time. Additionally, the routes of excretion of 11C-CLZ were determined by the AUC ratio method. The extrapolation was stopped at timepoint t′ = 17.1 h, when the percentage ratios of AUC0-t′,hepatobiliary and AUC0-t′,renal for AUC0-t′,source were 66.6% and 13.7%, respectively.
Figure 1.
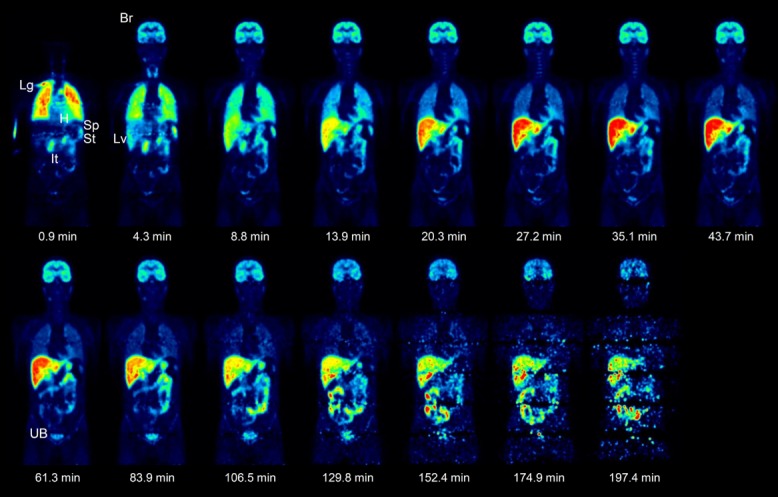
Time-course of 11C-CLZ distribution in the whole-body, represented by PET images of a subject at various timepoints. Images were normalized to the injected 11C-CLZ dose and subject body weight. Lg, lung; H, heart; Sp, spleen; St, stomach; It, intestine; Br, brain; Lv, liver; UB, urinary bladder.
Figure 2.
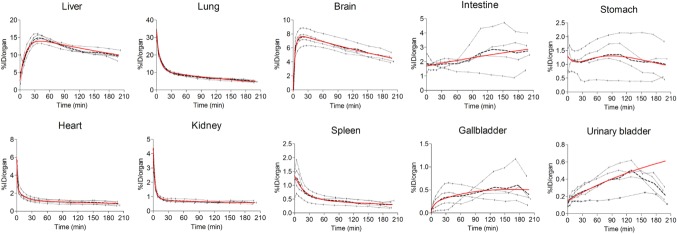
Time-course of 11C-CLZ distribution in various organs. Data are expressed as %ID/organ. The dotted lines represent individual subject data and the dashed line indicates mean data. Multiexponential fits to the means are shown as solid red lines. In the urinary bladder and intestine, the association model was used to fit the data. For the urinary bladder, only data from the first 120 minutes were used in the fitting procedure, due to scheduled voiding 2 hours after 11C-CLZ injection.
Tissue PK of 11C-CLZ in the brain regions
Figure 3 shows regional concentration profiles generated from brain PET images. PK parameters in each brain region calculated using the concentration profiles are shown in Table 2. The highest radioactivity in the brain was seen in the insula (AUC0-t = 586.0 ± 14.8%ID/g × min) and striatum (AUC0-t = 582.0 ± 5.9%ID/g × min), but high amounts were also observed in other cortical regions. The cerebellum exhibited a relatively lower peak (Cmax = 5.0 ± 0.4%ID/g) and faster clearance (T1/2 = 173.8 ± 23.5; AUC0-t = 422.2 ± 30.5%ID/g × min). It is noteworthy that limbic structures, such as the amygdala and hippocampus, showed distinct PK patterns from those of striatal and cortical regions. The amygdala and hippocampus had lower peaks (Cmax = 4.6 ± 0.0 and 4.4 ± 0.1%ID/g, respectively) and very slow clearance of 11C-CLZ (T1/2 = 401.4 ± 151.6 and 367.1 ± 92.2 minutes, respectively), while faster clearance after higher peak levels was seen in the striatum (T1/2 = 236.4 ± 40.9 minutes; Cmax = 5.8 ± 0.3%ID/g) and various cerebral cortices.
Figure 3.
