

Abstract
Recent metagenomic studies of environments, such as marine and soil, have significantly enhanced our understanding of the diverse microbial communities living in these habitats and their essential roles in sustaining vast ecosystems. The increase in the number of publications related to soil and marine metagenomics is in sharp contrast to those of air, yet airborne microbes are thought to have significant impacts on many aspects of our lives from their potential roles in atmospheric events such as cloud formation, precipitation, and atmospheric chemistry to their major impact on human health. In this review, we will discuss the current progress in airborne metagenomics, with a special focus on exploring the challenges and opportunities of undertaking such studies. The main challenges of conducting metagenomic studies of airborne microbes are as follows: 1) Low density of microorganisms in the air, 2) efficient retrieval of microorganisms from the air, 3) variability in airborne microbial community composition, 4) the lack of standardized protocols and methodologies, and 5) DNA sequencing and bioinformatics-related challenges. Overcoming these challenges could provide the groundwork for comprehensive analysis of airborne microbes and their potential impact on the atmosphere, global climate, and our health. Metagenomic studies offer a unique opportunity to examine viral and bacterial diversity in the air and monitor their spread locally or across the globe, including threats from pathogenic microorganisms. Airborne metagenomic studies could also lead to discoveries of novel genes and metabolic pathways relevant to meteorological and industrial applications, environmental bioremediation, and biogeochemical cycles.
Keywords: airborne microorganisms, culture-independent studies, microbial diversity, metagenomics, 16S rRNA sequencing, metabolic potential
Introduction
The impact of airborne microbes and their overall contributions to global ecosystem is disproportionally understudied, even though microorganisms are ubiquitously present in the air. Airborne microbes were considered passive dwellers moving with the wind; however, several studies strongly suggest that atmospheric microbes are metabolically active. Microorganisms metabolize the organic matter in cloud water and potentially contribute to the biogeochemical cycles of earth (Amato et al. 2005, 2007; Hill et al. 2007). Some authors suggest that microbes affect atmospheric chemistry and the precipitation cycles of earth (Delort et al. 2010; Morris et al. 2011) implicating airborne microbes in atmospheric processes, such as ice nucleation and cloud formation. The increase in cloud formation may potentially be responsible for earth’s climate changes (Rosenfeld et al. 2014). Beyond these studies, relatively little data exist on the potential impact of airborne microbes upon our planet.
The current understanding of airborne microorganisms comes from culture-based studies; however, the majority of environmental microbes cannot be cultured in this way. Moreover, as pure cultures of microorganisms contain a single type of microbes, culture-based approaches miss the opportunity to study the interactions among different microbes and their surrounding environment; therefore, they cannot uncover the genomic variances and biological functions associated with such interactions. The main advantage of culture-dependent studies is the ability to grow microorganisms in bulk and study them at the both molecular and cellular levels. Their main disadvantage is that they disproportionally favor some microbes over others, providing an inaccurate representation of the microbial community as a whole.
In recent years, our knowledge of airborne microbial diversity and their potential metabolic impact has increased significantly through molecular analysis of microbial genetic material through culture-independent studies of environmental samples. Two common approaches to investigate microbial diversity and/or their metabolic potential in the environment are 1) the polymerase chain reaction (PCR)-based rRNA (16S and 18S) gene sequencing approach for assessment of microbial diversity in which a single rRNA gene is used as a phylogenetic marker to compare relatedness between microorganisms; and 2) the whole-genome shotgun metagenomics approach for assessment of microbial diversity and function, in which the entire microbial genome is sheared into smaller fragments, sequenced, and reconstructed to assess their diversity and metabolic potential (fig. 1).
Fig. 1.—
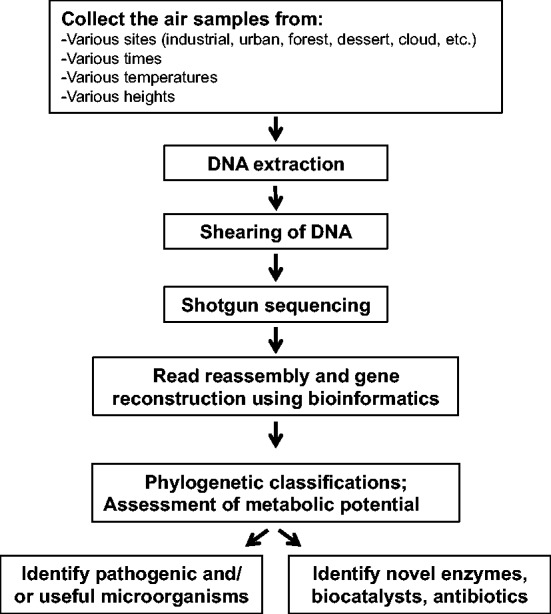
Flow chart of the process involved in shotgun metagenomic sequencing of airborne microbes from collection of air samples to analysis of its microbial content.
In this article, we use the term metagenomics in its original sense to mean the application of whole-genome shotgun sequencing to environmental samples. With the advent of next-generation sequencing (NGS), shotgun metagenomic studies of various environments, including soil (Fierer et al. 2012; Sangwan et al. 2012), marine (La Cono et al. 2011), and human biome (Methe et al. 2012), have expanded significantly as evidenced by the large data sets available for these ecosystems. In contrast, progress in air metagenomic research has been relatively slow (fig. 2). Here, we will explore current progress in airborne metagenomics with a particular emphasis on the challenges associated with these studies and their potential opportunities.
Fig. 2.—

Relative number of publications related to metagenomic studies of soil, marine, and air. The graph was generated with the data obtained from Scopus using the search words: “soil metagenomic,” “marine metagenomic,” and “air/airborne metagenomic,” respectively.
Airborne Microorganisms Play Active Roles in the Atmosphere; Exploration of This Seemingly Untouched Frontier Has Great Potential
A number of studies conducted in culture suggest that airborne microorganisms play active roles in atmospheric events and/or are metabolically active. For example, airborne bacteria were shown to metabolize glucose (Dimmick et al. 1975). More recently, Hill et al. (2007) used the uptake of tetrazolium dye by bacteria as an indicator of metabolic activity in cells and showed that 76% of the bacteria in cloud water were active. High concentrations of inorganic and dissolved organic nitrogen were found in cloud waters that contained nitrifying bacteria, suggesting that the bacteria in cloud water play a role in the cycling of organic nitrogen in the atmosphere (Hill et al. 2007). Amato et al. (2005) used the increase in ATP concentration in cloud water as an indicator of cell metabolic activity and showed that the bacteria in cloud water were growing exponentially and metabolically active. The bacteria isolated from cloud water metabolized the main carboxylic compounds (formate, acetate, lactate, and succinate) in cloud water; the end products of these metabolic activities were commonly found in cloud water, suggesting that these bacteria were actively involved in transformation of organic compounds in the atmosphere (Amato et al. 2007; Vaitilingom et al. 2010). Durand et al. (2006) identified a bacterial strain capable of biodegrading the herbicide mesotrione. Through biodegradation of organic matters in the atmosphere, airborne microorganisms could play a role in the bioremediation of environmental pollutions.
