

Abstract
Objective:
To explore the imaging features of meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumours (pPNETs).
Methods:
The imaging features and pathological characteristics of eight cases of surgically and pathologically confirmed Ewing sarcoma/pPNET were analysed retrospectively in light of recent literature on the disease.
Results:
The peak age was between 10 and 20 years. The lesions tended to be spindle shaped and dural based, usually widely so. CT showed that the lesions had slightly uneven high density in five cases and iso–low mixed density in three cases; marked heterogeneous enhancement was seen in all cases after contrast injection. MRI of the lesions showed varying proportions of isointense and hypointense signal in all cases on unenhanced T1 weighted imaging and varying proportions of isointense and hyperintense signal on T2 weighted imaging. After contrast injection, marked heterogeneous enhancement was seen in all cases; three cases showed a short and nodular dural tail and five cases showed adjacent skull erosion and osteolysis. Pathological results included high cell density, haemorrhage and necrosis. The cells resembled lymphocytes and spindle cells with transparent cytoplasm. CD99 and vimentin were expressed by all tumour cells.
Conclusion:
Features of meningeal Ewing sarcoma/pPNETs include peak incidence at 10–20 years of age, a broad connection to the meninges, a thick dural tail involved with tumour, skull and scalp erosion and early metastasis. Necrosis and cystic changes are the common histological findings.
Advances in knowledge:
The imaging features of meningeal Ewing sarcoma/pPNETs have not been reported. The study helps to identify meningeal Ewing sarcoma/pPNETs and meningioma.
Ewing sarcoma/peripheral primitive neuroectodermal tumours (pPNETs) are a group of highly malignant small round cell neoplasms, which include Ewing sarcoma, Askin tumour and pPNET.1 We collected eight cases of meningeal Ewing sarcoma/pPNET confirmed by pathology and immunohistochemistry to review the imaging and pathological findings in order to deepen our understanding and improve the diagnosis of this disease, and to provide more accurate clinical information.
METHODS AND MATERIALS
Eight cases of meningeal Ewing sarcoma/pPNET were confirmed by surgery and pathology in the Second Hospital of Lanzhou University, Lanzhou, China, between November 2005 and December 2011. There were five males and three females aged between 7 and 23 years (mean age, 15 years). Presenting symptoms had been of 2 weeks to 18 months of duration. There were four cases presenting with headache, two cases with headache, dizziness and vomiting and two cases presenting with fever. One case had right proptosis, accompanied by visual disturbances. Two cases were accompanied by physical disabilities. Two cases had non-tender masses. Pre-operative imaging diagnoses were meningioma in four cases, metastatic tumour in one case, hemangiopericytoma in one case and skull osteosarcoma in one case.
MRI scans were performed with a 1.0-T scanner (Magnetom Harmony; Siemens Medical Systems, Erlangen, Germany) and a 3.0-T scanner (Siemens Verio). The Siemens Magnetom Harmony 1.0-T MR scanner protocol included conventional spin-echo (SE) and fast SE sequences with axial, sagittal and coronal imaging. Scanning parameters were as follows: T1 weighted imaging (T1WI), repetition time (TR)/echo time (TE) 550 ms/12 ms; T2 weighted imaging (T2WI), TR/TE, 2200 ms/90 ms; thickness, 5.0 mm; spacing, 1.5 mm; field of view (FOV) 320 × 320 mm; matrix, 256 × 256; sagittal and coronal slice 8.0 mm; and layer spacing, 2.0 mm. For the enhanced scans, a bolus of gadolinium-diethylene triamine pentaacetic acid was given intravenously at a dose of 0.1 mmol kg−1 with a flow rate of 3 ml s−1. The 3.0-T superconducting MR scanner protocol included a three-dimensional T1 fluid-attenuated inversion recovery sequence: TR/TE, 2200 ms/25.2 ms; FOV, 240 mm; thickness, 4.0 mm; spacing, 0.5 mm; matrix, 256 × 256, and diffusion tensor imaging with single-shot SE echo-planar imaging: TR/TE 10000 ms/112 ms; FOV, 240 mm; thickness, 4.0 mm; spacing, 0.5 mm; and matrix, 128 × 128.
