

Abstract
Background:
Transcriptional repression is a key mechanism driving leukaemogenesis. In acute promyelocytic leukaemia (APL), the fusion protein promyelocytic leukaemia-retinoic acid receptor-α fusion (PML-RARα) recruits transcriptional repressors to myeloid differentiation genes. All-trans-retinoic acid (ATRA) induces the proteasomal degradation of PML-RARα and granulocytic differentiation. Histone deacetylases (HDACs) fall into four classes (I–IV) and contribute to the transcription block caused by PML-RARα.
Methods:
Immunoblot, flow cytometry, and May-Grünwald–Giemsa staining were used to analyze differentiation and induction of apoptosis.
Results:
A PML-RARα- and ATRA-dependent differentiation programme induces granulocytic maturation associated with an accumulation of the myeloid transcription factor CCAAT/enhancer binding protein (C/EBP)ɛ and of the surface protein CD11b. While this process protects APL cells from inhibitors of class I HDAC activity, inhibition of all Zinc-dependent HDACs (classes I, II, and IV) with the pan-HDACi (histone deacetylase inhibitor(s)) LBH589 induces apoptosis of immature and differentiated APL cells. LBH589 can eliminate C/EBPɛ and the mitochondrial apoptosis regulator B-cell lymphoma (BCL)-xL in immature and differentiated NB4 cells. Thus, BCL-xL and C/EBPɛ are newly identified molecular markers for the efficacy of HDACi against APL cells.
Conclusions:
Our results could explain the therapeutic limitations occurring with ATRA and class I HDACi combinations. Pro-apoptotic effects caused by pan-HDAC inhibition are not blunted by ATRA-induced differentiation and may provide a clinically interesting alternative.
Keywords: apoptosis, ATRA, C/EBPɛ, differentiation, HDAC, LBH589
Leukaemia, cancer of the haematopoietic system, originates from the bone marrow or the lymphatic system (Heidel et al, 2011). Leukaemic cells are characterised by a block in functional differentiation. Depending on the cell type affected, leukaemia fluctuates strongly in frequency, treatment options, and prognosis (Heidel et al, 2011).
Acute promyelocytic leukaemia (APL belongs to the French-American-British M3 subtype of acute myeloid leukaemia (AML) (Grignani et al, 1998). In over 95% of APL cases, the translocation t(15;17) (q22;q11-12) is present. This aberration fuses the genes promyelocytic leukaemia (PML) and retinoic acid receptor-α (RARα). The PML-RARα causally contributes to leukaemogenesis through recruiting transcriptional corepressors and associated histone deacetylases (HDACs) to RARα-dependent myeloid differentiation genes (Lin et al, 1998). Promyelocytic leukaemia-retinoic acid receptor-α-positive cells are insensitive to physiological concentrations of retinoic acid (∼10−9 M), but they differentiate in response to pharmacological concentrations (∼10−6 M) (Grignani et al, 1998; Altucci and Gronemeyer, 2001). Such doses reactivate RAR target genes via the recruitment of transcriptional coactivators and the degradation of PML-RARα (Duprez et al, 2000; Leiva et al, 2012). Moreover, pharmacological doses of all-trans-retinoic acid (ATRA) alter histone acetylation at PML-RARα/RXR binding sites (Martens et al, 2010). Survival rates of APL patients significantly increase after treatment with ATRA (Tallman et al, 1997; Müller and Krämer, 2010), and PML-RARα degradation appears to be therapeutically relevant (Raelson et al, 1996; Nasr et al, 2008; Leiva et al, 2012).
Histone deacetylase inhibitor(s) (HDACi) are epigenetic drugs. They promote cellular differentiation, senescence, and apoptosis. They increase protein acetylation and alter transcriptomes and proteomes (Spange et al, 2009). Of note, transformed cells respond to HDACi up to 10 times more sensitive than normal cells, suggesting that such drugs act selectively (Dokmanovic and Marks, 2005). The molecular players and pathways responsible for the increased robustness of normal cells are largely unclear. For example, it is not known whether the differentiation status of normal cells determines their sensitivity to HDACi.
Histone deacetylase inhibitor(s) induce the proteasomal degradation of PML-RARα (Krämer et al, 2013), and murine APL models suggest a superior effect of ATRA/HDACi co-treatment (He et al, 2001; Leiva et al, 2012). However, variable results were obtained in AML patients treated with ATRA and HDACi (Raffoux et al, 2005, 2010; Müller and Krämer, 2010; Kuendgen et al, 2011). Encouraging effects were achieved when female patients (13/29 years old), who were suffering from ATRA-resistant APL, were treated with HDACi. These patients were treated with the HDACi phenylbutyrate (Warrell et al, 1998), or valproic acid (VPA) (McMullin et al, 2005). However, out of five relapsed APL patients, who were co-treated with ATRA and phenylbutyrate, only one underwent clinical remission (Zhou et al, 2002). Due to these very limited numbers of patients and due to variable treatment schedules applied, it cannot be judged at present whether HDACi are useful to treat APL. Because the schedule of treatment with ATRA and cytotoxic chemotherapy (including DNA-damaging drugs and arsenic trioxide) determines therapy success in AML patients (Jing et al, 2001; Schlenk et al, 2004; Xia et al, 2006), the timing of HDACi and ATRA application as well as differences between class I-specific and pan-HDACi may also determine the outcome of ATRA/HDACi treatment.
Since these important parameters have not been defined, we analysed them in human NB4 APL cells; these are the only PML-RARα-positive cell line available (Duprez et al, 2000). We tested whether immature and terminally differentiated APL cells have variable requirements for HDACs. In addition, we addressed if such cells are sensitive to the inhibition of certain HDAC classes. Our analyses demonstrate that HDACi induce a B-cell lymphoma (BCL)-xL-sensitive mitochondrial apoptosis programme in APL cells. Strikingly, their granulocytic differentiation, which is associated with an accumulation of CCAAT/enhancer binding protein (C/EBP)ɛ and of CD11b, renders cells independent from the activity of class I HDACs. Nevertheless, such cells still depend on the combined activity of class I and class II HDACs.
Materials and methods
Chemicals
LBH589 was from Novartis (Basel, Switzerland). MS-275 was from Selleck Chemicals (Houston, TX, USA). Valproic acid (VPA), ATRA, and propidium iodide (PI) were from Sigma-Aldrich (St Louis, MO, USA).