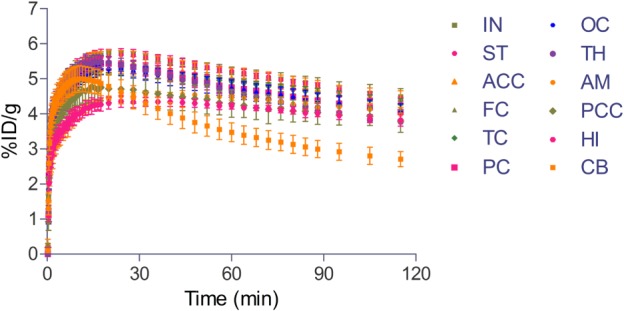
Time-course of the 11C-CLZ distribution profile in various brain regions. Data are expressed as %ID/g. Error bars indicate SD. IN, insula; ST, striatum; ACC, anterior cingulate cortex; FC, frontal cortex; TC, temporal cortex; PC, parietal cortex; OC, occipital cortex; TH, thalamus; AM, amygdala; PCC, posterior cingulate cortex; HI, hippocampus; CB, cerebellum.
Table 2.
PK parameters of brain regions
| Parameter | Tmax (min) | Cmax (%ID/g) | T1/2 (min) | AUC0-t (%ID/g × min) |
|---|---|---|---|---|
| Insula | 19.1 ± 1.8 | 5.8 ± 0.0 | 274.4 ± 79.9 | 586.0 ± 14.8 |
| Striatum | 22.9 ± 5.3 | 5.8 ± 0.3 | 236.4 ± 40.9 | 582.0 ± 5.9 |
| Anterior cingulate cortex | 21.1 ± 3.6 | 5.5 ± 0.4 | 330.1 ± 159.1 | 564.3 ± 31.7 |
| Frontal cortex | 17.1 ± 2.1 | 5.5 ± 0.1 | 261.7 ± 55.3 | 554.3 ± 11.8 |
| Temporal cortex | 26.0 ± 2.3 | 5.3 ± 0.1 | 290.8 ± 54.1 | 549.7 ± 11.4 |
| Parietal cortex | 18.9 ± 3.4 | 5.5 ± 0.1 | 321.1 ± 135.8 | 541.7 ± 23.8 |
| Occipital cortex | 19.8 ± 3.1 | 5.3 ± 0.1 | 330.2 ± 60.9 | 532.8 ± 23.0 |
| Thalamus | 16.4 ± 3.2 | 5.5 ± 0.3 | 221.8 ± 41.9 | 530.2 ± 8.6 |
| Amygdala | 40.0 ± 7.3 | 4.6 ± 0.0 | 401.4 ± 151.6 | 489.5 ± 7.3 |
| Posterior cingulate cortex | 14.2 ± 3.5 | 4.9 ± 0.5 | 408.2 ± 124.2 | 488.0 ± 38.7 |
| Hippocampus | 33.6 ± 19.7 | 4.4 ± 0.1 | 367.1 ± 92.2 | 468.3 ± 12.8 |
| Cerebellum | 11.2 ± 1.8 | 5.0 ± 0.4 | 173.8 ± 23.5 | 422.2 ± 30.5 |
Values are mean ± SD.
DISCUSSION
In the present study, whole-body organ and brain regional PKs of 11C-CLZ were successfully examined using PET. The in vivo time-course of 11C-CLZ tissue distribution in the organs and brain regions were generated from PET data and used to quantify key PK parameters. To the best of our knowledge, this is the first study to investigate tissue PKs of 11C-CLZ in this manner.
PET studies using radiolabeled drugs can provide PK information that cannot be obtained from conventional blood PK studies. Despite the important knowledge gained from tissue distribution of central nervous system (CNS) drugs, little is known about PK properties in brain tissue owing to methodological limitations. In the present study, we found that the brain accommodated 18.6% of the total radioactivity derived from 11C-CLZ injection during the scan. Since the specific activity of the injected mass (4.3 ± 1.1 μg) is known, we derived by a simple calculation that ∼0.8 μg of intravenously injected CLZ approached the brain in the present whole-body PET study. The concentration of CLZ in the brain peaked after 18.7 minutes and decreased by half at 274.3 minutes after the injection (Table 1). Tissue concentration of a drug measured by PET can be directly associated with its blood concentration. In the presence of blood PK data, this type of tissue–blood PK modeling may help optimize the clinical dosage of investigational drugs for clinical trials, under the assumption that there exists dose linearity between tracer and therapeutic doses of a drug.