The eight patients underwent total or subtotal resection. The resected tumour specimens were fixed in 4% formaldehyde for 24 h, dehydrated and embedded in paraffin. Each fixed sample was then cut into slices of 3–4 µm thickness to make five to six sections. The sections were separately stained with haematoxylin–eosin, periodic acid schiff, CD99, synaptophysin (Syn), neuron-specific enolase (NSE), vimentin (Vim) and S-100. The tumour tissue sections were reviewed by two neuropathologists who were not aware of the MRI appearance.
RESULTS
The primary tumour sites were the right side of the tentorium, the right side of the frontal, right temporal pole, right occipital dura, right top, left frontal dura, left temporal dura, left occipital dura each in one case. Three patients had distant metastases. Seven of the eight cases were connected to the dura with a wide base and were fusiform shaped. CT showed that the lesions had slightly uneven high density in five cases and iso–low mixed density in three cases; marked heterogeneous enhancement was seen in all cases after contrast injection. On MRI, all the lesions showed mixed signal. On T1WI, six lesions showed mixed isointense-to-hypointense signal and two cases had mixed hypointense signal. All cases showed mixed isointense-to-hyperintense or diffuse, slightly hyperintense signal on T2WI. There were varying degrees of cystic degeneration and necrosis (Figures 1–9). One case showed multiple septa (Figure 3). There was significant heterogeneous enhancement of the masses; three cases showed a short and nodular dural tail. Five cases had clear boundaries with brain tissue and a pseudocapsule. Only three cases showed mild peritumoral oedema (Figures 2 and 5). The tumours were otherwise aggressive; five cases showed distinct adjacent skull erosion and osteolysis. One lesion located on the right side of the tentorium displaced the right temporal (Figures 4–6) and one lesion located in the right frontal dura displaced the orbit anteriorly (Figures 7–9). Two cases broke through the outer table of the skull to invade the scalp. The case of metastatic disease involved a vertebra, which resulted in vertebral body destruction (Table 1).
Figure 1.

A 14-year-old male with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the left temporal region. Coronal T1 weighted imaging showed mixed low signal.
Figure 9.

A 13-year-old female with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the right frontal region. Enhanced coronal T1 weighted imaging showed significant heterogeneous enhancement.
Figure 3.

A 14-year-old male with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the left temporal region. Axial enhanced T1 weighted imaging showed significant enhancement; multiple septa could be seen inside the tumour.
Figure 2.

A 14-year-old male with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the left temporal region. Axial T2 weighted imaging showed mixed high signal. The tumour showed a wide base, adjacent skull erosion and soft tissue invasion under the scalp.
Figure 5.

An 18-year-old female with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the right tentorium. Axial T2 weighted imaging showed heterogeneous hyperintensity. The lesion border was clear.
Figure 4.

An 18-year-old female with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the right tentorium. Unenhanced axial T1 weighted imaging showed mixed isointense-to-hypointense signal.
Figure 6.

An 18-year-old female with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the right tentorium. Enhanced sagittal T1 weighted imaging showed significant enhancement and prominent vessels.
Figure 7.

A 13-year-old female with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the right frontal region. Unenhanced axial T1 weighted imaging showed mixed hypointense-to-isointense signal. FLAIR, fluid-attenuated inversion recovery.
Table 1.
Imaging features of meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumours
| Imaging features | Cases (n) | Proportion (%) |
|---|---|---|
| Fusiform shaped | 7 | 87.5 |
| CT appearance | ||
| Mixed slightly high density | 5 | 62.5 |
| Mixed iso–low density | 3 | 37.5 |
| MR appearance | ||
| Mixed isointense-to-hypointense signal on T1WI | 6 | 75.0 |
| Mixed hypointense signal on T1WI | 2 | 25.0 |
| Mixed isointense-to-hyperintense on T2WI | 8 | 100.0 |
| Degree of enhancement | ||
| Significantly heterogeneous enhancement | 8 | 100.0 |
| Cystic degeneration and necrosis | 8 | 100.0 |
| Short and nodular dural tail | 3 | 37.5 |
| Peritumoral oedema | ||
| Mild | 3 | 37.5 |
| No significant | 5 | 62.5 |
| Connected to the dura with a wide base | 7 | 87.5 |
| Adjacent skull erosion and osteolysis | 5 | 62.5 |
T1WI, T1 weighted imaging; T2WI, T2 weighted imaging.