Cell culture
NB4 and NB4-R2 cells were maintained in RPMI medium containing 10% FCS and 1% penicillin/streptomycin. Cells were cultured at 37 °C in a 5% CO2 atmosphere. The identity of NB4 cells was verified by DNA fingerprint at the Leibniz Institute, DSMZ GmbH, Braunschweig. NB4 cells are derived from the bone marrow of a 23-year-old female APL patient in relapse (Duprez et al, 2000). All-trans-retinoic acid-resistant NB4-R2 cells carry PML-RARα with a mutated ligand binding domain; this protein has a dominant negative effect on the wild-type protein (Duprez et al, 2000; Roussel and Lanotte, 2001). Cells were treated with ATRA, and/or HDACi for the time points and concentrations indicated. Cell counts were determined with a Z1 COULTER COUNTER (Beckman Coulter, Inc., Pasadena, CA, USA).
Flow cytometry
CD11b and PI staining were done as described (Krämer et al, 2008; Pietschmann et al, 2012b). Supplementary Figures S1 and S2 show examples of FACS analyses of CD11b-stained cells as well as examples of cell-cycle analyses done with fixed and PI-stained NB4 cells. Antibodies against CD11b were from BIOZOL Diagnostica (Eching, Germany) (CD11b-RPE (DAK-R084101-8)).
Immunoblot and antibodies
Experiments were performed as described (Krämer et al, 2008). Antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA): anti-C/EBPɛ #sc-158, anti-GAPDH #sc-137179 and anti-BCL-2 #sc-492; anti-acetylated-α-Tubulin #sc-23950; Cell Signaling Technology (Danvers, MA, USA): anti-cleaved caspase-3 #9664 and anti-cleaved caspase-9 #9501; Millipore/Upstate (Billerica, MA, USA): anti-acetyl-Histone H3 #06-599 and anti-HDAC1 #05-100; Sigma-Aldrich: anti-α-tubulin #T5168; BD Pharmingen (Heidelberg, Germany): anti-cleaved PARP #552596 and anti-BCL-x #551022; Abcam (Cambridge, MA, USA): anti-PML-RARα #ab43152.
Transduction of NB4 cells
Cells were retrovirally transduced with MSCV vector constructs co-expressing EGFP. Retroviral particles were preloaded on retronectin. Transduced cells were sorted for EGFP-positive cells with the FACS Aria cell sorter (BD Biosciences, Heidelberg, Germany).
Pappenheim May-Grünwald–Giemsa staining
There are classic haematological staining methods according to Giemsa and May-Grünwald as well as the combination of both (Pappenheim). For Pappenheim staining, slides were fixed with Methanol (Sigma-Aldrich) for 10 min. After this, the first staining step was performed using May-Grünwald's solution (Merck Millipore, Billerica, MA, USA) for 8 min. Afterwards, incubation in Giemsa's solution (Merck Millipore) was done for 20 min. Before the slides had to dry a washing step with buffer (K2PO4, Applichem GmbH, Darmstadt, Germany) had been done for 5 min; for further details see (Binder et al, 2012).
Results
ATRA prevents HDACi-induced caspase activation and cell death of NB4 cells
We analysed how the clinically tested HDACi VPA, MS-275, and LBH589 (Bug et al, 2005; Hess-Stumpp et al, 2007; DeAngelo et al, 2013), alone and in combination with ATRA, affected NB4 cells (treatment scheme: see Figure 1A). We started our tests with VPA, which inhibits class I HDACs (Figure 1B) (Bradner et al, 2010). FACS analyses revealed that treatment with VPA for 24 h increased the number of dead cells. Valproic acid and ATRA/VPA co-treatment for 24 h caused similar rates of cell death (Figure 2A). A previous administration (pre-treatment, Figure 1A) of ATRA for 24 h blunted the anti-leukaemic effects of VPA (Figure 2A). We achieved these data when we probed for active caspase-3 (Figure 2B), which triggers apoptosis (Krämer et al, 2008).
Figure 1.

Treatment schedule and classical HDACs and their inhibitors. (A) Treatment schedule of NB4 cells. Co-treatment implies that cells were treated together with ATRA and HDACi for 24 h, for example, ATRA 24 h and VPA 24 h. Pre-treatment indicates that cells were incubated for 24 h with ATRA, followed by a 24-h VPA treatment, during which ATRA stayed on the cells, for example, ATRA 48 h and VPA 24 h. (B) The figure shows the three classes of HDACs (I, II, and IV) that depend on Zn2+ for their catalytic activity. Members of these HDACs can be inhibited by HDACi used in this study. The data are derived from a study by Bradner et al (2010). They tested HDACs 1–9, the inhibition of HDAC10 and HDAC11 is inferred from a literature data search.
Figure 2.

ATRA induces differentiation and prevents the induction of apoptosis by VPA. (A) Flow-cytometry analysis of NB4 cells treated with 1 μM ATRA and/or 1.5 mM VPA for the indicated period of time. The diagram shows the subG1 fractions of PI-stained cells (mean±s.e.m., n⩾3). Statistics were done with the two-tailed unpaired Student's t-test, *P⩽0.05, **P⩽0.01, ***P⩽0.001, n.s. not significant P⩾0.05. (B) Whole-cell protein extracts were prepared from treated NB4 cells. Immunoblot was used to detect the protein expression of C/EBPɛ, acetylated histones H3 (acetyl-H3), cleaved caspase-3 (cl., cleaved form) which is the active form of the caspase, and α-tubulin as a loading control. All samples were analysed on one membrane. (C) Flow-cytometry analysis of NB4 cells treated with 1 μM ATRA and/or 1.5 mM VPA for the indicated period of time. Treated NB4 cells were stained with an antibody against CD11b and analysed by flow cytometry. All treated samples show more CD11b-positive cells than control cells, significance ***P⩽0.001. Statistics were done with the two-tailed unpaired Student's t-test. (D) To test maturation at the morphological level, we analysed NB4 cells exposed to 1.5 mM VPA, 1 μM ATRA for 72 h and to 100 nM LBH589 (LBH) for 24 h by May-Grünwald–Giemsa staining. This approach revealed that only ATRA evokes cellular maturation. An at most partial induction of differentiation by VPA was also found in other studies (Müller and Krämer, 2010). A 63-fold magnification was used.
The possibility that the inhibition of apoptosis by ATRA is linked to cell-cycle arrest is unlikely, as HDACi also cause cell-cycle arrest of NB4 cells (Supplementary Figures S1 and S3).
