

ABSTRACT
Farnesol, produced by the polymorphic fungus Candida albicans, is the first quorum-sensing molecule discovered in eukaryotes. Its main function is control of C. albicans filamentation, a process closely linked to pathogenesis. In this study, we analyzed the effects of farnesol on innate immune cells known to be important for fungal clearance and protective immunity. Farnesol enhanced the expression of activation markers on monocytes (CD86 and HLA-DR) and neutrophils (CD66b and CD11b) and promoted oxidative burst and the release of proinflammatory cytokines (tumor necrosis factor alpha [TNF-α] and macrophage inflammatory protein 1 alpha [MIP-1α]). However, this activation did not result in enhanced fungal uptake or killing. Furthermore, the differentiation of monocytes to immature dendritic cells (iDC) was significantly affected by farnesol. Several markers important for maturation and antigen presentation like CD1a, CD83, CD86, and CD80 were significantly reduced in the presence of farnesol. Furthermore, farnesol modulated migrational behavior and cytokine release and impaired the ability of DC to induce T cell proliferation. Of major importance was the absence of interleukin 12 (IL-12) induction in iDC generated in the presence of farnesol. Transcriptome analyses revealed a farnesol-induced shift in effector molecule expression and a down-regulation of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor during monocytes to iDC differentiation. Taken together, our data unveil the ability of farnesol to act as a virulence factor of C. albicans by influencing innate immune cells to promote inflammation and mitigating the Th1 response, which is essential for fungal clearance.
IMPORTANCE
Farnesol is a quorum-sensing molecule which controls morphological plasticity of the pathogenic yeast Candida albicans. As such, it is a major mediator of intraspecies communication. Here, we investigated the impact of farnesol on human innate immune cells known to be important for fungal clearance and protective immunity. We show that farnesol is able to enhance inflammation by inducing activation of neutrophils and monocytes. At the same time, farnesol impairs differentiation of monocytes into immature dendritic cells (iDC) by modulating surface phenotype, cytokine release and migrational behavior. Consequently, iDC generated in the presence of farnesol are unable to induce proper T cell responses and fail to secrete Th1 promoting interleukin 12 (IL-12). As farnesol induced down-regulation of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor, desensitization to GM-CSF could potentially explain transcriptional reprofiling of iDC effector molecules. Taken together, our data show that farnesol can also mediate Candida-host communication and is able to act as a virulence factor.
INTRODUCTION
Quorum sensing (QS) is a major mechanism of intermicrobial communication mediated by molecules that are continuously released from growing microorganisms and accumulate in relation to microbial growth and replication (1–3). Candida albicans was the first fungal species for which a QS system was identified (4, 5). This polymorphic yeast is a common human commensal which frequently causes superficial and invasive opportunistic infections. C. albicans produces three different autoregulatory compounds: tyrosol, farnesoic acid, and the best investigated, farnesol (FOH) (5–7). C. albicans continuously secretes FOH at levels up to 55 µM; however, in local microenvironments, much higher concentrations may exist (8, 9). In addition to C. albicans, FOH can also be produced by other species, some of them closely related to C. albicans, like Candida dubliniensis (8). FOH efficiently controls the transition from yeast to filamentous growth in C. albicans (4). This also results in FOH-mediated inhibition of biofilm formation in C. albicans (10, 11). Furthermore, it has been suggested that FOH protects C. albicans from oxidative stress (12). Aside from the effects of FOH on C. albicans, this QS molecule has been shown to exert effects on other microbial species and host cells. In a mixed heterogeneous population of C. albicans and Pseudomonas aeruginosa, FOH leads to down-regulation of the P. aeruginosa quinolone signal and thus enables the coexistence of these two species (13). Furthermore, FOH enhances the permeability of Staphylococcus aureus and Escherichia coli to exogenous chemical compounds (14) and induces apoptosis in the fungal pathogens Aspergillus nidulans and Aspergillus flavus (15, 16). Studies on the effects of FOH on host cells indicate a promotion of apoptosis, e.g., in human gingival cells and oral and lung carcinoma cells, at concentrations ranging from 30 µM to 300 µM (17–19). Rennemeier et al. reported multiple types of damage in spermatozoa at a concentration of only 25 µM (20). In murine macrophages, FOH treatment resulted in a decreased phagocytic activity, while in epithelial cells, it enhanced interleukin 6 (IL-6) and β-defensin 2 secretion (21, 22). To experimentally address potential immunomodulatory effects, we investigated the impact of FOH on primary human polymorphonuclear neutrophilic granulocytes (PMN), monocytes, and monocyte-derived dendritic cells (DC). Our data show that whereas FOH is able to trigger low-grade activation in human neutrophils, it dramatically impairs functional differentiation of human monocytes into DC and reduces the capacity of DC to trigger protective T cell activation and expansion.