Figure 8.

A 13-year-old female with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the right frontal region. Axial T2 weighted imaging showed mixed hypointense signal. High signal was seen around the lesion. TSE, turbo spin echo.
The gross appearance of the tumours was yellow or grey–red and indurated. Five cases had a pseudocapsule. Haemorrhage, necrosis and liquefaction were seen inside the tumours. Histology showed rich uniform small round cells (Figure 10) with little cytoplasm and deep nuclear staining (Figure 11), which is the characteristic of a neuroectodermal neoplasm. There were large areas of coagulation necrosis; immunohistochemistry showed that all cases were CD99 positive and Vim positive (Figure 12). Three cases were Syn positive, one case was weakly Syn positive and two cases were weakly S-100 protein positive.
Figure 10.
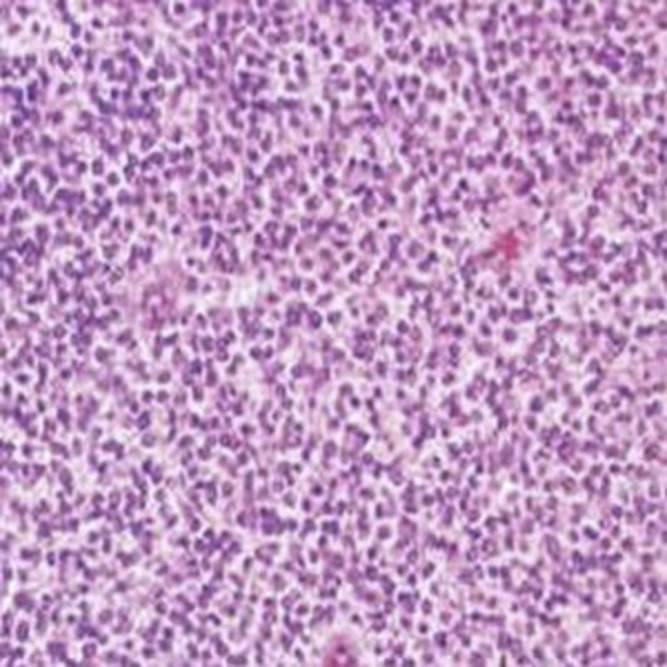
A 14-year-old male with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the left temporal region. Tumour was composed of extremely rich uniform small round cells (haematoxylin–eosin ×100).
Figure 11.
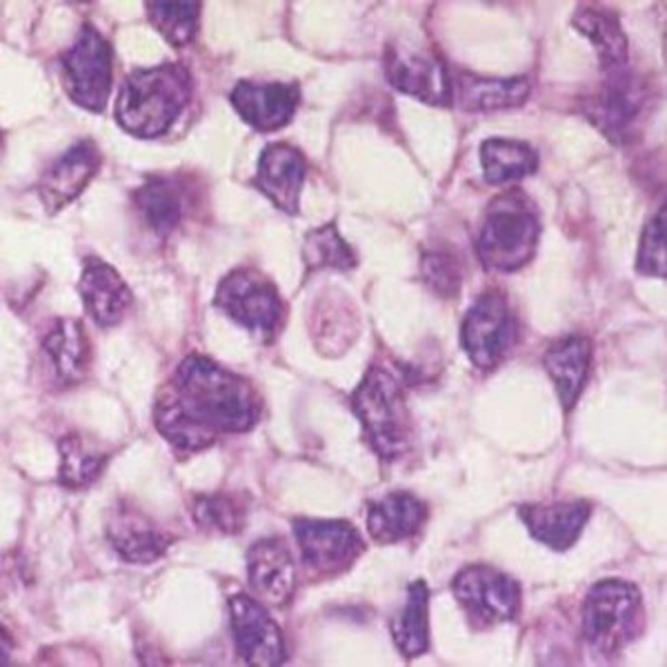
A 14-year-old male with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the left temporal region. Cell borders are unclear, with sparse transparent cytoplasm (haematoxylin–eosin ×400).
