

Abstract
Introduction
The transcription factor activating enhancer binding protein 2 epsilon (AP-2ε) was recently shown to be expressed during chondrogenesis as well as in articular chondrocytes of humans and mice. Furthermore, expression of AP-2ε was found to be upregulated in affected cartilage of patients with osteoarthritis (OA). Despite these findings, adult mice deficient for AP-2ε (Tfap2e−/−) do not exhibit an obviously abnormal cartilaginous phenotype. We therefore analyzed embryogenesis of Tfap2e−/− mice to elucidate potential transient abnormalities that provide information on the influence of AP-2ε on skeletal development. In a second part, we aimed to define potential influences of AP-2ε on articular cartilage function and gene expression, as well as on OA progression, in adult mice.
Methods
Murine embryonic development was accessed via in situ hybridization, measurement of skeletal parameters and micromass differentiation of mesenchymal cells. To reveal discrepancies in articular cartilage of adult wild-type (WT) and Tfap2e−/− mice, light and electron microscopy, in vitro culture of cartilage explants, and quantification of gene expression via real-time PCR were performed. OA was induced via surgical destabilization of the medial meniscus in both genotypes, and disease progression was monitored on histological and molecular levels.
Results
Only minor differences between WT and embryos deficient for AP-2ε were observed, suggesting that redundancy mechanisms effectively compensate for the loss of AP-2ε during skeletal development. Surprisingly, though, we found matrix metalloproteinase 13 (Mmp13), a major mediator of cartilage destruction, to be significantly upregulated in articular cartilage of adult Tfap2e−/− mice. This finding was further confirmed by increased Mmp13 activity and extracellular matrix degradation in Tfap2e−/− cartilage explants. OA progression was significantly enhanced in the Tfap2e−/− mice, which provided evidence for in vivo relevance. This finding is most likely attributable to the increased basal Mmp13 expression level in Tfap2e−/− articular chondrocytes that results in a significantly higher total Mmp13 expression rate during OA as compared with the WT.
Conclusions
We reveal a novel role of AP-2ε in the regulation of gene expression in articular chondrocytes, as well as in OA development, through modulation of Mmp13 expression and activity.
Introduction
Most components of the vertebrate skeleton are formed through a complex, multistep process termed endochondral ossification [1]. This process starts during early embryonic development, when mesenchymal cells condense at specific locations and prefigure future skeletal elements [2]. Subsequent differentiation of these cells results in chondrocytes that synthesize an abundance of extracellular matrix (ECM) proteins, including collagen type II and proteoglycans (for example, aggrecan). A premature skeletal template is thereby formed, entirely composed of cartilage [3]. After further differentiation steps, the cells become hypertrophic, attract blood vessels and ultimately undergo apoptosis, allowing osteoblasts to infiltrate the cartilaginous matrix to induce formation of trabecular bone [4, 5]. Endochondral ossification is completed in adults when most of the skeleton is replaced by bone tissue. However, in all diarthritic joints, a thin layer of permanent cartilage tissue remains intact throughout the organism’s lifetime. This “articular cartilage” acts as a shock absorber, minimizing peak pressures on the subchondral bone, and provides a smooth, low-friction gliding surface for efficient joint movement [6].
Chondrocytes are essential for physiological cartilage homeostasis. Under normal conditions, they maintain a constant, albeit low-level, equilibrium of matrix synthesis and degradation of ECM molecules. The latter is mediated by proteolytic enzymes such as matrix metalloproteinases (MMPs) and aggrecanases [7]. According to the prevailing hypothesis, disruption of this tightly controlled balance is one of the initial steps in osteoarthritis (OA) development [7–9]. OA is the most common joint disorder in Western populations. It is mainly described as an intrinsic, chronic disease of the articular cartilage (primary OA) [10, 11]. It is characterized by progressive degradation of the tissue, accompanied by biochemical and metabolic changes within the chondrocytes [12]. The exact molecular mechanisms that are responsible for OA onset are still poorly understood.
The initiation of OA often occurs on cartilage surface areas receiving the greatest mechanical forces. Eventually, chondrocytes begin to upregulate the production of ECM-degrading enzymes that mediate collagen and proteoglycan depletion. Here, members of the MMP family are attributed the most important role. MMPs comprise a group of zinc- and calcium-dependent endopeptidases that function as collagenases and aggrecanases. Of those, MMP13 (collagenase 3) is characterized as the central catabolic mediator that is overexpressed in nearly all cases of human OA, whereas other MMP variants only irregularly show enhanced expression [13–15]. In addition, several studies have demonstrated constitutive, albeit low-grade, expression of MMP13 in healthy human, rat and pig articular cartilage participating in physiologic ECM turnover [8, 14, 16, 17].
After a certain period, OA-induced damage of the articular cartilage becomes histologically obvious. Beginning with fissures and clefts in the cartilage surface, the defects quickly expand to the full depth of the tissue. Following staining with cationic dyes, local proteoglycan depletion is observed in the cartilage matrix bordering lesions [18]. Last, during end-stage OA, the hyaline cartilage is extensively eroded, and eburnated subchondral bone is formed. In addition, osteophyte formation at the joint margins can be observed [18, 19].
Recently, we revealed mRNA expression of the transcription factor activating enhancer binding protein 2 epsilon (AP-2ε) in chondrocytes and during chondrogenic differentiation of human mesenchymal stem cells. Additionally, we detected AP-2ε in vivo in murine hypertrophic chondrocytes during embryogenesis and in human articular cartilage via immunohistochemistry [20, 21]. AP-2ε is a member of the AP-2 transcription factor family, which consists of five isoforms (AP-2α to AP-2ε) and influences a vast number of physiological and pathogenic processes [22–24]. AP-2ε was also found to be upregulated in articular cartilage of OA patients [21]. Furthermore, in vitro, AP-2ε induced the expression of chemokine (C-X-C) motif ligand 1 (CXCL1) [25] and inhibited COL2A1 (encoding collagen type II) in human chondrocytic cells [26]. Despite these findings, adult mice deficient for AP-2ε (Tfap2e−/−) do not show an obvious cartilage phenotype under normal physiological conditions.
Therefore, in the present study, we analyzed possible roles of AP-2ε in embryonic skeletal development, as well as in articular cartilage function after the surgical induction of OA, using the Tfap2e−/− mouse. The obtained results will help to enhance understanding of the functional role of AP-2ε in cartilage homeostasis in health and disease, as well as knowledge of OA progression in general.
Methods
Transgenic mice, tissue preparation and animal studies
Tfap2e−/− mice were generated and kindly provided by Markus Moser (Max Planck Institute for Biochemistry, Munich, Germany). In those mice, a neomycin DNA construct containing a stop codon was inserted into the second exon of the Tfap2e gene between the ApaII and NotI restriction sites (unpublished observations). This modification leads to a premature termination of the translation process of subsequent AP-2ε mRNA transcripts, resulting in a complete absence of functional AP-2ε protein in animals homozygous for this genetic mutation.
Tfap2e−/− and the corresponding wild-type (WT) mice were bred at 26 °C and 70 % relative humidity under a 12-hour light/12-hour dark cycle at the University Hospital of Regensburg, Germany. The mice were fed a breeding and maintenance diet (Altromin Spezialfutter GmbH, Lage, Germany) and given water ad libitum. The mice were randomly housed in polypropylene cages with sawdust bedding, and the cages were sanitized twice weekly. Animal care and all experimental procedures were carried out in accordance with guidelines under the German law governing animal care. The OA surgery was approved by the Ethics Committee for Animal Research of the Bavarian government. For all other experiments in which murine tissue was used, it was sufficient to obtain supervision from the local animal welfare officer (Dr Thilo Spruss, University Hospital Regensburg) according to the German Animal Welfare Act 2006 (article 4, using mice for scientific purposes (including tissue, embryo and cell extraction) if no additional experimental procedures are carried out with the animals). Therefore, except of the OA model, no further notification or approval by the Ethics Committee for Animal Research of the Bavarian government was necessary for the mouse studies.
All adult mice were killed after anesthetization by isoflurane (2-chloro-2-(difluoromethoxy)-1,1,1-trifluoroethane) inhalation via cervical dislocation. Embryos were killed by decapitation. To generate histological sections, tissue and embryos were fixed for at least 3 days in phosphate-buffered saline (PBS) containing 4 % paraformaldehyde, decalcified in 20 % ethylenediaminetetraacetic acid (Sigma-Aldrich Chemie GmbH, Munich, Germany) if necessary and embedded in paraffin. Subsequently, 5-μm serial sections were cut. For genotyping, tail biopsies were used.
To obtain mouse embryos of a certain age, adult pubescent mice were coupled overnight, and the weight of the females was documented. At the desired time point, the pregnant mice were killed after successful pregnancy was determined on the basis of a clear weight increase. Embryos were harvested by carefully opening the abdominal wall and uterus.
To obtain adult articular cartilage of murine knee joints, the joints were dissected and the cartilage layer of the femoral condyles and the tibial plateau were carefully separated from the underlying bone using sharp scissors and a scalpel. Total RNA was isolated after pulverization of the cartilage using liquid nitrogen and a mortar.
