

ABSTRACT
Stress granules (SGs) are dynamic cytoplasmic repositories containing translationally silenced mRNAs that assemble upon cellular stress. We recently reported that the SG nucleating protein G3BP1 promotes antiviral activity and is essential in double-stranded RNA-dependent protein kinase (PKR) recruitment to stress granules, thereby driving phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). Here, we delineate the mechanism for SG-dependent PKR activation. We show that G3BP1 and inactive PKR directly interact with each other, dependent on both the NTF2-like and PXXP domains of G3BP1. The G3BP1-interacting protein Caprin1 also directly interacts with PKR, regulates efficient PKR activation at the stress granule, and is also integral for the release of active PKR into the cytoplasm to engage in substrate recognition. The G3BP1-Caprin1-PKR complex represents a new mode of PKR activation and is important for antiviral activity of G3BP1 and PKR during infection with mengovirus. Our data links stress responses and their resultant SGs with innate immune activation through PKR without a requirement for foreign double-stranded RNA (dsRNA) pattern recognition.
IMPORTANCE
Our previous work indicates that stress granules have antiviral activity and mediate innate immunity through functions of G3BP1; however, the mechanistic details of these functions were not resolved. We show that much of the antiviral activity of stress granules is contingent on the function of PKR in a complex with G3BP1 and Caprin1. The PKR-G3BP1-Caprin1 complex undergoes dynamic transitioning within and outside stress granules to accomplish PKR activation and translational repression. This mechanism appears to function distinctly from canonical pattern recognition of double-stranded RNA by PKR. Therefore, this mechanism bridges the stress response with innate immunity, allowing the cell to respond to many cellular stressors and amplify the pathogen pattern recognition systems of innate immunity.
INTRODUCTION
Stress granules (SGs) are dynamic cytoplasmic foci containing translationally silenced messenger RNP (mRNP) condensates which form in response to a wide range of cellular stresses that inhibit protein synthesis through the activation of any of four α subunit of eukaryotic initiation factor 2 (eIF2α) kinases (1, 2). SGs contain many translation initiation factors, 40S ribosomal subunits, mRNAs, and dozens of RNA binding proteins. Ras-GTPase-activating protein (SH3 domain) binding protein 1 (G3BP1) and Caprin1 are SG resident proteins that have been reported to form stable complexes with each other (3–5). Both G3BP1 and Caprin1 are also considered SG nucleating proteins, because expression of either protein results in assembly of SGs independent of exogenous stressors (2, 5–7). Further, depletion of G3BP1 inhibits SG formation in response to several stressors (8–10). Depletion of Caprin1 has not been extensively studied, although sequestration of Caprin1 by a viral protein does inhibit SG formation (11). The effects of Caprin1 or G3BP1 depletion may be caused by disruption of the G3BP1-Caprin1 protein complex, as genetic ablation of either G3BP1 or Caprin1 in mice causes similar neurological defects (12, 13).
Double-stranded RNA (dsRNA)-dependent protein kinase (PKR) is one of the four cellular eIF2α kinases and functions as an RNA sensor that can bind viral double-stranded RNA, autophosphorylate itself, and inhibit translation by phosphorylating translation factor eIF2α (14). PKR is also known to regulate cell fate decisions and innate immunity (14). Viruses employ a plethora of mechanisms to counter PKR activation and thus promote infection (9, 15–17). PKR was recently reported to localize to influenza virus-induced SGs (9, 18), but it is unclear if localization affects PKR function. Our previous data showed that PKR activation can occur after SG assembly, suggesting SGs may promote PKR-mediated innate immune responses (6).
SG assembly during virus infection is countered by many viruses; however, the reasons viruses oppose SGs are unclear. Viruses from different families disrupt or subvert SG proteins by a variety of approaches (8, 16, 19–24), suggesting that stress granules or components play antiviral roles against these viruses. In many cases, it is unclear if the SG per se is antiviral, or if the function of SG proteins is antiviral. G3BP1 is targeted by enteroviruses, alphaviruses, and flaviviruses to promote productive infection, suggesting that some aspect of G3BP1 function in SG assembly or elsewhere is antiviral (8, 21, 22, 25, 26). Caprin1 is also hijacked during infection with Japanese encephalitis virus and vaccinia virus (11, 17). Thus, disruption of either G3BP1 or Caprin1 alone or G3BP1-Caprin1 complexes is an important determinant for viral replication.
Recent reports have linked, but not clearly defined, a role for SGs in activation of innate immune pathways (9, 16, 25). We showed that G3BP1, but not G3BP2, is cleaved during poliovirus infection, resulting in the disassembly of SGs (8). Cleavage of G3BP1 may enhance viral translation by disassembling SGs and/or augment innate immune activation by SGs and/or G3BP1. We previously showed that PKR is activated as large SGs assemble, by using G3BP1-induced SGs as a model (6). Recently, we showed that G3BP1 mediates PKR recruitment to SGs and that G3BP1 is an antiviral protein whose antiviral functions require its PXXP domain (27). However, the mechanistic connections between G3BP1, SGs, and PKR activation that factor into G3BP1 antiviral activity have not been characterized. Together, these data suggest that poliovirus cleaves G3BP1 to disassemble SGs to aid viral translation but also inhibit activation of innate immunity.
Here, we sought to delineate the mechanism for G3BP1 and SG-mediated PKR activation in response to the assembly of large G3BP1-induced SGs. We found that PKR directly interacts with G3BP1 through the NTF2-like and PXXP domains of G3BP1. The recruitment of inactive PKR to SGs through this interaction correlates with its activation. We also show that Caprin1, in a complex with G3BP1, participates in PKR activation and regulates the release of PKR from the stress granule for interaction with its substrates. Importantly, we found that this mechanism restricts viral infection. These data indicate that innate immune defenses and the stress response are not just coincident but rather interdependent through G3BP1-mediated PKR activation. Further, virus-induced SGs provide a mechanism to elicit and amplify PKR and innate immunity without the need for specific pathogen recognition through the pathogen-associated molecular pattern (PAMP)--pattern recognition receptor (PRR) system.
RESULTS
The NTF2-like and PXXP domains of G3BP1 are important for PKR activation.
We recently showed that the antiviral properties of G3BP1 rely on G3BP1 nucleation of SGs and recruitment of PKR to G3BP1-induced SGs. PKR recruitment is dependent on the centrally located PXXP domain (Fig. 1A) (27). However, the mechanistic details of PKR recruitment to SGs, the effects of G3BP1 and SGs on PKR activation, and the importance of this pathway for viruses are as yet unclear. Because we are interested in SG-dependent signaling, we utilize G3BP1-induced SGs as a model to study stress granule function. G3BP1-induced SGs are compositionally similar to SGs that form in response to exogenous stressors (2, 6) and allow induction of stress granules in the absence of activation of many signaling pathways that complicate analysis of SG-dependent signaling pathways.
FIG 1 .

G3BP1 deletions modulate PKR activity. (A) A domain map of G3BP1-GFP fusion proteins used throughout this study, indicating the borders of each domain. (B) HeLa, G3BP1-KO MEF, and U2OS cells were transfected with G3BP1 constructs and stained for PKR. Images were captured using deconvolution microscopy. PKR is represented in red, and G3BP1 (expressed from pG3BP1-GFP) is shown in green. DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei are visible in blue. (C) The indicated GFP-tagged G3BP1 mutants were expressed in HeLa cells, and PKR was depleted with siRNAs. Total PKR, T446 phosphorylated PKR, eIF3C, and GFP were examined by Western blotting, as indicated. (D) Immunofluorescence was used to correlate P-eIF2α with PKR activation in HeLa cells. Previously published methodology was used to quantify P-eIF2α and is described in Materials and Methods (6).