Figure 12.
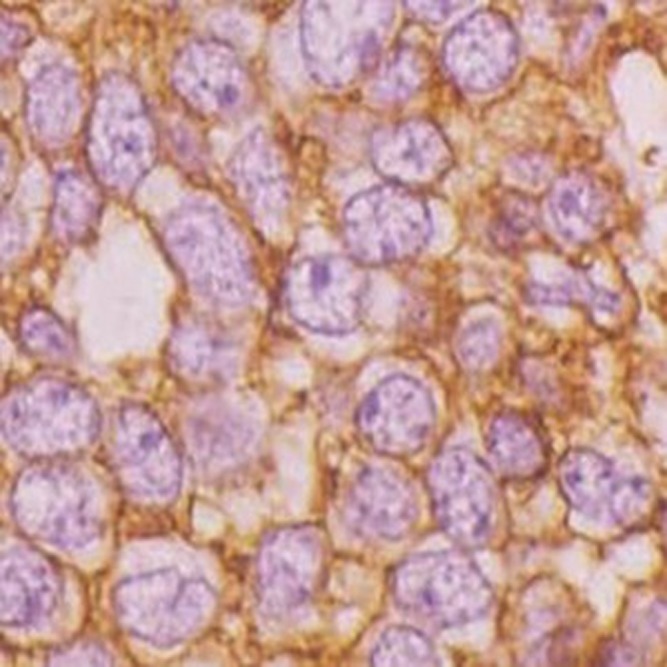
A 14-year-old male with meningeal Ewing sarcoma/peripheral primitive neuroectodermal tumour of the left temporal region. Immunohistochemical staining was positive for CD99 (×400).
Six patients died within 3 months, one patient died within 6 months and one patient was lost to follow-up. All patients underwent surgery and radiotherapy.
DISCUSSION
PNET was first reported occurring in the ulnar nerve by Stout in 1918;2 Ewing sarcoma was reported by Ewing in 1921.3 In 1973, Hart4 proposed the concept of origin was from primitive neuroectoderm, and it was composed of small round cell and was a rare, highly aggressive, malignant tumour.5,6 PNET was further divided into central type and peripheral type (pPNET) and the latter occurred outside the central and sympathetic nervous system.7 Meningeal Ewing sarcoma/pPNET was categorized among the meningeal mesenchymal tumours, and meningioma was categorized into meningeal epithelial tumours.8 The pathological diagnosis of Ewing sarcoma/pPNET requires the presence of Homer-Wright rosettes or immunohistochemical expression of at least two different neural markers, such as CD99, NSE, Syn, S-100 and Vim.9,10 In our series, all tumours expressed CD99 and Vim.
In our series, five cases of eight patients occurred at the usual age of onset of 10–20 years old. The mean age was 15 years. The tumours showed a male predominance with a male to female ratio of 5:3. The literature has reported that 75% of Ewing sarcoma/pPNET occurs before the age of 30 years, and males are affected more frequently than females.10,11 Those originating from the soft tissue or bone occur in older patients with an average age of 28 years.10,12 The mean age of onset in our group was slightly lower, but most likely not significant because of the small sample size. The clinical manifestations of the patients showed varying degrees of headache, dizziness and vomiting, which were most likely due to elevated intracranial pressure caused by oedema and mass effect. The main reason was the mass effect in our group. Seven patients died within 6 months and one patient was lost to follow-up. One case presented with vertebral metastasis and two more patients developed metastases. The majority of patients with brain and spinal Ewing sarcoma/pPNET reported in the literature died within 1 year. Ewing sarcoma/pPNET has a high degree of malignancy, the World Health Organization (WHO) grade is grade IV, and patients can readily relapse with resultant high mortality. The longest recorded survival was 72 months after three resections and chemo/radiotherapy.13
Intracranial meningeal Ewing sarcoma/pPNET imaging findings have similarities with those of meningioma and may mimic meningioma. Four of our cases were misdiagnosed as meningioma.
