

Abstract
Free fatty acid receptors (FFAR) belong to a family of five G-protein coupled receptors that are involved in the regulation of lipid metabolism, so that their loss of function increases the risk of obesity. The aim of this study was to determine the expansion of genes encoding paralogs of FFAR2 in the chicken, considered as a model organism for developmental biology and biomedical research. By estimating the gene copy number using quantitative polymerase chain reaction, genomic DNA resequencing, and RNA sequencing data, we showed the existence of 23 ± 1.5 genes encoding FFAR2 paralogs in the chicken genome. The FFAR2 paralogs shared an identity from 87.2% up to 99%. Extensive gene conversion was responsible for this high degree of sequence similarities between these genes, and this concerned especially the four amino acids known to be critical for ligand binding. Moreover, elevated nonsynonymous/synonymous substitution ratios on some amino acids within or in close-vicinity of the ligand-binding groove suggest that positive selection may have reduced the effective rate of gene conversion in this region, thus contributing to diversify the function of some FFAR2 paralogs. All the FFAR2 paralogs were located on a microchromosome in a same linkage group. FFAR2 genes were expressed in different tissues and cells such as spleen, peripheral blood mononuclear cells, abdominal adipose tissue, intestine, and lung, with the highest rate of expression in testis. Further investigations are needed to determine whether these chicken-specific events along evolution are the consequence of domestication and may play a role in regulating lipid metabolism in this species.
Keywords: FFAR, evolution, gene conversion, positive selection, chicken, duplication
Introduction
Free fatty acid receptors (FFAR) are members of the G-protein-coupled receptor (GPCR) superfamily and are activated by free fatty acids (FFAs). Five receptors of this subfamily have been yet identified: FFAR1 (GPR40), FFAR2 (GPR43), FFAR3 (GPR41), FFAR4 (GPR120), and GPR84. They are characterized by their respective ligands, their pattern of expression, and their biological functions. GPR84 and FFAR4 are activated by medium-chain and unsaturated long-chain FFAs, respectively, whereas FFAR1 is activated by both medium- and long-chain FFAs. In contrast, both FFAR2 and FFAR3 are selectively activated by short-chain FFAs (SCFAs) from one to six carbon chain length (Ulven 2012; Hudson et al. 2013). The pattern of expression of these receptors also differs, as previously shown in human and rodent species (Stoddart et al. 2008; Ulven 2012). FFAR1 expression has been mainly reported in pancreas (notably in β-cells), FFAR3 expression has been observed in many tissues with the highest level in white adipose tissue, and GPR84 is predominantly expressed in hematopoietic tissues and bone marrow (Wang et al. 2006). FFAR4 is widely expressed in various tissues and cell types including intestine, macrophages, adipose tissue, taste buds, brain, pancreas, lung, thymus, and pituitary (Ichimura et al. 2009). Finally, FFAR2 is highly expressed in immune cells such as neutrophils, monocytes and peripheral blood mononuclear cells (PBMC), but has been also detected in bone marrow, spleen, skeletal muscle, heart, adipose tissue, and intestine (Stoddart et al. 2008). Bovine FFAR2 was found in almost all tissues tested (Wang et al. 2009), with variations in adipose tissue according to the age of animals (Smith et al. 2012). Pig FFAR2 was also detected in adipose tissue (Hou and Sun 2008) and intestine (Colombo et al. 2012). To our knowledge, only one study reported the expression of a chicken FFAR, claimed to be FFAR1 in chicken hepatocytes in vitro (Suh et al. 2008).
Functional studies have highlighted different roles of FFAR family in human health. In particular, human loss-of-function variants of mouse FFAR2 and human FFAR4 have been shown to increase the risk to develop obesity (Ichimura et al. 2012; Kimura et al. 2013). FFAR2 deficiency protects from high-fat diet-induced obesity and dyslipidemia, at least partly through increased energy expenditure (Bjursell et al. 2011). Mice overexpressing FFAR2 specifically in adipose tissue remain lean even when fed a high-fat diet. Furthermore, SCFAs have been described as key molecules produced by gut microbial fermentation of soluble fibers and the activation of FFAR2 by SCFAs has been involved in the regulation of energy balance (Kimura et al. 2013). Taken together, it is suggested that FFAR2 may play a key role in lipid metabolism, glucose tolerance, immune regulation, and may be involved in the crosstalk between gut microbiota and whole-body homeostasis.
Beside rodents, other animal organisms are now recognized for their potential interest in a better understanding of developmental biology, physiology, and human diseases. The chicken was the first avian species and domestic animal selected for complete genome sequencing and assembly. Chicken exhibit “natural” hyperglycemia but no signs of insulin resistance (Simon 1989), making them a valuable model to understand the regulation of energy homeostasis (Burt 2007). Like humans, de novo synthesis of lipids occurs mainly in the liver, whereas fat is deposited mainly at the visceral (abdominal) location. As stated above, only one study has reported the expression in vitro of a chicken FFAR to date, which was claimed to be FFAR1 in chicken hepatocytes (Suh et al. 2008). Twenty-six chicken genes encoding paralogs of FFAR2 were accessible in old versions of Ensembl database (Ensembl release 70) but have been removed in the current version (Ensembl release 77).
In this article, we experimentally confirmed the existence of FFAR2 paralogs, examined their patterns of expression in different tissues, and studied their evolution by gene conversion and positive selection. Comparisons were made with pigs, where FFAR2 has been previously detected in adipose tissue (Hou and Sun 2008) and intestine (Colombo et al. 2012).
Materials and Methods
Animals and Experimental Procedures
Chicken
Broilers were reared together in a closed building at the experimental unit PEAT (Pôle d'Expérimentation Avicole de Tours, INRA) under standard condition. They were fed ad libitum using conventional diet for a minimum of 4 h after overnight fasting and then weighed and sacrificed by electrical stunning in the experimental processing plant. Following sacrifice, blood was collected from all animals for DNA extraction and tissues were collected, quickly frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Peripheral blood mononuclear cells (PBMC) were isolated from the blood using Ficoll Histopaque (Sigma, #10771) density gradient centrifugation according to standard protocols, before liquid nitrogen congelation.
In order to check the link between FFAR2 expression and adiposity, another animal protocol was used: Chickens from two experimental broiler lines divergently selected for abdominal fat (Leclercq et al. 1980) were fed with a conventional diet and slaughtered at 9 weeks of age for the reverse transcriptase (RT)-qPCR measure and at 14 weeks of age for the RNA-Seq study.
Concerning experiments on testis, seminiferous tubules (containing germ cells and Sertoli cells) and Sertoli cells preparations were obtained from 6-week-old (immature animals) chickens (ISA Brown, Institut de Sélection Animale, Saint Brieuc, France) and were adapted and modified from other methodologies published in rodents species (Ellingson and Yao 1970; Foster et al. 1984; Toebosch et al. 1989; Zwain and Cheng 1994; Staub et al. 2000; Guibert et al. 2011). At this age, the population of germ cells detectable is spermatogonia and type I spermatocytes. Testes were decapsulated, slightly minced, and incubated in a shaking water bath during 15 min at 37 °C with DNase I and collagenase (Sigma, l’Isle d’Abeau Chesnes, France). Cells were centrifuged to remove collagenase and then cells were allowed to sediment by gravity in order to separate seminiferous tubules, in the pellet, and Leydig cells, in the supernatant. The fragments of seminiferous tubules were slightly digested by a last collagenase bath. Chicken tubules were frozen at −80 °C. Then, Sertoli cells preparations were obtained by two successive digestion of seminiferous tubules (two collagenase bathes 0.6 and 0.8 mg/ml for 15 min at 37 °C each digestion) added with DNase (20 µg/ml) and followed by 0.1% hyaluronidase treatment (Sigma, l’Isle d’Abeau Chesnes, France) for 10 min at 37 °C, to reduce peritubular cell contamination. The purity of the Sertoli cell preparation was averaged 80–85%. Indeed, contamination of Sertoli cell preparations with germ cells was less than 10% and the percentage of peritubular myoid cells, evaluated by alkaline phosphatase staining, was close to 8% of the total cell population (Guibert et al. 2011). Sertoli cells were frozen at −80 °C.