To investigate the mechanism of G3BP1 antiviral activity and the role of PKR, we examined localization of PKR to SGs induced in several cell lines by G3BP1 expression. In HeLa and U2OS cells, we observed strong localization of PKR to G3BP1-induced SGs (Fig. 1B). PKR also colocalized with G3BP1-induced SGs produced during expression of G3BP1-green fluorescent protein (GFP) in mouse embryonic fibroblasts (MEFs) genetically ablated for endogenous G3BP1 expression (Fig. 1B). These results indicate that the signaling pathway linking SGs to PKR activation is likely intact in these cell lines, as we previously reported (6), and is not dependent on endogenous G3BP1. Our previous work implicated the PXXP domain of G3BP1 in PKR localization to SGs, so we sought to determine whether expression of the PXXP deletion mutant of G3BP1 (G3BP1ΔPXXP) correlates with PKR activation (27). We expressed the indicated G3BP1 deletion mutants in HeLa cells and analyzed PKR activation with a phospho-specific antibody against threonine 446 of PKR (Phospho T446 PKR; P-PKR). This site is autophosphorylated by PKR during activation and is therefore a good marker of PKR activity. Strikingly, we found that PKR is not activated when the PXXP deletion mutant of G3BP1 is expressed despite the assembly of granules (Fig. 1C, lane 4). To confirm that the band representing t446 P-PKR is indeed PKR, we depleted PKR with small interfering RNAs (siRNAs), which resulted in the complete loss of the P-PKR signal (Fig. 1C, compare lanes 2 and 3 with lanes 6 and 7).
We previously published a method for following PKR activation in cells containing large G3BP1-induced SGs using P-eIF2α intensity in immunofluorescence microscopy analysis (IFA) (6). G3BP1-induced SGs do not activate other eIF2α kinases (6). Therefore, monitoring eIF2α phosphorylation is a good indicator of PKR activation in cells with large G3BP1-induced SGs. To confirm the results presented in Fig. 1C, we examined HeLa cells expressing mutants of G3BP1 by using IFA (Fig. 1D). This approach confirmed that expression of the G3BP1ΔPXXP mutant does not activate PKR (Fig. 1D). Another G3BP1 deletion lacking the NTF2-like domain (G3BP1ΔNTF2 mutant) (Fig. 1A) does not induce SGs when expressed. We found that PKR is not activated in cells expressing high levels of the G3BP1ΔNTF2 mutant, suggesting that proper assembly of SGs is an important determinant for PKR activation (Fig. 1D). Together, these results indicate that both the NTF2 and PXXP domains are important in PKR activation at the stress granule.
G3BP1 directly interacts with PKR.
Our data suggest that G3BP1 may interact with PKR and regulate activation at the SG. To assess this possibility, we used a purified system with bacterially expressed proteins. PKR is known to autophosphorylate itself in bacterial cells, rendering it active; however, coexpression of λ phosphatase produces inactive PKR (28, 29). Amylose affinity chromatography was performed with maltose binding protein (MBP)-tagged MS2 as a control. MS2 is the coat protein from bacteriophage, which is not expressed in mammalian cells, and provides a negative control. In parallel, a series of MBP-tagged deletions of G3BP1 were also examined (Fig. 1A). Our results indicated that the full-length MBP-G3BP1 and the MBP-tagged G3BP1∆acidic truncation efficiently interacted with inactive PKR but did not interact with active PKR (Fig. 2A, lanes 7 and 9). However, neither the G3BP1ΔNTF2 mutant nor the G3BP1ΔPXXP mutant interacted with inactive PKR, consistent with our PKR activation experiments, illustrating that both the NTF2-like and PXXP domains are required to activate PKR (Fig. 1D). These results indicate that activation of PKR by G3BP1 is more complicated than simple recruitment of PKR to the SG. To further characterize the mechanism of G3BP1-mediated regulation of PKR, amylose chromatography was repeated in the presence of ATP with varied amounts of poly(I:C) in the reaction mixture. Indeed, poly(I:C) caused a reduction in the amount of PKR-G3BP1 complexes, indicating that conformational changes in PKR induced by poly(I:C) binding are sufficient to dissociate or prevent the association of PKR and G3BP1 (Fig. 2B, compare lanes 3 and 4). Alternatively, it is possible that PKR binding to G3BP1 is a lower-affinity interaction than that of PKR and poly(I:C).
FIG 2 .
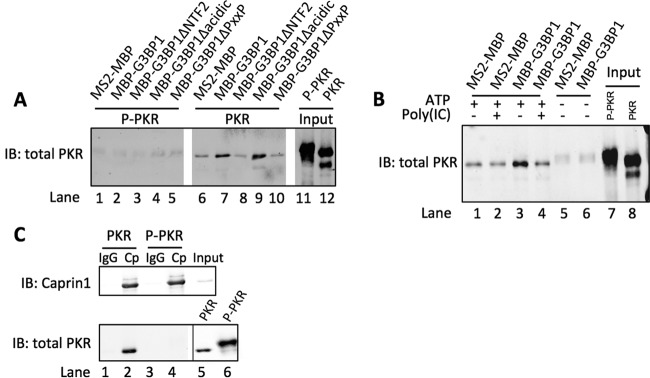
G3BP1 and Caprin1 directly interact with PKR. (A) Purified inactive PKR (PKR) or active PKR (P-PKR) were incubated in the presence of MBP-tagged MS2 or MBP-tagged G3BP1 deletion mutants, as indicated, and precipitated with amylose resin. Precipitated material was examined with Western blotting for total PKR. (B) MBP precipitations were performed as described for panel A with either MBP-tagged MS2 or G3BP1. Reactions were performed in the presence of ATP with or without poly(I:C) to induce a conformational change in PKR protein. (C) Purified Caprin1 and either inactive PKR (PKR) or active PKR (P-PKR) were incubated together. IPs were performed with protein A-Sepharose and either nonspecific IgG or Caprin1 antibodies, as indicated. Precipitates were analyzed by Western blotting for Caprin1 or total PKR. Relative amounts of PKR and P-PKR inputs are shown with different Western blot exposures (separated by a black line).
The G3BP1 binding protein Caprin1 associates with PKR.
Several studies have documented a protein complex containing G3BP1 and Caprin1 (3, 5, 30). Because PKR and Caprin1 binding to G3BP1 requires the NTF2-like domain of G3BP1, we considered whether Caprin1 also interacts with PKR and performed immunoprecipitation (IP) experiments with purified proteins. In this case, we used protein A-Sepharose to precipitate Caprin1-containing complexes with either nonspecific IgG or Caprin1 antibodies. When Caprin1 was incubated with either inactive PKR (PKR) or active PKR (P-PKR), we observed robust IP of inactive PKR but not active PKR (Fig. 2C, compare lanes 2 and 4). These results are similar to what we observed in MBP-G3BP1 pulldown experiments, indicating that Caprin1 directly interacts with PKR.
G3BP1, Caprin1, and PKR coprecipitate with each other.
To investigate the relative strength of the interactions between G3BP1 or Caprin1 and PKR, we performed competition experiments with constant amounts of wild-type MBP-G3BP1, inactive PKR, and increasing concentrations of purified Caprin1. We found that even at concentrations exceeding the concentration of MBP-G3BP1 in the reaction, Caprin1 was unable to dissociate inactive PKR from G3BP1 (Fig. 3A, compare lanes 3 to 6). Interestingly, increasing concentrations of Caprin1 caused a dose-dependent increase in the G3BP1-Caprin1 complexes, indicating that (i) the purified proteins are functional at least at the level of complex formation and (ii) Caprin1 and PKR likely have different binding sites on G3BP1.