Airborne bacteria could also affect atmospheric conditions by engaging in ice-nucleating activities, cloud formation, or precipitation (Bauer et al. 2003; Mohler et al. 2008; Morris et al. 2011). Ice-nucleating proteins were identified on the outer membrane of plant associated Gram-negative bacteria such as Pseudomonas syringae, Pseudomonas fluorescens, and Erwinia herbicola (Deininger et al. 1988; Kozloff et al. 1991; Turner et al. 1991). Ice-nucleation active bacteria are present in the atmosphere (Constantinidou et al. 1990; Bowers et al. 2009). Some strains of fungi including Fusarium avenaceum and related genera also possess ice-nucleating abilities (Pouleur et al. 1992; Hasegawa et al. 1994). Biological ice-nuclei can catalyze ice formation at temperatures as high as −2 °C, higher than the temperature required for ice-nucleation by nonbiological particles (Mohler et al. 2007). Knowledge of the components involved in ice-nucleating activities of airborne microbes is paramount to understanding their roles in atmospheric events.
Most assays examining the metabolic activities of airborne microbes were conducted under laboratory conditions, prone to bias as the exact atmosphere environment is not replicated. These studies were also mainly conducted on samples taken from cloud water. Cloud-borne microbes are not necessarily the same microbes that thrive in dry or humid air; therefore, they do not paint a full picture of airborne microbial ecosystems. However, these discoveries underscore the potential importance of airborne microbes and the need to characterize them in their entirety. As culture-based studies represent only a minority of the microbial population, they are not up to this task. Culture-independent studies are required.
Exploring Diversity of Microorganisms in the Atmosphere through Culture-Independent Analysis of Their rRNA Gene
It is estimated that in a cubic meter of air there can be hundreds of thousands of individual microbes (Burrows et al. 2009), with a diversity of taxa comparable to what has been found in soil (Franzetti et al. 2011). “Who” are these microbes and “what” do they do? In recent years, through use of culture-independent approaches and in particular with PCR-based applications such as rRNA gene sequencing, we have managed to answer the first question about who these microbes are through their phylogenetic diversity (table 1). However, the answer to the second question regarding the function of airborne microbes remains largely incomplete.
Table 1.
Microorganisms (bacteria, fungi, and viruses) Detected in the Atmosphere Using Culture-Independent Methodologies
| Bacteria | Fungi/Plant | Viruses | Method of Identification | Location | Reference |
|---|---|---|---|---|---|
| Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria | NA | NA | 16S rRNA microarray | Mountain (USA) | Smith et al. (2013) |
| Firmicutes, Proteobacteria | Ascomycota, Streptophyta | Pseudomonas phage F10 | Metagenomic sequencing (NGS) | Urban area (USA) | Be et al. (2014) |
| Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria | NA | NA | 16S rRNA sequencing (NGS) | Mushroom cultivation facility (South Korea) | Chun et al. (2012) |
| Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria | NA | NA | 16S rRNA sequencing (Sanger)/metagenomic sequencing | shopping center (Singapore) | Tringe et al. (2008) |
| Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Firmicutes, Nitrospira, Protobacteria, Verrucomicrobiae | NA | NA | 16S rRNA microarray | Urban area (USA) | Brodie et al. (2007) |
| Acidobacteria, Actinobacteria, Bacteroidetes, Firmicutes, Protobacteria | NA | NA | 16S rRNA sequencing (NGS) | Upper Troposphere (international) | Deleon-Rodriguez et al. (2013) |
| Acidobacteria, Actinobacteria, Cyanobacteria, Fermicutes, Flavobacteria, Gemmatimonadetes, Planctomycetacia, Proteobacteria, Verrucomicrobia, Sphingobacteria | NA | NA | 16S rRNA sequencing (Sanger) | Rural area (France) | Maron et al. (2005) |
| NA | Ascomycota, Basidiomycota, Chytridiomycota | NA | LSU rRNA sequencing (NGS) | Urban area (South Korea) | Oh et al. (2014) |
| Acidobacteria, Actinobacteria, Bacteroidetes, Cyanobacteria, Firmicutes, Protobacteria | Ascomycota, Basidiomycota, Zygomycota, Streptophyta | NA | 16S rRNA sequencing (Sanger)/16S rRNA bar-coded pyrosequencing | Mountain (USA) | Bowers et al. (2009) |
| Actinobacteria, Bacteriodetes, Chloroflexi, Firmicutes, Proteobacteria | Ascomycota | Pseudomonas Phage F116, Human adenovirus C, Enterobacteria Phage P1 | Metagenomic sequencing (NGS) | Urban area (China) | Cao et al. (2014) |
| NA | NA | Geminivirues-related viruses, Circoviruses, Nanoviruses, Microphage-related viruses | Metagenomic sequencing (NGS) | Forest, urban, coastal area (South Korea) | Whon et al. (2012) |
| Actinobacteria, Bacteriodetes, Firmicutes, Proteobacteria | NA | NA | 16S rRNA sequencing (NGS) | Sand storm (China and South Korea) | An et al. (2014) |
Note.—We used phylum name to represent the organisms except for viruses. NA, not applicable.
The PCR-based rRNA gene sequencing studies successfully demonstrated that air can harbor vastly diverse microbes. The composition of these microbes continuously changes in response to meteorological, regional (coastal, urban, and forest), seasonal, and diurnal patterns (Gandolfi et al. 2013). Gram-positive as well as Gram-negative spore and nonspore forming bacteria are both present in the air. The most common bacterial groups identified were Protobacteria, Firmicutes, Bacteriodetes, Actinobacteria, Acidobacteria, and Cyanobacteria (table 1), some of which contain gene sequences classically found in bacteria dwelling in soil, water, or plants (Maron et al. 2005; Brodie et al. 2007). Known ice nucleating plant-associated bacteria including P. syringae, P. fluorescens, E. herbicolas, and Sphingomonas spp were also present in the atmosphere (Bowers et al. 2009). A portion of microorganisms in the air was found to be potentially pathogenic to plants, animals, and humans (Brodie et al. 2007). Various fungal genera including those of Cladosporium, Aspergillus, and Penicillium also dwell in the air, many of which are potential allergens (Oh et al. 2014). Although various studies identified plant, animal, and human pathogens in the air, a systematic approach to monitoring airborne pathogens and examining their impact on global ecosystems is lacking.