Seven of the cases featured a wide meningeal base and a fusiform growth pattern along the meninges on MRI. A wide meningeal base showed the tumour was close to the meninges, and fusiform growth pattern might be due to the meninges, blood–brain barrier and skull affect meningeal Ewing sarcoma/pPNET growth pattern. Skull invasion was seen in five cases, and two patients suffered destruction of the outer table of the skull, and these regions could be seen as rich tumour cells in the microscope. This is consistent with reports that pPNET can spread along the spinal nerves or the neighbouring tissue, causing bone and soft tissue destruction.12,14–16 However, the fact that the lesions and brain tissue boundaries were clear might be due to the meninges and blood–brain barrier that played a role in infringing the tumour of the brain.
Meningeal Ewing sarcoma/pPNET mainly displayed slightly uneven high density on CT and mainly heterogeneous signal on MRI. Pathological examination of these regions showed abundant small round cells and high cell density, and some regions showed a large amount of protein-rich mucus associated with haemorrhage and necrosis. Therefore, meningeal Ewing sarcoma/pPNET mainly showed uneven density or signal. An obvious heterogeneous enhancement because of a rich blood supply, similar to pPNET and bone Ewing sarcoma, is reported in the literature; many authors argued that pPNET and bone Ewing sarcoma showed obvious heterogeneous enhancement, commonly with necrosis on histology, usually without ossification, calcification or periosteal reaction.17–20 This may be due to the short course of the disease, with little time for calcification. Most authors report that peritumoral oedema usually indicates a high degree of malignancy, although in our group of eight cases only three cases of mild peritumoral oedema occurred. A thick dural tail was seen in three cases in our group, most likely because the tumour undermined the surrounding normal meninges. The “dural tail sign” is generally believed to be a benign response to adjacent tumour, but appeared as a thickened atypical tail in these cases because of the aggressive nature of Ewing sarcoma/pPNET.
DIFFERENTIAL DIAGNOSIS
Meningiomas are common extra-axial tumours, usually occurring in middle age, affecting females more than males, with a slow course and a low degree of malignancy. MRI shows isointense signal and little mass effect. Enhancement is usually homogeneous and bright. The dural tail sign is commonly seen. Especially microcapsules meningioma, a pathololgical subtype of meningiomas, which shows scattered size distribution of microcapsule structure is easily confused with meningeal Ewing sarcoma/pPNET. However, microcapsule meningioma shows significant even hypointense signal on T1WI, significant even hyperintense signal on T2WI and a grid-like change after enhanced scan. Moreover, the WHO grade is grade I, and no obvious signs of invasion are in the surrounding areas. For intracranial hemangiopericytoma, MRI mainly shows iso-to-hypointense signal on T1WI and iso-to-hyperintense heterogeneous signal on T2WI. Bleeding is common. Enhancement is heterogeneous. It has a lobulated shape and a narrow base connected to the dura mater. Skull osteosarcoma is rare. Tumour bone formation with a radial appearance is common, and the age of onset is older.
In summary, meningeal Ewing sarcoma/pPNETs was typified by an age of onset of 10–20 years, rapid disease progression, wide meningeal base and fusiform shape. It displayed slightly uneven high density on CT, mainly heterogeneous signal on MRI, with frequent cystic necrosis, significant heterogeneous enhancement, a nodular dural tail indicating tumour infiltration and a tendency to invade adjacent skull and soft tissue.