DNA samples from wild jungle fowls from Thailand (Gallus gallus and Gallus spadiceus) (one individual per breed) were also kindly provided by Michèle Tixier-Boichard from UMR GABI, INRA, AgroParisTech, 78352 Jouy-en-Josas.
All experiments were conducted under Licence No. 37-123 from the Veterinary Services (Indre et Loire, France) and in accordance with guidelines for care and use of animals in Agricultural Research and Teaching (French Agricultural Agency and Scientific Research Agency).
Pig
Male (n = 4) and female (n = 4) pigs of a crossbred genotype (Pietrain × (Large White × Landrace)) were considered. From 40 kg body weight to slaughter, pigs were reared in isolated cages in a temperature-controlled room, and fed ad libitum a standard cereal-based diet. At 98.2 ± 0.9 kg body weight (i.e., 151 ± 12 days of age), pigs were killed 2 h after their last meal intake, by exsanguination following electronarcosis. The liver and perirenal adipose tissue (visceral location) were taken off and weighed. Samples were immediately prepared, frozen in liquid nitrogen, and stored at −76 °C until analysis.
Gene Dosage Analysis by qPCR
Variable amounts of chicken genomic DNA (6.25, 3.125, 1.563, 0.781, and 0.391 ng/µl) were used to quantify FFAR2 copy number in a 96-well plate. PCR primers were determined to amplify all the chicken FFAR2 sequences and were forward (F)-GCCCCATAGCAAACTTCT and reverse (R)-GGGCAGCCATAAAGAGAG. As a control, four single-copy genes were amplify: FADS1, ACOX1, CPT1A, and ETS1. The primers were FADS1_F-CAGCACCACGCGAAACC, FADS1_R-TCTACAGAGAGCTTCTTTCCCAAAG, ACOX1_F-TCATCCGGTCTCTGATTGTAGGA, ACOX1_R-GCACTATAGCGGATGGCAATG, CPT1A_F-CCCTGAAAATGCTGCTTTCCTA, CPT1A_R-TGGTGCCTGCAGAAAGTTTG, ETS1_F-CAGCATCAGCACAAAGCAG, and ETS1_R-CAGCCAACCCAACCAAAG. Quantitative real-time PCR was performed using SsoFast EvaGreen Supermix on a CFX96 Touch Real-Time PCR system (Bio-Rad) in 15 μl final volume (comprising 1× SsoFast EvaGreen supermix, 0.3 μM forward and reverse primers, and 5 µl of DNA). PCR cycling program consisted of 5 min at 98 °C, followed by 44 cycles of 5 s at 98 °C and 10 s at 59 °C. An additional step was used (from 65 to 95 °C during 10 s, with 0.5 °C increment) for dissociation curve analysis. A same fluorescence threshold was fixed for all the plate. For each PCR, post-PCR melting curves confirmed the specificity of single-target amplification. The amplification efficiencies were similar for the five reactions, so the 2−ΔΔCq method (Livak and Schmittgen 2001) could be used with the average Cq of the four control genes as calibrator. Cq is defined as the number of cycles required for the fluorescent signal to cross the threshold, which is when the signal exceeds the background level. ΔCq then shows the difference of expression between two genes.
RNA Isolation
For all tissue samples except the pancreas, total RNA was extracted with TRIzol reagent (Invitrogen, Cergy Pontoise, France) according to the manufacturer’s instructions. Pancreatic total RNA was prepared by the guanidinium thiocyanate extraction procedure (Chirgwin et al. 1979). The quantity and quality of RNA extracted were evaluated by 1% agarose gel electrophoresis and spectrophotometrically using a nanodrop measuring system. No sign of RNA degradation was observed. The TURBO DNA-free kit (Ambion) was used for the DNase treatment.
Real-Time RT PCR
One microgram of total RNA was reverse-transcribed using iScript cDNA Synthesis Kit (Bio-Rad). cDNA templates (diluted in a 1:10 ratio) were used to perform PCR quantification (utilizing conditions described above). For the measurement of tissue expression, three biological replicates were used. Data were normalized to levels of 18 S rRNA (primers sequences: F-TTAAGTCCCTGCCCTTTGTACAC; R-CGATCCGAGGACCTCACTAAAC). Average Cq of the samples with weak expression was used as calibrator. Specific primers able to target five FFAR2 paralogs were F-CTCTTTATGGCTGCCCTCAG and R-GTAGCCCAGGCTTGGTTGG. They amplified the genes ENSGALG00000022595, ENSGALG00000022164, ENSGALG00000022608, ENSGALG00000022422, and ENSGALG00000022399 (supplementary data S1, Supplementary Material online).
In tissue-specific expression measure, reference genes were either GAPDH for FFAR2 quantification in adipose tissue of fat and lean lines (primers sequences: F-GCTAAGGCTGTGGGGAAAGT; R-TCAGCAGCAGCCTTCACTAC) or RPL15 (Ribosomal Protein L15) for expression measure in Sertoli cells, seminiferous tubules, and testis (F-TGTGATGCGTTTCCTCCTTGG; R-CCATAGGTTGCACCTTTTGGG).
RH Mapping
PCR amplifications were carried out for each marker (FFAR2: primers used above, MAG: F-GGCAGCCCATACCCTAAAAG, R-GCTCGCTGAAGCTGTACTGG, USF2: F-GCGCAGGAGGGATAAAATC, R-CCCCGTCTTGCTGTTGTC) in 15 µl reactions containing 25 ng DNA from the chickRH6 panel (Morisson et al. 2002), 0.4 µM of each primer, 0.25 units Taq polymerase (GoTaq, Promega), 1.5 mM MgCl2, and 0.2 mM dNTP on a GeneAmp PCR System 9700 thermocycler (Applied Biosystems). The first 5-min denaturation step was followed by 35 cycles, each consisting of denaturation at 94 °C for 30 s, annealing at Tm for 30 s and elongation at 72 °C for 30 s. PCR products were analyzed on 2% agarose gels, electrophoresed in 1× TBE (Tris/Borate/EDTA) buffer, and visualized by staining with ethidium bromide. Mapping of the markers on the RH panel was performed through the ChickRH server (http://chickrh.toulouse.inra.fr/, last accessed May 7, 2015). Distances and two-point LODs (logarithm of odds) were calculated through the Carthagene software (De Givry et al. 2005). Maps were drawn with MapChart 2.0 (Voorrips 2002). FFAR2 fragment from each positive hybrid was sequenced on an ABI 3730 sequencer (Applied Biosystem).
Genome DNA Resequencing Data
Genome DNA resequencing was performed from 18 birds belonging to European broiler lines. The 18 DNA-Seq libraries were prepared using the TruSeq DNA Sample Preparation Kit (Illumina, San Diego, CA) according to the manufacturer’s instructions. Briefly, paired-end libraries with a 250 bp insert size were generated. The libraries were quantified using QPCR Library Quantification Kit (Agilent), checked on a High Sensitivity DNA Chip (Agilent), and sequenced in pair end 2 × 100 bp on a HiSeq 2000 (Illumina) using a TruSeq v3 Kit. Sequencing produced per animal an average 20.4 Gb corresponding to a sequencing depth of 19.7×. For each animal, reads were aligned against the 26 gene sequences encoding paralogs of FFAR2 identified in 2011 by Ensembl, using BWA software (Li and Durbin 2009). Only reads with a unique mapping hit and a minimal quality score of 30 were kept.