RESULTS
FOH triggers low-grade activation of human innate immune cells.
FOH provided by Sigma used for all experiments was analyzed using high-performance liquid chromatography (HPLC), HPLC-high-resolution electrospray ionisation mass spectrometry (HRESI)-mass spectrometry (MS), and matrix-assisted laser desorption/ionization (MALDI)-MS. These analyses confirmed the molecular formula and excluded any contamination with lipopolysaccharide (LPS) (see Materials and Methods). As FOH has been shown to induce apoptosis in fungal and host cells, we examined the effects of FOH on viability of human monocytes, DC and PMN (17, 23). For PMN, no cell death effects could be observed at concentrations up to 200 µM, whereas monocytes and DC showed significant cell death at concentrations of 200 µM (see Fig. S1A to C in the supplemental material). In control experiments, the Gram-negative QS molecule 3-oxo-homoserine lactone (HSL), which is known for its potent proapoptotic activity, induced significant cell death at a concentration of 25 µM in monocytes and DC (see Fig. S1D and E in the supplemental material). In accordance with previously published data, the presence of serum was found to protect cells against FOH induced cell death (24) (see Fig. S1 in the supplemental material). As lower concentrations of FOH did not induce cell death in human PMN and monocytes, we evaluated whether the phenotype of these cells was altered. After incubation with FOH an activated surface phenotype was observed for both PMN and monocytes, indicating low-grade activation (see Fig. S2A and B in the supplemental material). PMN displayed slightly enhanced surface levels of CD66b, CD11b, and decreased expression of CD16 and CD62L. Furthermore, activation of PMN in the presence of FOH could be demonstrated by a concentration-dependent increase in reactive oxygen species (ROS) (Fig. 1A) and release of myeloperoxidase (MPO), elastase 2 (both derived from azurophilic granules), and lactoferrin (derived from specific granules) (Fig. 1B). To assess whether FOH treatment affects PMN fungicidal activity, phagocytosis and killing of C. albicans were quantified after coincubation for 1 h (Fig. 1C and D). Interestingly and despite the activated phenotype, the ability of FOH-treated neutrophils to phagocytose and kill Candida was not enhanced (Fig. 1C and D). Comparable results could be observed for monocytes. FOH treatment of monocytes resulted in increased levels of CD86 and HLA-DR (see Fig. S2B in the supplemental material) as well as the release of proinflammatory cytokines IL-6, IL-8, MIP-1β, GRO, MIP-1α, tumor necrosis factor alpha (TNF-α), and MCP-1 (Fig. 1E). In contrast, FOH had no impact on IL-1α, IL-1β, and the anti-inflammatory cytokine IL-10 at the tested time points (data not shown). No significant effects could be observed on phagocytosis and killing of C. albicans by monocytes (Fig. 1F and G). These data indicate that despite inducing activation and release of proinflammatory mediators, FOH does not trigger enhanced fungicidal activity of monocytes and neutrophils.
FIG 1 .