FIG 3 .

G3BP1 outcompetes Caprin1 for binding PKR. (A) Competition experiments were performed with constant amounts of MBP-G3BP1 and inactive PKR and increasing concentrations of Caprin1 as indicated. Precipitates were Western blotted for Caprin1, total PKR, and G3BP1. (B) Competition experiments were conducted as described for panel A, except in the presence of increasing amounts of MBP-G3BP1, as indicated. Inputs displayed are for both panels A and B. Panels A and B are from the same gel and exposure conditions. Bands were quantified and normalized to intensity values for lane 2. (C) Caprin1 (siCap1) was depleted from HeLa cells, and cells were subsequently transfected with either GFP alone or G3BP1-GFP. GFP-tagged protein was immunoprecipitated, and precipitates were analyzed by Western blotting with antibodies against GFP, Caprin1, and total PKR. Bands for total PKR were quantified and normalized against lanes 5 and 7 for siCon and siCap1, respectively. (D) G3BP1 was depleted from HeLa cells (sig 1), and cells were transfected with either GFP alone or GFP-Caprin1 followed by IP of GFP-tagged protein as described for panel C. Immunoprecipitates were analyzed by Western blotting with antibodies against GFP, total PKR, t446 P-PKR, and endogenous G3BP1, as indicated. ImageJ was used to quantify total PKR and P-PKR, which were normalized to the respective GFP control, and values for each condition are indicated. siCon indicates cells treated with nontargeting siRNAs for panels C and D. Results for all panels are representative of experiments repeated in triplicate.
Since both G3BP1 and Caprin1 interacted with inactive PKR, we conducted another competition experiment to examine the dynamics of the Caprin1-PKR interaction. Constant amounts of Caprin1 and inactive PKR were added to reactions with increasing concentrations of G3BP1. In this case, increasing concentrations of G3BP1 caused dissociation of inactive PKR from Caprin1 (Fig. 3B; compare lanes 2 to 5). Decreased levels of Caprin1-PKR complexes coincided with increasing G3BP1-Caprin1 complexes. These results suggest that G3BP1 and PKR bind the same site on Caprin1. Together, these results clearly indicate that PKR can bind both G3BP1 and Caprin1, and each component of the complex likely causes allosteric changes that could be important for regulation within cells. An alternative explanation is that the Caprin1-G3BP1 interaction has a higher affinity than that of Caprin1-PKR, causing the observed reduction in inactive PKR precipitated with Caprin1 (Fig. 3B).
Since our experiments indicate that purified G3BP1 or Caprin1 protein can interact with inactive PKR, we considered whether either G3BP1 or Caprin1 protein is required to interact with PKR to gain insight into the interdependence of each component of the protein complex cells. Thus, Caprin1 was depleted from HeLa cells, and then overexpressed G3BP1-GFP was immunoprecipitated. There was no reduction in G3BP1-bound PKR when Caprin1 was depleted (Fig. 3C, compare lanes 6 and 8). In this experiment, we utilized a C-terminal GFP-tagged G3BP1 (G3BP1-GFP) construct and found that fold enrichment of PKR by G3BP1-GFP in immunoprecipitates was less than that observed for the N-terminal GFP-G3BP1 tag (compare Fig. 3C, lanes 5 and 6, to Fig. 4A, lanes 7 and 8). Additional pulldown experiments with the C-terminal G3BP1-GFP construct indicated that G3BP1 with a C-terminal GFP tag consistently enriches for PKR over GFP alone but less efficiently than the N-terminally tagged GFP-G3BP1 construct (see Fig. S1A and B in the supplemental material, which show low enrichment of PKR with G3BP1-GFP). Therefore, an N-terminally tagged GFP-G3BP1 construct was used for most experiments in this study.
FIG 4 .

G3BP1 dimerization is not required for PKR activation. (A) The indicated GFP alone, GFP-G3BP1, the NTF2 domain (amino acids 1 to 135), the GFP-G3BP1 F33W mutant, GFP-G3BP1 Δ1-11aa (lacking the first 11 amino acids of G3BP1), or GFP-G3BP2a was transfected into HeLa cells and precipitated with anti-GFP Sepharose. Precipitated material was analyzed by Western blotting against GFP, Caprin1, total PKR, P-PKR, and endogenous G3BP1. The arrow beside the endogenous G3BP1 blot indicates the correct band, while the asterisk highlights an unknown product originating from expression of the GFP-G3BP1 Δ1-11aa protein. ImageJ was used to quantify induction of P-PKR in the input, total PKR immunoprecipitated, and endogenous G3BP1 precipitated with the indicated GFP-tagged proteins. Band intensity for GFP alone was set to 1 in both cases. Nonessential lanes were removed from the gels in panel A (indicated by white space). (B) HeLa cells were transfected with GFP alone or G3BP1-GFP (green), and cells were stained with antibodies against t446 P-PKR (red). Untransfected (white “U”) and transfected (yellow “T”) cells are indicated. Results for all panels are representative of experiments repeated in triplicate.
When the reciprocal experiment, in which endogenous G3BP1 was depleted and overexpressed GFP-Caprin1 was immunoprecipitated, was conducted, a decrease in Caprin1-bound PKR was observed (Fig. 3D, compare lanes 6 and 8). This decrease was similar to the reduction in G3BP1 in complex with Caprin1 (Fig. 3D, compare lanes 6 and 8). We also found that active PKR was capable of interacting with Caprin1 in cells and was affected by G3BP1 depletion (Fig. 3D, compare lanes 6 and 8). Since direct interaction was not observed with purified PKR and Caprin1 (Fig. 2C), this suggests that a third unknown factor in cells is required for this interaction. Together, these results indicate that G3BP1 is an integral cofactor in the G3BP1-Caprin1-PKR complex, and G3BP1 contributes to the integrity of the complex, promoting PKR activation.
The NTF2-like domain of G3BP1 is critical for PKR activation independent of G3BP1 dimerization.
To further investigate the mechanism of SG-dependent PKR activation and the role of G3BP1 in activation, we expressed the NTF2-like domain of G3BP1 in HeLa cells followed by IP. Surprisingly, we do not observe coprecipitation of PKR with the NTF2-like domain by itself (Fig. 4A, lane 9). These results are consistent with a role for an additional domain, likely the PXXP domain of G3BP1 (Fig. 2A) (27), to stabilize the interaction between G3BP1 and PKR. Indeed, pulldown of G3BP1 deletion mutants from cells shows that both the PXXP and NTF2-like domains are determinants of the strength of the G3BP1-PKR interaction (see Fig. S1C in the supplemental material).
To investigate the sites of G3BP1 and PKR interaction in more detail, we transfected HeLa cells with G3BP1 bearing mutations in the NTF2-like domain (F33W and Δ1-11aa), which are critical for binding FGDF motifs in other proteins, although FGDF motifs do not exist in PKR (31). Strikingly, we found that the Δ1-11aa deletion was still capable of homodimerization with other G3BP1 monomers, while the F33W mutation is less efficient (Fig. 4A, compare lanes 10 and 11 to lane 8). We rationalized that since PKR dimerization is critical for activity (32), G3BP1 dimerization may be important for bringing two molecules of PKR into proximity to promote PKR dimerization and activation. However, since both the Δ1-11aa and F33W mutants displayed reduced interaction with PKR and activation, we conclude that G3BP1 dimerization is not an important function in the PKR activation scheme (Fig. 4A, compare lanes 4 and 5 to lane 2).