OA in 6- to 8-week-old WT and Tfap2e−/− mice was induced by detaching the medial meniscus (DMM) from the tibial plateau as described previously [27]. Briefly, with the mice under general anesthesia using a 0.9 % sodium chloride solution containing 0.75 % ketamine and 0.16 % xylazine (10-μl intraperitoneal injection per gram of body weight), the hind limbs were prepared for aseptic surgery. The right knee capsule was exposed following a medial incision without transection of the patellar ligament. Next, the medial meniscus was detached from the tibial plateau using a fine cannula and a surgical microscope. As a result, the meniscus translocated freely into the joint space. Afterward, the skin was closed with a metal clamp. During the procedure, close attention was paid not to injure the articular cartilage surface. In the left knee joint, the capsule was opened without further treatment and served as a control (sham). The animals were killed 10 or 17 days after surgery, and the knee joints were prepared for histological sectioning or RNA isolation from articular cartilage. The progress of OA was scored by stage and grade according to the scoring system proposed by Pritzker et al. [18] using sections stained with Safranin O/Fast Green and hematoxylin and eosin, respectively. In a preliminary test, no significant differences in OA development between male and female mice could be observed. Thus, both sexes were used in equal numbers.
Preparation of genomic DNA and genotyping
Whole genomic DNA extracts were isolated from tail biopsies as well as from hip cartilage explants using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) as described by the manufacturer. The DNA concentration and purity were measured using a NanoDrop device (Peqlab Biotechnologie GmbH, Erlangen, Germany). Tfap2e genotyping was performed using a PTC-200 thermocycler (MJ Research, Waltham, MA, USA). The following PCR program was used: 5 minutes at 95 °C (initial denaturation); 30 seconds at 95 °C (denaturation), 30 seconds at x °C (annealing) (Table 1) and 30 seconds at 72 °C (elongation), repeated 10 times, beginning with an annealing temperature of 63 °C and 1 °C reduction per each subsequent cycle in a total volume of 50 μl containing 2 μl of genomic DNA template, 1.5 μl of a mixture of three specific primers (20 μM; Sigma-Aldrich Chemie GmbH) (Table 1), which bound to the Tfap2e genomic locus and the neomycin insert, as well as 25 μl of FailSafe PCR 2× PreMix D (Epicentre, Madison, WI, USA) and 0.5 μl of Taq DNA polymerase (Roche Diagnostics GmbH, Mannheim, Germany). The initial PCR was followed by 30 additional cycles of 30 seconds at 95 °C, 30 seconds at 53 °C and 30 seconds at 72 °C. The PCR products were evaluated by performing gel electrophoresis in a 1.5 % agarose gel. Running the genomic DNA of homozygous Tfap2e−/− mice on the gel resulted in a 300-bp band; DNA from homozygous WT mice resulted in a 100-bp band; and DNA of heterozygous animals produced both bands.
Table 1.
Primers used for genotyping of wild-type and Tfap2e−/− micea
| Primer | Sequence (5′-3′) |
|---|---|
| mAP-2ε gen anti2 | CTACGTCGCCCTGGACTTCGAGC |
| mAP2ε gen sense2 | TGGAATCCTGTGGCATCCATGAAAC |
| pGK polyA down | GGCTCTCCAGAACATCATCCCTGC |
aSequences were obtained from M Moser
RNA isolation, reverse transcription and quantitative real-time PCR
Total RNA of murine tissues was isolated using a E.Z.N.A. MicroElute Total RNA Kit (Omega Bio-Tek, Norcross, GA, USA) as described by the manufacturer. The purity and concentration of the RNA were measured using a NanoDrop device (Peqlab Biotechnologie GmbH), and cDNA was generated by reverse transcription as described elsewhere [28]. Each reaction was performed in a total volume of 20 μl containing at least 150 ng of total RNA.
Quantitative RT-PCR (qRT-PCR) was carried out using the LightCycler 480 system from Roche Diagnostics GmbH. Volumes of 1 μl of cDNA template, 0.5 μl of forward and reverse primers (20 mM), 10 μl of SYBR Green Premix (Roche Diagnostics GmbH) and 8 μl of water were combined in a total volume of 20 μl. Primers were obtained from Sigma-Aldrich Chemie GmbH and are listed in Table 2. The following PCR program was used: 95 °C for 10 minutes (initial denaturation); 4.4 °C per second temperature transition rate up to 95 °C for 10 seconds, x °C for 10 seconds (annealing) and 72 °C for 20 seconds (elongation), and y °C acquisition mode single, repeated 45 times (amplification). The annealing (x) and acquisition (y) temperatures were optimized for each primer set. The PCR product was evaluated using melting curve analysis, and each sample was analyzed at least in duplicate. The expression level of the analyzed genes was normalized to the expression level of the housekeeping gene Actb (encoding β-actin).
Table 2.
Murine primer pairs used for quantitative real-time PCR
| Gene | Primer | Product | Sequence (5′-3′) |
|---|---|---|---|
| Actb | mβ-Act_885for | 348 bp | TGGAATCCTGTGGCATCCATGAAAC |
| mβ-Act_1233rev | TAAAACGCAGCTCAGTAACAGTCCG | ||
| Acan | mAggrecan_1922for | 206 bp | CAGTTCACCTTCCAGGAAG |
| mAggrecan_2128rev | GTAGAGGTAGACCGTTCTCACG | ||
| Col2a1 | mColl2_2657for | 261 bp | CTACTGGAGTGACTGGTCCTAAGG |
| mColl2_2918rev | GGACCATCATCTCCAGGTTCTCC | ||
| Col10a1 | mCol10_38for | 287 bp | CTGCCCCACGCATCTCCCAG |
| mCol10_325rev | GCTTGCCTGGCGGTCCTGAG | ||
| Mmp1a/Mmp1b | mMMP1_17for | 175 bp | CTGTTGCTTCTCTGGGCTGC |
| mMMP1_192rev | CTGCATTTGCCTCAGCTTTTC | ||
| Mmp3 | mMMP3_362for | 104 bp | GTTCCTGATGTTGGTGGCTTCAG |
| mMMP3_466rev | CTGTCTTGGCAAATCCGGTGTA | ||
| Mmp13 | mMMP13_899for | 80 bp | CCCAGCCCTATCCCTTGATGCCA |
| mMMP13_979rev | TGCAGGCGCCAGAAGAATCTGT | ||
| Tfap2a | mAP-2α_1170for | 69 bp | GCGGCCCAATCCTATCCT |
| mAP-2α_1238rev | CCATGGGAGATGAGGTTGAAG | ||
| Tfap2b | mAP-2β_1355for | 67 bp | AAAGCTGTCTCACGCACTTCAGT |
| mAP-2β_1421rev | AGCGCAGCGCAAATGG | ||
| Tfap2c | mAP-2γ_1115for | 537 bp | ACCTAGCACGGGACTTCGCCT |
| mAP-2γ_1651rev | GGGCGGGCGGGTTGTAACTG | ||
| Tfap2e | mAP-2ε_388for | 154 bp | GCCGACCCTGGGGAGCTACAC |
| mAP-2ε_542rev | CACCTCCGGCGCCGCTTAAA | ||
| Timp1 | mTIMP1_147for | 270 bp | AGACACACCAGAGCAGATACC |
| mTIMP1_417rev | CCGGATATCTGCGGCATTTC | ||
| Timp2 | mTIMP2_442for | 280 bp | GCAGACGTAGTGATCAGAGCC |
| mTIMP2_722rev | TCCCAGGGCACAATGAAGTC | ||
| Timp3 | mTIMP3_255for | 315 bp | GACCCTTGGCCACTTAGTCC |
| mTIMP3_570rev | CGGATCACGATGTCGGAGTTG |
In situ hybridization
For in situ hybridization of paraffin-embedded sections, digoxigenin-labeled antisense RNA probes that were specific for mouse Col2a1 (collagen, type II, alpha 1) and Col10a1 (collagen, type X, alpha 1) mRNA transcripts were generated using reverse transcription. The corresponding vectors, which were kindly provided by Klaus von der Mark (University of Erlangen-Nürnberg, Erlangen, Germany), were linearized using EcoRI (for Col2a1) and BamHI (for Col10a1) restriction enzymes, and the antisense RNA probes were subsequently reverse-transcribed using T3 RNA polymerase (for Col2a1) or T7 RNA polymerase (for Col10a1) with digoxigenin-labeled deoxynucleoside triphosphates (Roche Diagnostics GmbH). Serial sections of the hind limbs from WT and Tfap2e−/− embryos were analyzed using in situ hybridization as described elsewhere [29]. RNA probes bound to the tissue were detected using an anti-digoxigenin alkaline phosphatase antibody and BM purple (both purchased from Roche Diagnostics GmbH) according to the manufacturer’s instructions. The sections were mounted with coverslips in Kaiser’s glycerol gelatin (Merck, Darmstadt, Germany).