RNA-Seq Data
RNA-Seq was performed on liver and adipose tissue mRNA of 14-week-old prepubertal chicken (n = 8: four males and four females; two issued from fat lines and two from lean lines per sex) and pig (4.5 months) (four males and four females) (SRA accession number SRP042257). RNA-Seq data from 4.5-day chick embryos (n = 20) were obtained from a previous study (SRA accession number SRP033603; Frésard et al. 2014).
The 32 mRNA-Seq libraries were prepared using the TruSeq RNA Sample Preparation Kit (Illumina) according to the manufacturer’s instructions. Briefly, Poly-A RNA was purified from 4 μg of total RNA using oligo(dT) magnetic beads, fragmented into 150–400 bp pieces using divalent cations at 94 °C for 8 min, and retrotranscribed to double-stranded cDNA using random primers. The resulting cDNA was purified using Agencourt AMPure XP beads (Beckman Coulter, Villepinte, France). Then, cDNA was subjected to end-repair and phosphorylation and subsequent purification was performed using Agencourt AMPure XP beads (Beckman Coulter). These repaired cDNA fragments were 3′-adenylated producing cDNA fragments with a single “A” base overhung at their 3′-ends for subsequent adapter-ligation. Illumina adapters containing indexing tags were ligated to the ends of these 3′-adenylated cDNA fragments followed by two purification steps using Agencourt AMPure XP beads (Beckman Coulter). Ten rounds of PCR amplification were performed to enrich the adapter-modified cDNA library using primers complementary to the ends of the adapters. The PCR products were purified using Agencourt AMPure XP beads (Beckman Coulter) and size-selected (200 ± 25 bp) on a 2% agarose Invitrogen E-Gel (Thermo Scientific). Libraries were then checked on an Agilent Technologies 2100 Bioanalyzer using the Agilent High Sensitivity DNA Kit and quantified by qPCR with the QPCR NGS Library Quantification kit (Agilent Technologies). After quantification, tagged cDNA libraries were pooled in equal ratios and a final qPCR check was performed postpooling. The pooled libraries were used for 2 × 100 bp paired-end sequencing on one lane of the Illumina HiSeq2000 with a TruSeq SBS v3-HS Kit (Illumina). After sequencing, the samples were demultiplexed and the indexed adapter sequences were trimmed using the CASAVA v1.8.2 software (Illumina). Sequencing produced an average of 6.67 Gb (i.e., 33 million of pair end reads) per animal. Reads were then mapped to the chicken reference transcript database (Ensembl cDNA release 64) using BWA software (bwa-0.5.9rc1; Li and Durbin 2009). The number of reads mapped to each transcript was determined using samtools (0.1.17) and an in house script (Li et al. 2009).
Gene Conversion
The 26 genes of the FFAR2 cluster used for the positive selection calculation were tested for intralocus gene conversion by using GENECONV version 1.81 (Sawyer 1989), which is a well-established method for detecting partial gene conversion (Posada 2002). The analysis was performed on the codon-based alignment performed by the Muscle algorithm (Edgar 2004) in the PhyleasProg pipeline (Busset et al. 2011) and permitted to the program to look for pair of sequences which are sufficiently similar to be suggestive of nonreciprocal transfer of genetic information between the different sequences. Three P values were calculated and compared to assess the significance of the results. Strong evidence of gene conversion was noted when a fragment had a P value that was less than 0.05 for at least two different types of statistical test. In each alignment, indels and missing data were ignored. All polymorphic sites were tested for evidence of gene conversion using adjusted mismatch penalties of 0 (g0) to detect recent gene conversion events. The figure for gene conversion events detected with the g0 option was made using Circos (Krzywinski et al. 2009).
Positive Selection Calculation
The inference of positive selection was performed on the tree of paralogous genes by PhyleasProg with branch-site and site models of codeml of the PAML package, with the fast option (Busset et al. 2011). The multiple sequence alignment was carefully examined to avoid all false positive results. In particular, amino acids predicted to be under positive selection that were at the boundary of the alignments were not considered because they are doubtful. Both branch-site and site models are designed to identify amino acids under positive selection. However, the site model allows the ω ratio to vary among sites (among amino acids in the protein). The branch-site model on the other side allows ω to vary both among sites in the protein and across branches on the tree and therefore aims to detect positive selection affecting a few sites along particular lineages, sites that would not be detected by using the site model. For the use of branch-site models, each branch of the phylogenetic tree was tested for positive selection. So we performed multiple test corrections by controlling for the false discovery rate using the R package QVALUE (Storey and Tibshirani 2003). Results are considered significant with a threshold of q = 10% of false positives. Sites with posterior probabilities of Bayes Empirical Bayes analyses superior to 95% or 99% were considered as positively selected. No overlap was found between the two models because the branch-site model is detecting positive selection on a selected branch whereas the site model is detecting positive selection affecting the whole phylogenetic tree.
3D Structure Modeling
A model of the 3D structure of FFAR2 has been constructed using Modeller (v9.10) (Martí-Renom et al. 2000), the alignment shown in supplementary data S7, Supplementary Material online, deduced from a HHpred search (Söding et al. 2005) and, as template, the 3D structure of proteinase-activated receptor 1 (pdb 3vw7; Zhang et al. 2012). The local reliability was evaluated using VERIFY3D (http://services.mbi.ucla.edu/Verify_3D/, last accessed May 6, 2015; Bowie et al. 1991), whereas the stereochemical quality was evaluated with PROCHECK (Laskowski et al. 1993), 3D structures were manipulated using Chimera (Pettersen et al. 2004), and the sequence alignment was presented using ESPript (Robert and Gouet 2014).
Results
FFAR2 Gene Was Highly Duplicated in the Chicken Genome
In the version 70 of Ensembl database, 26 chicken genes encoding paralogs of FFAR2 were accessible (Ensembl accession numbers are provided in supplementary data S1, Supplementary Material online). To check the existence of these multiple FFAR2 paralogs in the chicken genome, complementary analyses were performed in this study.
Validation by Quantitative Polymerase Chain Reaction
We first performed real-time polymerase chain reaction (PCR) using series of genomic DNA dilutions obtained from two French commercial broiler chickens. Primer pairs were designed to amplify all chicken FFAR2 sequences, so called “universal primers” of FFAR2. Four single copy genes were used as controls. The differences of quantitative cycle (ΔCq values) between the single-copy genes and FFAR2 genes were found between 4.27 and 4.70 (fig. 1). These results confirm the massive duplication of FFAR2 genes in the chicken genome, and estimate a relative copy number of 23.7 ± 1.2 paralogs (mean ± SEM; n = 5). A similar experiment was also performed using genomic DNA dilutions from two ancestral chicken lines from Thailand. For these populations, we also found a massive duplication of FFAR2 (fig. 1) with a copy number close-similar to that found in the European domestic line (relative copy number of 23.0 ± 1.5 (mean ± SEM; n = 4).
Fig. 1.—

FFAR2 has numerous copies in European broiler and in ancestral chicken genome. qPCR on genomic DNA shows clear difference in ΔCq between FFAR2 genes and the genes carrying only one copy per genome (four genes in European lines and three genes in Ancestral lines). qPCR was performed using “universal” primers able to amplify the 26 sequences of FFAR2 (see supplementary data S1, Supplementary Material online). FFAR2 error bars indicate standard deviation between two individual chickens. For the single copy genes, error bars represent standard deviation between the Cq measures for the three or four genes from two individual chickens.