Research on the source of airborne microorganisms reveals that although the majority of these microbes originate from local sources (soil, plants, and marine), some are unique to the local atmosphere. These microbes could originate from distant sources transported by strong winds, sandstorms, or hurricane. A recent study by An et al. (2014) using amplicon-based 16S rRNA gene sequencing demonstrated that the atmospheric sand-associated bacterial composition was modified during sandstorm events in China and South Korea. In particular, they found an increase in the level of putative human bacterial pathogens in samples obtained during sandstorms (An et al. 2014). Microbes from sandstorm events, such as the ones originating from Gobi desert, can be transported very large distances, even reaching across the Pacific Ocean to North America (Bishop et al. 2002). This has significant implications for the dispersal of pathogenic microbes around the globe. Deleon-Rodrigues et al. (2013) show that hurricanes, similar to sandstorms, dramatically change atmospheric bacterial composition, even at high altitudes (10 km above the sea level), suggesting that hurricanes aerosolize and transport bacterial cells over long distances (DeLeon-Rodriguez et al. 2013).
Diverse and viable communities of bacteria reside high in the troposphere despite the extreme cold, high UV irradiation, and thin air, suggesting adaptive mechanisms. Surprisingly, viable bacteria form up to 20% of the total number of particles found in the troposphere (DeLeon-Rodriguez et al. 2013). Some atmospheric regions have extreme conditions, but microorganisms already live under even harsher environments with extremes of pH, temperatures, and radiation (Pikuta et al. 2007). The roles of high-altitude dwelling bacteria are not well understood. It is likely that these microbes affect meteorological events, such as cloud formation, precipitation, or atmospheric chemistry.
The PCR-based rRNA gene sequencing approach has provided a glimpse into the phylogenetic diversity of microorganisms in the atmosphere. However, this approach has many limitations, as follows: 1) It is challenging to identify novel microorganisms with this approach; the universal rRNA primers used to amplify the rRNA genes are designed based on preexisting rRNA sequences in the gene database, and these primers might not amplify new genes (Rajendhran and Gunasekaran 2011); 2) characterization of microorganisms based on a single rRNA gene is difficult due to potential horizontal gene transfer of the rRNA gene between distally related microorganisms (Acinas et al. 2004); 3) due to a high degree of similarity in the rRNA gene among related species and strains, this approach is limited to characterizing microbes mostly at higher taxonomic levels, such as family or genus where the difference in the rRNA gene is more significant, but not so at lower taxonomic levels, such as species or strains where more similarities in the gene are observed (Bowers et al. 2009; DeLeon-Rodriguez et al. 2013); 4) amplicon-based methods in general result in bias due to preferential amplification and generation of artifacts and erroneous products introduced during PCR steps (Hong et al. 2009; Wylie et al. 2012; Logares et al. 2014; Sharpton 2014); 5) As this approach is based on the analysis of a single rRNA gene, it cannot provide information on the function of other microbial genes and their metabolic potential; and 6) it cannot be used to study viruses because they lack common phylogenetic markers such as the rRNA gene.
Whole-genome shotgun metagenomics, on the other hand, provides a more powerful approach to direct sequencing of microbial genomes, and to the assessment of their diversity, and metabolic potential (Sharpton 2014). This approach is relatively new but has revolutionized environmental microbiology by providing new insights into the diversity and metabolic potential of microbes in their natural habitat, such as marine, soil, and human biome. Due to its unique properties, air could be another promising frontier for discovery of unexplored microbial ecosystems with unique genes, biomolecules, and novel enzymes waiting to be discovered.
Current Progress in Airborne Metagenomics and Its Potential Opportunities
Although progress in air metagenomics has been slow relative to other environments, the little progress that has been made is promising. In a recent study, Cao et al. (2014) used metagenomics to analyze the microbial composition of air samples during a severe smog event in China and successfully identified various airborne microorganisms including double-stranded DNA viruses. Their work identified the sequences of several respiratory pathogens and allergens in the air, and showed that their relative abundance increased with increased air pollution. Yooseph et al. (2013) established a metagenomic framework for studying the microbial composition of air samples from several indoor and outdoor environments, and successfully identified highly diverse microbial communities with genes involved in metabolism, transport, translation, and signal transduction. Tringe et al. (2008) studied the airborne metagenome in an indoor environment and identified genes for potentially adaptive mechanisms involved in resistance to desiccation and oxidative damage. Whon et al. (2012) investigated viral diversity in the near-surface atmosphere over three distinct land locations (residential, forest, and industrial), and showed the presence of diverse airborne viruses including those that infect plants and animals. They also revealed seasonal changes in the abundance of airborne viral communities with higher viral abundance toward winter and lower abundance toward spring. The increase in viral abundance during winter could explain higher rates of influenza viral transmission during cold and dry season (Lowen et al. 2007; Shaman and Kohn 2009). With continuous progress and advances in the field, metagenomic approaches could become convenient tools for monitoring airborne viral, bacterial, and fungal pathogens and studying their global distribution patterns.
There are also strong indications, based on culture-dependent studies, that airborne microorganisms can transform organic matters in clouds (Durand et al. 2006; Amato et al. 2007), or contribute to atmospheric events such as ice nucleation and cloud formation (Gurian-Sherman and Lindow 1993; Mohler et al. 2007, 2008; DeLeon-Rodriguez et al. 2013), but the extent of microbial involvement and the mechanisms involved are unclear. Metagenomic studies can help identify the airborne microbes, and their genes and metabolic pathways that potentially contribute to these events. These studies have great potential for novel and advanced discoveries with applications in atmospheric transformation, meteorological applications, environmental bioremediation, and health. Among the challenges of metagenomic studies are unraveling this vast potential and finding ways to incorporate these finding into novel applications.
Current airborne metagenomic research demonstrates, in principle, that it can identify and characterize microorganisms in the air. However, considerable advances in the field are required to enable sufficient characterization of the entire airborne microbiome.
Challenges in Conducting Airborne Metagenomics
The Low Density of Microorganisms in the Air
Microorganisms including bacteria, viruses, and fungi are scattered throughout the atmosphere. The bacterial concentration in air is estimated to range between 104 and 106 microbes/m3 (Lighthart et al. 2000) much lower than those of marine and soil environments, where 1 g of topsoil is believed to contain approximately 108–109 prokaryotes, and 1 ml of marine water is believed to contain approximately 104–107 microorganisms (Whitman et al. 1998).