In vitro micromass cultivation of embryonic limb bud cells
Twelve to twenty-four limb buds that were derived from littermates of pregnant homozygous mice were pooled and dissolved in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 (1:1) (PAA, Pasching, Austria) containing dispase (1 U/ml) (Life Technologies, Carlsbad, CA, USA), 10 % fetal calf serum (FCS; PAN Biotech GmbH, Aidenbach, Germany), penicillin (100 U/ml; Sigma-Aldrich Chemie GmbH) and streptomycin (10 μg/ml; Sigma-Aldrich Chemie GmbH) at 37 °C for 30 minutes. Single cells were collected by passaging through a 40-μm filter and centrifugation at 280 × g for 4 minutes. Afterward, the cells were resuspended in DMEM/Ham’s F-12 (1:1) containing 10 % FCS and penicillin-streptomycin at a concentration of 20,000 cells/μl. Ten microliters of this suspension were applied to each well of a 24-well plate, which was then incubated for 3 hours at 37 °C and 5 % CO2 for cell attachment. Subsequently, each well was carefully flooded with 800 μl of DMEM/Ham’s F-12 (1:1) supplemented with 10 % FCS, penicillin-streptomycin, l-ascorbic acid 2-phosphate (100 mM; Sigma-Aldrich Chemie GmbH) and β-glycerophosphate (100 mM; Sigma-Aldrich Chemie GmbH). The cultures were incubated for up to 4 days, and the media were changed daily. Directly after establishment (d0) and on each following day, the cells of two wells were harvested for RNA isolation. Additionally, on each day, a culture was stained with 1 % Alcian Blue (Sigma-Aldrich Chemie GmbH) in 0.1 M HCl for 2 hours after fixation with a solution containing 30 % EtOH, 0.37 % formaldehyde and 3.9 % acetic acid. After removal of excessive Alcian Blue with PBS and 70 % EtOH, the characteristic nodules that resembled chondrocytic differentiation became visible.
In vitro cultivation of articular cartilage explants
To obtain cartilage explants from the hip joints, the pan-shaped articular cartilage that coated the femoral head was carefully pulled off the underlying bone using a fine forceps. From each mouse, two explants were isolated, and their weights were determined. The explants were cultured in 24-well plates at 37 °C and 5 % CO2 in 500 μl of DMEM/high glucose (PAA) containing 10 % FCS (PAN Biotech GmbH), penicillin (100 U/ml; Sigma-Aldrich Chemie GmbH), streptomycin (10 μg/ml; Sigma-Aldrich Chemie GmbH) and amphotericin B (0.5 μg/ml; Sigma-Aldrich Chemie GmbH). After 2 days, the medium was exchanged to serum-free medium (supplemented as before), and a specific Mmp13 inhibitor (pyrimidine-4,6-dicarboxylic acid, bis-(4-fluoro-3-methyl-benzylamide); Merck) was added to a number of the explants at a concentration of 100 nM (one explant with and one without inhibitor per mouse). After 4 days at 37 °C and 5 % CO2, the supernatants were collected, and Mmp13 activity and glycosaminoglycan (GAG) concentration were measured. Additionally, genomic DNA of the remaining explants was isolated, and their concentrations were measured to indicate the number of chondrocytes in the cartilage fragments. This value and the weight of the explants were used to normalize the absolute GAG concentration and Mmp13 activity in the supernatants.
Measurement of matrix metalloproteinase 13 activity and glycosaminoglycan concentration in media
Analysis of Mmp13 activity in media was performed using the SensoLyte 520 MMP-13 Assay Kit *Fluorimetric* (AnaSpec, Fremont, CA, USA) according to the manufacturer’s instructions. The fluorescence intensity, which directly correlated with Mmp13 activity, was determined using a FLUOstar Omega fluorescence reader (BMG LABTECH, Ortenberg, Germany) at excitation/emission wavelengths of 485/520 nm.
For the detection of GAG, the WIESLAB sGAG quantitative kit was used (Euro-Diagnostica, Malmö, Sweden) as suggested by the manufacturer. The color intensity, which represented the GAG concentration, was measured using a plate reader (MWG Biotech, Ebersberg, Germany) at 650 nm.
Immunohistochemical staining of collagen cleavage neoepitopes
Decalcified, paraffin-embedded sections of murine knee joints were deparaffinized, rehydrated and incubated with chondroitinase ABC (Sigma-Aldrich Chemie GmbH) at 0.025 U/100 μl in 0.1 M Tris-HCl (pH 8.0) containing 60 mM sodium acetate and 0.02 % bovine serum albumin for 90 minutes at 37 °C to remove GAG. Endogenous peroxidase activity was quenched by treating the sections with 1 % H2O2 for 3 minutes. For the detection of collagenase-induced generation of collagen type 2 neoepitopes in the cartilage matrix, a rabbit antiserum raised against the C1,2C (COL2 3/4Cshort) neoepitope was used (IBEX Technologies, Montreal, QC, Canada). Sections were incubated with the primary antibody (1:100) for 30 minutes at room temperature. After extensive rinsing, the secondary antibody Histofine Simple Stain MAX PO anti-rabbit (Nichirei Biosciences, Tokyo, Japan) was incubated for 30 minutes at room temperature and subsequently visualized using diaminobenzidine chromogen (Dako, Hamburg, Germany). Finally, the tissue was counterstained with hematoxylin. The specificity of the staining was controlled with sections of human OA cartilage as positive controls and sections without primary antibody as negative controls.
Light and transmission electron microscopy
Light microscopy was performed using a Zeiss Axiovert200 microscope, which was equipped with an AxioCam MRc camera (Carl Zeiss, Jena, Germany). AxioVs40 V 4.5.0.0 software (Carl Zeiss) was applied to quantify the zones of gene expression after in situ hybridization, as well as to measure skeletal parameters.
Transmission electron microscopy was conducted in collaboration with Josef Schroeder and Heiko Siegmund (University Hospital of Regensburg, Institute for Pathology, Regensburg, Germany). For this purpose, the femora and tibiae of 6- to 7-week-old WT and Tfap2e−/− mice were isolated and chemically fixed for 16 hours in Karnovsky’s reagent (2.5 % glutaraldehyde/1 % paraformaldehyde). Articular cartilage of the knee joint was processed as described earlier [30, 31]. The transmission electron micrographs were qualitatively evaluated by Ernst B Hunziker. For this purpose, pictures derived from equal articular cartilage zones were compared.
Statistical analysis
The results are expressed as the mean ± standard error of the mean or as box-and-whisker plot with minimum and maximum. Comparison between groups was made using Student’s paired or unpaired t-test, and a P-value <0.05 was considered statistically significant. All calculations were performed using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA).
Results
Tfap2e−/− mice exhibit only minor abnormalities in cartilage and skeletal development during embryogenesis
AP-2ε expression was recently detected in human articular cartilage and in hypertrophic chondrocytes of embryonic day 14.5 (E14.5) WT mouse embryos [20, 21]. However, adult Tfap2e−/− mice showed no obvious phenotype, and the exact role of the transcription factor in cartilage development and homeostasis was still unknown. Therefore, in the first part of this study, we examined the embryonic development of the Tfap2e−/− mouse for potential transient abnormalities that might be compensated later and could provide information about the influence of the transcription factor on chondrogenesis.
We initially concentrated on E15.5 and E16.5, as these stages are particularly suited to assessment of chondrocyte differentiation and endochondral ossification in the murine limbs. For this purpose, heterozygous adult mice were coupled overnight, and homozygous littermates derived from the same female mouse were compared. This ensured that all Tfap2e−/− and WT embryos were of identical age and avoided unequal development due to divergent mother animals. No significant differences in total weight or size between the two genotypes could be observed (Fig. 1a). Additionally, the hind limbs of the embryos were embedded in paraffin, and sections were stained for Col2a1 and Col10a1 mRNA expression via in situ hybridization to more closely analyze limb development (Fig. 1b). The expression zones of the two genes were measured and normalized to the femur length. As depicted in Fig. 1b, no significant differences between the femora of WT and Tfap2e−/− specimens in total length, Col2a1 expression zone or calcified region (bone) could be observed. However, the Col10a1 expression zone at E16.5 in Tfap2e−/− embryos was slightly but significantly enhanced.
Fig. 1.