Validation by Genomic DNA Resequencing
Whole-genome resequencing data were generated from 18 chickens with a 20× sequencing depth per individual. For each animal, reads were then mapped against the 26 gene sequences encoding paralogs of FFAR2 identified by Ensembl, v70 (supplementary data S1, Supplementary Material online), providing that only reads which had a unique hit after mapping were kept. For each animal, we were able to map reads to all 26 gene sequences, thus confirming existence of FFAR2 paralogs in the chicken genome. The percentages of identity between FFAR2 nucleotide sequences were found to range from 87.2% to 99.0% (supplementary data S3, Supplementary Material online). Examination of pairwise alignments of nucleotide sequences of FFAR2 paralogs that shared an identity rate above 98.5% (supplementary data S3, Supplementary Material online, in red) allowed detecting from 9.8 to 11.9 single nucleotide polymorphisms (SNP)/kb. Importantly, Basic Local Alignment Search Tool (BLAST) searches against turkey, finch, and quail genome and expressed sequence tags (ESTs) database did not detect any duplication of FFAR2 genes.
Validation by RNA Sequencing Data in Whole Embryos and Prepupertal Tissues
The availability of RNA sequencing (RNA-Seq) data from whole body or specific tissues can be valuable to prove expression of particular genes. We produced on average 47 million reads per chicken whole embryos and 20 and 16 million reads for liver and adipose tissue, respectively, per prepupertal chickens. Reads were then mapped to the chicken reference transcript database (Ensembl cDNA release 64). Among the 26 Ensembl FFAR2 paralogs, 11 were observed with at least 10 reads in each tissue (supplementary data S2, Supplementary Material online). These results confirm existence of many expressed FFAR2 paralogs in chicken. It remains however to determine whether other paralogs could be detected when other tissues or other physiological conditions are considered.
Chicken FFAR2 Paralogs Have Highly Similar Sequences and Are Located in the Same Genomic Region
Amino Acid and Protein Sequences
The percentages of identity shared by the chicken FFAR2 amino acid sequences reached 83.1–98.6% (table 1), these identities being much greater than for other more distant chicken paralogs of the same (F2R, F2Rl1, F2RL2, F2RL3, P2RY8, from 21.2% to 49.3%). FFAR1 and FFAR3 genes were not found in the chicken genome. In order to compare with other species, percentages of identity analysis were determined on nucleic and protein sequences of FFAR paralogs (FFAR1, FFAR2, FFAR3, FFAR4, and GPR84) in human, mouse, and pig species (supplementary data S4–S6, Supplementary Material online). The percentages of identity between FFAR1, -2, -3, and -4 were below 58% and 48% for nucleic and protein sequences, respectively (in green in supplementary data S5 and S6, Supplementary Material online). Moreover, these different FFAR genes are shared by most of mammals, suggesting that they have duplicated before divergence between these species, and that the duplications that led to FFAR2 chicken paralogs are more recent.
Table 1.
Percentage of Identity of Amino Acids Sequences between FFAR-2 Chicken Paralogs
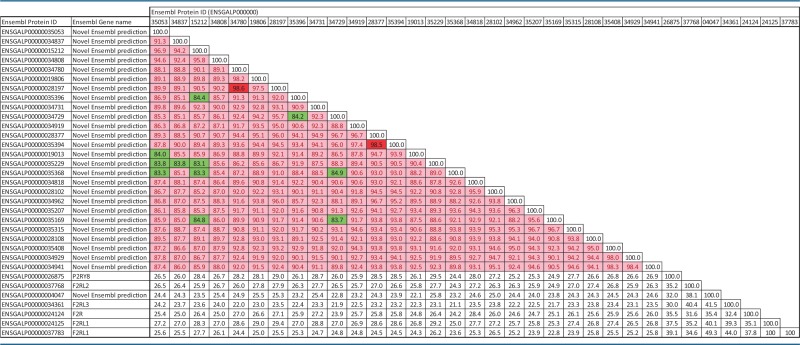 |
The construction of the phylogenetic tree of the 26 FFAR2 amino acid sequences in chicken confirmed that they belonged to the FFAR2 subclass of FFAR encoding genes (fig. 2).
Fig. 2.—

Phylogenetic tree of FFAR2 paralogous genes. The phylogenetic tree was reconstructed using PhyML (Guindon and Gascuel 2003). Bootstrap values are given when nodes are strongly supported (>70%). The scale represents the substitution rate. The sequence with an * has a specific insertion of eight amino acids (DNGSEADG) at the following positions: ENSGALP00000034780 (163-170), ENSGALP00000028197 (152-159), and ENSGALP00000019806 (158-165).
Alignment of the chicken protein sequences of FFAR2 (supplementary data S7, Supplementary Material online) showed that the two arginine and two histidine, previously described as central for the binding of fatty acid ligands (Sum et al. 2007; Stoddart et al. 2008; Hudson et al. 2013), are well conserved in chicken FFAR2, as in human FFAR2 but also human FFAR1 and FFAR3 sequences (fig. 3). The 3D fold is well conserved between FFAR1, FFAR2 and FFAR3, as highlighted by the overall conservation of amino acids over the whole alignment (fig. 3).
Fig. 3.—

Amino acids that are critical for ligand binding are conserved within the FFAR gene family. Amino acid sequences of one of the chicken FFAR2 paralogs, and FFAR1, -2 and -3 human sequences were aligned. The well-conserved amino acids that are critical of ligand binding are indicated with colored stars. Sequence identities are reported white on a black background, whereas similarities are boxed. Secondary structures, as deduced from our modeling (supplementary data S7, Supplementary Material online, and fig. 8), are reported above the sequences.
Hydrophobic amino acids, which make up the core of the fold, are especially well conserved. The first arginine residue (Arg180 in human FFAR2) was found in the 26 sequences (located in position 172 of the alignment as indicated by a blue star, supplementary data S7, Supplementary Material online, and fig. 3), whereas the second one (Arg255 in human FFAR2) was present in 25 out of 26 sequences (position 247 of the alignment indicated by a light blue star). The protein ENSGALP00000034818 is truncated before the arginine residue, which strongly suggests that the receptor function of this gene is altered. One histidine residue (His242 in human FFAR2), considered as playing an important role in fatty acid length selectivity, was conserved in chicken FFAR2 (position 234 of the alignment as indicated by a red star). This histidine was present only in human FFAR2 and human FFAR3, but was replaced by an asparagine in human FFAR1. The other histidine residue that contributes to recognition of the carboxylate (His140 in human FFAR2) was found in 24 out of 26 sequences (position 133 of the alignment, green star); noticeable exceptions concerned proteins ENSGALP00000035229 and ENSGALP00000034731, which could exhibit differences in their specificity/affinity for ligands. Analysis of the sequences obtained after DNA resequencing of chickens confirmed the lack of this His133 in the ENSGALP00000035229 sequence. Of note, an SNP was found at this position in the ENSGALP00000034731 with either a proline or a histidine (50% of both variants in the studied populations).
Chromosomal Localization
“Universal” primers were used to amplify a consensus fragment of FFAR2, giving rise to a unique band on agarose gels (supplementary data S8, Supplementary Material online). Sequencing of this band confirmed that we amplified several FFAR2 fragments. These fragments were mapped near the SEQ0067 marker (accession number AJ862640) and the ROS0264 marker (31.7 cR, LOD = 10.8, accession number XM_003643130; fig. 4), which is located inside the predicted LIG1 gene (ligase I, DNA, ATP-dependent). These two markers are located on linkage group E64 (ROS0264: chrLGE64:721094 + 721330, SEQ0067: chrLGE64:709088 + 709284, UCSC Galgal4).