A number of studies investigated the concentration of airborne microorganisms, but it is difficult to relate their data because they used different methodologies. Traditional methods use culture-based techniques, which measure the concentration of microorganisms as colony-forming units per cubic meter of air (CFU/m3). As the vast majority of environmental microbes cannot be cultured, culture-based methods likely underestimate microbial concentrations (Li and Huang 2006). In recent years, microorganisms have been primarily quantified directly from the environment using techniques such as quantitative PCR (qPCR), epifluorescence microscopy, and flow cytometry. These techniques provide different estimates of microbial density in air, sometimes by a few orders of magnitude (Gandolfi et al. 2013). Airborne bacterial concentrations, for example, were reported over the following ranges: Approximately 0–104 CFU/m3 using culture-based methods (Fahlgren et al. 2010; Ravva et al. 2012; Haas et al. 2013), approximately 103–106 cells/m3 by epiflourescence microscopy (Bowers et al. 2009, 2011; Vaitilingom et al. 2012), and approximately 101–106 cells/m3 by qPCR (Lee et al. 2010; Cho and Hwang 2011; Bertolini et al. 2013). Unfortunately, without base-line comparative studies, these numbers can only be understood in isolation. A precise comparison between various quantification techniques is only possible when samples are collected from the same location using the same collection device. This is important as microbial concentration varies significantly depending on the location where the air samples were taken and the type of air samplers used. Current air sampling devices often do not capture microorganisms at maximum efficiency. It is likely that the abundance of microorganisms in the air is greater than what has been reported. If that is the case, the bottleneck lies with current technology.
Whole-genome shotgun sequencing requires large quantities of the starting genetic material, which entails collecting thousands of liters of air. In practice, capturing large quantities of microorganisms from air is challenging. Improving the collection efficiency of air sampling devices would help mitigate this. To circumvent the problem of low DNA yield from air, some metagenomic protocols incorporate an additional step of amplifying the environmental DNA prior to sequencing using methods such as Multiple Displacement Amplification (Lasken 2009; Yooseph et al. 2013). These amplification steps can produce micrograms of DNA from femtograms of the starting genetic material (Lasken 2009), ensuring that sufficient amounts of DNA are available for sequencing. However, amplification can generate primer mismatches, chimeras, and sequence biases—the degree of which depends upon the quantity and quality of the starting genetic material, and the number of required amplification cycles.
Metagenomic studies require high quantities of DNA, which could potentially be obtained through efficient collection of microorganisms from the air, and the development of unbiased methodologies to enhance DNA yield following extraction.
Efficient Collection of Microorganisms from the Air
Due to the lower density of microorganisms in air, it is paramount that air-sampling devices collect microorganisms from the air with high efficiency. Typical air samplers draw air containing various particles into an airflow nozzle, which then directs the air toward a collection surface. The sampling efficiency of such devices is affected by a combination of factors including intake air velocity, the shape and diameter of the airflow nozzles, the distance between the nozzle and the collection surface, the properties of the collection surface, the particle cut-off diameter, and collection times (Nevalainen et al. 1992; Whyte et al. 2007). The sampling efficiency is also influenced by the inertial properties of microorganisms, related to their size and density (Nevalainen et al. 1992).
Two air sampling strategies are commonly used to collect microorganisms from air for molecular analysis: Filtration-based air samplers work by drawing air, either through pumps or vacuum lines, through filters of varying pore sizes, shapes, and composition (Lundholm 1982; Brodie et al. 2007; Bowers et al. 2009; Fahlgren et al. 2010, 2011). These samplers are the most commonly used devices for aerosol collection. They are relatively easy to use and less expensive, but have shortcomings. The low porosity of the filters can dramatically reduce the airflow rates, increasing sample collection time. Long collection times on dried filters lead to desiccation of the collected microbes. Furthermore, these devices are not suitable for quantification-type studies due to difficulty in retrieving intact microorganisms from filters (Fahlgren et al. 2011).
Liquid impinger-based samplers work by drawing high-flow-rate, high-velocity air through a nozzle into a liquid collection medium. The particles in the air stream are impacted on the collection plates, wetted, and retained in the collection medium (Burge and Solomon 1987; Fierer et al. 2008; Fahlgren et al. 2011). Due to their high air-flow rate, they can collect larger biomass in shorter time, relative to filter-based devices. As the microbes are collected in liquid medium, their viability is retained. These devices are typically designed for relatively short sample collection times, which is required to prevent evaporation of the liquid collection medium and loss of cell viability (Fahlgren et al. 2011). This could prevent collection of sufficient biomass for studies requiring large concentrations of nucleic acids. Modifications, such as replenishing liquids, could lengthen sample collection times and overcome this problem.
Specific types of studies necessitate the use of particular types of air samplers. For example, culture-based studies require air samplers that can maintain cell viability during the collection period. Metagenomic studies of the entire microbial communities require devices that can provide sufficient microbial biomass representing the sampled community. Studies examining specific types of microorganisms (viruses vs. bacteria vs. fungi) need devices that preferentially and efficiently collect microbes based on size. Once the capturing strategy is decided, that is, using filtration versus impingement, then the efficiency of the various devices using the same strategy must be thoroughly investigated before embarking on a large-scale study.
Variability in Airborne Microbial Community Composition
The composition of airborne microbes continuously changes due to meteorological, spatial, and temporal patterns (Brodie et al. 2007; Frohlich-Nowoisky et al. 2009; Bowers et al. 2011; Franzetti et al. 2011; Bertolini et al. 2013; Gandolfi et al. 2013). Bowers et al. (2011) investigated the spatial variability in airborne microbial composition in the near surface atmosphere, and showed that bacterial composition significantly varied depending on the land-use type, suggesting that bacteria from the local land sources contributed to this variability. Bertolini et al. (2013) demonstrated temporal variability in airborne bacterial community composition in an urban atmosphere, showing that bacterial composition varied significantly depending on the season. Even within the same season, bacterial composition was significantly different between consecutive days.
This inherent variability makes studies of airborne microbial composition challenging. It is difficult, for example, to study the possible contribution of environmental pollution or anthropogenic factors to microbial dynamics, if a background level cannot be established, due to variability (Brodie et al. 2007). Without the reproducible background level, monitoring of environmental pathogens becomes complicated. The continuous change in composition of microbes in the atmosphere also makes it difficult to compare across different studies as the outcome of these studies could vary significantly depending on location, time of day, season, altitude, and other environmental factors.