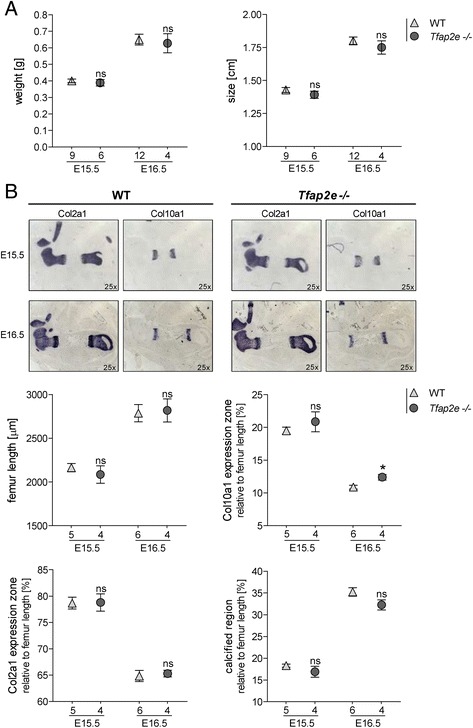
Comparison of the embryonic skeletal development in embryonic days 15.5 and 16.5 wild-type and Tfap2e −/− mice. a The total weight and size of 15.5- and 16.5-day-old (E15.5 and E16.5, respectively) wild-type (WT) and Tfap2e −/− embryos derived from three independent heterozygote litters per time point were approximately identical. b In situ hybridization against collagen, type II, alpha 1 (Col10a1) and collagen, type X, alpha 1 (Col10a1) mRNA on the hind limbs of the embryos and subsequent measurement of the total femur length, the Col2a1 and Col10a1 expression zones and the bone region was performed. The latter three were normalized to total femur length; the distal and proximal expression zones of Col2a1 and Col10a1 were combined before normalization. A slight but significant increase in the Col10a1 expression zone could be observed in the Tfap2e −/− fetuses at E16.5. Otherwise, no evidence for abnormal development in the Tfap2e −/− mice could be determined. The data are given as the means ± standard error of the mean. ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. *P < 0.05. Numbers indicate individual embryos used for each group
To define the impact of AP-2ε on earlier steps of embryonic cartilage development, we used mesenchymal cells isolated from limb buds of E11.5 WT and Tfap2e−/− mouse embryos in high-density micromass cultures. The production and accumulation of sulfated GAG was determined by staining the cultures with Alcian Blue solution. Formation of the typical nodules could be observed 2 days after initiation of the cultures (Fig. 2a). Subsequently, the number and color intensity of the nodules increased, but no definite discrepancy between the two genotypes was observed. Additionally, the expression patterns of prominent differentiation marker genes in cartilage (aggrecan, Col2a1, Col10a1 and Mmp13) were compared by qRT-PCR (Fig. 2b). No differences between Tfap2e−/− and WT cells could be observed for aggrecan or Col2a1; however, expression of Mmp13 and Col10a1 was increased in Tfap2e−/− cultures in late stages of differentiation.
Fig. 2.

Micromass in vitro differentiation of mesenchymal limb bud cells. Micromass cultures of mesenchymal cells isolated from the limb buds of embryonic day 11.5 (E11.5) wild-type (WT) and Tfap2e −/− embryos were maintained for 4 days in vitro. a To compare the differentiation behavior of WT and Tfap2e −/− cells, each day, a culture was treated with Alcian Blue solution to stain for secreted glycosaminoglycans. The nodules that are characteristic for this assay became visible at the second day of the culture period. Their number, size and color intensity increased in both genotypes by approximately the same amount. b Directly after initiation of the cultures (d0) and on each following day, mRNA expression of the cartilage differentiation markers aggrecan, collagen, type II, alpha 1 (Col10a1) and collagen, type X, alpha 1 (Col10a1) and matrix metalloproteinase (Mmp13) was analyzed by quantitative RT-PCR. A definite increase in the expression level of all four genes during the differentiation process was detectable in cells of both genotypes. The expression of Col10a1 was enhanced, and expression of Mmp13 tended to be enhanced, at day 4 in Tfap2e −/− cells compared with WT cells. The data are given as the means ± standard error of the mean. ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. *P < 0.05. The assay was carried out four times with cells of four individual WT litters and three times with cells of three individual Tfap2e −/− litters
In sum, these data imply that AP-2ε most likely does not play a major role during embryonic cartilage development in mice. Alternatively, other factors may effectively compensate for the loss of AP-2ε in the knockout animals during embryogenesis. Nevertheless, the results suggest that expression of the genes for Mmp13 and Col10a1 could be affected by the loss of AP-2ε in differentiated chondrocytes. To further support this hypothesis, we expanded our investigation of the Tfap2e−/− mouse to postnatal articular cartilage tissue in the second part of this study.
Expression and activity of the proteinase matrix metalloproteinase 13 is upregulated in articular cartilage of Tfap2e−/− mice
Measuring the AP-2ε mRNA levels in articular chondrocytes of the knee joints of adult WT mice revealed a strong induction in expression compared with the mesenchymal limb bud cells used for micromass differentiation (Fig. 3a). This strongly hinted at a role of AP-2ε in the homeostasis of articular cartilage and/or the regulation of gene expression in this tissue type. However, initially, no evident abnormalities in the articular cartilage layer of 10.5-week-old Tfap2e−/− mice could be determined at the histological or ultrastructural level based on a qualitative analysis (Fig. 3b and c). To analyze whether abnormal expression of another AP-2 isoform might effectively compensate for the loss of AP-2ε in articular cartilage of the knockout mice, expression of AP-2α, AP-2β and AP-2γ was determined, but no differences from WT animals could be measured (data not shown). Next, we analyzed Mmp13 and Col10a1 expression, as these two genes showed differential expression when we compared highly differentiated mesenchymal cells of WT and Tfap2e−/− mice (see Fig. 2b). Interestingly, in articular chondrocytes deficient for AP-2ε, expression of Mmp13 was significantly enhanced, whereas this was not the case for Col10a1 (Fig. 4a).
Fig. 3.
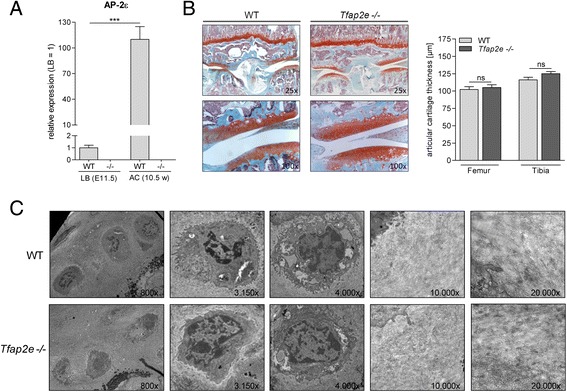
Activating enhancer binding protein 2, epsilon, expression in articular cartilage and analysis of the cartilage layer in wild-type and Tfap2e −/− mice. a Activating enhancer binding protein 2, epsilon (AP-2ε) mRNA expression was measured in mesenchymal limb bud cells (LB) of embryonic day 11.5 (E11.5) embryos and in articular chondrocytes (AC) from the knee joints of 10.5-week-old (10.5 w) mice via quantitative RT-PCR. In the wild-type (WT) samples, a strong induction in the expression rate of the transcription factor was observed. As expected, when we used genotype-specific primers, normal AP-2ε mRNA without the neomycin insert could not be detected in corresponding Tfap2e −/− samples, serving as a negative control. b Histological sections through the knee joints of 10.5-week-old WT and Tfap2e −/− mice after staining with Safranin O/Fast Green (left). Morphologically, no obvious abnormalities between the two genotypes could be detected at this age. Furthermore, the thickness of the articular cartilage layer in the tibia and femur was similar in each genotype (right). c Exemplary pictures from the ultrastructural analysis of the articular cartilage layer in the knee joints of adult WT and Tfap2e −/− mice depicting chondrocytes (left) and the extracellular matrix (right). No obvious differences could be determined in a qualitative evaluation. The data are given as the means ± standard error of the mean. ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. ***P < 0.001. In (A), cells of four individual litters were used for LB and nine individual animals for AC. In (B), 14 WT and 15 Tfap2e −/− mice were compared. In (c), two mice were used for each genotype. Micrographs shown are derived from the middle and deep zones of the cartilage layer and were adjusted to equal magnification
Fig. 4.
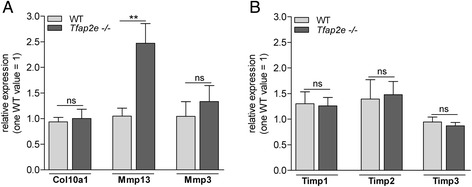
Gene expression analysis in articular chondrocytes of wild-type and Tfap2e −/− mice. a The expression of collagen, type X, alpha 1 (Col10a1), matrix metalloproteinase 13 (Mmp13) and Mmp3 was analyzed in the articular cartilage layer of knee joints of wild-type (WT) and Tfap2e −/− mice (10.5 w) by quantitative RT-PCR. Mmp13, but not Col10a1 or Mmp3, was significantly overexpressed in Tfap2e −/− mice. b mRNA expression of tissue inhibitor of metalloproteinase 1 (Timp1), Timp2 and Timp3 was analyzed in the articular cartilage samples, but no differences in the expression rates of the three molecules could be detected when we compared WT and Tfap2e −/− animals. The data are given as the means ± standard error of the mean. ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. **P < 0.01. Nine WT mice and eight Tfap2e −/− mice were used for this experiment
To evaluate the specificity of the detected Mmp13 dysregulation, expression of Mmp1 and Mmp3, two other metalloproteinases that are known to play critical roles in cartilage development and in human OA, was also examined [7, 15]. Two highly conserved genes for the interstitial collagenase Mmp1, Mmp1a and Mmp1b, exist in mice, but the mRNA of neither was detectable in articular chondrocytes of WT and Tfap2e−/− mice (data not shown). Mmp3 was expressed, but no differences between the two genotypes could be determined (Fig. 4a). In addition, no differential expression of the genes for tissue inhibitors of metalloproteinase 1, 2 and 3 (Timp1, Timp2 and Timp3, respectively) was determined (Fig. 4b). Taken together, this suggested that primarily Mmp13 is dysregulated in articular chondrocytes of Tfap2e−/− mice.