Fig. 4.—

Localization on human chromosome 19 of genes from the FFAR2 region. (A) Fragment of human chromosome 19 from 35.9 to 48.6 Mb and (B) FFAR2 region on human chromosome 19. Maps are given in Mb, from assembly version GRCh37/hg19. At the left of each map are shown blocks of conserved synteny between HSA19 and the chicken genome, obtained through RH mapping. Genes with an asterisk are not found in the current version of the chicken genome (Galgal4).
Two other markers in the vicinity of FFAR2 in the human genome were analyzed on the RH panel: MAG (myelin-associated glycoprotein) and USF2 (upstream transcription factor 2, c-fos interacting). Both map to the same linkage group, not linked to E64 (fig. 4). This suggests that, contrary to the human genome, MAG and USF2 are not located near FFAR2 genes in the chicken genome, suggesting a genomic rearrangement.
Tissue Expression of FFAR2 Paralogs
Chicken FFAR2 Genes Are Highly Expressed in the Testis
The pattern of expression of FFAR2 mRNA was analyzed by quantitative PCR (qPCR), using “universal” primers able to amplify all the 26 mRNA paralogs on 18 tissues sampled from chickens. FFAR2 was expressed at low levels in all these tissues (Cq between 27 and 36). The highest expression level was observed in testis, spleen, PBMC, abdominal adipose tissue, intestine, and lung (fig. 5A). Despite the high percentage of sequence identity between paralogs, we were also able to design primer pairs able to specifically amplify five different paralogs. The specificity in amplification was confirmed by sequencing. No significant difference in expression levels between the subgroup of these five paralogs was detected, and the higher expression levels of these paralogs were confirmed in testis, spleen, PBMC, and adipose tissue.
Fig. 5.—

FFAR2 mRNA expression in different tissues in chicken (A), in chicken Sertoli cells, seminiferous tubules, and whole testis (B), in adipose tissue of lean and fat divergent lines (C). Relative tissue abundances of FFAR2 genes were examined by RT-qPCR using “universal” primers able to amplify the 26 sequences of FFAR2. The ribosomal subunit 18 S, RPL15, and GAPDH were used as reference genes. Y axis indicates relative level of mRNA expressed in comparison to the lowest level observed (Uropygial gland [A], seminiferous tubules [B], and fat chickens [C]). Error bars indicate standard deviation between three biological replicates (A), three independent experiments performed in triplicate (B), and ten animals (C). Different letters indicate significant expression difference (P < 0.05).
A particular attention was then paid to testis, which was divided in seminiferous tubules and Sertoli cells. A similar expression level of FFAR2 was observed in Sertoli cells and seminiferous tubules, which are composed of Sertoli cells and germ cells (spermatogonia and type I spermatocyte stage). Expression level is higher in the whole testis (fig. 5B).
Expression of FFAR2 Genes Is Inversely Related to Adiposity
To explore whether the expression of FFAR2 paralogs can be dependent of body adiposity, FFAR2 expression levels were measured using qPCR, in abdominal (visceral) adipose tissue from two experimental chicken lines divergently selected for abdominal fatness (Leclercq et al. 1980). Expression of FFAR2 genes was 2.7-fold higher (P = 0.008) in adipose tissue of the lean line compared with the fat line (fig. 5C). Similar results were obtained using adipose chicken RNA-Seq data, for which total FFAR2 read numbers were markedly higher (3.78 vs. 1.34; P = 0.034) in the lean line than in the fat line.
Species-Specific Expression of FFAR Genes in Tissues Involved in Lipid Metabolism
We compared the expression of chicken FFAR genes with porcine FFAR genes. Expression level of the genes of the FFAR family in pig is interesting on several aspects. First, pig is another domesticated species that show divergence along the phylogenetic tree with chicken, and second, although de novo lipid synthesis occurs in the liver in chicken, it takes place in the adipose tissue in the pig. Expression levels of the FFAR family genes were compared between chicken and pig in both liver and adipose tissues (fig. 6 and supplementary data S4, Supplementary Material online). Although we observed a higher expression of FFAR2 paralogs in adipose tissue (39 reads, 2.56 fragments per kilobase of exon per million fragments mapped [FPKM]) compare with the liver (12 reads, 0.7 FPKM) in the chicken, the expression of the porcine FFAR2 was low in both tissues (<10 reads). Expression levels for other members of the FFAR family revealed that FFAR4 was highly expressed in the pig (632 reads, 39 FPKM). Finally, very low expression levels were observed for porcine FFAR1 and FFAR3 in both adipose tissue (0 and 0.1 reads, respectively) and liver (0 and 0.4 reads, respectively).
Fig. 6.—

FFAR expression in chicken and pig using RNA-Seq in adipose tissue (A) and liver (B). Values correspond to the mean of eight individual expression counts (four males and four females). Chicken FFAR2 reads are the sum of all the paralog expression counts. Errors bars indicate standard deviation between individual reads.
Estimation of Gene Conversion and Positive Selection in FFAR2 Gene
To determine the selective pressures that may have triggered the evolution of the FFAR2 gene clusters in the chicken genome, we conducted two different analyses, the gene conversion events detection and the positive selection calculation. Both analyses were conducted using the multiple amino acid sequence alignment and the phylogenetic tree (fig. 2 and supplementary data S7, Supplementary Material online).
Gene conversion can occur between sequences if they present enough sequence identity. By using GENECONV tool, we searched for statistical evidence of this phenomenon. We used the option g0 that allows no mismatch between sequences involved in a gene conversion event, so fragments resulting from gene conversion events contain no polymorphism. The control of “randomize sites,” which randomizes the order of polymorphic sites before analysis, detected no gene conversion events in the cluster, so that the results of subsequent GENECONV analyses for FFAR2 are reliable. Each ribbon on figure 7 represents a unique conversion event between FFAR2 paralogs in chicken (supplementary data S9, Supplementary Material online). We found that gene conversion is a pervasive evolutionary mechanism in the FFAR2 cluster in the chicken genome with 24 out to 26 paralogs involved in at least one recent gene conversion (fig. 7 and supplementary data S9, Supplementary Material online). Moreover, by using the site models implemented in PhyleasProg, which allows the ω ratio to vary among sites, we detected eight different positions under positive selection scattered along the sequence of the FFAR2 paralogs. By using the branch-site models, which estimate different ω values among branches and among sites, we identified three amino acids specifically under positive selection for one of the paralogs (Table 2).
Fig. 7.—

Gene conversion events in the FFAR2 cluster. Each line represents a gene and each ribbon represents a gene conversion event between two genes of the FFAR2 cluster. The thickness of the ribbon represents the significance of each identified fragment. The biggest, medium and smallest ribbons represent the fragments identified with three, two and one significant P values, respectively. For clarity, Ensembl gene IDs are shortened. The correspondence between reduced gene ID, gene ID, and protein ID can be found in the supplementary data S1, Supplementary Material online. The coordinates of each fragment can be found in the supplementary data S9, Supplementary Material online. Note that only the 24 genes involved in at least one gene conversion event are represented.
Table 2.