Lack of Standardized Methodologies in Sample Collection and Processing
The procedures and protocols used to obtain nucleic acids from the air play a significant role on the outcome of metagenomic studies. As metagenomic sequencing is conducted on the nucleic acids extracted directly from the environment, the quantity and quality of these nucleic acids determine the outcome of these studies. If sufficient DNA is not obtained, the metagenomic data will not sufficiently represent the sampled community. Similarly, if the composition of the extracted DNA does not sufficiently represent that of the sampled community, neither will the metagenomic data.
Air samplers can affect the outcome of metagenomic studies, as their collection efficiencies can differ significantly. Although many studies have investigated sampling efficiencies of various types of air samplers (Fabian et al. 2009; Griffin et al. 2011; Li 2011; Dybwad et al. 2014; Hoisington et al. 2014), no consensus exists on standardized sampling approaches across various studies. Li (2011) compared performances of different air samplers using qPCR and fingerprinting by denaturing gradient gel electrophoresis. The authors showed that although impactor-based samplers provided higher efficiency for culturable bacteria in terms of their count and diversity, impinger- and filter-based devices both demonstrated highest efficiency for total bacteria (Li 2011). Fahlgren et al. (2011) used 16S rRNA gene sequencing to examine whether different air samplers, including an impinger-, an impactor-, and a filter-based sampler, affected outcomes. They showed that all three air-samplers provided similar representations of higher abundant bacterial species and their overall composition in the atmosphere; however, the bacterial composition and diversity were considerably different for the lower abundant species depending on the sampler (Fahlgren et al. 2011). Hoisington et al. (2014) used pyrosequencing to examine bacterial and fungal diversity in indoor air using four different air samplers, and showed that only 14% of bacterial operational taxonomic units (OTUs) and 44% of the fungal OTUs were similar in samples collected by the four different bioaerosol samplers. These studies stress the use of standardized air-sampling techniques and devices to generate data that sufficiently represent the sampled community, and to compare between different studies.
The protocols used to extract nucleic acids from the collected microbes also significantly affect the outcome of studies (Morgan et al. 2010; Momozawa et al. 2011; Yuan et al. 2012). However, no consensus exists across laboratories to use standardized protocols for extraction. Microbes have varying degrees of susceptibility to the chemicals used during DNA extraction procedures. For example, Gram-positive bacteria with thicker peptidoglycan layers in their cell walls are less susceptible to certain chemicals or treatments compared with Gram-negative bacteria. In other words, certain DNA extraction procedures might preferentially lyse specific types of cells but be biased against others. This could result in DNA samples that are not representative of the microbial communities in the environment (Yuan et al. 2012). Yuan et al. (2012) evaluated six commonly used DNA extraction methods on a mock microbial community that contained 11 different human-associated bacteria, and showed that each of the six methods provided significantly different 16S rRNA gene sequencing profiles of the mock microbial community. These profiles were also different than those of the expected mock community profile. In other words, none of the methods provided an accurate representation of the mock microbial community. The optional steps that are added to the DNA extraction protocols to help with cell lysis also contribute to additional variability. For example, protocols that use bead beating (Brodie et al. 2007; Yamamoto et al. 2012) to mechanically disrupt cells appear to provide better representations of microbial community profiles compared with those that do not (Yuan et al. 2012).
In summary, studies that use different approaches and protocols generate different outcomes concerning microbial quantity, diversity, and composition. Establishment of standardized strategies and methodologies across studies can eliminate experimental variability, and make comparison of data between studies feasible.
DNA Sequencing and Bioinformatics Related Challenges
With rapid advances in the NGS technologies, metagenomics has become one of the fastest growing disciplines in the field of environmental microbiology. In spite of well-deserved success, metagenomics still faces many challenges that need to be addressed. We will briefly review some of the relevant challenges. For more detailed descriptions, refer to the following subject reviews: Wooley et al. (2010), Teeling and Glockner (2012), and Thomas et al. (2012).
Although metagenomics has a potential to identify and characterize the entire microbial communities and their genome from the environment, the identification and characterization of potentially millions of genes in a soup of nucleic acids remains a daunting task. In contrast to cultured-based studies where the genomic contents of single known microorganisms are examined, environmental samples typically contain diverse communities of unknown species of high and low abundance whose genomes need to be sorted and identified. Theoretically, metagenomic data should be able to provide an exact representation of both high and low abundant microorganisms in the environment; however, the higher abundant species are generally overrepresented in metagenomic data, which leads to bias. This problem can be remediated, somewhat, through deep sequencing of DNA and by increasing the depth of coverage (reads per nucleotide) to confirm that every nucleotide in the sample is sequenced at least once or preferably numerous times to ensure that low abundant microorganisms have a representation in the environmental metagenome. Deep sequencing, however, requires high quantities of starting genetic material, which is challenging to obtain from an environment such as the air with relatively low concentrations of microorganisms.
As current DNA sequencing technologies are incapable of sequencing long intact genomes, environmental DNA needs to be sheared into small fragments before metagenomic sequencing. The millions of small reads that are subsequently generated need to be reassembled de novo using advanced bioinformatics tools and software. The reassembly of these reads into contiguous fragments (contigs) is a substantial computational challenge for metagenomics. Although great progress has been made in the development of bioinformatics software and tools, the complete reconstruction of the entire genome of microorganisms in environmental samples, in particular those from complex environments, remains difficult. The read lengths obtained from the current NGS platforms typically range between 75 and 1,000 bp, depending on the platform used (Henson et al. 2012). When the read lengths are short and the depth of coverage is shallow, large gaps are introduced in the assembled contigs. Depending on the length and number of these gaps, accurate assembly of the contigs, and thus reconstruction of the entire gene sequences, can become cumbersome or even impossible. Assembly of short reads becomes even more challenging when many long repeat sequences are present in the genes, which is usually the case for bacterial and archael genomes. It is often impossible to reconstruct the entire genome of microbial communities. These challenges are surmountable through development and refinement of high throughput gene sequencing technologies, and the establishment of methodologies used to sequence longer reads with greater depth and accuracy. For example, the third generation sequencing platform, Single Molecule Real Time Sequencing technology, is the latest innovation developed by Pacific Biosciences (PacBio), which has the potential to overcome the limitations of the current gene sequencing platforms by providing longer reads, up to 30 kb with the PacBio RS II platform, with an average read length of near 3,000 bp (Wu et al. 2014). Long overlapping reads, together with increased depth of coverage, make genome reconstruction less challenging.