To provide evidence for enhanced catabolic activity in articular cartilage tissue of Tfap2e−/− mice due to the observed overexpression of Mmp13, we performed in vitro culture of femoral head cartilage explants. After cultivation, Mmp13 activity in the supernatants was measured using a fluorescence-based assay. We detected significantly higher activity of the proteinase in medium derived from explants deficient for AP-2ε relative to WT explants (Fig. 5a, left). Furthermore, addition of a specific Mmp13 inhibitor resulted in a slight reduction of Mmp13 activity in the WT and a significant reduction nearly to the WT level in the Tfap2e−/− explants.
Fig. 5.
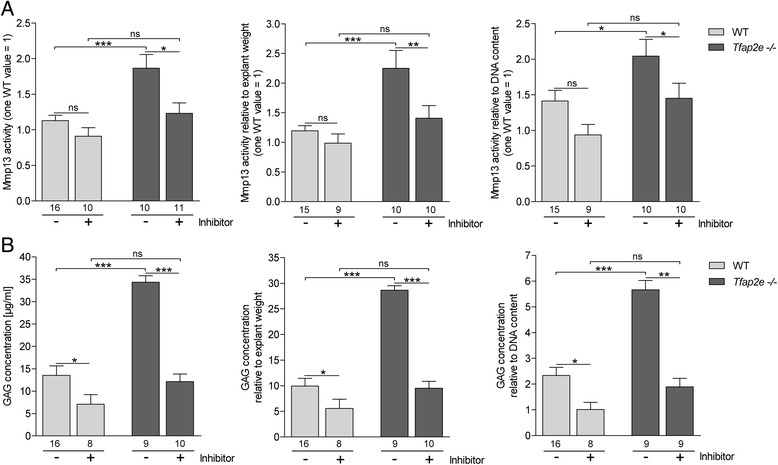
Enhanced matrix metalloproteinase 13 activity and matrix degradation in articular cartilage explants deficient for activating enhancer binding protein 2, epsilon. Articular cartilage of the femoral head of adult wild-type (WT) and Tfap2e −/− mice (two per animal) was isolated and subjected to in vitro cultivation. Two days after establishment, a specific matrix metalloproteinase (Mmp13) inhibitor was added to a subset of the explants. Another 3 days later, the supernatants were collected, and Mmp13 activity (a) and glycosaminoglycan (GAG) concentrations (b) were determined. Both were significantly enhanced in media from the Tfap2e −/− explants compared with the WT explants (left). Treatment with the Mmp13 inhibitor resulted in a significant reduction in Mmp13 activity and GAG release in the Tfap2e −/− explants. This was also detectable in the supernatants of WT explants, albeit at a lower level. In addition, the absolute values for both parameters were normalized to the total wet weight (middle), as well as to the DNA content (right), of the corresponding explants. Here, similar results were discovered. The data are given as the means ± standard error of the mean. ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. *P < 0.05; **P < 0.01; ***P < 0.001. Numbers indicate individual explants used for each group
Additionally, the GAG content in the supernatants, which is an indicator of proteoglycan and ECM degradation, was determined. Interestingly, the absolute GAG concentration was also significantly higher in the supernatants of the Tfap2e−/− cartilage explants compared with WT cartilage explants (Fig. 5b, left). Furthermore, the Mmp13 inhibitor significantly reduced GAG release compared with the untreated explants. Similar results were obtained after normalization of the Mmp13 activity and the GAG concentration to the weight of the explants before cultivation, as well as to their DNA content, representing the number of cells in the cartilage fragments (Fig. 5a and b, middle and right). In sum, these in vitro data strongly support that expression and activity of Mmp13 is upregulated in articular chondrocytes of Tfap2e−/− mice, resulting in enhanced matrix degradation.
Enhanced osteoarthritis development in Tfap2e−/− mice
To confirm this hypothesis in vivo, WT and Tfap2e−/− mice were subjected to a model of OA. As we were unable to observe any morphological or ultrastructural abnormalities in the articular cartilage layer of 10.5-week-old Tfap2e−/− mice under physiological conditions (see Fig. 3), we speculated that potential abnormalities in the AP-2ε-knockout mice become apparent under these conditions of stress. It is also known, on the basis of studying other mouse models deficient for cartilage-specific genes, that phenotypic variations sometimes become obvious only in pathological situations [27, 32–34].
OA was induced in the right hind limbs of 6- to 8-week-old animals by DMM from the tibial plateau; the left hind limbs served as controls (sham). The progression of OA was analyzed 10 and 17 days after surgery. As depicted in Fig. 6a, animals of both genotypes exhibited only minor signs of OA 10 days after disease onset. Seventeen days after surgery, the WT mice showed mild to moderate signs of OA, as expected on the basis of previous studies [27] (Fig. 6a). These included wear, lesions and inhomogeneous staining near the cartilage surface (Fig. 6b). In striking contrast, both the severity and the extent of the cartilaginous destruction were significantly enhanced in Tfap2e−/− mice at the corresponding juncture (Fig. 6a and b). In all of the sham joints, the articular cartilage layer had a smooth surface contour and manifested no signs of OA (Fig. 6b). To substantiate that the DMM-treated joints exhibit an OA phenotype and to confirm involvement of MMPs in this process, immunostaining for collagenase-induced collagen breakdown products was performed using an antiserum against the C1,2C cleavage neoepitope. A clear positive staining signal was detectable at the surface of the articular cartilage layer in the DMM joints of both WT and Tfap2e−/− animals (Fig. 6c). In contrast, no staining could be determined in the cartilage surface of the sham joints. The specificity of the staining was controlled with murine sections without primary antibody treatment used as negative controls and sections of human OA cartilage used as positive controls.
Fig. 6.
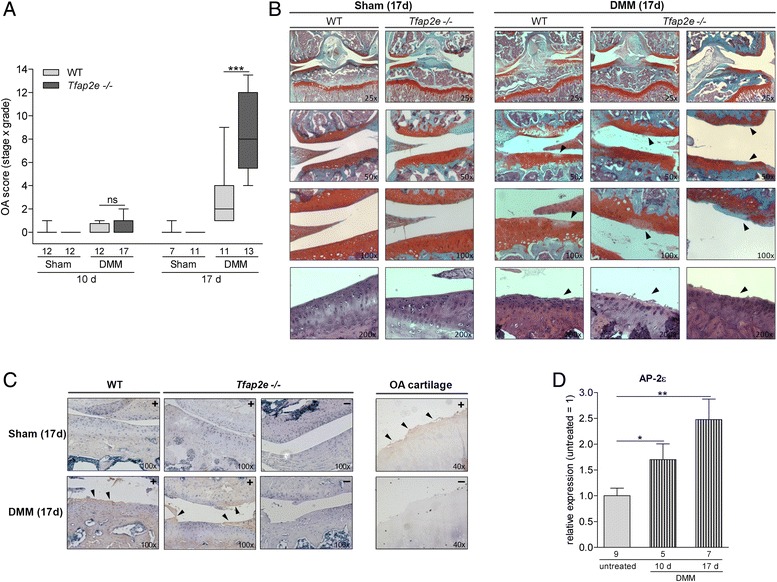
Enhanced osteoarthritis progression in Tfap2e −/− mice. a In 6- to 8-week-old wild-type (WT) and Tfap2e −/− mice, osteoarthritis (OA) was induced in the right knee joint by detaching the medial meniscus (DMM). In the corresponding left knee joint, a control surgery was performed (Sham). Ten days after surgery, all mice showed only minimal signs of OA. Seventeen days after disease onset, WT mice exhibited low to moderate OA scores, whereas OA severity in Tfap2e −/− mice was significantly enhanced at this time point. b Visible damage to the articular cartilage tissue at day 17 after DMM surgery included wear, lesions and inhomogeneous staining (right, arrowheads). Cartilage of the sham joints had a smooth surface and featured no signs of OA (left). Slides of the top three rows were stained with Safranin O/Fast Green. In the bottom row, further examples of the cartilage surface after staining with hematoxylin and eosin are shown. c Immunohistochemistry with an antiserum raised against the C1,2C collagen cleavage neoepitope was performed to confirm matrix metalloproteinase (MMP)-mediated extracellular matrix degradation and thus development of an OA-like phenotype in response to the DMM treatment. Here, positive staining at the articular cartilage surface could be detected in the DMM joints (arrowheads), whereas this was not the case in the sham joints. The specificity of the staining was controlled with Tfap2e −/− sections without primary antibody treatment (indicated by a “-”), and sections of human OA cartilage were used as positive controls. d Total RNA from the murine articular cartilage layer was isolated 10 and 17 days after OA induction, and expression of activating enhancer binding protein 2, epsilon (AP-2ε) was analyzed in knee joints of WT mice 10 and 17 days after OA onset relative to untreated WT knee joints. A significant induction in the expression rate of the transcription factor could be measured in the articular cartilage of the DMM joints. The data are given as box-and-whisker plots with minimum and maximum values in (a) and as the means ± standard errors of the mean in (d). ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. *P < 0.05; **P < 0.01; ***P < 0.001. Numbers indicate individual animals per group
In addition to histology, we isolated RNA from the articular cartilage of the knee joints of the mice in the OA model. As detected in patients with OA compared with healthy donors [21], a significant upregulation of AP-2ε expression was measured in the joints subjected to OA surgery compared with the untreated joints in WT mice (Fig. 6d). Mmp1 was not detectable in murine articular chondrocytes even after OA onset (data not shown). For Mmp3, we also did not observe differential expression in Tfap2e−/− mice compared with WT mice. Furthermore, no significant changes in Mmp3 expression in the DMM joints compared with the controls could be determined at either time point (Fig. 7a). For Mmp13, again, a significantly higher basal expression level in AP-2ε-deficient articular chondrocytes could be detected when we compared the sham joints of Tfap2e−/− mice with the sham joints of WT mice at both time points (Fig. 7b). In addition, in both genotypes, the expression of Mmp13 was significantly upregulated in articular cartilage derived from the DMM joints compared with the sham controls 17 days after disease onset. This was expected owing to the pathological conditions [13–15]. However, in combination with the higher basal Mmp13 expression rate, this resulted in a significantly increased total Mmp13 expression level at day 17 after OA onset in the Tfap2e−/− mice. This finding most likely explains the enhanced OA manifestation observed in these animals and shows that AP-2ε is involved in progression of the disease.