Parameter Estimates and Likelihood Scores for Branch-Site and Site Models
| Gene | Model | la | Estimates of Parametersb | 2Δl | Positively Selected Sites (BEB)c |
|---|---|---|---|---|---|
| Branch-site models: | Null | 2,170.978924 | ρ0 = 0.42803, (ρ1 = 0.17627), ω0 = 0.03667, (ω1 = 1) | 7.97** | Not allowed |
| ENSGALG 00000022004 | Alternative | 2,166.991593 | ρ0 = 0.66474, ρ1 = 0.27047, (ρ2 = 0.0648), ω0 = 0.03763, (ω1 = 1), ω2 = ∞ | Three sites P > 99%: 113E, 115H, and 201T | |
| Site models | M8a | 2,173.482314 | ρ0 = 0.70692, ρ1 = 0.29308, p = 4.58561 q = 99 | 20.45*** | Not allowed |
| M8a | 2,163.258223 | ρ0 = 0.85604, ρs = 0.14396, p = 0.24218, q = 1.07143, ωs = 2.58370 | Seven sites P > 95%: 80M, 108M, 132W, 142S, 182H, 241I, 246Y. One site with P > 99%: 209G | ||
aLog-likelihood values.
bNull and alternative models: ρ0, ρ1, and ρ2 are the proportions of codons subject to purifying selection, neutral evolution, and positive selection, respectively. ω0, ω1, and ω2 represented dN/dS for each class (purifying, neutral, and positive selection, respectively). M8 and M8a models: ωs, Average dN/dS ratio for sites subject to positive selection (models M2a and M8); p and q, shape parameters for the beta distribution of ω (models M7, M8, and MEC); ρ0, ρ1, and ρS are the proportions of codons subject to purifying selection, neutral evolution, and positive selection, respectively.
cSite numbers and amino acids refer to the ENSGALP00000034780 sequence (gene ID: ENSGALG00000022004).
**Significant at P < 0.01 and ***P < 0.001.
Taken together, this analysis of gene conversion events on FFAR2 cluster showed that extensive gene conversion led to sequence homogenization. However, the amino acids under positive selection may be involved in the diversification of function of the different paralogs.
Mapping of Amino Acids under Positive Selection on the 3D Structure of Chicken FFAR2
A model of the 3D structure of chicken FFAR2 (ENSGALP00000034780) was made, using the experimental 3D structure of the proteinase-activated receptor 1 as a template (pdb 3vw7, 25% identity, HH-Pred probability 100, E value 4.5 × 10−45). Despite the low levels of sequence identity, the protein sequences were well aligned, with clear anchor points within the transmembrane helices and insertions occurring within loops (see the corresponding alignment in supplementary data S10, Supplementary Material online). The four basic amino acids mentioned above to be crucial for receptor functionality were clearly located in the ligand-binding groove, at the top of the transmembrane assembly (fig. 8). One of the amino acids under positive selection (Y246) participates in this groove, whereas the other amino acids described here did not appear to be in critical positions. However, two of them (M80 and H182) lied in the extracellular loops, in the vicinity of the ligand-binding groove.
Fig. 8.—

Ribbon representation of the 3D model of FFAR2. The model was made by homology with the 3D structure of the proteinase-activated receptor 1 (pdb 3vw7). This last structure was obtained in complex with the antagonist vorapaxar, whose binding site is shown here light gray shaded. The four critical amino acids (corresponding to H140, R180, H242, and R255 in fig. 3 after correction of the alignment) are presented in yellow, whereas amino acids under positive selection, as reported in table 2, are indicated in orange, red, and purple.
Discussion
The chicken was the first avian species and domestic animal selected for complete genome sequencing and assembly (International Chicken Genome Sequencing Consortium [ICGSC] Hillier et al. 2004). The chicken genome (as other bird genomes) however displays a reduced size compared with mammals. Moreover, phylogenetic analyses of chicken, mammalian, and fish paralogs support the hypothesis that loss of paralogs occurred much more frequently in chicken than in mammals (Hughes and Friedman 2008). However, several gene duplications in chicken have also been described, such as genes encoding Toll-receptors (ICGSC 2004; Temperley et al. 2008). Specific epimutations have been also recently associated with genes related to the bone morphogenic protein, toll receptor, and melanogenesis signaling pathways in birds (Skinner et al. 2014). Beside these pathways, genes related to energy metabolism could have been affected differently in birds compared with mammals, because chicken exhibit normal hyperglycemia without signs for insulin resistance. Recently, the subfamily of FFAR has received a considerable interest in literature related to energy metabolism, obesity and diabetes in human and rodents (Hara et al. 2014; Ichimura et al. 2012), because of their potential interest as key therapeutic targets (Hara et al. 2014). A computational analysis of the chicken genome notably reported an expansion of eight homologs to human FFAR2 gene (GPR43; Lagerström et al. 2006). Here, we prove the existence of more than 20 genes encoding FFAR2 paralogs in the chicken genome. First, qPCR experiments on genomic DNA using different chicken populations estimated a relative copy number of 23 ± 1.5 of these paralogs in the chicken genome. Second, analysis of genome DNA resequencing and RNA-Seq data confirmed the massive duplication of FFAR2. Although having strong identity (above 98.5%), 9.8–11.9 substitutions between FFAR2 nucleotide sequences (each containing approximately 1,000 nucleotides) were detected. This is more than the 5 SNP/kb previously estimated for the entire chicken genome (Wong et al. 2004). This is also much more than the approximately 2.7 SNP/kb observed in coding regions between broilers and red jungle fowl (Wong et al. 2004). This suggests that true paralogs rather than allelic variants of FFAR2 genes exist in the chicken genome. Furthermore, sequence comparison of these FFAR2 chicken paralogs further showed that the four basic amino acids responsible of fatty acid binding (Hudson et al. 2013) were conserved among paralogs, suggesting that most of these paralogs are functional.
In mammalian genome, FFAR2 gene is located in a locus that also contains genes encoding two other types of the FFAR family (FFAR1, FFAR3), HAMP coding for hepcidin, involved in the regulation of iron metabolism, and USF2, the upstream transcription factor involved in lipid homeostasis. By exploring the chicken genome by reciprocal tBLASTn analysis on genome and EST database, we did not find traces of FFAR1 and FFAR3 genes in chicken. This suggests that the only one study that has reported the expression of a chicken FFAR to date, which was claimed to be FFAR1 (Suh et al. 2008), would refer rather to FFAR2. The hepcidin gene is absent from chicken, fish, and reptile genomes. Although chicken USF2 sequence is available for chicken (http://www.ncbi.nlm.nih.gov/protein/47420475, last accessed May 6, 2015), USF2 has not been still assigned to a chicken chromosome. The amplification pattern obtained here on the RH panel however indicates that the FFAR2 locus is not mapped near the USF2 linkage group: Very few hybrid clones were both positive for USF2 and the FFAR2 locus, indicating a different location for the FFAR2 locus and the portion of the genome orthologous to human HSA19 bearing USF2. Altogether, this suggests that this particular locus encompassing FFAR2 has been subjected to a specific rearrangement in chicken. We cannot discriminate between the 26 FFAR2 fragments, but sequencing of each hybrid clone amplicon proved that there are several different FFAR2 fragments in each clone, leading to the hypothesis that the ancestral FFAR2 gene has been duplicated in the same locus. Assuming a unique position for the FFAR2 locus, it belongs to chicken linkage group E64, a small group (799,899 bp) probably corresponding to chicken microchromosome 31 (Masabanda et al. 2004). Altogether, this confirms that there is no overall synteny conservation between human chromosome 19, where the FFAR2 gene is mapped, and the chicken genome, with only several small groups of synteny mainly localized on chicken microchromosomes or unknown linkage groups (Morisson et al. 2007). The duplication of the FFAR2 locus may have occurred during the rearrangements of these genomic fragments.