Next, we tested by immunoblot and by flow cytometry whether HDACi and ATRA evoke the expression of C/EBPɛ (Koeffler, 2010) and CD11b (Paietta, 2003). CCAAT/enhancer binding protein ɛ accumulates in leukaemic cells differentiating towards granulocytes (Morosetti et al, 1997; Park et al, 1999). Application of ATRA to NB4 cells caused the accumulation of C/EBPɛ and CD11b. While VPA also induced CD11b expression (Figure 2C), it failed to induce C/EBPɛ expression (Figure 2B). Using May-Grünwald–Giemsa staining, we tested for NB4 cell differentiation by ATRA and VPA at the cellular level. We noticed that only ATRA triggers terminal granulocytic maturation (Figure 2D). This result agrees with the data we collected for the expression of C/EBPɛ (Figure 2B) and it indicates that VPA causes only a very limited differentiation of NB4 cells. Moreover, these results illustrate that HDACi can induce CD11b without an increase in C/EBPɛ expression and without clear signs of cellular maturation. Thus, we demonstrate that C/EBPɛ is a more reliable marker for granulocytic differentiation than CD11b.
Granulocytic differentiation of NB4 cells and induction of C/EBPɛ are associated with protection from HDACi-induced cell death
Having found that ATRA antagonises pro-apoptotic effects of HDACi, we asked whether ATRA causes immediate molecular alterations neutralising pro-apoptotic effects. We observed that already a 6-h pre-exposure to ATRA programmed NB4 cells to resist the activation of caspase-3 by VPA. Again the appearance of C/EBPɛ inversely correlated with the accumulation of cleaved caspase-3 (Figure 3A).
Figure 3.
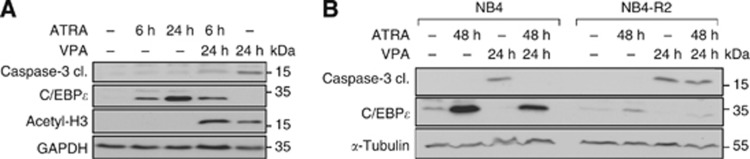
Pro-apoptotic effects of VPA are rapidly decreased by ATRA and inversely correlate with C/EBPɛ expression. (A) After stimulating the NB4 cells for 6 h with ATRA (1 μM), the medium was changed and cells were washed. Then the cells were exposed to VPA for 24 h or they were not further treated. NB4 cells treated for 24 h with ATRA were used as a control. Immunoblot analysis of NB4 whole-cell protein lysates was done to assess the levels of C/EBPɛ, acetylated histones H3 (acetyl-H3), cleaved caspase-3, and GAPDH as a loading control. (B) Whole-cell protein extracts were prepared from treated NB4 and NB4-R2 cells. Immunoblot was used to detect the protein expression of C/EBPɛ, cleaved caspase-3 (cl., cleaved form) which is the active form of the caspase, and α-tubulin as a loading control.
To solve whether the anti-apoptotic effects of ATRA rely on PML-RARα, we used the ATRA-unresponsive NB4 cell derivative NB4-R2. These cells carry mutant PML-RARα (Raelson et al, 1996; Duprez et al, 2000). CCAAT/enhancer binding protein ɛ was not induced by ATRA in NB4-R2 cells (Figure 3B). Of note, the lack of C/EBPɛ accumulation in NB4-R2 cells tied in with caspase-3 activation by VPA (Figure 3B). Hence, a lack of C/EBPɛ induction—which marks granulocytic maturation—indicates that VPA can cause apoptosis of ATRA-treated cells. Furthermore, these experiments illustrate that HDACi are effective against APL cells with mutated PML-RARα. Such tumours represent a clinical problem (Raelson et al, 1996; Duprez et al, 2000).
ATRA inhibits apoptosis induction by class I HDACi but not by pan-HDACi
To this end, our data demonstrate that the anti-apoptotic mechanisms induced by ATRA discredit its application before treatment with the class I-specific HDACi VPA (Figure 1B) (Bradner et al, 2010; Müller and Krämer, 2010). As an additional control, we used the benzamide HDACi MS-275, which only targets HDAC1-3 (Figure 1B) (Bradner et al, 2010). We found that the HDACi-induced cell death was significantly reduced when ATRA had been applied before MS-275 or VPA (Figure 4A). Similar results were obtained for ATRA and Butyrate (data not shown).
Figure 4.

Interaction of ATRA with pan-HDACi. (A) NB4 cells were treated with 1 μM ATRA for 48 h and/or VPA, MS-275 (MS) or LBH589 (LBH) with indicated concentrations for 24 h. Cells were pre-treated with ATRA or not. PI-stained cells were analysed by flow cytometry, the SubG1 fractions (mean±s.e.m., n=3) are shown. Statistics were done with the two-tailed unpaired Student's t-test, ***P⩽0.001, n.s. not significant P⩾0.05. (B) Lysates were made from cell populations that had been treated with 1 μM ATRA for 48 h and/or 1.5 mM VPA or 100 nM LBH589 for 24 h. Immunoblot was used to detect the protein expression of active/cleaved caspase-3, active/cleaved caspase-9, cleaved PARP, and α-tubulin as a loading control.
We next addressed whether cytoprotective effects of ATRA can be overcome through a broader inhibition of HDACs. LBH589, currently the most potent and clinically relevant hydroxamic acid-based HDACi (Pietschmann et al, 2012a; DeAngelo et al, 2013) blocks HDACs belonging to classes I, II, and IV (pan-HDACi, Figure 1B) (Bradner et al, 2010). Remarkably, LBH589 induced apoptosis in naïve as well as in NB4 cells pre-treated with ATRA (Figure 4A). Immunoblots detecting activation of the initiator caspase-9, the effector caspase-3, and the cleavage of the caspase substrate PARP1 verified that LBH589, but not VPA, can abrogate ATRA-induced cytoprotective effects (Figure 4B).
To ensure specificity of VPA and MS-275, the acetylation of α-tubulin was analysed. HDAC6, which is inhibited by LBH589 but not by VPA and MS-275 (Figure 1B) (Bradner et al, 2010) was shown to be the deacetylase of α-tubulin (Hubbert et al, 2002). We show that solely LBH589 increases the acetylation of α-tubulin, but all three HDACi induce hyperacetylation at histone 3 (Supplementary Figure S4).
Pan-HDACi prevents the accumulation of the differentiation marker C/EBPɛ
We asked whether this beneficial effect of LBH589 might be associated with the differentiation of NB4 cells. LBH589 did not cause significant accumulation of C/EBPɛ (Figures 2B, 3A, and 5A) and of CD11b (Figures 2D and 5C).
Figure 5.

LBH589 inhibits differentiation by ATRA and reduces BCL-xL. (A, B) Lysates were made from NB4 cells treated as noted in Figure 4A. Immunoblot was used to detect the protein expression of C/EBPɛ, BCL-xL, BCL-2, active/cleaved caspase-3, and HDAC1 as a loading control. (B) All samples were analysed on one membrane. (C) The results of FACS analyses are shown. NB4 cells were treated as stated in Figure 4A. Cells were stained for CD11b. (D) Immunoblot analysis of NB4 whole-cell protein lysates was used to assess the levels of PML-RARα. NB4 cells were treated with 100 nM LBH589 (LBH) for 24 h. (E) Lysates were made from NB4 cells treated as noted in Figure 4B. Immunoblot was used to detect the protein expression of BCL-2, BCL-xL, and HDAC1 as a loading control. Lysates treated with VPA and LBH589 were spotted onto one membrane for direct comparison.