FOH induces activation of PMN and monocytes. (A) Quantification of intracellularly generated ROS after FOH treatment for 30 min. Data are percentage of mean fluorescence intensity (MFI). (B) Culture supernatants of PMN treated with FOH at different concentrations for 2 h were analyzed for the release of myeloperoxidase (MPO), lactoferrin, and elastase 2 and revealed significantly increased levels compared to supernatant of mock-treated PMN. All bars in panels A and B show means and standard deviations (SD) from at least 4 independent experiments with PMN from different donors; data were normalized to an untreated control (white bar). (C) Phagocytosis of C. albicans by PMN pretreated with FOH at concentrations ranging from 25 µM to 200 µM was measured via flow cytometry. GFP-expressing C. albicans cells were used, and extracellular fungi were stained with a specific anti-Candida antibody after a 1-h confrontation. (D) Quantification of C. albicans viability after a 1-h confrontation with PMN using the XTT metabolic activity assay. Values were normalized to 100% metabolic activity of fungal cells in medium and are means ± SD from 7 independent experiments. (E) Analysis of cytokine levels in culture supernatants harvested after 24 h of incubation of monocytes with 50 µM FOH. Mock-treated monocytes served as controls for background secretion of cytokines. Data are means ± SD from 4 independent experiments with monocytes from different donors. (F) Phagocytosis of C. albicans by monocytes pretreated with FOH at concentrations ranging from 25 µM to 200 µM was measured via flow cytometry. GFP-expressing C. albicans cells were used, and extracellular fungi were stained with a specific anti-Candida antibody after a 1-h confrontation. (G) Quantification of C. albicans viability after a 1-h confrontation with monocytes using the XTT metabolic activity assay. Values were normalized to 100% metabolic activity of fungal cells in medium and are means and SD from 7 independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FOH impairs differentiation of monocytes into iDC.
To analyze a potential impact of FOH on differentiation of monocytes into immature DC (iDC), cells were exposed to FOH or the solvent control throughout the process of differentiation. After 6 days, we checked the viability of iDC generated in the presence of FOH at concentrations ranging from 10 µM to 100 µM (Fig. 2A and B). We observed no decrease in viability for FOH in concentrations up to 50 µM (viability at 50 µM FOH, 97.3 ± 13.1%; P is not significant) compared to the mock-treated control, whereas 100 µM FOH resulted in reduced viability (78.0 ± 15.7%; P < 0.01). Therefore, subsequent experiments were performed only with FOH concentrations up to 50 µM. To characterize the phenotype of DC after differentiation in the presence of FOH, we investigated the surface expression of several markers for DC differentiation and function (Fig. 2C). The most prominent effect was observed for the transmembrane glycoprotein CD1a. During differentiation to iDC, CD1a is strongly induced on the cell surface (25). However, in the presence of FOH, CD1a was not exposed on the cell surface (at 50 µM FOH, 1.3 ± 0.7%; P < 0.001). This effect could also be observed for exposure to lower concentrations of FOH (10 µM and 25 µM). For CD80, an important costimulatory molecule, we observed a reduction on the cell surface after 6 days of differentiation in the presence of 50 µM FOH (71.0 ± 21.5%; P < 0.01). In contrast, the activation marker CD86 and the major histocompatibility complex class II (MHC-II) molecule HLA-DR showed increased expression on the surfaces of iDC treated with 25 µM and 50 µM FOH (at 50 µM FOH, CD86 expression was 400.3 ± 184.5%, P < 0.001; and HLA-DR expression was 159.3 ± 31.2%, P < 0.001).
FIG 2 .

Impact of FOH on the differentiation process from monocytes to iDC. iDC generated in the presence of FOH at concentrations ranging from 10 µM to 50 µM showed no impairment of viability but alteration in surface marker expression important for maturation (CD83) and antigen presentation (CD1d, CD80, CD86, and HLA-DR). (A) Quantitative analysis of viability of monocyte-derived dendritic cells (iDC) after differentiation in the presence of FOH. (B) Representative scatter plots showing the distribution of annexin V and PI staining for iDC generated in the presence of FOH. (C) The immunophenotype of iDC generated in the presence of FOH was evaluated. For each surface marker (CD14, CD1a, HLA-DR, CD80, CD83, and CD86), quantitative analysis and a representative histogram profile after 6 days of differentiation (left) and additional 24-h stimulation with LPS (right) are shown. Quantitative data are means and SD from at least 4 independent experiments and normalized to a basal level of untreated DC (white bar) (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
iDC undergo a process of maturation after contact with pathogen-associated patterns, which is accompanied by a change of surface marker molecules. To test the ability of iDC generated in the presence of FOH to undergo maturation, these cells were stimulated with LPS for 24 h, which results in a mature DC (mDC) phenotype in control cells. In line with our data for iDC, CD1a also failed to be up-regulated after LPS-induced maturation of iDC to mature DC (mDC) (at 50 µM FOH, 1.7 ± 0.8%, P < 0.001). Levels of CD80 remained low after LPS-induced maturation (at 50 µM FOH, 47.7 ± 21.9%; P < 0.01), and FOH-treated DC failed to efficiently up-regulate CD83 after LPS-induced maturation (at 50 µM FOH, 31.4 ± 19.7%; P < 0.001). Furthermore, the myeloid marker CD14, which is characteristic of monocytes and typically down-regulated during differentiation, was retained on the cell surface after LPS stimulation (at 50 µM FOH, 387.7 ± 252.7%; P < 0.01). Altogether, the altered surface phenotype implied possible functional defects of iDC and mDC differentiated in the presence of FOH.