In CNS drug development, BBB penetration and residence time are of paramount importance because these factors directly determine the viability and potency of an investigational drug. Regardless of the mechanism of drug action, a drug must first reach its site of action at a sufficient concentration to produce an effect.8,14,16 This study also demonstrated the superb ability of PET studies using radiolabeled drugs to facilitate CNS drug research and development by visualizing and quantifying the excellent BBB penetration and brain residence of 11C-CLZ.
The liver showed the greatest accumulation of radioactivity. This may be attributable to the high hepatic blood flow and involvement of the hepatic P450 microsomal system (CYP1A2 and CYP3A4 enzymes) in CLZ metabolism.3,21 After rapidly attaining a high peak, the lung, kidney, and heart exhibited two phases of radioactivity clearance (fast and slow), with substantial radioactivity retention until the end of scanning. This pulmonary PK profile in humans was consistent with a previous preclinical study, showing prolonged localization of CLZ in the lungs of rats.22
We determined and quantified the routes of excretion of 11C-CLZ using PET-derived AUC data from excretory organs. Our analysis revealed that intravenously administered 11C-CLZ was predominantly excreted by the hepatobiliary tract (67%) and partly by the urinary tract (14%). In contrast, a study in which the radioactivity in urine and feces samples was measured after a single oral dose of 14C-CLZ demonstrated that excretion of total radioactivity was ∼50% in urine and 30% in feces.23 This discrepancy may be due to differences between the studies in terms of the drug administration route (intravenous vs. oral), the methodology employed (PET-derived AUC data from organs involved in the excretory pathways vs. scintillation counting of excreta), and/or the duration of data collection (hours vs. days). The short duration of the PET scan (3 hours in the present study) may also have contributed to this discrepancy, because whole excretion cycles could not be completed during the scan. This drawback could be overcome if the drug (or drugs distributed and eliminated over longer periods of time) can be labeled with a radioisotope that has a longer physical half-life, such as fluorine-18 (physical half-life = 110 minutes), thereby allowing a scanning time of up to 24 hours. Moreover, sampling the urine and feces during or after PET scanning would help assess the route of excretion more precisely.
11C-CLZ accumulated in various cerebral cortical and subcortical regions. The PK profiles in these regions were distinct from that in the cerebellum, an area devoid of binding sites for CLZ and its metabolites. Thus, radioactivity in the cortical and subcortical regions is likely associated with specific binding of 11C-CLZ and/or its radiolabeled metabolites to various receptors on which CLZ acts, including dopaminergic, serotonergic, α-adrenergic, cholinergic, and histaminergic receptors. The highest activity in the brain was seen in the striatum and insula—areas containing a high density of dopamine D1/D2 and D1 receptors, respectively.24,25 Interestingly, the concentration profiles in the amygdala and hippocampus showed a lower peak but longer residence of radioactivity, as compared with the striatum and cortical regions. This finding may be linked to the hypothesis that CLZ has preferential effects on limbic and cortical dopaminergic systems.
This brain region analysis of 11C-CLZ PK may improve our understanding of the activities of drugs used to treat the symptoms of schizophrenia. The insula demonstrated the highest uptake of 11C-CLZ. The insula is a cortical structure with extensive connections to many areas of the cortex and limbic system, including the hippocampus and amygdala. It integrates external sensory input with the limbic system and is integral to awareness of the state of a body, including processing of visual, auditory, and emotional input.26–28 Insular dysfunction may contribute to the positive and negative symptoms of schizophrenia, including hallucinations.29 Studies of specific neurotransmitter systems in the human insula showed that major targets of CLZ (dopamine D4 and serotonin 5-HT1A receptors) were enriched here, in comparison to those in the other regions of the cortex.30,31
Limitations
One of the most significant instrumental limitations of PET is that the parent drug cannot be distinguished from its radiolabeled metabolites in tissue, because both produce the same signal.12 For this reason, the present study provided no additional information about CLZ N-oxide and N-desmethylclozapine, which are the major metabolites found in human plasma.3 When a radiolabeled drug is extensively metabolized in vivo, tissue PK may be confused by the presence of radiolabeled metabolites. It is generally advisable to perform validation studies in animals that can be sacrificed, enabling evaluation of the fractions of radioactivity related to intact drug in the tissue of interest, to get a sense of the potential magnitude of the problem. If necessary, additional PET studies can be performed with a radiolabeled metabolite to characterize its tissue distribution and kinetics. With CNS-active drugs, metabolites are often more polar than the native lipophilic compound and these might not cross the BBB.