Fig. 7.
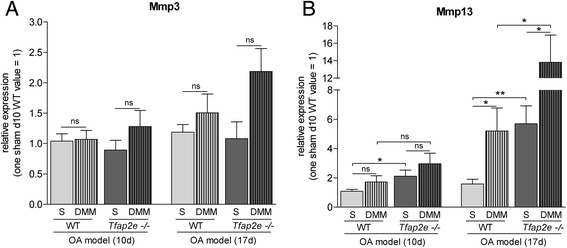
Analysis of matrix metalloproteinase expression during osteoarthritis progression. Total RNA from the articular cartilage layer was isolated 10 and 17 days after osteoarthritis (OA) induction by detaching the medial meniscus (DMM), and expression of the matrix metalloproteinase 3 (Mmp3) (a) and Mmp13 (b) was analyzed by quantitative RT-PCR. (A) Mmp3 mRNA expression was approximately identical in all sham joints (S) at both time points, and only a minor induction could be detected in the DMM joints compared with the sham joints. (b) In contrast, a significant higher basal level of Mmp13 expression could be detected in the sham joints of the Tfap2e −/− mice compared with the sham joints of the wild-type (WT) mice at both time points. In addition, the expression of Mmp13 was significantly upregulated in the DMM joints compared with the sham joints 17 days after OA induction in both genotypes. In combination with the higher basal expression rate, this resulted in a significantly increased total Mmp13 expression level in articular chondrocytes of the Tfap2e −/− mice at day 17 after OA onset. The data are given as the means ± standard error of the mean. ns, Not significant; Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon. *P < 0.05; **P < 0.01. Five to eight animals were used for each group
Discussion
Recently, we discovered physiological expression of the transcription factor AP-2ε in hypertrophic chondrocytes as well as in adult articular cartilage [20, 21]. Adult mice deficient for AP-2ε, however, do not exhibit an apparent cartilaginous or skeletal phenotype compared with WT mice. Nevertheless, as observed in other murine knockout models of molecules important for cartilage development, it could be possible that abnormalities arise only in the embryo during chondrogenesis and are compensated later in development via redundancy or irregular cellular differentiation and proliferation. For instance, mice with a cartilage-specific knockout of Mmp13 show a significantly increased width of the hypertrophic zone in the growth plate during embryonic and early postnatal development [35]. This phenotype completely resolves at approximately 12 weeks of age, and adult Mmp13−/− mice display no phenotypic abnormalities [35]. Another example for redundancy during chondrogenesis is the mouse deficient for melanoma inhibitory activity/cartilage-derived, retinoic acid–sensitive protein (MIA/CD-RAP), which also does not manifest strong abnormalities [31]. Nevertheless, recent studies by our group revealed enhanced proliferation and delayed differentiation in MIA−/− mouse embryos at E15.5/E16.5, resulting in abnormal growth plate architecture. This is compensated shortly afterward, however, possibly via reduced cAMP response element-binding protein and activator protein 1 (AP-1) activity [36].
Thus, in the first part of this study, we compared the embryonic skeletal development of Tfap2e−/− and WT mice to find transient differences in chondrogenesis and endochondral ossification, respectively. Those differences, in turn, could provide valuable information about the influence of the transcription factor in these processes. Advanced stages of skeletal development (E15.5/E16.5) were assessed in vivo via measurement of the embryos’ weight, size and femoral lengths, as well as by determination of the femoral expression zones of Col2a1 and Col10a1. In addition, early chondrogenesis was monitored in vitro via micromass cultivation of mesenchymal cells that were isolated from limb buds of E11.5 WT and Tfap2e−/− mouse embryos. Taken together, no fundamental abnormalities in the embryonic development of Tfap2e−/− mice became apparent in these experiments. However, at E16.5, a small, albeit significant, increase in the Col10a1 expression zone, which is specific for highly differentiated hypertrophic chondrocytes [37–39], was observed. Similarly, the Col10a1 transcription rate was upregulated in AP-2ε-deficient limb bud cells at very late stages of micromass differentiation. Likewise, the expression of Mmp13 tended to be enhanced in those cells, which suggests that loss of AP-2ε results in enhanced expression of these hypertrophic markers [37, 39, 40]. Again, the observed discrepancies were of rather low magnitude, and no differences could be observed earlier during chondrogenesis, although the mRNA expression of AP-2ε was clearly detectable in the mesenchymal cells. One possible explanation for these findings is that the loss of AP-2ε is compensated by an enhanced activity of redundant factors. Here, other AP-2 isoforms expressed during chondrogenesis come into consideration. For instance, Huang et al. provided evidence that AP-2α is a negative regulator of chondrocyte differentiation [41]. Likewise, AP-2β expression was detected in the developing mouse limb [42]. However, when AP-2ε is highly expressed in differentiated chondrocytes, the compensatory mechanisms may not be sufficient to fully negate the loss of the transcription factor. Alternatively, it could also be possible that AP-2ε does not impact gene expression in mesenchymal cells and early chondroblasts, because of either a lack of additional factors that are essential for AP-2ε function or the general low level of expression of AP-2ε in these cells. Regarding the latter, we determined a strong induction of AP-2ε expression in articular cartilage tissue isolated from adult mice compared with mesenchymal limb buds in WT animals.
We therefore concentrated on AP-2ε in adult joint cartilage. The focus was on Col10a1 and Mmp13, as these genes already showed differential expression during late stages of in vitro differentiation of mesenchymal cells of WT and Tfap2e−/− mice. Indeed, we found Mmp13 expression to be significantly upregulated in articular cartilage isolated form Tfap2e−/− mice compared with WT animals, whereas Col10a1 was not deregulated. Although we detected Col10a1 mRNA, the latter is most likely attributable to the fact that expression of Col10a1 in articular cartilage is restricted to single cells in the calcified deep zone and thus is generally very low in this tissue type [38, 43].
Mmp13 is known to be constitutively expressed in articular cartilage at low levels during physiologic ECM turnover [8, 14, 16, 17] and is capable of degrading native collagen type II [14, 44, 45] and aggrecan [46–48]. In addition, the family of matrix metalloproteinases comprises crucial mediators of cartilage destruction in OA. Of those, MMP13 is the most important, being overexpressed in the affected tissue of most patients with OA. Other variants, such as MMP1 and MMP3, also were shown to play a role in some cases of human OA [7, 13, 14, 19]. Furthermore, Mmp activity is strongly modulated at the posttranscriptional level via catalytic processing and Timp1, Timp 2 and Timp 3 [45]. Expression of the latter was unchanged in Tfap2e−/− mice, revealing that Timps do not counterbalance the enhanced Mmp13 mRNA expression [49, 50].
Furthermore, Mmp13 activity was directly measured in supernatants of articular cartilage explants and was indeed significantly enhanced in Tfap2e−/− mice. To determine the functional correlation, GAG release was assessed, which we found to be significantly higher in media from AP-2ε-deficient explants. A specific Mmp13 inhibitor confirmed that Mmp13 was mainly responsible for these observations because, after addition of the compound, the prior significant difference between the two genotypes was abolished.
Unexpectedly, in vivo the articular cartilage tissue of untreated 10.5-week-old Tfap2e−/− mice exhibited no evident abnormalities. In striking contrast, profound differences compared with WT mice became obvious after OA induction via DMM surgery. In one of our previous studies, the same OA model was carried out with MIA/CD-RAP deficient mice of a similar age. Here, slight signs of OA were detectable 10 days after surgery, and moderate signs were detectable 21 days after surgery [27]. Accordingly, comparable OA development could be observed in this study after 10 and 17 days. Again, in this model, a significantly higher basal level of Mmp13 expression in AP-2ε-deficient articular chondrocytes could be confirmed when we compared the sham joints of both genotypes. This, in combination with the characteristic OA-dependent induction of Mmp13 expression, resulted in a significantly increased total Mmp13 expression level in the Tfap2e−/− mice at day 17 after disease onset, which most likely is responsible for enhanced OA severity in these animals. Other examples of genetically manipulated mice that were deficient for cartilage-associated molecules and did not exhibit phenotypic alterations in the adult stage, but nevertheless responded to pathologic conditions or tissue stress differently from the WT mice, including mice deficient for Adamts5 (a disintegrin and metalloproteinase with thrombospondin motifs 5), MIA/CD-RAP and Mmp13 [27, 32–34]. In indirect correlation with our results, OA severity was significantly reduced in the latter mice. Further analyses in Mmp13 transgenic mice revealed that a constant overexpression of Mmp13 in articular chondrocytes could indeed promote disease progression [51], thus confirming that a dysregulation of Mmp13, as observed in the Tfap2e−/− mouse, strongly influences OA development.