DC generated in the presence of FOH show altered patterns of transcriptome regulation.
To get a more detailed view of the impact of FOH on DC differentiation, we investigated transcriptomic changes during differentiation under the influence of FOH. For this, cells differentiated in the presence of 50 µM FOH from 4 independent donors (4 biological replicates) were harvested after 3 days and 6 days of differentiation and after exposure to LPS for 6 h (at day 6) and analyzed on an Illumina HumanHT-12 v4 Expression BeadChip. Upon comparison of gene expression of FOH-treated cells with that of mock-treated cells, a total of 704 differentially expressed genes (DEG) were identified, indicating large-scale alterations of the transcriptome by FOH. Distinguishing between the initial differentiation process from monocytes to immature DC (0 days to 6 days) and the final maturation to mature DC (after LPS stimulation), DEG at the 3 day and 6 day time points (“differentiation”) were grouped and compared to DEG at the 6 day time point plus a 6 h LPS stimulation (“maturation”). A total of 381 genes were differentially regulated during differentiation (25 genes at day 3 and 356 additional genes at day 6), of which 196 genes were up-regulated and 185 genes down-regulated. A set of 516 differentially expressed genes (an additional 323 genes after LPS exposure) was identified during maturation of iDC to mDC, including 239 up-regulated and 277 down-regulated genes. Gene correlation networks were constructed for both differentiation and maturation, with genes being grouped into functional categories. Networking visualization revealed differential regulation of genes involved in fungal recognition, cell adhesion and migration at early time points followed by deviated regulation of chemokine and cytokine signaling genes (Fig. 3A and B). Pathway analysis confirmed that a high number of differentially regulated genes are involved in processes like hematopoietic cell lineage differentiation, cytokine-cytokine receptor interaction, antigen processing and presentation, and cell adhesion, indicating broad functional implications of FOH treatment (Fig. 3C and D). After 3 days we could detect down-regulated expression levels of genes encoding proteins involved in antigen processing and presentation (CD74, fold change [FC] of −2.6; CD1e, FC of −2.86) and C-type lectin receptors (CLEC1A, FC of −2.85) and an up-regulation of genes encoding the chemokine MCP-1 (FC of +4.99) and the surface molecule CD1d (FC of +4.47). The number of differentially regulated genes involved in antigen processing and presentation increased throughout the differentiation and maturation process of DC, including up-regulation of CD1d and CD14 (FC of +17.6 and FC of +13.4, respectively) and down-regulation of CD1a and CD1e (FC of −20.08 and −11.46, respectively). An increased expression of genes coding for the chemokines MCP-1, MIP-4α, and eotaxin 2 (FC of +17.85, +9.77, and +6.0, respectively) was detected after 6 days of DC differentiation in the presence of 50 µM FOH. After LPS-induced maturation, expression levels of genes involved in chemotactic activity for neutrophils (CXCL1, FC of −4.01), monocytes (CCL1, FC of −25.46), or activated T cells (CXCL9, FC of −19.84) were significantly reduced. A number of genes encoding C-type lectin receptors, including dectin-1 (FC of −2.32), which is of crucial importance for recognition of fungal pathogens, showed decreased expression levels in the presence of FOH during both differentiation and maturation. Furthermore, we found that several genes with functions in cell adhesion and migration (AMICA1, FC of −3.05; MMP12, FC of −44.67; MMP25, FC of −7.6) were down-regulated and the gene coding for the matrix metallopeptidase (MMP) inhibitor TIMP1 was up-regulated (FC of +4.12). To confirm a functional impact of microarray findings, we first investigated the alteration of surface marker CD1d that was found to be strongly up-regulated at all time points for FOH-treated cells. In our experimental setting, freshly isolated human monocytes showed a low but measurable expression level of CD1d on the cell surface. During normal differentiation, CD1d levels were not affected. However, after FOH treatment, DC showed a strongly increased expression of CD1d at all time points (Fig. 4A). Even at the early differentiation stage (3 days), a 78.9% higher expression was detectable (176.0 ± 105.4%; P < 0.01). This increase was maintained during the maturation process from iDC to mDC. Maintained expression of CD14 for FOH-treated cells could be confirmed by showing significantly increased levels of CD14 on the cell surface of DC generated in the presence of FOH after LPS treatment (387.5 ± 252.7%; P < 0.01) (Fig. 2C) and an 18-fold-higher level of soluble CD14 in cell culture supernatants (Fig. 4B).