CONCLUSION
In the present study, the tissue PKs of intravenously injected 11C-CLZ was assessed using PET; the data showed good BBB penetration, hepatobiliary metabolism, and hepatobiliary excretion of 11C-CLZ. Brain regional PK data reflected the multiple receptor binding characteristics of CLZ. This is, to the best of our knowledge, the first publication of 11C-CLZ tissue PK performed in humans, even though CLZ has been in use for more than half a century.
PET has a very important role in drug research and development because it enables direct measurement of tissue drug concentrations with good temporal and spatial resolution and exceptional sensitivity. Although the use of PET for PK assessment of radiolabeled drugs has issues that remain to be resolved, the growing availability of PET (e.g., PET devices, cyclotrons, and radiopharmaceuticals) will make this a central approach to target validation, drug candidate selection, PK characterization, and clinical evaluation of CNS drugs. This study also demonstrated the unique value of PET and radiolabeled drugs for PK studies, particularly for early clinical trials during CNS drug development.
Acknowledgments
This research was supported by grants from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C1072, HI09C1444) and the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT and Future Planning, Republic of Korea (2014000478).
Conflict of Interest
The authors declared no conflict of interest.
Author Contributions
S.E.K., H.S.P. and E.K. wrote the article; S.E.K., H.S.P. and E.K. designed the research; H.S.P. and N.H.L. performed the research; S.E.K., H.S.P. and E.K. analyzed the data; B.S.M. and B.C.L. contributed new reagents/analytical tools.
References
- Brunello N, Masotto C, Steardo L, Markstein R. Racagni G. New insights into the biology of schizophrenia through the mechanism of action of clozapine. Neuropsychopharmacology. 1995;13:177–213. doi: 10.1016/0893-133X(95)00068-O. [DOI] [PubMed] [Google Scholar]
- Choc MG, et al. Multiple-dose pharmacokinetics of clozapine in patients. Pharm. Res. 1987;4:402–405. doi: 10.1023/a:1016434312388. [DOI] [PubMed] [Google Scholar]
- Eiermann B, Engel G, Johansson I, Zanger UM. Bertilsson L. The involvement of CYP1A2 and CYP3A4 in the metabolism of clozapine. Br. J. Clin. Pharmacol. 1997;44:439–446. doi: 10.1046/j.1365-2125.1997.t01-1-00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ereshefsky L, Watanabe MD. Tran-Johnson TK. Clozapine: an atypical antipsychotic agent. Clin. Pharm. 1989;8:691–709. [PubMed] [Google Scholar]
- Alvir JM, Lieberman JA, Safferman AZ, Schwimmer JL. Schaaf JA. Clozapine-induced agranulocytosis. Incidence and risk factors in the United States. N. Engl. J. Med. 1993;329:162–167. doi: 10.1056/NEJM199307153290303. [DOI] [PubMed] [Google Scholar]
- Alvir JM. Lieberman JA. A reevaluation of the clinical characteristics of clozapine-induced agranulocytosis in light of the United States experience. J. Clin. Psychopharmacol. 1994;14:87–89. [PubMed] [Google Scholar]
- Bergstrom M. Langstrom B. Pharmacokinetic studies with PET. Prog. Drug Res. 2005;62:279–317. doi: 10.1007/3-7643-7426-8_8. [DOI] [PubMed] [Google Scholar]
- Gillette J. The importance of tissue distribution in pharmacokinetics. J. Pharmacokinet. Biopharm. 1973;1:497–520. [Google Scholar]
- Fowler JS, Volkow ND, Wang GJ, Ding YS. Dewey SL. PET and drug research and development. J. Nucl. Med. 1999;40:1154–1163. [PubMed] [Google Scholar]
- Farde L. The advantage of using positron emission tomography in drug research. Trends Neurosci. 1996;19:211–214. doi: 10.1016/0166-2236(96)40002-9. [DOI] [PubMed] [Google Scholar]
- Wong DF. Imaging in drug discovery, preclinical, and early clinical development. J. Nucl. Med. 2008;49:26N–28N. [PubMed] [Google Scholar]