We further found that LBH589 suppresses the ATRA-induced, differentiation-associated induction of C/EBPɛ. This loss of C/EBPɛ tied in with caspase-3 activation (Figure 5A). Of note, whereas LBH589 blocked the ATRA-dependent accumulation of C/EBPɛ, C/EBPɛ was still induced by ATRA in NB4 cells exposed to VPA or MS-275 (Figure 5A and B). The lack of differentiation and C/EBPɛ induction in NB4 cells plated with LBH589 and ATRA correlated with cell death (Figures 4A and 5A). These results are reminiscent of our previous data, which demonstrated that ATRA-resistant NB4-R2 cells are unable to mount C/EBPɛ expression and to become resistant to HDACi (Figure 3B).
As LBH589 did not promote any cellular maturation (Figures 2D, 5A and C), we tested whether LBH589 can though evoke an elimination of PML-RARα. In NB4 cells treated with LBH589 we noticed a strong depletion of PML-RARα (Figure 5D). Apparently, granulocytic maturation of NB4 cells requires more than the loss of PML-RARα.
HDACi target the mitochondrial apoptosis regulator BCL-xL
In Eμ-myc lymphoma cells, the anti-apoptotic proteins BCL-xL and BCL-2 can antagonise cell death caused by the pan-HDACi LBH589 and LAQ824 (Ellis et al, 2009; Michels et al, 2013). We found that HDACi reduced the levels of BCL-xL and of the HDACi we tested LBH589 had the strongest impact on BCL-xL (Figure 5A, B, and E). In contrast to BCL-xL, the levels of BCL-2 were reduced in cells exposed to ATRA and neither HDACi had an effect on BCL-2 (Figure 5A, B and E).
BCL-xL can protect APL cells from apoptosis induced by HDACi
Using the BCL protein inhibitor ABT-737 (Lee et al, 2007), we observed that NB4 cells relied on the activity of BCL proteins (data not shown). Since BCL-xL was reduced by HDACi in NB4 cells (Figure 5A, B, and E), we asked whether BCL-xL overexpression could rescue their viability after HDACi exposure. Indeed, NB4 cells overexpressing BCL-xL were protected from VPA-induced apoptosis and were significantly less sensitive to LBH589 (Figure 6A). This finding was confirmed by a lesser occurrence of caspase-3 conversion (Figure 6B). These data and the results, which are shown in Figures 2A–C, 3, 5A, B, and E, identify BCL-xL and C/EBPɛ as markers for the efficacy of HDACi against APL cells.
Figure 6.

BCL-xL rescues NB4 cells from LBH589-induced apoptosis. (A) NB4 cells transduced with BCL-xL or the empty vector (mock) were treated with 1.5 mM VPA or 100 nM LBH589 (LBH) for 24 h. PI-stained cells were analysed by flow cytometry, the subG1 fractions (mean±s.e.m., n⩾3) are shown. Statistics were done with the two-tailed unpaired Student's t-test, *P⩽0.05, **P⩽0.01. (B) Transduced NB4 cells were treated as noted in (A). Immunoblot was probed with antibodies recognising BCL-xL, cleaved caspase-3, to ensure equal sample loading α-tubulin and HDAC1 were analysed.
Discussion
Our results challenge that a combined application of HDACi and ATRA generates superior effects against APL cells. All-trans-retinoic acid-treated APL cells even resist HDACi targeting class I HDACs. These results are supported by the observation that a pre-treatment of NB4 cells with ATRA also blunts apoptosis induced by the class I HDACi sodium butyrate (Licht et al, 2014). Experiments with the pan-HDACi LBH589 further demonstrate that class I, II, and IV HDAC activity is required for the survival of APL cells that differentiated into CD11b- and C/EBPɛ-positive granulocytes. The BCL-xL and C/EBPɛ levels become strongly reduced in NB4 cells treated with ATRA and LBH589 compared with ATRA single treatment. However, C/EBPɛ remained stably expressed when ATRA was combined with VPA or MS-275 (Figures 2B, 3A, B, 5A, B, and E). Thus, the levels of BCL-xL and C/EBPɛ indicate whether an HDACi can abrogate ATRA's cytoprotective effects (Figure 7). While we show that chronic BCL-xL overexpression can significantly reduce pro-apoptotic HDACi effects (Figure 6), it should be kept in mind that the overexpression of an anti-apoptotic BCL protein can generally shut down caspase cascades. In contrast to BCL-xL and C/EBPɛ, the levels of BCL-2 and CD11b do not correlate with the extent of apoptosis induced by HDACi (Figures 4 and 5). While VPA induced CD11b expression (Figure 2C), it could not induce C/EBPɛ expression (Figure 2B). These data were confirmed by quantitative real-time PCR experiments (data not shown) and others also found that HDACi can cause the accumulation of CD11b in leukaemic cells (Sunami et al, 2013).
Figure 7.

Model of the supposed molecular mechanism. Pan-HDACi (LBH589) can deplete BCL-xL and inhibit C/EBPɛ induction by ATRA. Consequently, cells show caspase activation and undergo cell death.
Since a pre-exposure to ATRA antagonises the efficacy of chemotherapeutics triggering DNA damage (Schlenk et al, 2004), pan-HDACi may also abrogate this unwanted process. Furthermore, it was shown that ATRA pre-treatment inhibits apoptosis induced by arsenic trioxide, which is used in the therapy of APL (Jing et al, 2001; Xia et al, 2006). Also for this treatment strategy the application of pan-HDACi may reduce the unwanted, protective side effects of ATRA.
It will also be interesting to see how HDACi affects other types of cellular differentiation, for example, those evoked by 1α,25-hydroxycholecalciferol, by the recently identified interplay between vitamins and EGF-receptor inhibitors (Lainey et al, 2013), or by the class III deacetylase inhibitor Tenovin-6 (Sunami et al, 2013).