In summary, the obtained data suggest that AP-2ε effectively represses basal Mmp13 expression under normal, non-OA conditions in articular chondrocytes of WT mice and that this control mechanism is lost in Tfap2e−/− animals. However, despite this inhibitory effect, an induction of Mmp13 expression after OA onset could be measured not only in Tfap2e−/− mice but also in WT animals. It is known that changes in the gene expression profile of the affected chondrocytes take place during OA, including upregulation of factors that drive expression of various effector molecules such as Mmps. For example, Mmp13 expression was shown to be upregulated by Runx2 (runt-related transcription factor 2), AP-1, HIF-2α (hypoxia-inducible factor 2, alpha) and Ets (E26 transformation-specific) family members [52–56]. As Mmp13 expression is similarly induced in both genotypes after OA onset (about 3.2-fold in WT mice and 2.4-fold in Tfap2e−/− mice), the inhibitory effect of AP-2ε on Mmp13 in the WT animals must be overbalanced by one or more of these activating factors. However, owing to the significantly higher basal expression, the induction results in a significantly higher Mmp13 expression level in articular chondrocytes of Tfap2e−/− mice compared with WT mice also at day 17 after OA surgery (Fig. 8). As demonstrated in the OA model, this increases the susceptibility of the cartilage tissue to destruction after joint overload, resulting in enhanced OA severity in the AP-2ε-deficient animals.
Fig. 8.
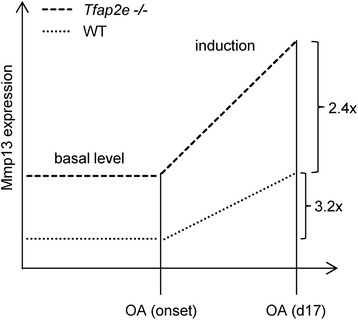
Graphical illustration of the results. Our data suggest that the loss of activating enhancer binding protein 2 epsilon (AP-2ε) results in an enhanced basal matrix metalloproteinase 13 (Mmp13) expression level in articular chondrocytes of Tfap2e −/− mice as compared with wild-type (WT) mice. After osteoarthritis (OA) onset, expression of Mmp13 is induced in both genotypes by a comparable amount (3.2-fold in WT mice and 2.4-fold in Tfap2e −/− mice), which is most likely due to the effect of other OA-associated transcription factors. However, because of the higher basal expression level, a similar induction rate leads to a significantly higher Mmp13 expression level in chondrocytes of Tfap2e −/− mice compared with WT mice 17 days after OA surgery. Tfap2e −/−, Deficient for activating enhancer binding protein 2, epsilon
As determined in vitro, the higher basal expression and activity of Mmp13 in articular chondrocytes of Tfap2e−/− mice shifts physiologic matrix turnover to the catabolic side also under non-OA conditions. Hence, it is possible that Tfap2e−/− mice develop OA-like symptoms with advanced age. In Mmp13 transgenic mice that strongly overexpress Mmp13 in articular chondrocytes, cartilage destruction was apparent at 5 months of age. However, it could also be possible that the loss of AP-2ε is compensated in the Tfap2e−/− animals via unknown mechanisms later during development. For this study, all experiments were carried out with mice up to 12 weeks of age, and further investigations with older mice of 6 to 12 months of age will be very interesting.
The expression of AP-2ε was induced in joints subjected to OA surgery compared with untreated joints in WT mice, which was similarly observed in humans [21]. This induction may constitute an attempt of chondrocytes to prevent the activation of Mmp13 after initial OA onset. However, at the same time, other regulatory factors drive the expression of Mmp13 and eventually predominate over AP-2ε. Thus, the increase of AP-2ε expression might only be sufficient to delay OA progression, but not to fully prevent cartilage destruction, which is in line with our observations in the OA model.
Conclusions
Despite the publication of large numbers of scientific reports, numerous aspects of cartilage development, destruction and regeneration are still not fully understood. Hence, studies introducing new players or interactions influencing these complex processes are of great importance. In this work, we reveal a novel role of the transcription factor AP-2ε in the control of articular chondrocyte gene expression and OA development by influencing Mmp13 expression and activity in vivo. Thus, this finding adds to the understanding of regulatory mechanisms underlying OA and might influence the development of future treatment options.
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (to AKB). Special thanks go to M Moser (Max Planck Institute for Biochemistry, Munich, Germany) for the Tfap2e −/− mouse, K von der Mark (University of Erlangen-Nürnberg, Erlangen, Germany) for vectors containing sequences for Col2a1- and Col10a1-specific RNA probes, as well as J Schroeder and H Siegmund (both from the University Hospital of Regensburg, Institute for Pathology, Regensburg, Germany). Additionally, we thank S Hofmeister and M Waeber for histological sections (both from the University Hospital of Regensburg, Institute for Pathology, Regensburg, Germany).
Abbreviations
- AC
Articular chondrocytes
- AP-1
Activator protein 1
- AP-2
Activating enhancer binding protein 2
- Col2a1
Collagen, type II, alpha 1
- Col10a1
Collagen, type X, alpha 1
- CXCL1
Chemokine (C-X-C) motif ligand 1
- DMEM
Dulbecco’s modified Eagle’s medium
- DMM
Detaching the medial meniscus
- E
Embryonic day
- ECM
Extracellular matrix
- FCS
Fetal calf serum
- GAG
Glycosaminoglycan
- LB
Limb bud cells
- MIA/CD-RAP
Melanoma inhibitory activity/cartilage-derived, retinoic acid–sensitive protein
- Mmp
Matrix metalloproteinase
- OA
Osteoarthritis
- PBS
Phosphate-buffered saline
- qRT-PCR
Quantitative real-time polymerase chain reaction
- Tfap2e
Transcription factor activating enhancer binding protein 2 epsilon gene
- Timp
Tissue inhibitor of metalloproteinase
- WT
Wild type
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SN carried out the molecular experiments, as well as most of the animal studies, and drafted the manuscript. TS carried out the OA model with the assistance of SN. TS evaluated OA development, interpreted and discussed the data and helped to revise the manuscript. EBH surveyed photomicrographs, interpreted, discussed the data and revised the manuscript. AKB designed the study, interpreted and discussed the data and participated in writing and revising the manuscript. All authors made substantive intellectual contributions to the study and read and approved the final manuscript.
Contributor Information
Stephan Niebler, Email: stephan.niebler@ukr.de.
Thomas Schubert, dr.t.schubert@googlemail.com.
Ernst B Hunziker, Email: ernst.hunziker@dkf.unibe.ch.
Anja K Bosserhoff, Phone: +49-(0)9131/85-24910, Email: anja.bosserhoff@fau.de.