Interestingly, this expansion among FFAR2 genes seems to be chicken-specific, because tBLASTn analysis on ESTs from finch, turkey, or quail species found only one significant match in this study. We cannot totally rule out the possibility that further sequencing of other bird genomes may reveal FFAR2 duplication similar to that in chicken. However, as we found no trace of a massive duplication of FFAR2 in neither turkey nor quail but found FFAR2 duplicates in the ancestral line from Thailand, we can date the massive duplication event of FFAR2 between 30 Ma (estimated divergence between chicken and turkey) and 8,000 years ago (chicken domestication) (Dimcheff et al. 2002; Jarvis et al. 2014).
Although all FFAR2 paralogs seem to encode full-length proteins, the functionality of those genes remains unknown. Further investigations are needed to understand physiological significance of such a massive duplication in chicken. In human and rodents, FFAR2 has been described as a receptor specific of small-chain fatty acids (1–6 carbon chain length), so that it is activated by physiological concentrations of volatile FFAs acetate, propionate, and butyrate which are gut-microbiote-derived bacterial fermentation products (for review, Hara et al. 2013). Whether chicken produce concentrations of small-chain fatty acids that differ from mammals would be interesting to investigate. The FFAR2 has been shown to mediate the beneficial effects associated with high soluble fiber diet and protect from high-fat diet-induced obesity and dyslipidemia. In particular, in vivo in the mouse, acetate is able to reduce levels of nonesterified fatty acids in serum, these effects being abolished in FFAR2-/- mice (Ge et al. 2008). Because chicken in modern breeding are usually fed cereal-based diets without major sources of fibers, the physiological significance of the massive FFAR2 duplication remains questionable. Activation of FFAR2 by short-chain fatty acids may lead to the inhibition of lipolysis and the decrease of FFA levels in serum, a phenomenon which may be amplified by the presence of these numerous FFAR2 paralogs in chicken.
Overall, whether the duplication of FFAR2 gene is involved in a particular regulation of energy metabolism in chicken compared with mammals has to be investigated. In human, specific duplications of amylase gene were described, and copy number of this gene was shown to be positively correlated with salivary amylase protein level (Perry et al. 2007). Moreover, individuals from populations with high-starch diets have more copies than those with traditionally low-starch diets. Thus, the number of copies of amylase genes may be selected to adapt populations to their traditional diet. An adaptation to diet is then an interesting hypothesis to explain FFAR2 gene numerous duplications.
To gain insights into the functions of FFAR2 in chicken, we decided to investigate the tissue-expression of FFAR2 and its different paralogs. Considering the very high percentage of identity between nucleotide sequences of the FFAR2 paralogs, it is however impossible to produce primers that are specific of each of them. We demonstrated for the first time the expression of FFAR2 gene in testis, spleen, PBMC, adipose tissue, intestine, and lung of the chicken, with testis expressing the highest levels of these genes. Therefore, we also evaluated FFAR2 expression in different compartments of the chicken testis. We found equivalent mRNA levels in seminiferous tubules and Sertoli cell. Whether these genes are predominantly expressed in Sertoli cells or other somatic or germ cells remains questionable. Testis is known for permissive transcription (transcription of DNA caused by biochemical happenstance rather than usefulness) (Soumillon et al. 2013), so the physiological significance of expression of FFAR2 genes still remains to be experimentally explored. Further investigations of the role of FFAR2 paralogs in testis require specific experimental designs modulating male fertility after meal supplementation, together with in vitro modulation of Sertoli and/or Leydig cells steroidogenesis.
Importantly, FFAR2 was expressed in chicken and pig adipose tissue, as already described in mouse in this tissue (Hong et al. 2005; Kimura et al. 2013). However, because there is an evidence of an implication of FFAR2 on obesity (Dewulf et al. 2011; Kimura et al. 2013), we looked at the expression levels of FFAR2 in visceral adipose tissue between chicken lines divergently selected for abdominal fatness. We showed that FFAR2 was significantly overexpressed in the lean line compared with the fat line. This result is consistent with previous study in mouse (Kimura et al. 2013), showing that FFAR2 knockout mice are obese, whereas transgenic mice overexpressing adipose tissue-specific FFAR2 are lean.
Suh et al. (2008) reported the expression of a chicken FFAR claimed to be FFAR1 in primary cultured chicken hepatocytes. In their study, FFAR1 was activated with linoleic acid (C18:2), and FFAR1 protein expression was blocked by “FFAR1-specific siRNA” in vitro. However the corresponding sequence is not available, so it is not possible to check this experiment. Whether FFAR1 does exist in the chicken remains to be investigated.
Algorithms designed to detect recombination between nucleotide sequences indicate that gene conversion has a homogenized part of the length of the chicken FFAR2 paralogs, suggesting that extensive gene conversion is responsible for the particularly high degree of sequence similarity between these genes, in particular the high conservation of the four amino acids known to be critical of ligand binding (Hudson et al. 2013). Moreover, elevated nonsynonymous/synonymous substitution ratios on some amino acids within (Y248) or in the vicinity (Met80, H182) of the ligand-binding groove suggest that positive selection may have reduced the effective rate of gene conversion in this region, and may contribute to diversify/specialize the function of some FFAR2 paralogs in chicken.
In conclusion, this work brings significant new data on the evolution in the FFAR2 subfamily in chicken, by massive duplication, gene conversion, and positive selection. These dominant processes of their sequence evolution might have led to a sort of mixing of homogenization and diversification of protein functions. Further investigations are needed to study whether these recent chicken-specific events are the consequence of domestication. It is important to determine which might be the functions of these FFAR2 paralogs in chicken. Namely, it would be interesting to test whether those paralogs, if actually functional GPCRs, activate specific pathways and by which agonists they can be activated. Present data suggest roles of FFAR2 in testis and adipose tissue expansion in chicken.
Supplementary Material
Supplementary data S1–S10 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by INRA (Chick-seq and SOSrnaSEQ programs) and by French ANR (Programs FATINTEGER and EpiBird). The authors thank Mireille Morisson for providing the ChickRH panel.