- Wagner CC. Langer O. Approaches using molecular imaging technology — use of PET in clinical microdose studies. Adv. Drug Del. Rev. 2011;63:539–546. doi: 10.1016/j.addr.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin RH. Fischman AJ. Positron emission tomography in drug development. Q. J. Nucl. Med. 1997;41:171–175. [PubMed] [Google Scholar]
- Bauer M, Claudia Christina W. Langer O. Microdosing studies in humans: the role of positron emission tomography. Drugs R&D. 2008;9:73–81. doi: 10.2165/00126839-200809020-00002. [DOI] [PubMed] [Google Scholar]
- Wagner CC, Muller M, Lappin G. Langer O. Positron emission tomography for use in microdosing studies. Curr. Opin. Drug Discov. Dev. 2008;11:104–110. [PubMed] [Google Scholar]
- Bergstrom M, Grahnen A. Langstrom B. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur. J. Clin. Pharmacol. 2003;59:357–366. doi: 10.1007/s00228-003-0643-x. [DOI] [PubMed] [Google Scholar]
- Ding YS, et al. Synthesis and PET studies of fluorine-18-BMY 14802: a potential antipsychotic drug. J. Nucl. Med. 1993;34:246–254. [PubMed] [Google Scholar]
- Klimas MT. Positron emission tomography and drug discovery: contributions to the understanding of pharmacokinetics, mechanism of action and disease state characterization. Mol. Imaging Biol. 2002;4:311–337. doi: 10.1016/s1536-1632(02)00017-3. [DOI] [PubMed] [Google Scholar]
- Bender D, Holschbach M. Stöcklin G. Synthesis of n.c.a. carbon-11 labelled clozapine and its major metabolite clozapine-N-oxide and comparison of their biodistribution in mice. Nucl. Med. Biol. 1994;21:921–925. doi: 10.1016/0969-8051(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Pirmohamed M, Williams D, Madden S, Templeton E. Park BK. Metabolism and bioactivation of clozapine by human liver in vitro. J. Pharmacol. Exp. Ther. 1995;272:984–990. [PubMed] [Google Scholar]
- Gardiner TH, Lewis JM. Shore PA. Distribution of clozapine in the rat: localization in lung. J. Pharmacol. Exp. Ther. 1978;206:151–157. [PubMed] [Google Scholar]
- Dain JG, Nicoletti J. Ballard F. Biotransformation of clozapine in humans. Drug Metab. Dispos. 1997;25:603–609. [PubMed] [Google Scholar]
- Hall H. Sedvall G, Magnusson O, Kopp J, Halldin C. Farde L. Distribution of D1- and D2-dopamine receptors, and dopamine and its metabolites in the human brain. Neuropsychopharmacology. 1994;11:245–256. doi: 10.1038/sj.npp.1380111. [DOI] [PubMed] [Google Scholar]
- Hurd YL, Suzuki M. Sedvall GC. D1 and D2 dopamine receptor mRNA expression in whole hemisphere sections of the human brain. J. Chem. Neuroanat. 2001;22:127–137. doi: 10.1016/s0891-0618(01)00122-3. [DOI] [PubMed] [Google Scholar]
- Phillips ML, et al. The effect of negative emotional context on neural and behavioural responses to oesophageal stimulation. Brain. 2003;126:669–684. doi: 10.1093/brain/awg065. [DOI] [PubMed] [Google Scholar]
- Flynn FG. Anatomy of the insula functional and clinical correlates. Aphasiology. 1999;13:55–78. [Google Scholar]
- Stein MB, Simmons AN, Feinstein JS. Paulus MP. Increased amygdala and insula activation during emotion processing in anxiety-prone subjects. Am. J. Psychiatry. 2007;164:318–327. doi: 10.1176/ajp.2007.164.2.318. [DOI] [PubMed] [Google Scholar]
- Wylie KP. Tregellas JR. The role of the insula in schizophrenia. Schizophr. Res. 2010;123:93–104. doi: 10.1016/j.schres.2010.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti RA, et al. Direct determination of dopamine D4 receptors in normal and schizophrenic postmortem brain tissue: a [3H]NGD-94-1 study. Mol. Psychiatry. 1998;3:528–533. doi: 10.1038/sj.mp.4000423. [DOI] [PubMed] [Google Scholar]
- Rabiner EA, et al. A database of [(11)C]WAY-100635 binding to 5-HT(1A) receptors in normal male volunteers: normative data and relationship to methodological, demographic, physiological, and behavioral variables. Neuroimage. 2002;15:620–632. doi: 10.1006/nimg.2001.0984. [DOI] [PubMed] [Google Scholar]