Both the G3BP1 F33W and Δ1-11aa mutants also showed reduced binding of G3BP1 to Caprin1 (Fig. 4A, lanes 10 and 11). These results indicate that two determinants of G3BP1 regulate PKR activation: (i) a functional NTF2-like domain in G3BP1, which is also required for SG formation, and (ii) the PXXP domain of G3BP1, as previously reported (Fig. 1C and D) (27). These results also indicate that Caprin1 is important in the G3BP1-Caprin1-PKR complex for PKR activation, since both NTF2 mutants of G3BP1, which are impaired for Caprin1 binding, are deficient in PKR interaction and activation (Fig. 4A).
G3BP1 is 64% identical to G3BP2a, and the NTF2-like domain is 82% identical between the two proteins. Furthermore, G3BP2a contains five PXXP motifs within the disordered PXXP domain, compared to one PXXP motif within G3BP1. Since our data suggest both of these domains are important for PKR interaction, we compared G3BP1 and G3BP2a for PKR binding. We found that G3BP2a is similarly capable of PKR binding (Fig. 4A, compare lanes 8 and 12) and activation (Fig. 4A, compare lanes 2 and 6). Furthermore, G3BP2a strongly interacted with Caprin1 (Fig. 4A, lane 12). Immunofluorescence analysis of individual cells indicated that large G3BP2a-induced granules also induced PKR activation (see Fig. S2 in the supplemental material). Western blotting of lysates from G3BP1 KO and HeLa cells depleted for G3BP1 indicated that G3BP2 expression increased in the absence of G3BP1 expression, suggesting compensatory regulation (see Fig. S2; 3.4- and 2-fold upregulation of G3BP2, respectively). Although this study did not focus on G3BP2, these results are important considerations for levels of PKR activation and response to viral infection in siRNA knockdown experiments presented here and elsewhere.
Caprin1 but not G3BP1 interacts with active PKR in cells.
We noted that Caprin1 was not a strong inducer of PKR activation when expressed by itself (Fig. 3D, compare lanes 1 and 2), while G3BP1 potently induces PKR activation (Fig. 4A, compare lanes 1 and 2). To our surprise, G3BP1 does not efficiently interact with active PKR (Fig. 4A, compare lane 7 with lanes 8 to 11). However, Caprin1 can interact with active PKR in cells (Fig. 3D, compare lanes 5 and 6). These results differ from in vitro pulldown experiments with purified proteins and likely reflect the presence of an additional cofactor(s) that aids Caprin1 binding to active PKR protein. Because our results indicated that G3BP1 is important for recruitment and activation of PKR but does not interact with active PKR, we examined the subcellular localization of active PKR in IFA. As expected, the P-PKR signal was elevated in cells with large G3BP1-induced granules but not in untransfected cells or cells transfected with GFP alone (Fig. 4B). We found that active PKR did not concentrate in G3BP1-induced stress granules (Fig. 4B). Together with previous data implicating SGs and the PXXP domain of G3BP1 in PKR activation, we suggest that active PKR is initially recruited to SGs, activated, and then exits stress granules. This would result in most of the P-PKR signal concentrating outside the SG, which generally coincides with eIF2α localization in the cells used in this study (6).
G3BP1 and Caprin1 coregulate PKR activation within the stress granule during virus infection.
Our results thus far indicate that the G3BP1-Caprin1-PKR interaction is important during G3BP1-mediated PKR activation, but we were interested to know whether this mechanism is an important component of G3BP1 antiviral activity when virus-induced SGs are present. We previously reported that mengovirus, a strain of encephalomyocarditis virus (EMCV), can inhibit SG formation (16). However, a mutant mengovirus containing two point mutations in the Zn finger domain of the L protein (Mengo-Zn) induces PKR activation (Fig. 5A and B) (16). In these experiments, because PKR can be activated by traditional pattern recognition by binding viral dsRNA, we sought to determine if SGs could augment this response. To confirm that G3BP1 is indeed antiviral against Mengo-Zn as it is in other picornaviruses (27), we expressed G3BP1-GFP in HeLa cells and examined PKR activation and viral replication. Similar to that in poliovirus, enterovirus 70, coxsackievirus B3, and coxsackievirus B5, G3BP1 expression suppressed Mengo-Zn replication during ectopic expression (27) (Fig. 5D). Because these cells were infected with a mutant virus, active PKR was present in all cells, including the GFP controls. However, G3BP1-GFP expression induced further elevation of active P-PKR (Fig. 5C, compare lanes 1 and 2). Depletion of G3BP1 also increased viral replication, as predicted (see Fig. S3 in the supplemental material). Deletion of either the NTF2-like domain or PXXP domains ablated PKR activation during infection, whereas deletion of the acidic domain did not, consistent with our protein interaction results (Fig. 5C, compare lanes 3 to 5 with lane 2). The increase in PKR activation from the G3BP1 and G3BP1Δacidic expression was coupled with repressed viral replication. This indicates Mengo-Zn is sensitive to effects of G3BP1-mediated PKR activation (Fig. 5D). Overall, these data demonstrate that mengovirus is repressed by G3BP1 expression and Mengo-Zn infection can be used to study the antiviral effects of the G3BP1-Caprin1-PKR complex.
FIG 5 .

G3BP1 restricts Mengo-Zn infection. (A) HeLa cells were infected with Mengo-Zn at an MOI of 10 for the indicated times, and cells were harvested for Western blotting against total and t446 P-PKR, as indicated. (B) HeLa cells were infected as described for panel A, and cells were harvested at 10 hpi for IF analysis. Cells were stained with antibodies that recognize either P-eIF2α (red) or endogenous G3BP1 (green), as indicated. (C) GFP alone or GFP-tagged G3BP1 mutants, as indicated, were transfected into HeLa cells, and cells were subsequently infected by Mengo-Zn for 14 h prior to the harvesting of viral supernatants and cells for analysis. Lysates were Western blotted for either GFP, t446 P-PKR, total PKR, or eIF3C, as indicated. (D) Plaque assays were performed on viral supernatants from panel C. Results are presented as PFU × 107 per ml. The lower band present in total PKR blots (panels A and C, *) is a nonspecific band appearing with this antibody. Results for all panels are representative of experiments repeated in triplicate.
To study the antiviral effects of SGs and the G3BP1-Caprin1-PKR complex on Mengo-Zn replication, we depleted either G3BP1 or Caprin1 from HeLa cells (Fig. 6A). Subsequently, cells were infected with Mengo-Zn at a multiplicity of infection (MOI) of 10 and harvested at 14 h postinfection (hpi), when PKR was activated (Fig. 5A). Immunofluorescence analysis was performed to determine SG formation and PKR recruitment in individual cells by using antibodies to Tia1, a canonical SG component, and PKR. We observed a statistically significant decrease in Tia1 SGs per cell when G3BP1 was depleted and a slight increase when Caprin1 was depleted (see Fig. S3) (11). We found that depletion of either G3BP1 or Caprin1 reduced the amount of PKR localizing to SGs (Fig. 6B). However, Caprin1 depletion resulted in a more robust and statistically significant reduction in PKR concentration in stress granules (Fig. 6B). We hypothesize that depletion of G3BP1 did not score a statistically significant effect in many cells because of the presence of G3BP2, which is both capable of PKR activation and compensates for G3BP1 expression (Fig. 5A and 2).
FIG 6 .