FIG 3 .

Networking and analysis of FOH-induced differentially expressed DC genes. (A and B) Networking visualization showed differential regulation of genes involved in fungal recognition, cell adhesion and migration at early time points followed by deviated regulation of chemokine and cytokine signaling genes. A gene correlation network using Cytoscape was constructed based on the whole-genome expression data. Nodes represent up-regulated (green) or down-regulated (red) genes. Edges represent positive (brown) or negative (orange) correlation. Significant correlations of ≥0.85 are displayed distinguishing between the initial differentiation process from monocytes to iDC (3 days and 6 day time point) (A) and the final maturation step to mDC (6 days + 6 h LPS) (B). Genes were grouped into functional categories. For ease of visualization, a smaller core network was edited from the full subnetwork of specific time points and grouped into functional categories. (C) Enrichment of KEGG pathways within the set of differentially expressed genes was carried out to associate genes with known cellular functions. Analysis showed that a high number of differentially regulated genes are involved in processes like hematopoietic cell lineage, cytokine-cytokine receptor interaction, antigen processing and presentation and cell adhesion molecules. Enrichment scores are shown as −log10(P) (P < 0.05).
FIG 4 .

Impact of FOH on dendritic cell characteristics and effector mechanisms. (A) CD1d is enhanced on the surface after FOH treatment. Surface marker CD1d was analyzed during generation of iDC in the presence of FOH (50 µM) at day 3, day 6 and after stimulation with LPS for 24 h. Representative histograms (left) and quantified levels of CD1d (right) are shown. Graphs show means ± SD normalized to an untreated control (white bar). (B and C) Release of soluble CD14 (sCD14) (B) and secretion of cytokines/chemokines (C) were measured in cell culture supernatants after 24 h LPS stimulation of mock-treated iDC and iDC generated in the presence of 50 µM FOH. Bars show means and SD from at least 4 independent experiments with cells from different donors. (D) IL-12 release of iDC after stimulation with LPS (24 h) alone or in addition to FOH, shown as means and SD. (E) Changes in cytokine transcript expression level (6 days + 6 h LPS) and cytokine release in supernatants (after 24 h LPS stimulation) are plotted. Data are displayed as fold change of FOH (50 µM)-treated cells relative to mock-treated cells (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FOH modulates cytokine release by DC and prevents induction of a protective Th1 response.