One current issue with the analysis of metagenomic data is the limited availability of reference sequences in genome databases, particularly for airborne microorganisms. Very few databases for environmental microbes currently exist (for a review of metagenomic databases, see Thomas et al. 2012; Kim et al. 2013; Sharpton 2014). This complicates identification of novel genes. The solution is more metagenomic study. Metagenomic studies lead to larger genomic databases, whereas larger databases make metagenomic analysis easier.
Another issue is the current state of the art with respect to computational software and tools. New tools that can identify genome-specific markers within metagenomic data, for example, could be useful in identifying related species or strains that share high sequence similarity (Tu et al. 2014). In general, the more complex a data set, the more advanced the computational software and analytical tools needed to make sense of the data. Tremendous progress has been made in the area and further advances in analytical software and tools will make metagenomics a more practical and desirable approach to studies of environmental microbes in general and those of air in particular. For a review on the list of available software and tools for metagenomics, please refer to Bahassi and Stambrook (2014), Kim et al. (2013), and Sharpton (2014).
With continued advances in gene sequencing platforms, more complete genomic databases, and advancements in bioinformatics, many of these constraints should resolve in the near future. These new capabilities would enable fuller understanding of the potential impact of airborne microorganisms on global ecosystems.
Conclusions
Metagenomics is a powerful approach to screening microbial diversity and metabolic capabilities in any environment. The approach has proven effective in characterizing the genomic diversity and metabolic potential of many environments, including soil, marine, and the human guts, and has unraveled the vast impact of microorganisms on these ecosystems. The air harbors vastly diverse microorganisms with a potentially diverse range of metabolic activities. This frontier of microbial discoveries has largely been unexplored due to a number of challenges, which are due to the lower density of microorganisms in the air; inefficient retrieval of microbes and their nucleic acids from the air; lack of standardized approaches and methodologies; and bioinformatics-related challenges of de novo genome reconstruction. Overcoming these challenges could potentially pave the way to discoveries of novel pathways and genes important in meteorological and industrial applications, and environmental bioremediation. Metagenomic studies can additionally facilitate monitoring of airborne microorganisms through identifying pathogenic microbes and their distribution patterns and involvement in disease outbreaks that impact plant, animal, and human health.
We propose a consortium of interested parties to establish uniform approaches and methodologies designed to improve the collection efficiency, protocol reproducibility, and subsequent comparative analysis of airborne microbes. Conferences or meetings specifically designed for airborne metagenomics would provide the necessary forums for interested parties to discuss potential solutions to the present challenges.
Acknowledgment
This work was supported by King Abdullah University of Science and Technology (KAUST), Saudi Arabia.
Literature Cited
- Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol. 2004;186:2629–2635. doi: 10.1128/JB.186.9.2629-2635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato P, et al. Microbial population in cloud water at the Puy de Dome: implications for the chemistry of clouds. Atmos Environ. 2005;39:4143–4153. [Google Scholar]
- Amato P, et al. A fate for organic acids, formaldehyde and methanol in cloud water: their biotransformation by micro-organisms. Atmos Chem Phys. 2007;7:4159–4169. [Google Scholar]
- An S, Sin HH, DuBow MS. Modification of atmospheric sand-associated bacterial communities during Asian sandstorms in China and South Korea. Heredity. 2014;114:460–7. doi: 10.1038/hdy.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahassi E, Stambrook PJ. Next-generation sequencing technologies: breaking the sound barrier of human genetics. Mutagenesis. 2014;29:303–310. doi: 10.1093/mutage/geu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer H, et al. Airborne bacteria as cloud condensation nuclei. J Geophys Res Atmos. 2003;108:4658. [Google Scholar]
- Be NA, et al. Metagenomic Analysis of the Airborne Environment in Urban Spaces. Microbial Ecology. 2014;69:346–355. doi: 10.1007/s00248-014-0517-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini V, et al. Temporal variability and effect of environmental variables on airborne bacterial communities in an urban area of Northern Italy. Appl Microbiol Biotechnol. 2013;97:6561–6570. doi: 10.1007/s00253-012-4450-0. [DOI] [PubMed] [Google Scholar]
- Bishop JKB, Davis RE, Sherman JT. Robotic observations of dust storm enhancement of carbon biomass in the North Pacific. Science. 2002;298:817–821. doi: 10.1126/science.1074961. [DOI] [PubMed] [Google Scholar]
- Bowers RM, et al. Characterization of airborne microbial communities at a high-elevation site and their potential to act as atmospheric ice nuclei. Appl Environ Microbiol. 2009;75:5121–5130. doi: 10.1128/AEM.00447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RM, McLetchie S, Knight R, Fierer N. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J. 2011;5:601–612. doi: 10.1038/ismej.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie EL, et al. Urban aerosols harbor diverse and dynamic bacterial populations. Proc Natl Acad Sci U S A. 2007;104:299–304. doi: 10.1073/pnas.0608255104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge HA, Solomon WR. Sampling and analysis of biological aerosols. Atmos Environ. 1987;21:451–456. [Google Scholar]
- Burrows SM, Elbert W, Lawrence MG, Pöschl U. Bacteria in the global atmosphere—Part 1: Review and synthesis of literature data for different ecosystems. Atmos Chem Phys. 2009;9:9263–9280. [Google Scholar]
- Cao C, et al. Inhalable microorganisms in Beijing’s PM2.5 and PM10 pollutants during a severe smog event. Environ Sci Technol. 2014;48:1499–1507. doi: 10.1021/es4048472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho BC, Hwang CY. Prokaryotic abundance and 16S rRNA gene sequences detected in marine aerosols on the East Sea (Korea) FEMS Microbiol Ecol. 2011;76:327–341. doi: 10.1111/j.1574-6941.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- Chun SC, et al. The microbial population in the air of cultivation facility of oyster mushrooms. Journal of Microbiology. 2012;50:1053–1057. doi: 10.1007/s12275-012-2195-1. [DOI] [PubMed] [Google Scholar]