Interestingly, expression of C/EBPɛ is not only linked to granulocytic differentiation. CCAAT/enhancer binding protein ɛ is also required for the clonal growth of NB4 and HL60 cells (Morosetti et al, 1997). It should be considered that on the one hand PML-RARα suppresses the C/EBPɛ gene. On the other, PML-RARα accentuated the induction of C/EBPɛ in response to ATRA (Park et al, 1999). This ambivalent regulation was also found for C/EBPβ (Duprez et al, 2003). CCAAT/enhancer binding protein ɛ was not induced by ATRA in NB4-R2 cells (mutant PML-RARα, Figure 3B). These data additionally verify that C/EBPɛ is an RAR-induced gene (Chih et al, 1997; Morosetti et al, 1997; Park et al, 1999; Duprez et al, 2003) and that PML-RARα can suppress the expression of C/EBPɛ (Hu and Saunthararajah, 2012).
Promyelocytic leukemia-retinoic acid receptor-α fusion represses RAR target genes, but due to its oligomerisation via the PML domain the ligated PML-RARα promotes RAR-dependent target gene expression. It is tempting to speculate that such a mechanism prevents the elimination of APL cells via C/EBP upregulation and a consequently blocked induction of apoptosis by endogenous HDACi, such as butyrate (Müller and Krämer, 2010).
Our report is the first that describes C/EBPɛ as a target of HDACi (Figures 2B and 5A). We additionally show that an expression of C/EBPɛ in cells differentiated into granulocytes is eliminated by a pan-HDACi, but not by class I-specific HDACi (Figure 5A and B). Apparently, LBH589 causes molecular alterations that class I HDACi cannot induce. Concentrations of VPA (1.5–5 mM), MS-275 (5–15 μM), and LBH589 (30–100 nM) used in our experiments inhibit HDAC1-3, to an approximately similar level; based on the IC50 values of the inhibitors (Bradner et al, 2010). In a clinical study involving patients with advanced haematologic malignancies, maximum plasma concentrations of 56–200 nM LBH589 were readily achieved (DeAngelo et al, 2013). Hence, the main difference between these HDACi is that LBH589 inhibits additional HDAC isoenzymes (Figure 1B). For example, LBH589 inhibits HDAC6 (Figure 1B) (Bradner et al, 2010), a deacetylase that stabilises several client proteins of HSP90. Such proteins are frequently degraded in the presence of pan-HDACi and this might be associated with the acetylation of HSP90 (Boyault et al, 2007). Further studies are underway to resolve molecular mechanisms by which LBH589 affects C/EBPɛ. Additional work is also required to clarify whether the inhibition of a certain HDAC by LBH589 breaks the ATRA-induced resistance. This task is hampered by the existence of 18 deacetylases in human cells and by the fact that HDACi also cause the loss of certain HDACs in leukaemic cells (Krämer et al, 2013).
One key aspect of our study is that LBH589 causes apoptosis but no signs of cell differentiation. This is not due to a failure to cause PML-RARα degradation. Hence, PML-RARα degradation and HDAC inhibition can be separable from differentiation. Poor induction of differentiation was also noticed when NB4 cells were treated with other pan-HDACi (Kosugi et al, 2001; Bradner et al, 2010). Because PML-RARα enhances the responsiveness of leukaemic cells to pharmacological concentrations of ATRA (Duprez et al, 2003), it may be that the very strong depletion of PML-RARα by LBH589 prevents NB4 cell maturation. Such effects could be clinically relevant as the depletion of PML-RARα is critical for the elimination of PML-RARα-positive leukaemic stem cells (Nasr et al, 2008). Of note, clinically successful ATRA/arsenic trioxide combinations also synergise in PML-RARα elimination and apoptosis, but they do not enhance differentiation (Nasr et al, 2009; Lo-Coco et al, 2013).
In a clinical setting, the radical elimination of APL cells by LBH589 (Figure 4A) may equally prevent the advent of secondary mutations and resistance. Moreover, the inhibition of class I HDACs can accelerate PML-RARα-driven tumorigenesis at the pre-leukaemic stage (Santoro et al, 2013), and pan-HDACi may circumvent this problem. Our results further demonstrate that HDACi are effective against NB4 cells with a mutated PML-RARα protein unresponsive to ATRA (Figure 3B). Others reported similar findings (Altucci and Gronemeyer, 2001; Côté et al, 2002; Fazi et al, 2005). Additionally, our data have implications for treatment schemes including pan-/class-specific HDACi together with retinoids for cancer therapy. We provide evidence that ATRA should not be applied before an HDACi. Nonetheless, the simultaneous addition of HDACi and ATRA does not antagonise apoptosis induction by the HDACi (Figure 2A and B). This finding might explain why a hybrid molecule consisting of ATRA and the 2-aminoanilide tail of MS-275 can induce caspase-8 activation and apoptosis of NB4 cells (De Bellis et al, 2014). Further, it was shown that MS-275 (non-toxic concentrations, 500 nM) sensitised non-PML-RARα cells and primary AML blasts for ATRA-induced differentiation (Blagitko-Dorfs et al, 2013). To our knowledge, our study is the first one systematically analyzing whether and how the sequence of HDACi/ATRA treatment affects the efficacy of these drugs and the fate of APL cells (with wild type or mutant PML-RARα). Moreover, our work reveals significant differences between the efficacy of class I-specific and pan-HDACi against ATRA-treated APL cells. While ATRA and all HDACi tested attenuate the growth of NB4 cells (Supplementary Figure S3), ATRA blunts only pro-apoptotic effects of class I HDACi but not of pan-HDACi (Figures 2, 3, 4, 5). Specifically the path of intrinsic apoptosis, which is controlled by BCL proteins, appears to be relevant for the control of HDACi-induced cell death (Figure 6).
The ATRA/HDACi combinations were tested in several clinical trials with patients suffering from AML or myelodysplastic syndrome (MDS) (Müller and Krämer, 2010; DeAngelo et al, 2013). Twenty-six patients with AML or MDS received VPA/ATRA in combination. One AML patient experienced a minor response and two patients underwent partial remission and blast clearance (Bug et al, 2005). Valproic acid alone or in combination with ATRA was also tested in patients with MDS, de novo AML and AML secondary to MDS (sAML/MDS). Eighteen patients with MDS and sAML/MDS received VPA. Whereas responses including one partial remission were observed in eight patients only treated with VPA, five patients who had VPA in combination with ATRA experienced no beneficial responses (Kuendgen et al, 2004). Similar to our in vitro analysis, Kuendgen et al (2005) found that ATRA should rather be applied after VPA to achieve a benefit for patients. In a phase II study, 75 patients with MDS or relapsed/refractory AML were treated with VPA, alone or with ATRA. Response rates for AML were 16%. Sixty-six patients started on VPA monotherapy and ATRA was given to relapsed patients and non-responders. While haematological improvement was observed in 18 patients, ATRA gave no additional benefits and could not ameliorate a lack of responsiveness to VPA. A further study enrolled 32 sAML/MDS, 22 de novo AML, and 4 sAML/MDS patients. Thirty-one patients obtained VPA as monotherapy and ATRA was added later in 13 patients. Twenty-seven patients received both drugs from the beginning. Response rates were 5% for AML and 16% for MDS (Kuendgen et al, 2006). This study also found no beneficial effect when ATRA was added to VPA. In a further trial, 20 AML patients were treated with VPA combined with ATRA. Out of 20 patients enrolled, there was one complete and two partial remissions (Khanim et al, 2009). One trial assessed the activities of VPA followed by the addition of ATRA in eight high-risk AML patients (Cimino et al, 2006). Five out of eight patients showed stable disease for up to 180 days. In summary, most of these trials failed to show ATRA/VPA co-treatment benefits (Kuendgen et al, 2004, 2005, 2006; Bug et al, 2005;Cimino et al, 2006;Khanim et al, 2009; Müller and Krämer, 2010). We provide results demonstrating that applying ATRA before VPA/MS-275 can produce antagonistic effects. In a patient situation, this would translate into side effects without any benefit. LBH589 might overcome this caveat (Figure 7).