G3BP1 and Caprin1 regulate PKR recruitment to antiviral SGs. (A) Western blots for Caprin1, G3BP1, and GAPDH were performed on control, G3BP1, or Caprin1 siRNA-treated HeLa cells, as indicated. (B) PKR localization was detected by IF in HeLa cells treated with siControl, siG3BP1, and siCaprin1 and infected for 14 h with Mengo-Zn. PKR intensity was normalized to Tia1 intensity for each granule and presented as a relative value. (C) HeLa cells treated with control (Con), G3BP1, or Caprin1 (Cap1) siRNAs and infected as described for panel B were stained and quantified for P-eIF2α intensity and are presented in a box and whisker plot. (D) HeLa cells were treated with either control or Caprin1 (Cap1)-specific siRNAs and then transfected with GFP-tagged G3BP1 (green) to induce P-PKR. Cells were fixed and stained for P-eIF2α (red) and DAPI (blue). (E) Levels of P-eIF2α were quantified from panel D as previously described (6). All plots with statistics were analyzed with a Student t test (**, P < 0.01; ***, P < 0.001).
We monitored eIF2α phosphorylation as a proxy for PKR activation (6) during Mengo-Zn infection under G3BP1 and Caprin1 knockdown conditions in which PKR localization to SGs is reduced (Fig. 6B). Despite the expected activation of PKR by viral dsRNA, depletion of either G3BP1 or Caprin1 caused a statistically significant reduction in PKR-dependent eIF2α phosphorylation consistent with the reduction in PKR localization to SGs (Fig. 6C). This finding supports prior results that indicate that the accumulation of inactive PKR to SGs, which is dependent on the G3BP1-Caprin1 complex, is important for PKR activation and release prior to eIF2α phosphorylation.
Because G3BP1 promotes activation of PKR together with Caprin1, we reasoned that depletion of Caprin1 during conditions of G3BP1 overexpression would restrict G3BP1-dependent PKR activation. We examined PKR activation by monitoring eIF2α phosphorylation in cells with large G3BP1-induced stress granules as previously published (6). Using this system, we observed a significant reduction in intensity of eIF2α phosphorylation when Caprin1 was depleted (Fig. 6D) and in cells with large G3BP1-induced stress granules (approximately 50%) (Fig. 6E). Taken together, using either G3BP1 expression or Mengo-Zn infection to activate PKR, we have shown that the G3BP1-Caprin1-PKR complex within SGs is important for PKR activation and subsequent eIF2α phosphorylation.
DISCUSSION
We report here details of a novel mechanism of PKR activation involving interaction with G3BP1 and Caprin1 within SGs, which contributes to the previously reported antiviral function of G3BP1 against several picornaviruses (27). This mechanism shows that SG formation, through G3BP1 and Caprin1, can induce innate immune activation. These results link activation of the cellular stress responses that assemble SGs with innate immune activation.
Taken together, our data suggest a mechanism wherein PKR is recruited to SGs in a complex with G3BP1 and Caprin1 in response to cellular stress. Dynamic interplay between these proteins may not require RNA ligands and mediates PKR activation and release from the SG. Release allows PKR to interact with downstream effectors, such as eIF2α, and likely other innate immune activators (Fig. 7). G3BP1 interacts with inactive PKR through both the NTF2-like and PXXP domains. Deletion of either of these domains abolishes in vitro binding, and point mutations that disrupt the function of the NTF2-like domain reduce PKR binding and activation. These point mutants are unlikely to significantly affect folding of G3BP1 such that a nonfunctional protein is produced. These G3BP1 mutants are also deficient in SG induction due to exogenous expression, in contrast to wild-type G3BP1 (data not shown). Since SGs are dynamic and intrinsically unstable, SGs are unlikely to remain assembled under the dilute conditions of our pulldown experiments. Thus, Caprin1 is likely part of a complex with G3BP1 and inactive PKR when SGs are not assembled (Fig. 7).
FIG 7 .
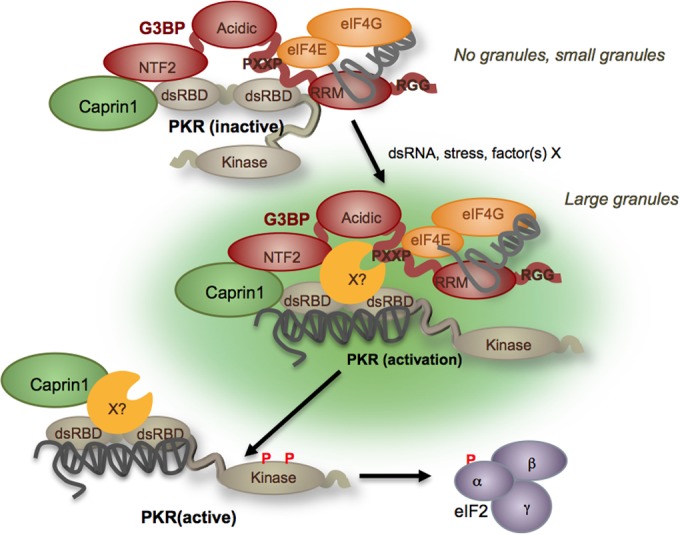
Model for G3BP1-Caprin1 regulation of PKR and eIF2α phosphorylation. Caprin1, G3BP1, and PKR exist in a complex in the cell when PKR is inactive and SGs are not induced. The NTF2-like and PXXP domains are pictured in contact with PKR based on our data. The PXXP domain is involved in interactions with mRNPs containing translation initiation factors. Upon encountering a stress and activation of some stress signaling pathways, G3BP1 mediates aggregation and the appearance of small SGs containing PKR and Caprin1. Our data indicate that these small SGs do not activate PKR, and eIF2α phosphorylation is not observed (6). When small SGs coalesce into large SGs, cellular dsRNA or an unknown factor(s) (X?) activates PKR, causing reorganization of the G3BP1-Caprin1-PKR complex. Subsequently, active PKR and Caprin1 are ejected from the stress granule, where PKR seeks out the PKR substrate eIF2α. It is unclear from our data whether active PKR released from SGs is a monomer (depicted) or dimer (not depicted).
Once cells encounter a stressor that assembles the SG, such as G3BP1 overexpression or mengovirus infection, G3BP1 concentrates PKR in the granule or dynamically recruits it to the stress granule to be activated. In concordance with the importance of the PXXP domain for the interaction, expression of the G3BP1ΔPXXP mutant does assemble defective stress granule-like structures, but PKR is never concentrated nor activated in cells containing G3BP1ΔPXXP mutant granules (27). During Mengo-Zn infection, depletion of either G3BP1 or Caprin1 decreases PKR localization to SGs coincident with decreased eIF2α phosphorylation, which serves as a proxy for PKR activation (6). When Caprin1 is depleted from cells prior to induction of G3BP1 stress granules, levels of P-eIF2α are reduced in cells with large G3BP1-induced granules (approximately 50%). These data, using two inducers of SG assembly, indicate that PKR recruitment to SGs is a prerequisite for activation by this mechanism (Fig. 7). It is likely that P-PKR is released from the SG to seek out its substrates, because our IFA data indicate there is no enrichment of the P-PKR signal in G3BP1-induced SGs (Fig. 2). Whether or not active PKR is released as a monomer or dimer, typically associated with PKR activation, is not clear from our data (32). The process of release of active PKR likely involves Caprin1, which we show can interact with active PKR in cells. The notion that active PKR is released from SGs following activation fits observations that eIF2α, the most well-characterized substrate, is rarely observed in SGs (1, 6).