- Constantinidou HA, Hirano SS, Baker LS, Upper CD. Atmospheric dispersal of ice nucleation-active bacteria—the role of rain. Phytopathology. 1990;80:934–937. [Google Scholar]
- Deininger CA, Mueller GM, Wolber PK. Immunological characterization of ice nucleation proteins from Pseudomonas syringae, Pseudomonas fluorescens, and Erwinia herbicola. J Bacteriol. 1988;170:669–675. doi: 10.1128/jb.170.2.669-675.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon-Rodriguez N, et al. Microbiome of the upper troposphere: species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc Natl Acad Sci U S A. 2013;110:2575–2580. doi: 10.1073/pnas.1212089110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delort A-M, et al. A short overview of the microbial population in clouds: potential roles in atmospheric chemistry and nucleation processes. Atmos Res. 2010;98:249–260. [Google Scholar]
- Dimmick RL. Evidence for metabolic activity of airborne bacteria. J Aerosol Sci. 1975;6:387–393. [Google Scholar]
- Durand S, et al. First isolation and characterization of a bacterial strain that biotransforms the herbicide mesotrione. Lett Appl Microbiol. 2006;43:222–228. doi: 10.1111/j.1472-765X.2006.01923.x. [DOI] [PubMed] [Google Scholar]
- Dybwad M, Skogan G, Blatny JM. Comparative testing and evaluation of nine different air samplers: end-to-end sampling efficiencies as specific performance measurements for bioaerosol applications. Aerosol Sci Technol. 2014;48:282–295. [Google Scholar]
- Fabian P, McDevitt JJ, Houseman EA, Milton DK. Airborne influenza virus detection with four aerosol samplers using molecular and infectivity assays: considerations for a new infectious virus aerosol sampler. Indoor Air. 2009;19:433–441. doi: 10.1111/j.1600-0668.2009.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren C, Bratbak G, Sandaa RA, Thyrhaug R, Zweifel UL. Diversity of airborne bacteria in samples collected using different devices for aerosol collection. Aerobiologia. 2011;27:107–120. [Google Scholar]
- Fahlgren C, Hagstrom A, Nilsson D, Zweifel UL. Annual variations in the diversity, viability, and origin of airborne bacteria. Appl Environ Microbiol. 2010;76:3015–3025. doi: 10.1128/AEM.02092-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, et al. Short-term temporal variability in airborne bacterial and fungal populations. Appl Environ Microbiol. 2008;74:200–207. doi: 10.1128/AEM.01467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci U S A. 2012;109:21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzetti A, Gandolfi I, Gaspari E, Ambrosini R, Bestetti G. Seasonal variability of bacteria in fine and coarse urban air particulate matter. Appl Microbiol Biotechnol. 2011;90:745–753. doi: 10.1007/s00253-010-3048-7. [DOI] [PubMed] [Google Scholar]
- Frohlich-Nowoisky J, Pickersgill DA, Despres VR, Poschl U. High diversity of fungi in air particulate matter. Proc Natl Acad Sci U S A. 2009;106:12814–12819. doi: 10.1073/pnas.0811003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandolfi I, Bertolini V, Ambrosini R, Bestetti G, Franzetti A. Unravelling the bacterial diversity in the atmosphere. Appl Microbiol Biotechnol. 2013;97:4727–4736. doi: 10.1007/s00253-013-4901-2. [DOI] [PubMed] [Google Scholar]
- Griffin DW, et al. Observations on the use of membrane filtration and liquid impingement to collect airborne microorganisms in various atmospheric environments. Aerobiologia. 2011;27:25–35. [Google Scholar]
- Gurian-Sherman D, Lindow SE. Bacterial ice nucleation: significance and molecular basis. FASEB J. 1993;7:1338–1343. doi: 10.1096/fasebj.7.14.8224607. [DOI] [PubMed] [Google Scholar]
- Haas D, et al. The concentrations of culturable microorganisms in relation to particulate matter in urban air. Atmos Environ. 2013;65:215–222. [Google Scholar]
- Hasegawa Y, Ishihara Y, Tokuyama T. Characteristics of ice-nucleation activity in Fusarium avenaceum IFO-7158. Biosci Biotechnol Biochem. 1994;58:2273–2274. doi: 10.1271/bbb.58.2273. [DOI] [PubMed] [Google Scholar]
- Henson J, Tischler G, Ning ZM. Next-generation sequencing and large genome assemblies. Pharmacogenomics. 2012;13:901–915. doi: 10.2217/pgs.12.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KA, et al. Processing of atmospheric nitrogen by clouds above a forest environment. J Geophys Res Atmos. 2007;112:1–16. [Google Scholar]
- Hoisington AJ, Maestre JP, King MD, Siegel JA, Kinney KA. Impact of sampler selection on the characterization of the indoor microbiome via high-throughput sequencing. Build Environ. 2014;80:274–282. [Google Scholar]
- Hong SH, Bunge J, Leslin C, Jeon S, Epstein SS. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009;3:1365–1373. doi: 10.1038/ismej.2009.89. [DOI] [PubMed] [Google Scholar]
- Kim M, et al. Analytical tools and databases for metagenomics in the next-generation sequencing era. Genomics Inform. 2013;11:102–113. doi: 10.5808/GI.2013.11.3.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozloff LM, Turner MA, Arellano F. Formation of bacterial-membrane ice-nucleating lipoglycoprotein complexes. J Bacteriol. 1991;173:6528–6536. doi: 10.1128/jb.173.20.6528-6536.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Cono V, et al. Unveiling microbial life in new deep-sea hypersaline Lake Thetis. Part I: Prokaryotes and environmental settings. Environ Microbiol. 2011;13:2250–2268. doi: 10.1111/j.1462-2920.2011.02478.x. [DOI] [PubMed] [Google Scholar]
- Lasken RS. Genomic DNA amplification by the multiple displacement amplification (MDA) method. Biochem Soc Trans. 2009;37:450–453. doi: 10.1042/BST0370450. [DOI] [PubMed] [Google Scholar]
- Lee SH, et al. Identification of airborne bacterial and fungal community structures in an urban area by T-RFLP analysis and quantitative real-time PCR. Sci Total Environ. 2010;408:1349–1357. doi: 10.1016/j.scitotenv.2009.10.061. [DOI] [PubMed] [Google Scholar]
- Li CS, Huang TY. Fluorochrome in monitoring indoor bioaerosols. Aerosol Sci Technol. 2006;40:237–241. [Google Scholar]
- Li KJ. Molecular comparison of the sampling efficiency of four types of airborne bacterial samplers. Sci Total Environ. 2011;409:5493–5498. doi: 10.1016/j.scitotenv.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Lighthart B. Mini-review of the concentration variations found in the alfresco atmospheric bacterial populations. Aerobiologia. 2000;16:7–16. [Google Scholar]
- Logares R, et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ Microbiol. 2014;16:2659–2671. doi: 10.1111/1462-2920.12250. [DOI] [PubMed] [Google Scholar]