Acknowledgments
Antibody against PML-RARα was a gift from Dr Gronemeier, Straßbourg. MSCV and MSCV-BCL-xL were kindly provided by R Moriggl. Excellent technical assistance was provided by S Reichardt, A Gompf, S Scheiding, M Schreiber, G Greiner, and A Vogel. We thank B Kaina, T Wagner, and C Kosan for helpful discussions. This work was supported by grants from the Deutsche Krebshilfe (#110125 to OHK and GB; 110909 to OHK), from the Wilhelm Sander foundation (#2010.078.2 to OHK) and Deutsche Forschungsgemeinschaft (to OHK #KR 2291/4-1).
Gesine Bug has received honoraria and travel grants from Novartis Pharma GmbH and from Celgene GmbH.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Material
References
- Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1 (3:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- Binder T, Diem H, Fuchs R, Gutensohn K, Nebe T. Pappenheim -Färbung: Beschreibung einer hämatologischen Standardfärbung – Geschichte, Chemie, Durchführung, Artefakte und Problemlösungen. Lab Med. 2012;36 (5:293–309. [Google Scholar]
- Blagitko-Dorfs N, Jiang Y, Duque-Afonso J, Hiller J, Yalcin A, Greve G, Abdelkarim M, Hackanson B, Lübbert M. Epigenetic priming of AML blasts for all-trans retinoic acid-induced differentiation by the HDAC class-I selective inhibitor entinostat. PLoS One. 2013;8 (10:e75258. doi: 10.1371/journal.pone.0075258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26 (37:5468–5476. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- Bradner JE, Mak R, Tanguturi SK, Mazitschek R, Haggarty SJ, Ross K, Chang CY, Bosco J, West N, Morse E, Lin K, Shen JP, Kwiatkowski NP, Gheldof N, Dekker J, DeAngelo DJ, Carr SA, Schreiber SL, Golub TR, Ebert BL. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci USA. 2010;107 (28:12617–12622. doi: 10.1073/pnas.1006774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Krämer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104 (12:2717–2725. doi: 10.1002/cncr.21589. [DOI] [PubMed] [Google Scholar]
- Chih DY, Chumakov AM, Park DJ, Silla AG, Koeffler HP. Modulation of mRNA expression of a novel human myeloid-selective CCAAT/enhancer binding protein gene (C/EBP epsilon) Blood. 1997;90 (8:2987–2994. [PubMed] [Google Scholar]
- Cimino G, Lo-Coco F, Fenu S, Travaglini L, Finolezzi E, Mancini M, Nanni M, Careddu A, Fazi F, Padula F, Fiorini R, Spiriti MA, Petti MC, Venditti A, Amadori S, Mandelli F, Pelicci PG, Nervi C. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Res. 2006;66 (17:8903–8911. doi: 10.1158/0008-5472.CAN-05-2726. [DOI] [PubMed] [Google Scholar]
- Côté S, Rosenauer A, Bianchini A, Seiter K, Vandewiele J, Nervi C, Miller WH., Jr Response to histone deacetylase inhibition of novel PML/RARalpha mutants detected in retinoic acid-resistant APL cells. Blood. 2002;100 (7:2586–2596. doi: 10.1182/blood-2002-02-0614. [DOI] [PubMed] [Google Scholar]
- De Bellis F, Carafa V, Conte M, Rotili D, Petraglia F, Matarese F, Francoijs KJ, Ablain J, Valente S, Castellano R, Goubard A, Collette Y, Mandoli A, Martens JH, de The H, Nebbioso A, Mai A, Stunnenberg HG, Altucci L. Context-selective death of acute myeloid leukemia cells triggered by the novel hybrid retinoid-HDAC inhibitor MC2392. Cancer Res. 2014;74 (8:2328–2339. doi: 10.1158/0008-5472.CAN-13-2568. [DOI] [PubMed] [Google Scholar]
- DeAngelo DJ, Spencer A, Bhalla KN, Prince HM, Fischer T, Kindler T, Giles FJ, Scott JW, Parker K, Liu A, Woo M, Atadja P, Mishra KK, Ottmann OG. Phase Ia/II, two-arm, open-label, dose-escalation study of oral panobinostat administered via two dosing schedules in patients with advanced hematologic malignancies. Leukemia. 2013;27 (8:1628–1636. doi: 10.1038/leu.2013.38. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96 (2:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- Duprez E, Benoit G, Flexor M, Lillehaug JR, Lanotte M. A mutated PML/RARA found in the retinoid maturation resistant NB4 subclone, NB4-R2, blocks RARA and wild-type PML/RARA transcriptional activities. Leukemia. 2000;14 (2:255–261. doi: 10.1038/sj.leu.2401683. [DOI] [PubMed] [Google Scholar]
- Duprez E, Wagner K, Koch H, Tenen DG. C/EBPbeta: a major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO J. 2003;22 (21:5806–5816. doi: 10.1093/emboj/cdg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, Scott CL, Strasser A, Atadja P, Lowe SW, Johnstone RW. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood. 2009;114 (2:380–393. doi: 10.1182/blood-2008-10-182758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazi F, Travaglini L, Carotti D, Palitti F, Diverio D, Alcalay M, McNamara S, Miller WH, Jr, Lo Coco F, Pelicci PG, Nervi C. Retinoic acid targets DNA-methyltransferases and histone deacetylases during APL blast differentiation in vitro and in vivo. Oncogene. 2005;24 (11:1820–1830. doi: 10.1038/sj.onc.1208286. [DOI] [PubMed] [Google Scholar]
- Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391 (6669:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- He LZ, Tolentino T, Grayson P, Zhong S, Warrell RP, Jr, Rifkind RA, Marks PA, Richon VM, Pandolfi PP. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest. 2001;108 (9:1321–1330. doi: 10.1172/JCI11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel FH, Mar BG, Armstrong SA. Self-renewal related signaling in myeloid leukemia stem cells. Int J Hematol. 2011;94 (2:109–117. doi: 10.1007/s12185-011-0901-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess-Stumpp H, Bracker TU, Henderson D, Politz O. MS-275, a potent orally available inhibitor of histone deacetylases—the development of an anticancer agent. Int J Biochem Cell Biol. 2007;39 (7-8:1388–1405. doi: 10.1016/j.biocel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Hu Z, Saunthararajah Y. CEBPE activation in PML-RARA cells by arsenic. Blood. 2012;119 (9:2177–2179. doi: 10.1182/blood-2011-12-395350. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417 (6887:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Jing Y, Wang L, Xia L, Chen GQ, Chen Z, Miller WH, Waxman S. Combined effect of all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia cells in vitro and in vivo. Blood. 2001;97 (1:264–269. doi: 10.1182/blood.v97.1.264. [DOI] [PubMed] [Google Scholar]