In contrast to G3BP1 overexpression experiments, many of our G3BP1 depletion experiments showed minor differences in PKR activation. This results from incomplete knockdown but also likely from the continual presence of G3BP2, which also interacts with PKR, can induce PKR activation, and compensates for G3BP1 expression in several cell lines (see Fig. S2). G3BP2 can also bind Caprin1, an important component of this activation mechanism (Fig. 4). Finally, it is important to consider the relative concentrations of the components of this activation mechanism, because each protein interacts directly with the others, as shown by in vitro experiments. For example, overexpression of Caprin1 may not strongly activate PKR by itself because there is insufficient G3BP1 available to accomplish recruitment of PKR to the SG. In the context of virus infection, we propose that the SG-dependent activation by the G3BP1-Caprin1 mechanism is distinct from the canonical dsRNA-binding activation mechanism of PKR activation and further amplifies total PKR activation.
It is also interesting to note that other PKR-interacting proteins have been described that either activate PKR (PACT) or inhibit PKR (MDA7, hDUS2, and p58IPK) (33–38). IPS1 is an innate immune protein (also known as MAVS) and has been shown to promote SG formation and PKR activation (39). However, the exact mechanism of activation of PKR by MAVS and the importance of SGs in that pathway have not been completely elucidated. Another interesting SG antagonist is NF45, which appears to bind G3BP1 (3). Strikingly, NF45 also interacts with Caprin1 and NF90 (also known as NFAR1/2). NFAR1/2 regulates PKR activation in response to the PKR activator poly(I:C), but it is unknown whether that is through G3BP1, Caprin1, or SGs (40). Finally, cyclophilin A is involved in PKR activation. PKR activation by cyclophilin A is likely through a direct interaction and is important in SG assembly, since depletion of cyclophilin A reduces SG assembly (41). Future work will determine if any of these factors play prominent roles in PKR activation in G3BP1-induced granules.
Many viruses have been shown to prevent or disassemble SGs during infection (42, 43). These viruses represent diverse taxonomies, indicating the importance of SGs in a general antiviral response. Within the picornavirus family, poliovirus, coxsackievirus B3, EMCV, and mengovirus have all been shown to block or disassemble SGs (9, 16, 24–26, 44). Our work indicates that viruses can impair innate immune activation, at least through PKR, by disassembling SGs and highlights the importance of this function for infectivity. Because G3BP1 is an important component of both SGs and innate immune activation through PKR, viruses that target G3BP1 for destruction can “kill two birds with one stone.”
The bulk of the innate immune system is dependent on recognition of pathogen patterns via a network of pathogen pattern recognition receptors. Viruses have many mechanisms to counter pattern recognition receptors, and picornavirus proteases cleave many components (e.g., RIG-I, MDA5, and MAVS; 45). However, sensing of pathogen invasion through activation of stress responses is not dependent on pattern recognition, providing a useful alternative sensor for viral invaders. It is also possible that SGs regulate substrate specificity of PKR, which is itself a pattern recognition receptor, by assembling PKR complexes with distinct substrate specificities. In this way, the type of SG could influence downstream PKR-mediated signal transduction in a stress-specific manner.
MATERIALS AND METHODS
Plasmids and cloning.
All expression constructs containing G3BP1 are derivatives of human G3BP1 wild-type coding sequences. C-terminal G3BP1-GFP-λN-tagged constructs are referred to as “GFP-tagged” throughout this study. The F33W was constructed in the peGFP-C1-G3BP1 backbone and kindly provided by Gerald McInerney (Karolinska Institute, Solna, Sweden), and extensive characterization has been reported elsewhere (31). N-terminal GFP-tagged constructs are peGFP-C1-G3BP1, in the peGFP-C1 backbone (Clontech). Caprin1 was expressed from the pcDNA4/TO vector (Life Technologies).
Maltose binding protein bacterial expression constructs for G3BP1 purification were generated by subcloning G3BP1 into pMal-c2e to generate pMal-G3BP1. pMS2-MBP was kindly provided by Tom Cooper (BCM, Houston, TX).
pTYB2-PKR and pTYB2-PKR (PP) were used to generate the active and inactive PKR proteins, respectively. These constructs were kindly provided by Graeme Conn (Emory University, Atlanta, GA) (28).
Virus.
All Mengo-Zn infections were single-cycle infections at an MOI of 10 conducted in 2% fetal bovine serum (FBS)-Dulbecco’s modified Eagle’s medium (DMEM). Mengo-Zn was added to cells and incubated for 4 h prior to cells being washed with PBS and the addition of fresh 2% FBS-DMEM. After the completion of the infection judged by the appearance of cytoplasmic effect in 80% of cells, supernatants were removed and 50% tissue culture infectious dose (TCID50) assays were performed to quantify virus titers. TCID50 assays were performed on each sample in 96-well plates in duplicate on BHK21 cells. Each assay was repeated at least three times.
Cell culture and transfections.
Cells were cultured under standard conditions of 10% FBS in DMEM. G3BP1-knockout (KO) mouse embryonic fibroblasts (MEFs) were a kind gift from Sophy Martin and Jamal Tazi (CNRS, France) (46). For analysis of G3BP1-induced stress granules, G3BP1 expression constructs were transfected using Fugene HD under conditions optimized for SG induction, as previously described (Promega) (6). For expression of other transgenes, cells were transfected with Fugene HD in 2% FBS-DMEM overnight and harvested the following day for analysis. This procedure typically yielded greater than 70% transfection efficiency for GFP expression constructs. siRNAs were performed with a neon electroporation device (Life Technologies) in accordance with the manufacturer’s instructions. siRNAs were transfected at 500 pmol per 3 million cells. siRNA experiments were harvested 48 or 72 h posttransfection. Arsenite was used at 500 µM for 30 min at 37°C for experiments in which arsenite was used as a control for eIF2α phosphorylation.
Immunofluorescence microscopy.
Both epifluorescent and deconvolution microscopy (IF) were performed in the Integrated Microscopy Core at Baylor College of Medicine. Epifluorescence microscopy was performed using a Nikon TE2000 microscope, and deconvolution microscopy experiments were performed using an Applied Precision DeltaVision image restoration microscope with conservative deconvolution algorithms. Microscopy was performed essentially as described previously (6). Primary antibodies were bound either for 2 h at room temperature or overnight at 4°C. All secondary antibodies (Molecular Probes) were used at 1:1,000 for 30 min at 25°C. All images were processed using Adobe Photoshop CS4.
Quantification.
For quantification of eIF2α phosphorylation to determine the percentage of cells with P-eIF2α, microscope parameters were calibrated relative to arsenite-treated control cells containing SGs. Two hundred to 300 cells were counted for each condition. eIF2α phosphorylation was scored as 0 when eIF2α phosphorylation was absent and 1 when present. For the intensity of P-eIF2α measurements, ImageJ was used to set thresholds to quantify P-eIF2α in at least 3 fields with at least 25 cells per condition. For virus infections, more than 150 cells per condition were used. For PKR quantification in SGs, staining was performed with an anti-PKR antibody (ProSci), and at least 50 Tia1-positive SGs were scored in at least 10 cells. PKR intensity was normalized to Tia1 intensity obtained using ImageJ for each granule prior to population analysis.
ImageJ was used to quantify pulldown experiments after Western blotting by using standard densitometry procedures. An internal control (typically total PKR or the corresponding band in a GFP control pulldown) was used to normalize values obtained by quantifying bands in Western blots.
Protein purification.