REFERENCES
- 1.Kim JT, Chung DS, Han YM, Park YS, Kang JK. Extraskeletal cervical epidural Ewing’s sarcoma: case report and review of the literature. J Korean Neurosurg Soc 2002; 32: 48–51. [Google Scholar]
- 2.Stout AP. A tumor of the ulnar nerve. Proc NY Pathol Soc 1918; 12: 2–12. [Google Scholar]
- 3.Ewing J. Diffuse endothelioma of bone. Proc NY Pathol Soc 1921; 21: 17–24. [Google Scholar]
- 4.Hart MN, Earle KM. Primitive neuroectodermal tumors of the brain in children. Cancer 1973; 32: 890–7. [DOI] [PubMed] [Google Scholar]
- 5.Stout AP, Murray MD. Neuroepithelioma of the radial nerve with a study of behavior in vitro. Rev Can Biol 1942; 1: 651–2. [Google Scholar]
- 6.Dehner LP. Peripheral and central primitive neuroectodermal tumors. A nosologic concept seeking a consensus. Arch Pathol Lab Med 1986; 110: 997–1005. [PubMed] [Google Scholar]
- 7.Carvajal R, Meyers P. Ewing’s sarcoma and primitive neuroectodermal family of tumors. Hematol Oncol Clin North Am 2005; 19: 501–25, vi–vii. [DOI] [PubMed] [Google Scholar]
- 8.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinkhuis M, Wijnaendts LC, van der Linden JC, van Unnik AJ, Voûte PA, Baak JP, et al. Peripheral primitive neuroectodermal tumor and extra-osseous Ewing’s sarcoma; a histological, immunohistochemical and DNA flow cytometric study. Virchows Arch 1995; 425: 611–16. [DOI] [PubMed] [Google Scholar]
- 10.Qian X, Kai X, Shaodong L, Gaohong C, Hong M, Jingjing L. Radiological and clinicopathological features of pPNET. Eur J Radiol 2013; 82: e888–93. [DOI] [PubMed] [Google Scholar]
- 11.Kransdorf MJ. Malignant soft-tissue tumors in a large referral population: distribution of diagnosis by age, sex and location. AJR Am J Roentgenol 1995; 164: 129–34. [DOI] [PubMed] [Google Scholar]
- 12.Huang WY, Tan WL, Geng DY, Zhang J, Wu G, Zhang BY, et al. Imaging findings of the spinal peripheral Ewing's sarcoma family of tumours. Clin Radiol 2014; 69: 179–85. [DOI] [PubMed] [Google Scholar]
- 13.Perry R, Gonzales I, Finlay J, Zacharoulis S. Primary peripheral primitive neuroectodermal tumors of the spinal cord: report of two cases and review of the literature. J Neurooncol 2007; 81: 259–64. doi: 10.1007/s11060-006-9178-1 [DOI] [PubMed] [Google Scholar]
- 14.Zhang WD, Chen YF, Li CX, Zhang L, Xu ZB, Zhang FJ. Computed tomography and magnetic resonance imaging findings of peripheral primitive neuroectodermal tumors of the head and neck. Eur J Radiol 2011; 80: 607–11. doi: 10.1016/j.ejrad.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 15.Lin YY, Lin CJ, Ho DM, Guo WY, Chang CY. Primary intramedullary spinal cord lymphoma. Spine J 2012; 12: 527–8. doi: 10.1016/j.spinee.2012.05.028 [DOI] [PubMed] [Google Scholar]
- 16.Ng SH, Ko SF, Cheung YC, Wong HF, Jung SM. Extraskeletal Ewing's sarcoma of the parapharyngeal space. Br J Radiol 2004; 77: 1046–9. doi: 10.1259/bjr/16676268 [DOI] [PubMed] [Google Scholar]
- 17.Alexander HS, Koleda C, Hunn MK. Peripheral Primitive Neuroectodermal Tumour (pPNET) in the cervical spine. J Clin Neurosci 2010; 17: 259–61. [DOI] [PubMed] [Google Scholar]
- 18.Khong PL, Chan GC, Shek TW, Tam PK, Chan FL. Imaging of peripheral PNET: common and uncommon locations. Clin Radiol 2002; 57: 272–7. doi: 10.1053/crad.2001.0807 [DOI] [PubMed] [Google Scholar]
- 19.Kim MS, Kim B, Park CS, Song SY, Lee EJ, Park NH, et al. Radiologic findings of peripheral primitive neuroectodermal tumor arising in the retroperitoneum. AJR Am J Roentgenol 2006; 186: 1125–32. doi: 10.2214/AJR.04.1688 [DOI] [PubMed] [Google Scholar]
- 20.Hari S, Jain TP, Thulkar S, Bakhshi S. Imaging features of peripheral primitive neuroectodermal tumours. Br J Radiol 2008; 81: 975–83. doi: 10.1259/bjr/30073320 [DOI] [PubMed] [Google Scholar]