References
- 1.Michigami T. Regulatory mechanisms for the development of growth plate cartilage. Cell Mol Life Sci. 2013;70:4213–4221. doi: 10.1007/s00018-013-1346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartmann C. Transcriptional networks controlling skeletal development. Curr Opin Genet Dev. 2009;19:437–443. doi: 10.1016/j.gde.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem. 2006;97:33–44. doi: 10.1002/jcb.20652. [DOI] [PubMed] [Google Scholar]
- 4.Goldring MB. Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis. 2012;4:269–285. doi: 10.1177/1759720X12448454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/S1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 6.Bhosale AM, Richardson JB. Articular cartilage: structure, injuries and review of management. Br Med Bull. 2008;87:77–95. doi: 10.1093/bmb/ldn025. [DOI] [PubMed] [Google Scholar]
- 7.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 8.Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 2007;56:1854–1868. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- 9.Pulsatelli L, Addimanda O, Brusi V, Pavloska B, Meliconi R. New findings in osteoarthritis pathogenesis: therapeutic implications. Ther Adv Chronic Dis. 2013;4:23–43. doi: 10.1177/2040622312462734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy L, Schwartz TA, Helmick CG, Renner JB, Tudor G, Koch G, et al. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Rheum. 2008;59:1207–1213. doi: 10.1002/art.24021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felson DT. Osteoarthritis of the knee. N Engl J Med. 2006;354:841–8. A published erratum appears in. N Engl J Med. 2006;354:2520. doi: 10.1056/NEJMcp051726. [DOI] [PubMed] [Google Scholar]
- 13.Billinghurst RC, Dahlberg L, Ionescu M, Reiner A, Bourne R, Rorabeck C, et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest. 1997;99:1534–1545. doi: 10.1172/JCI119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shlopov BV, Lie WR, Mainardi CL, Cole AA, Chubinskaya S, Hasty KA. Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum. 1997;40:2065–2074. doi: 10.1002/art.1780401120. [DOI] [PubMed] [Google Scholar]
- 15.Cawston TE, Wilson AJ. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006;20:983–1002. doi: 10.1016/j.berh.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Forsyth CB, Cole A, Murphy G, Bienias JL, Im HJ, Loeser RF., Jr Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J Gerontol A Biol Sci Med Sci. 2005;60:1118–1124. doi: 10.1093/gerona/60.9.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes: a role in osteoarthritis. J Clin Invest. 1996;97:2011–2019. doi: 10.1172/JCI118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14:13–29. doi: 10.1016/j.joca.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 19.van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage. 2012;20:223–232. doi: 10.1016/j.joca.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Wenke AK, Rothhammer T, Moser M, Bosserhoff AK. Regulation of integrin α10 expression in chondrocytes by the transcription factors AP-2ε and Ets-1. Biochem Biophys Res Commun. 2006;345:495–501. doi: 10.1016/j.bbrc.2006.04.123. [DOI] [PubMed] [Google Scholar]
- 21.Wenke AK, Grässel S, Moser M, Bosserhoff AK. The cartilage-specific transcription factor Sox9 regulates AP-2ε expression in chondrocytes. FEBS J. 2009;276:2494–2504. doi: 10.1111/j.1742-4658.2009.06973.x. [DOI] [PubMed] [Google Scholar]
- 22.Tummala R, Romano RA, Fuchs E, Sinha S. Molecular cloning and characterization of AP-2ε, a fifth member of the AP-2 family. Gene. 2003;321:93–102. doi: 10.1016/S0378-1119(03)00840-0. [DOI] [PubMed] [Google Scholar]
- 23.Feng W, Williams T. Cloning and characterization of the mouse AP-2ε gene: a novel family member expressed in the developing olfactory bulb. Mol Cell Neurosci. 2003;24:460–475. doi: 10.1016/S1044-7431(03)00209-4. [DOI] [PubMed] [Google Scholar]
- 24.Hilger-Eversheim K, Moser M, Schorle H, Buettner R. Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene. 2000;260:1–12. doi: 10.1016/S0378-1119(00)00454-6. [DOI] [PubMed] [Google Scholar]
- 25.Wenke AK, Niebler S, Grässel S, Bosserhoff AK. The transcription factor AP-2ε regulates CXCL1 during cartilage development and in osteoarthritis. Osteoarthritis Cartilage. 2011;19:206–212. doi: 10.1016/j.joca.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Niebler S, Bosserhoff AK. The transcription factor activating enhancer-binding protein epsilon (AP-2ε) regulates the core promoter of type II collagen (COL2A1) FEBS J. 2013;280:1397–1408. doi: 10.1111/febs.12130. [DOI] [PubMed] [Google Scholar]
- 27.Schmid R, Schiffner S, Opolka A, Grässel S, Schubert T, Moser M, et al. Enhanced cartilage regeneration in MIA/CD-RAP deficient mice. Cell Death Dis. 2010;1 doi: 10.1038/cddis.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiffner S, Zimara N, Schmid R, Bosserhoff AK. p54nrb is a new regulator of progression of malignant melanoma. Carcinogenesis. 2011;32:1176–1182. doi: 10.1093/carcin/bgr103. [DOI] [PubMed] [Google Scholar]
- 29.Schmidl M, Adam N, Surmann-Schmitt C, Hattori T, Stock M, Dietz U, et al. Twisted gastrulation modulates bone morphogenetic protein-induced collagen II and X expression in chondrocytes in vitro and in vivo. J Biol Chem. 2006;281:31790–31800. doi: 10.1074/jbc.M603419200. [DOI] [PubMed] [Google Scholar]
- 30.Tatzel J, Poser I, Schroeder J, Bosserhoff AK. Inhibition of melanoma inhibitory activity (MIA) expression in melanoma cells leads to molecular and phenotypic changes. Pigment Cell Res. 2005;18:92–101. doi: 10.1111/j.1600-0749.2005.00212.x. [DOI] [PubMed] [Google Scholar]
- 31.Moser M, Bosserhoff AK, Hunziker EB, Sandell L, Fässler R, Buettner R. Ultrastructural cartilage abnormalities in MIA/CD-RAP-deficient mice. Mol Cell Biol. 2002;22:1438–1445. doi: 10.1128/MCB.22.5.1438-1445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ, et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res Ther. 2013;15:R5. doi: 10.1186/ar4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, et al. Matrix metalloproteinase 13–deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmid R, Bosserhoff AK. Redundancy in regulation of chondrogenesis in MIA/CD-RAP-deficient mice. Mech Dev. 2014;131:24–34. doi: 10.1016/j.mod.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 37.Castagnola P, Moro G, Descalzi-Cancedda F, Cancedda R. Type X collagen synthesis during in vitro development of chick embryo tibial chondrocytes. J Cell Biol. 1986;102:2310–2317. doi: 10.1083/jcb.102.6.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linsenmayer TF, Eavey RD, Schmid TM. Type X collagen: a hypertrophic cartilage-specific molecule. Pathol Immunopathol Res. 1988;7:14–19. doi: 10.1159/000157085. [DOI] [PubMed] [Google Scholar]
- 39.Reichenberger E, Aigner T, von der Mark K, Stoss H, Bertling W. In situ hybridization studies on the expression of type X collagen in fetal human cartilage. Dev Biol. 1991;148:562–572. doi: 10.1016/0012-1606(91)90274-7. [DOI] [PubMed] [Google Scholar]
- 40.D’Angelo M, Yan Z, Nooreyazdan M, Pacifici M, Sarment DS, Billings PC, et al. MMP-13 is induced during chondrocyte hypertrophy. J Cell Biochem. 2000;77:678–693. doi: 10.1002/(SICI)1097-4644(20000615)77:4<678::AID-JCB15>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 41.Huang Z, Xu H, Sandell L. Negative regulation of chondrocyte differentiation by transcription factor AP-2α. J Bone Miner Res. 2004;19:245–255. doi: 10.1359/jbmr.2004.19.2.245. [DOI] [PubMed] [Google Scholar]
- 42.Rock JR, Lopez MC, Baker HV, Harfe BD. Identification of genes expressed in the mouse limb using a novel ZPA microarray approach. Gene Expr Patterns. 2007;8:19–26. doi: 10.1016/j.modgep.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 43.Gannon JM, Walker G, Fischer M, Carpenter R, Thompson RC, Jr, Oegema TR., Jr Localization of type X collagen in canine growth plate and adult canine articular cartilage. J Orthop Res. 1991;9:485–494. doi: 10.1002/jor.1100090404. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, et al. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97:761–768. doi: 10.1172/JCI118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krane SM, Inada M. Matrix metalloproteinases and bone. Bone. 2008;43:7–18. doi: 10.1016/j.bone.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 46.Mercuri FA, Maciewicz RA, Tart J, Last K, Fosang AJ. Mutations in the interglobular domain of aggrecan alter matrix metalloproteinase and aggrecanase cleavage patterns: evidence that matrix metalloproteinase cleavage interferes with aggrecanase activity. J Biol Chem. 2000;275:33038–33045. doi: 10.1074/jbc.275.42.33038. [DOI] [PubMed] [Google Scholar]
- 47.Fosang AJ, Last K, Knäuper V, Murphy G, Neame PJ. Degradation of cartilage aggrecan by collagenase-3 (MMP-13) FEBS Lett. 1996;380:17–20. doi: 10.1016/0014-5793(95)01539-6. [DOI] [PubMed] [Google Scholar]
- 48.Singer II, Kawka DW, Bayne EK, Donatelli SA, Weidner JR, Williams HR, et al. VDIPEN, a metalloproteinase-generated neoepitope, is induced and immunolocalized in articular cartilage during inflammatory arthritis. J Clin Invest. 1995;95:2178–2186. doi: 10.1172/JCI117907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knäuper V, Cowell S, Smith B, López-Otin C, O’Shea M, Morris H, et al. The role of the C-terminal domain of human collagenase-3 (MMP-13) in the activation of procollagenase-3, substrate specificity, and tissue inhibitor of metalloproteinase interaction. J Biol Chem. 1997;272:7608–7616. doi: 10.1074/jbc.272.12.7608. [DOI] [PubMed] [Google Scholar]
- 50.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang S, Kim J, Ryu JH, Oh H, Chun CH, Kim BJ, et al. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med. 2010;16:687–693. doi: 10.1038/nm.2153. [DOI] [PubMed] [Google Scholar]
- 53.Tardif G, Pelletier JP, Dupuis M, Hambor JE, Martel-Pelletier J. Cloning, sequencing and characterization of the 5′-flanking region of the human collagenase-3 gene. Biochem J. 1997;323:13–16. doi: 10.1042/bj3230013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiménez MJ, Balbín M, López JM, Alvarez J, Komori T, López-Otín C. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol. 1999;19:4431–4442. doi: 10.1128/mcb.19.6.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otero M, Plumb DA, Tsuchimochi K, Dragomir CL, Hashimoto K, Peng H, et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J Biol Chem. 2012;287:3559–3572. doi: 10.1074/jbc.M111.265744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hess J, Porte D, Munz C, Angel P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J Biol Chem. 2001;276:20029–20038. doi: 10.1074/jbc.M010601200. [DOI] [PubMed] [Google Scholar]