MS2-MBP, MBP-G3BP1, and MBP-G3BP1 deletion mutants were purified as previously described. Briefly, protein was expressed in Rosetta cells (Novagen) for 3 h at 30°C with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were then lysed by sonication for 1 min in column buffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl, 1 mM EDTA, 10% glycerol), and lysates were cleared by centrifugation and incubated with amylose resin at 4°C with tumbling (New England Biolabs). Subsequently, amylose Sepharose-immobilized MBP-tagged proteins were washed and eluted with 10 mM maltose in column buffer prior to dialysis into buffer containing 0.1 M phosphate-buffered saline (PBS) and 50% glycerol. MBPs were analyzed by SDS-PAGE for protein integrity, and protein concentrations were quantified by comparison to bovine serum albumin (BSA) standards. PKR protein was generated as previously described (28). Caprin1 purified from HEK cells is commercially available (OriGene).
MBP pulldown studies.
For in vitro MBP pulldown studies, MBP-tagged G3BP1 protein (~1 µg) was incubated with PKR protein (~1 µg) for 2 h at 4°C with tumbling at a final volume of 500 µl. Equilibrated amylose resin was then added for an additional 30 min to 1 h with tumbling. Samples were then washed 4 times with NETN (20 mM Tri-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol, 0.1% NP-40) buffer prior to elution, SDS-PAGE, and Western blotting.
Immunoprecipitations.
HeLa cells were harvested in RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM KCl, 2 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate). Cleared lysates were quantified using a bicinchoninic acid (BCA) assay (Pierce). Either 500 µg or 2 mg of protein lysate was then aliquoted into 60% RIPA-40% NETN buffer for pulldown experiments at 4°C. GFP-nAb magnetic Sepharose resin (Allele Biotech) was blocked with 0.5% BSA in NETN for 1 h at 4°C. Blocked resin was added to pulldown reaction mixtures for no more than 1 h at 4°C with tumbling, and reaction mixtures were washed for 5 to 10 min with 0.5 to 1 ml NETN buffer prior to elution with 2× Laemmli sample buffer.
Western blotting.
SDS-PAGE was performed and proteins were transferred to a nitrocellulose membrane in accordance with standard procedures. Membranes were blocked with either 5% BSA in TBST (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 0.1% Tween 20) or Sea Block (Pierce), and the primary antibody was incubated for 3 h at room temperature or overnight at 4°C. Secondary antibodies were incubated for 1 h at room temperature in TBST prior to detection with ECL reagents (Thermo) or Odyssey infrared imaging.
Plaque assays.
Plaque assays were performed in triplicate on supernatants from HeLa cells transfected with G3BP1-GFP and infected with Mengo-Zn at an MOI of 10 for 14 h. Plaque assays were done with half-log dilutions over the range of the titer using BHK cells with a 0.5% methylcellulose overlay. Plaque assays were allowed to develop for 2 days before plaques were stained with 1% crystal violet and counted.
SUPPLEMENTAL MATERIAL
Quantification of G3BP1-PKR interactions and G3BP1 domain dependence in cells. (A) Either GFP alone or G3BP1-GFP was expressed in HeLa cells under conditions that induce SG formation and immunoprecipitated with anti-GFP antibodies. Immunoprecipitates were then analyzed by Western blotting with GFP or total PKR antibodies, as indicated. (B) Since PKR nonspecifically binds anti-GFP Sepharose, we quantified enrichment of PKR in G3BP1 immunoprecipitates from Western blots from three independent experiments (**, P < 0.01). (C) The indicated GFP-tagged G3BP1 deletion mutants were expressed in HeLa cells and precipitated as described for panel A. Western blots were conducted to examine enrichment of Caprin1, total PKR, and t446 P-PKR in immunoprecipitates. Download
G3BP2a activates PKR and compensates for G3BP1 depletion. (A) HeLa cells were transfected with GFP-G3BP2a (green) and stained with antibodies against t446 P-PKR (red). Untransfected and transfected cells in the same field are indicated with a white “U” or yellow “T,” respectively. (B) Wild-type (WT) and G3BP1 KO MEFs or HeLa cells treated with either control or G3BP1-specific siRNAs were analyzed for G3BP2, GAPDH, and G3BP1 expression, as indicated, by using Western blotting. Download
Mengo-Zn-induced SGs per cell during siRNA-mediated depletion of either G3BP1 or Caprin1. (A) Plaque assays were performed on a Mengo-Zn-infected control or G3BP1 siRNA-treated HeLa cells at either 11 or 14 hpi. Uninfected (un) cells were also examined as a control. (B) HeLa cells were treated with control, G3BP1, or Caprin1-specific siRNAs prior to infection with Mengo-Zn at an MOI of 10 and fixed at 14 hpi. Cells were stained with antibodies against Tia1 and G3BP1 and counted. SGs were quantified for each siRNA treatment indicated. SGs are presented as the number of Tia1-positive SGs per cell in a box and whisker plot. The box represents the first and third quintiles, the bar represents the mean, and the error bars represent the maximum and minimum values within the data set (*, P < 0.05; **, P < 0.01). Download
ACKNOWLEDGMENTS
This work was funded by NIH Public Health Service grants AI50237 (R.E.L.) and CA168872 and GM111700-01 (N.K.) and supported by the Integrated Microscopy Core at Baylor College of Medicine with funding from the NIH (HD007495, DK56338, and CA125123), the Dan L. Duncan Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. Work in the lab of Frank J. M. van Kuppeveld is supported by an ECHO grant from the Netherlands Organization for Scientific Research (NWO-CW-700.59.007). Martijn A. Langereis is funded by a personal grant from the Netherlands Organization for Scientific Research (NWO-VENI-863.13.008).
Footnotes
Citation Reineke LC, Kedersha N, Langereis MA, van Kuppeveld FJM, Lloyd RE. 2015. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. mBio 6(2):e02486-14. doi:10.1128/mBio.02486-14.