- Khanim FL, Bradbury CA, Arrazi J, Hayden RE, Rye A, Basu S, MacWhannell A, Sawers A, Griffiths M, Cook M, Freeman S, Nightingale KP, Grimwade D, Falciani F, Turner BM, Bunce CM, Craddock C. Elevated FOSB-expression; a potential marker of valproate sensitivity in AML. Br J Haematol. 2009;144 (3:332–341. doi: 10.1111/j.1365-2141.2008.07449.x. [DOI] [PubMed] [Google Scholar]
- Koeffler HP. Is there a role for differentiating therapy in non-APL AML. Best Pract Res. 2010;23 (4:503–508. doi: 10.1016/j.beha.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi H, Ito M, Yamamoto Y, Towatari M, Ito M, Ueda R, Saito H, Naoe T. In vivo effects of a histone deacetylase inhibitor, FK228, on human acute promyelocytic leukemia in NOD/Shi-scid/scid mice. Jpn J Cancer Res. 2001;92 (5:529–536. doi: 10.1111/j.1349-7006.2001.tb01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer OH, Müller S, Buchwald M, Reichardt S, Heinzel T. Mechanism for ubiquitylation of the leukemia fusion proteins AML1-ETO and PML-RARalpha. FASEB J. 2008;22 (5:1369–1379. doi: 10.1096/fj.06-8050com. [DOI] [PubMed] [Google Scholar]
- Krämer OH, Stauber RH, Bug G, Hartkamp J, Knauer SK. SIAH proteins: critical roles in leukemogenesis. Leukemia. 2013;27 (4:792–802. doi: 10.1038/leu.2012.284. [DOI] [PubMed] [Google Scholar]
- Kuendgen A, Bug G, Ottmann OG, Haase D, Schanz J, Hildebrandt B, Nachtkamp K, Neukirchen J, Dienst A, Haas R, Germing U, Gattermann N. Treatment of poor-risk myelodysplastic syndromes and acute myeloid leukemia with a combination of 5-azacytidine and valproic acid. Clin Epigenet. 2011;2 (2:389–399. doi: 10.1007/s13148-011-0031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, Germing U, Haas R, Gattermann N. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Ann Hematol. 2005;84 (Suppl 1:61–66. doi: 10.1007/s00277-005-0026-8. [DOI] [PubMed] [Google Scholar]
- Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106 (1:112–119. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- Kuendgen A, Strupp C, Aivado M, Bernhardt A, Hildebrandt B, Haas R, Germing U, Gattermann N. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood. 2004;104 (5:1266–1269. doi: 10.1182/blood-2003-12-4333. [DOI] [PubMed] [Google Scholar]
- Lainey E, Wolfromm A, Sukkurwala AQ, Micol JB, Fenaux P, Galluzzi L, Kepp O, Kroemer G. EGFR inhibitors exacerbate differentiation and cell cycle arrest induced by retinoic acid and vitamin D3 in acute myeloid leukemia cells. Cell Cycle (Georgetown, Tex) 2013;12 (18:2978–2991. doi: 10.4161/cc.26016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EF, Czabotar PE, Smith BJ, Deshayes K, Zobel K, Colman PM, Fairlie WD. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007;14 (9:1711–1713. doi: 10.1038/sj.cdd.4402178. [DOI] [PubMed] [Google Scholar]
- Leiva M, Moretti S, Soilihi H, Pallavicini I, Peres L, Mercurio C, Dal Zuffo R, Minucci S, de The H. Valproic acid induces differentiation and transient tumor regression, but spares leukemia-initiating activity in mouse models of APL. Leukemia. 2012;26 (7:1630–1637. doi: 10.1038/leu.2012.39. [DOI] [PubMed] [Google Scholar]
- Licht V, Noack K, Schlott B, Forster M, Schlenker Y, Licht A, Krämer OH, Heinzel T. Caspase-3 and Caspase-6 cleave STAT1 in leukemic cells. Oncotarget. 2014;5 (8:2305–2317. doi: 10.18632/oncotarget.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391 (6669:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, Specchia G, Sica S, Divona M, Levis A, Fiedler W, Cerqui E, Breccia M, Fioritoni G, Salih HR, Cazzola M, Melillo L, Carella AM, Brandts CH, Morra E, von Lilienfeld-Toal M, Hertenstein B, Wattad M, Lubbert M, Hanel M, Schmitz N, Link H, Kropp MG, Rambaldi A, La Nasa G, Luppi M, Ciceri F, Finizio O, Venditti A, Fabbiano F, Dohner K, Sauer M, Ganser A, Amadori S, Mandelli F, Dohner H, Ehninger G, Schlenk RF, Platzbecker U. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. New Engl J Med. 2013;369 (2:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F, Altucci L, Stunnenberg HG. PML-RARalpha/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia. Cancer Cell. 2010;17 (2:173–185. doi: 10.1016/j.ccr.2009.12.042. [DOI] [PubMed] [Google Scholar]
- McMullin MF, Nugent E, Thompson A, Hull D, Jones FG, Grimwade D. Prolonged molecular remission in PML-RARalpha-positive acute promyelocytic leukemia treated with minimal chemotherapy followed by maintenance including the histone deacetylase inhibitor sodium valproate. Leukemia. 2005;19 (9:1676–1677. doi: 10.1038/sj.leu.2403883. [DOI] [PubMed] [Google Scholar]
- Michels J, Kepp O, Senovilla L, Lissa D, Castedo M, Kroemer G, Galluzzi L. Functions of BCL-X L at the interface between cell death and metabolism. Int J Cell Biol. 2013;2013:705294. doi: 10.1155/2013/705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morosetti R, Park DJ, Chumakov AM, Grillier I, Shiohara M, Gombart AF, Nakamaki T, Weinberg K, Koeffler HP. A novel, myeloid transcription factor, C/EBP epsilon, is upregulated during granulocytic, but not monocytic, differentiation. Blood. 1997;90 (7:2591–2600. [PubMed] [Google Scholar]