- Lowen AC, Mubareka S, Steel J, Palese P. Influenza virus transmission is dependent on relative humidity and temperature. PLoS Pathog. 2007;3:1470–1476. doi: 10.1371/journal.ppat.0030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholm IM. Comparison of methods for quantitative determinations of airborne bacteria and evaluation of total viable counts. Appl Environ Microbiol. 1982;44:179–183. doi: 10.1128/aem.44.1.179-183.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron PA, et al. Assessing genetic structure and diversity of airborne bacterial communities by DNA fingerprinting and 16S rDNA clone library. Atmos Environ. 2005;39:3687–3695. [Google Scholar]
- Methe BA, et al. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler O, DeMott PJ, Vali G, Levin Z. Microbiology and atmospheric processes: the role of biological particles in cloud physics. Biogeosciences. 2007;4:1059–1071. [Google Scholar]
- Mohler O, et al. Heterogeneous ice nucleation activity of bacteria: new laboratory experiments at simulated cloud conditions. Biogeosciences. 2008;5:1425–1435. [Google Scholar]
- Momozawa Y, Deffontaine V, Louis E, Medrano JF. Characterization of bacteria in biopsies of colon and stools by high throughput sequencing of the V2 region of bacterial 16S rRNA gene in human. PLoS One. 2011;6:e16952. doi: 10.1371/journal.pone.0016952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JL, Darling AE, Eisen JA. Metagenomic sequencing of an in vitro-simulated microbial community. PLoS One. 2010;5(4):e10209. doi: 10.1371/journal.pone.0010209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CE, et al. Microbiology and atmospheric processes: research challenges concerning the impact of airborne micro-organisms on the atmosphere and climate. Biogeosciences. 2011;8:17–25. [Google Scholar]
- Nevalainen A, Pastuszka J, Liebhaber F, Willeke K. Performance of bioaerosol samplers—collection characteristics and sampler design considerations. Atmos Environ. 1992;26:531–540. [Google Scholar]
- Oh SY, Fong JJ, Park MS, Chang L, Lim YW. Identifying airborne fungi in Seoul, Korea using metagenomics. J Microbiol. 2014;52:465–472. doi: 10.1007/s12275-014-3550-1. [DOI] [PubMed] [Google Scholar]
- Pikuta EV, Hoover RB, Tang J. Microbial extremophiles at the limits of life. Crit Rev Microbiol. 2007;33:183–209. doi: 10.1080/10408410701451948. [DOI] [PubMed] [Google Scholar]
- Pouleur S, Richard C, Martin JG, Antoun H. Ice nucleation activity in Fusarium acuminatum and Fusarium avenaceum. Appl Environ Microbiol. 1992;58:2960–2964. doi: 10.1128/aem.58.9.2960-2964.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendhran J, Gunasekaran P. Microbial phylogeny and diversity: small subunit ribosomal RNA sequence analysis and beyond. Microbiol Res. 2011;166:99–110. doi: 10.1016/j.micres.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Ravva SV, Hernlem BJ, Sarreal CZ, Mandrell RE. Bacterial communities in urban aerosols collected with wetted-wall cyclonic samplers and seasonal fluctuations of live and culturable airborne bacteria. J Environ Monit. 2012;14:473–481. doi: 10.1039/c1em10753d. [DOI] [PubMed] [Google Scholar]
- Rosenfeld D, Sherwood S, Wood R, Donner L. Climate effects of aerosol-cloud interactions. Science. 2014;343:379–380. doi: 10.1126/science.1247490. [DOI] [PubMed] [Google Scholar]
- Sangwan N, et al. Comparative metagenomic analysis of soil microbial communities across three hexachlorocyclohexane contamination levels. PLoS One. 2012;7(9):e46219. doi: 10.1371/journal.pone.0046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaman J, Kohn M. Absolute humidity modulates influenza survival, transmission, and seasonality. Proc Natl Acad Sci U S A. 2009;106:3243–3248. doi: 10.1073/pnas.0806852106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpton TJ. An introduction to the analysis of shotgun metagenomic data. Front Plant Sci. 2014;5:209. doi: 10.3389/fpls.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, et al. Intercontinental Dispersal of Bacteria and Archaea by Transpacific Winds. Appl Environ Microbiol. 2013;79:1134–1139. doi: 10.1128/AEM.03029-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling H, Glockner FO. Current opportunities and challenges in microbial metagenome analysis-a bioinformatic perspective. Brief Bioinform. 2012;13:728–742. doi: 10.1093/bib/bbs039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Gilbert J, Meyer F. Metagenomics—a guide from sampling to data analysis. Microb Inform Exp. 2012;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tringe SG, et al. The airborne metagenome in an indoor urban environment. PLoS One. 2008;3:e1862. doi: 10.1371/journal.pone.0001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu QC, He ZL, Zhou JZ. Strain/species identification in metagenomes using genome-specific markers. Nucleic Acids Res. 2014;42:e67. doi: 10.1093/nar/gku138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MA, Arellano F, Kozloff LM. Components of ice nucleation structures of bacteria. J Bacteriol. 1991;173:6515–6527. doi: 10.1128/jb.173.20.6515-6527.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaitilingom M, et al. Contribution of microbial activity to carbon chemistry in clouds. Appl Environ Microbiol. 2010;76:23–29. doi: 10.1128/AEM.01127-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaitilingom M, et al. Long-term features of cloud microbiology at the puy de Dome (France) Atmos Environ. 2012;56:88–100. [Google Scholar]
- Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whon TW, et al. Metagenomic characterization of airborne viralDNA diversity in the near-surface atmosphere. J Virol. 2012;86:8221–8231. doi: 10.1128/JVI.00293-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W, Green G, Albisu A. Collection efficiency and design of microbial air samplers. J Aerosol Sci. 2007;38:97–110. [Google Scholar]
- Wooley JC, Godzik A, Friedberg I. A primer on metagenomics. PLoS Comput Biol. 2010;6(2):e1000667. doi: 10.1371/journal.pcbi.1000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, et al. A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: insight into the plastid evolution of basal eudicots. BMC Plant Biol. 2014;14:289. doi: 10.1186/s12870-014-0289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie KM, et al. Novel bacterial taxa in the human microbiome. PLoS One. 2012;6(2):e1000667. doi: 10.1371/journal.pone.0035294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, et al. Particle-size distributions and seasonal diversity of allergenic and pathogenic fungi in outdoor air. ISME J. 2012;6:1801–1811. doi: 10.1038/ismej.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph S, et al. A metagenomic framework for the study of airborne microbial communities. PLoS One. 2013;8:e81862. doi: 10.1371/journal.pone.0081862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SQ, Cohen DB, Ravel J, Abdo Z, Forney LJ. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One. 2012;7(3):e33865. doi: 10.1371/journal.pone.0033865. [DOI] [PMC free article] [PubMed] [Google Scholar]