REFERENCES
- 1.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. 2002. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiina N, Nakayama K. 2014. RNA granule assembly and disassembly modulated by nuclear factor associated with dsRNA 2 and nuclear factor 45. J Biol Chem 289:21163–21180. doi: 10.1074/jbc.M114.556365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. 2007. Distinct structural features of Caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol 27:2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reineke LC, Dougherty JD, Pierre P, Lloyd RE. 2012. Large G3BP-induced granules trigger eIF2α phosphorylation. Mol Biol Cell 23:3499–3510. doi: 10.1091/mbc.E12-05-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol 160:823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Onomoto K, Jogi M, Yoo J-S, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, Fujii M. 2013. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol Cell Biol 33:815–829. doi: 10.1128/MCB.00763-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katoh H, Okamoto T, Fukuhara T, Kambara H, Morita E, Mori Y, Kamitani W, Matsuura Y. 2013. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with caprin-1 and facilitates viral propagation. J Virol 87:489–502. doi: 10.1128/JVI.02186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiina N, Yamaguchi K, Tokunaga M. 2010. RNG105 deficiency impairs the dendritic localization of mRNAs for Na+/K+ ATPase subunit isoforms and leads to the degeneration of neuronal networks. J Neurosci 30:12816–12830. doi: 10.1523/JNEUROSCI.6386-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zekri L, Chebli K, Tourrière H, Nielsen FC, Hansen TV, Rami A, Tazi J. 2005. Control of fetal growth and neonatal survival by the RasGAP-associated endoribonuclease G3BP. Mol Cell Biol 25:8703–8716. doi: 10.1128/MCB.25.19.8703-8716.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. 2006. Inhibition of PKR by RNA and DNA viruses. Virus Res 119:100–110. doi: 10.3201/eid1204.051181. [DOI] [PubMed] [Google Scholar]
- 16.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. Mda5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J Virol 87:6314–6325. doi: 10.1128/JVI.03213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, Connor JH. 2011. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J Virol 85:1581–1593. doi: 10.1128/JVI.02247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoo J-S, Takahasi K, Ng CS, Ouda R, Onomoto K, Yoneyama M, Lai JC, Lattmann S, Nagamine Y, Matsui T, Iwabuchi K, Kato H, Fujita T. 2014. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog 10:e1004012. doi: 10.1371/journal.ppat.1004012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. 2014. Influenza A virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog 10:e1004217. doi: 10.1371/journal.ppat.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pager CT, Schütz S, Abraham TM, Luo G, Sarnow P. 2013. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 435:472–484. doi: 10.1016/j.virol.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog 10:e1004242. doi: 10.1371/journal.ppat.1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khaperskyy DA, Hatchette TF, McCormick C. 2012. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J 26:1629–1639. doi: 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- 24.Borghese F, Michiels T. 2011. The leader protein of cardioviruses inhibits stress granule assembly. J Virol 85:9614–9622. doi: 10.1128/JVI.00480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J Virol 87:9511–9522. doi: 10.1128/JVI.03248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fung G, Ng CS, Zhang J, Shi J, Wong J, Piesik P, Han L, Chu F, Jagdeo J, Jan E, Fujita T, Luo H. 2013. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during cvb3 infection. PLoS One 8:e79546. doi: 10.1371/journal.pone.0079546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reineke LC, Lloyd RE. 2015. The stress granule protein G3BP1 recruits PKR to promote multiple innate immune antiviral responses. J Virol 89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conn GL. 2003. Expression of active RNA-activated protein kinase (PKR) in bacteria. BioTechniques 35:682–684, 686. [DOI] [PubMed] [Google Scholar]
- 29.Lemaire PA, Lary J, Cole JL. 2005. Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J Mol Biol 345:81–90. doi: 10.1016/j.jmb.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 30.Katsafanas GC, Moss B. 2004. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem 279:52210–52217. doi: 10.1074/jbc.M411033200. [DOI] [PubMed] [Google Scholar]
- 31.Panas MD, Schulte T, Thaa B, Sandalova T, Kedersha N, Achour A, McInerney GM. 2015. Viral and cellular proteins containing FGDF motifs bind G3BP to block stress granule formation. PLoS Pathog 11:e1004659. doi: 10.1371/journal.ppat.1004659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole JL. 2007. Activation of PKR: an open and shut case? Trends Biochem Sci 32:57–62. doi: 10.1016/j.tibs.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee TG, Tomita J, Hovanessian AG, Katze MG. 1990. Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of Mr 68,000 from influenza virus-infected cells. Proc Natl Acad Sci U S A 87:6208–6212. doi: 10.1073/pnas.87.16.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee TG, Tomita J, Hovanessian AG, Katze MG. 1992. Characterization and regulation of the 58,000-dalton cellular inhibitor of the interferon-induced, dsRNA-activated protein kinase. J Biol Chem 267:14238–14243. [PubMed] [Google Scholar]
- 35.Mittelstadt M, Frump A, Khuu T, Fowlkes V, Handy I, Patel CV, Patel RC. 2008. Interaction of human tRNA-dihydrouridine synthase-2 with interferon-induced protein kinase PKR. Nucleic Acids Res 36:998–1008. doi: 10.1093/nar/gkm1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pataer A, Vorburger SA, Chada S, Balachandran S, Barber GN, Roth JA, Hunt KK, Swisher SG. 2005. Melanoma differentiation-associated gene-7 protein physically associates with the double-stranded RNA-activated protein kinase PKR. Mol Ther 11:717–723. doi: 10.1016/j.ymthe.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters GA, Hartmann R, Qin J, Sen GC. 2001. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol 21:1908–1920. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang P, Li Y, Xia J, He J, Pu J, Xie J, Wu S, Feng L, Huang X, Zhang P. 2014. IPS-1 plays an essential role in dsRNA-induced stress granule formation by interacting with PKR and promoting its activation. J Cell Sci 127:2471–2482. doi: 10.1242/jcs.139626. [DOI] [PubMed] [Google Scholar]
- 40.Wen X, Huang X, Mok BW, Chen Y, Zheng M, Lau S-Y, Wang P, Song W, Jin D-Y, Yuen K-Y, Chen H. 2014. NF90 exerts antiviral activity through regulation of PKR phosphorylation and stress granules in infected cells. J Immunol 192:3753–3764. doi: 10.4049/jimmunol.1302813. [DOI] [PubMed] [Google Scholar]
- 41.Daito T, Watashi K, Sluder A, Ohashi H, Nakajima S, Borroto-Esoda K, Fujita T, Wakita T. 2014. Cyclophilin inhibitors reduce phosphorylation of RNA-dependent protein kinase to restore expression of IFN-stimulated genes in HCV-infected cells. Gastroenterology 147:463–472. doi: 10.1053/j.gastro.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 42.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White JP, Lloyd RE. 2012. Regulation of stress granules in virus systems. Trends Microbiol 20:175–183. doi: 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White JP, Reineke LC, Lloyd RE. 2011. Poliovirus switches to an eIF2-independent mode of translation during infection. J Virol 85:8884–8893. doi: 10.1128/JVI.00792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Q, Langereis MA, van Kuppeveld FJ. 2014. Induction and suppression of innate antiviral responses by picornaviruses. Cyt Growth Factor Rev 25(5):577–585. doi: 10.1016/j.cytogfr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin S, Zekri L, Metz A, Maurice T, Chebli K, Vignes M, Tazi J. 2013. Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. J Neurochem 125:175–184. doi: 10.1111/jnc.12189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantification of G3BP1-PKR interactions and G3BP1 domain dependence in cells. (A) Either GFP alone or G3BP1-GFP was expressed in HeLa cells under conditions that induce SG formation and immunoprecipitated with anti-GFP antibodies. Immunoprecipitates were then analyzed by Western blotting with GFP or total PKR antibodies, as indicated. (B) Since PKR nonspecifically binds anti-GFP Sepharose, we quantified enrichment of PKR in G3BP1 immunoprecipitates from Western blots from three independent experiments (**, P < 0.01). (C) The indicated GFP-tagged G3BP1 deletion mutants were expressed in HeLa cells and precipitated as described for panel A. Western blots were conducted to examine enrichment of Caprin1, total PKR, and t446 P-PKR in immunoprecipitates. Download
G3BP2a activates PKR and compensates for G3BP1 depletion. (A) HeLa cells were transfected with GFP-G3BP2a (green) and stained with antibodies against t446 P-PKR (red). Untransfected and transfected cells in the same field are indicated with a white “U” or yellow “T,” respectively. (B) Wild-type (WT) and G3BP1 KO MEFs or HeLa cells treated with either control or G3BP1-specific siRNAs were analyzed for G3BP2, GAPDH, and G3BP1 expression, as indicated, by using Western blotting. Download
Mengo-Zn-induced SGs per cell during siRNA-mediated depletion of either G3BP1 or Caprin1. (A) Plaque assays were performed on a Mengo-Zn-infected control or G3BP1 siRNA-treated HeLa cells at either 11 or 14 hpi. Uninfected (un) cells were also examined as a control. (B) HeLa cells were treated with control, G3BP1, or Caprin1-specific siRNAs prior to infection with Mengo-Zn at an MOI of 10 and fixed at 14 hpi. Cells were stained with antibodies against Tia1 and G3BP1 and counted. SGs were quantified for each siRNA treatment indicated. SGs are presented as the number of Tia1-positive SGs per cell in a box and whisker plot. The box represents the first and third quintiles, the bar represents the mean, and the error bars represent the maximum and minimum values within the data set (*, P < 0.05; **, P < 0.01). Download