Literature Cited
- Bjursell M, et al. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. J Physiol Endocrinol Metab. 2011;300(1):E211–E220. doi: 10.1152/ajpendo.00229.2010. [DOI] [PubMed] [Google Scholar]
- Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Burt D. Emergence of the chicken as a model organism: implications for agriculture and biology. Poult Sci. 2007;86:1460–1471. doi: 10.1093/ps/86.7.1460. [DOI] [PubMed] [Google Scholar]
- Busset J, Cabau C, Meslin C, Pascal G. PhyleasProg: a user-oriented web server for wide evolutionary analyses. Nucleic Acids Res. 2011;39:W479–W485. doi: 10.1093/nar/gkr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- Colombo M, Trevisi P, Gandolfi G, Bosi P. Assessment of the presence of chemosensing receptors based on bitter and fat taste in the gastrointestinal tract of young pig. J Anim Sci. 2012;90:128–130. doi: 10.2527/jas.53793. [DOI] [PubMed] [Google Scholar]
- De Givry S, Bouchez M, Chabrier P, Milan D, Schiex T. Carh ta Gene: multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics. 2005;21:1703–1704. doi: 10.1093/bioinformatics/bti222. [DOI] [PubMed] [Google Scholar]
- Dewulf EM, et al. Inulin-type fructans with prebiotic properties counteract GPR43 overexpression and PPARγ-related adipogenesis in the white adipose tissue of high-fat diet-fed mice. J Nutr Biochem. 2011;22:712–722. doi: 10.1016/j.jnutbio.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Dimcheff DE, Drovetski SV, Mindell DP. Phylogeny of Tetraoninae and other galliform birds using mitochondrial 12S and ND2 genes. Mol Phylogenet Evol. 2002;24:203–215. doi: 10.1016/s1055-7903(02)00230-0. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingson D, Yao K. Growth and observations of Chinese hamster seminiferous epithelium in vitro. J Cell Sci. 1970;6:195–205. doi: 10.1242/jcs.6.1.195. [DOI] [PubMed] [Google Scholar]
- Foster P, Creasy DM, Foster JR, Gray T. Testicular toxicity produced by ethylene glycol monomethyl and monoethyl ethers in the rat. Environ Health Perspect. 1984;57:207. doi: 10.1289/ehp.8457207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frésard L, et al. Transcriptome-wide investigation of genomic imprinting in chicken. Nucleic Acids Res. 2014;42:3768–3782. doi: 10.1093/nar/gkt1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, et al. Activation of G protein-coupled receptor 43 in adipocytes leads to inhibition of lipolysis and suppression of plasma free fatty acids. Endocrinology. 2008;149:4519–4526. doi: 10.1210/en.2008-0059. [DOI] [PubMed] [Google Scholar]
- Guibert E, Brière S, Pelletier R, Brillard J, Froment P. Characterization of chicken Sertoli cells in vitro. Poult Sci. 2011;90:1276–1286. doi: 10.3382/ps.2010-01081. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hara T, et al. Role of free fatty acid receptors in the regulation of energy metabolism. Biochim Biophys Acta Mol Cell Biol Lipids. 2014;1841:1292–1300. doi: 10.1016/j.bbalip.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Hara T, Kimura I, Inoue D, Ichimura A, Hirasawa A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev Physiol Biochem Pharmacol. 2013;164:77–116. doi: 10.1007/112_2013_13. [DOI] [PubMed] [Google Scholar]
- Hillier LW, et al. International Chicken Genome Sequencing Consortium: Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432(7018):69–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Hong Y-H, et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology. 2005;146:5092–5099. doi: 10.1210/en.2005-0545. [DOI] [PubMed] [Google Scholar]
- Hou Z, Sun C. Transcriptional expression of GPR43 gene in adipose tissue and primary cultured adipocytes of pig. Chin J Biotechnol [Sheng wu gong cheng xue bao]. 2008;24:1361–1366. [PubMed] [Google Scholar]
- Hudson BD, Murdoch H, Milligan G. Minireview: the effects of species ortholog and SNP variation on receptors for free fatty acids. Mol Endocrinol. 2013;27:1177–1187. doi: 10.1210/me.2013-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Friedman R. Genome size reduction in the chicken has involved massive loss of ancestral protein-coding genes. Mol Biol Evol. 2008;25:2681–2688. doi: 10.1093/molbev/msn207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura A, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483(7389):350–354. doi: 10.1038/nature10798. [DOI] [PubMed] [Google Scholar]
- Ichimura A, Hirasawa A, Hara T, Tsujimoto G. Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins other Lipid Mediat. 2009;89:82–88. doi: 10.1016/j.prostaglandins.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Jarvis ED, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–1331. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström MC, et al. The G protein-coupled receptor subset of the chicken genome. PLoS Comput Biol. 2006;2:e54. doi: 10.1371/journal.pcbi.0020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Leclercq B, Blum J, Boyer J. Selecting broilers for low or high abdominal fat: initial observations. Br Poult Sci. 1980;21:107–113. [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martí-Renom MA, et al. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Masabanda JS, et al. Molecular cytogenetic definition of the chicken genome: the first complete avian karyotype. Genetics. 2004;166:1367–1373. doi: 10.1534/genetics.166.3.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisson M, et al. ChickRH6: a chicken whole-genome radiation hybrid panel. Genet Sel Evol. 2002;34:521. doi: 10.1186/1297-9686-34-4-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisson M, et al. The chicken RH map: current state of progress and microchromosome mapping. Cytogenet Genome Res. 2007;117:14–21. doi: 10.1159/000103160. [DOI] [PubMed] [Google Scholar]
- Perry GH, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Posada D. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Mol Biol Evol. 2002;19:708–717. doi: 10.1093/oxfordjournals.molbev.a004129. [DOI] [PubMed] [Google Scholar]
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(W1):W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S. Statistical tests for detecting gene conversion. Mol Biol Evol. 1989;6:526–538. doi: 10.1093/oxfordjournals.molbev.a040567. [DOI] [PubMed] [Google Scholar]
- Simon J. Chicken as a useful species for the comprehension of insulin action. Crit Rev Poult Biol. 1989;2(2):121–148. [Google Scholar]
- Skinner MK, et al. Epigenetics and the Evolution of Darwin s Finches. Genome Biol Evol. 2014;6(8):1972–1989. doi: 10.1093/gbe/evu158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, et al. Adipogenic gene expression and fatty acid composition in subcutaneous adipose tissue depots of Angus steers between 9 and 16 months of age. J Anim Sci. 2012;90:2505–2514. doi: 10.2527/jas.2011-4602. [DOI] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 2013;3:2179–2190. doi: 10.1016/j.celrep.2013.05.031. [DOI] [PubMed] [Google Scholar]
- Staub C, et al. The whole meiotic process can occur in vitro in untransformed rat spermatogenic cells. Exp Cell Res. 2000;260:85–95. doi: 10.1006/excr.2000.4998. [DOI] [PubMed] [Google Scholar]
- Stoddart LA, Smith NJ, Jenkins L, Brown AJ, Milligan G. Conserved polar residues in transmembrane domains V, VI, and VII of free fatty acid receptor 2 and free fatty acid receptor 3 are required for the binding and function of short chain fatty acids. J Biol Chem. 2008;283:32913–32924. doi: 10.1074/jbc.M805601200. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HN, Huong HT, Song CH, Lee JH, Han HJ. Linoleic acid stimulates gluconeogenesis via Ca2+/PLC, cPLA2, and PPAR pathways through GPR40 in primary cultured chicken hepatocytes. Am J Physiol Cell Physiol. 2008;295:C1518–C1527. doi: 10.1152/ajpcell.00368.2008. [DOI] [PubMed] [Google Scholar]
- Sum CS, et al. Identification of residues important for agonist recognition and activation in GPR40. J Biol Chem. 2007;282(40):29248–29255. doi: 10.1074/jbc.M705077200. [DOI] [PubMed] [Google Scholar]
- Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW. Evolution of the chicken Toll-like receptor gene family: a story of gene gain and gene loss. BMC Genomics. 2008;9:62. doi: 10.1186/1471-2164-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toebosch A, Brussée R, Verkerk A, Grootegoed J. Quantitative evaluation of the maintenance and development of spermatocytes and round spermatids in cultured tubule fragments from immature rat testis. Int J Androl. 1989;12:360–374. doi: 10.1111/j.1365-2605.1989.tb01325.x. [DOI] [PubMed] [Google Scholar]
- Ulven T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol. 2012;3:111. doi: 10.3389/fendo.2012.00111. doi: 10.3389/fendo.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorrips R. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93:77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- Wang A, Gu Z, Heid B, Akers R, Jiang H. Identification and characterization of the bovine G protein-coupled receptor GPR41 and GPR43 genes. J Dairy Sci. 2009;92:2696–2705. doi: 10.3168/jds.2009-2037. [DOI] [PubMed] [Google Scholar]
- Wang J, Wu X, Simonavicius N, Tian H, Ling L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J Biol Chem. 2006;281:34457–34464. doi: 10.1074/jbc.M608019200. [DOI] [PubMed] [Google Scholar]
- Wong GK, et al. A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature. 2004;432:717–722. doi: 10.1038/nature03156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwain IH, Cheng CY. Rat seminiferous tubular culture medium contains a biological factor that inhibits Leydig cell steroidogenesis: its purification and mechanism of action. Mol Cell Endocrinol. 1994;104:213–227. doi: 10.1016/0303-7207(94)90124-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.