- Müller S, Krämer OH. Inhibitors of HDACs—effective drugs against cancer. Curr Cancer Drug Targets. 2010;10 (2:210–228. doi: 10.2174/156800910791054149. [DOI] [PubMed] [Google Scholar]
- Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C, Rousselot P, Robledo-Sarmiento M, Lallemand-Breitenbach V, Gourmel B, Vitoux D, Pandolfi PP, Rochette-Egly C, Zhu J, de The H. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat Med. 2008;14 (12:1333–1342. doi: 10.1038/nm.1891. [DOI] [PubMed] [Google Scholar]
- Nasr R, Lallemand-Breitenbach V, Zhu J, Guillemin MC, de The H. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin Cancer Res. 2009;15 (20:6321–6326. doi: 10.1158/1078-0432.CCR-09-0209. [DOI] [PubMed] [Google Scholar]
- Paietta E. Expression of cell-surface antigens in acute promyelocytic leukaemia. Best Pract Res. 2003;16 (3:369–385. doi: 10.1016/s1521-6926(03)00042-2. [DOI] [PubMed] [Google Scholar]
- Park DJ, Chumakov AM, Vuong PT, Chih DY, Gombart AF, Miller WH, Jr, Koeffler HP. CCAAT/enhancer binding protein epsilon is a potential retinoid target gene in acute promyelocytic leukemia treatment. J Clin Invest. 1999;103 (10:1399–1408. doi: 10.1172/JCI2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietschmann K, Bolck HA, Buchwald M, Spielberg S, Polzer H, Spiekermann K, Bug G, Heinzel T, Böhmer FD, Krämer OH. Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol Cancer Ther. 2012;11 (11:2373–2383. doi: 10.1158/1535-7163.MCT-12-0129. [DOI] [PubMed] [Google Scholar]
- Pietschmann K, Buchwald M, Müller S, Knauer SK, Kogl M, Heinzel T, Krämer OH. Differential regulation of PML-RARalpha stability by the ubiquitin ligases SIAH1/SIAH2 and TRIAD1. Int J Biochem Cell Biol. 2012;44 (1:132–138. doi: 10.1016/j.biocel.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Raelson JV, Nervi C, Rosenauer A, Benedetti L, Monczak Y, Pearson M, Pelicci PG, Miller WH., Jr The PML/RAR alpha oncoprotein is a direct molecular target of retinoic acid in acute promyelocytic leukemia cells. Blood. 1996;88 (8:2826–2832. [PubMed] [Google Scholar]
- Raffoux E, Chaibi P, Dombret H, Degos L. Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia. Haematologica. 2005;90 (7:986–988. [PubMed] [Google Scholar]
- Raffoux E, Cras A, Recher C, Boelle PY, de Labarthe A, Turlure P, Marolleau JP, Reman O, Gardin C, Victor M, Maury S, Rousselot P, Malfuson JV, Maarek O, Daniel MT, Fenaux P, Degos L, Chomienne C, Chevret S, Dombret H. Phase 2 clinical trial of 5-azacitidine, valproic acid, and all-trans retinoic acid in patients with high-risk acute myeloid leukemia or myelodysplastic syndrome. Oncotarget. 2010;1 (1:34–42. doi: 10.18632/oncotarget.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MJ, Lanotte M. Maturation sensitive and resistant t(15;17) NB4 cell lines as tools for APL physiopathology: nomenclature of cells and repertory of their known genetic alterations and phenotypes. Oncogene. 2001;20 (49:7287–7291. doi: 10.1038/sj.onc.1204863. [DOI] [PubMed] [Google Scholar]
- Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, Barozzi I, Senese S, Fornasari L, Moretti S, Altucci L, Pelicci PG, Chiocca S, Johnstone RW, Minucci S. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121 (17:3459–3468. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]
- Schlenk RF, Fröhling S, Hartmann F, Fischer JT, Glasmacher A, del Valle F, Grimminger W, Götze K, Waterhouse C, Schoch R, Pralle H, Mergenthaler HG, Hensel M, Koller E, Kirchen H, Preiss J, Salwender H, Biedermann HG, Kremers S, Griesinger F, Benner A, Addamo B, Döhner K, Haas R, Döhner H, AML Study Group Ulm Phase III study of all-trans retinoic acid in previously untreated patients 61 years or older with acute myeloid leukemia. Leukemia. 2004;18 (11:1798–1803. doi: 10.1038/sj.leu.2403528. [DOI] [PubMed] [Google Scholar]
- Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41 (1:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Sunami Y, Araki M, Hironaka Y, Morishita S, Kobayashi M, Liew EL, Edahiro Y, Tsutsui M, Ohsaka A, Komatsu N. Inhibition of the NAD-dependent protein deacetylase SIRT2 induces granulocytic differentiation in human leukemia cells. PLoS One. 2013;8 (2:e57633. doi: 10.1371/journal.pone.0057633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, Shepherd L, Willman C, Bloomfield CD, Rowe JM, Wiernik PH. All-trans-retinoic acid in acute promyelocytic leukemia. New Engl J Med. 1997;337 (15:1021–1028. doi: 10.1056/NEJM199710093371501. [DOI] [PubMed] [Google Scholar]
- Warrell RP, Jr, He LZ, Richon V, Calleja E, Pandolfi PP. Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J Natl Cancer Inst. 1998;90 (21:1621–1625. doi: 10.1093/jnci/90.21.1621. [DOI] [PubMed] [Google Scholar]
- Xia L, Wurmbach E, Waxman S, Jing Y. Upregulation of Bfl-1/A1 in leukemia cells undergoing differentiation by all-trans retinoic acid treatment attenuates chemotherapeutic agent-induced apoptosis. Leukemia. 2006;20 (6:1009–1016. doi: 10.1038/sj.leu.2404198. [DOI] [PubMed] [Google Scholar]
- Zhou DC, Kim SH, Ding W, Schultz C, Warrell RP, Jr, Gallagher RE. Frequent mutations in the ligand-binding domain of PML-RARalpha after multiple relapses of acute promyelocytic leukemia: analysis for functional relationship to response to all-trans retinoic acid and histone deacetylase inhibitors in vitro and in vivo. Blood. 2002;99 (4:1356–1363. doi: 10.1182/blood.v99.4.1356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.