Transcriptomic profiles had shown differences in the expression profile of various cytokine encoding genes. To confirm a functional relevance of this regulation, we tested a broad set of cytokines to determine if the presence of FOH during differentiation to dendritic cells has an impact on the cytokine profile. Our data reveal that FOH induces significantly elevated levels of proinflammatory cytokines as well as chemokines (Fig. 4C). We detected a >2-fold up-regulation of IL-6, IL-8, MIP-1α, and MIP-1β and >4-fold-higher levels for granulocyte-macrophage colony-stimulating factor (GM-CSF), G-CSF, IL-1 receptor antagonist (IL-1RA), and TNF-α. With a 15-fold up-regulation, MCP-1 (CCL2) showed the most pronounced increase, reflecting the transcriptomic data, which showed that the MCP-1 gene belonged to the most up-regulated genes (12-fold). In contrast, we determined significantly reduced levels for IL-12 (IL-12p40/p70) and the chemokines RANTES, MIG, and I-309 (CCL1). For IL-12, we found a >12-fold-lower release compared to that in mock-treated DC. Interestingly, we also observed a substantial reduction in the secretion of IL-12 for iDC treated with FOH after differentiation (Fig. 4D). For the anti-inflammatory cytokine IL-10 we could determine significantly higher levels for DC differentiated in the presence of 50 µM FOH. These data clearly indicate that iDC generated in the presence of FOH release cytokines, which promote inflammation but dampen the Th1 response, which is crucial for fungal clearance. Furthermore, both MCP-3 and eotaxin 2 were up-regulated in DC by FOH on the transcriptomic level, and elevated concentrations of MCP-3 and eotaxin-2 could be detected in culture supernatants (Fig. 4C). These chemokines are known to be chemoattractants for eosinophils (26). Overall, the observed changes in cytokine release reflect very well the observed changes in transcript level (Fig. 4E).
FOH-treated dendritic cells are restricted in migration.
DC need to be mobile to fulfill their immune function. Spreading on the extracellular matrix displays a functional actin cytoskeleton of the cells, which is important for cell migration, the ability to establish contact with T cells, and the induction of T cell responses (27). After observing a strong down-regulation of genes encoding metalloproteinases (MMP12, MMP25, and ADAM9), which are important for migration of DC, we assumed that iDC generated in the presence of FOH might show a defect in migration and cell spreading. In line with the transcriptomic data, DC generated in the presence of FOH showed 6-times-lower levels of MMP12 in the supernatant than mock-treated cells after stimulation with LPS (Fig. 5A). To elucidate if FOH had an impact on iDC spreading, the ability to spread on fibronectin was investigated (Fig. 5B). After differentiation of monocytes to iDC in the presence of FOH, we allowed the cells to spread on fibronectin-coated slides for 40 min and determined differences using a microscope. iDC generated in the presence of FOH showed impaired spreading compared to the mock-treated control. After 40 min, FOH-treated iDC showed mostly a rounded morphology instead of protrusions; less than 20% of cells (18.2 ± 5.3%; P < 0.001) showed a spreading phenotype, in contrast to mock-treated cells (88.4 ± 9.9%).
FIG 5 .

Altered migrational behavior of iDC generated in the presence of FOH. (A) Elevated levels of MMP12 in the supernatants of DC differentiated in the presence of FOH. Supernatants after 24 h LPS stimulation of mock-treated and FOH-treated iDC were analyzed for MMP12 secretion. (B) FOH-treated iDC showed limited ability to spread. After day 6 of differentiation, mock-treated and FOH-treated iDC were plated on fibronectin-coated plates and allowed to attach for 40 min. Representative images of cell spreading are shown (top, mock-treated cells; bottom, iDC generated in the presence of 50 µM FOH). The percentage of spread cells was scored. Data are means and SD normalized to the level of untreated cells (white bars) (***, P < 0.001).
Impact of FOH on the ability of dendritic cells to act as professional antigen-presenting cells.
All data reported so far suggested a defect of DC generated in the presence of FOH to induce proper T cell responses. To investigate if FOH had an impact on the function of DC to act as antigen-presenting cells (APC), we determined the ability of DC generated in the presence of FOH to induce T cell proliferation. For this purpose, we performed a mixed allogeneic lymphocyte reaction using 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE) labeling. On day 6, mock-treated and FOH-treated mDC were cocultured with allogeneic naive CFSE-labeled CD3+ T cells for 4 days. The percentage of CFSElow T cells was determined by flow cytometry as shown in Fig. 6. While mock-treated mature DC induced T cells to undergo multiple cell division, FOH-treated mDC were significantly less potent in inducing T cell proliferation. For all tested donors, FOH-treated mDC showed a >50%-reduced percentage of CFSElow T cells (49.8 ± 24.9%; P < 0.01) compared to mock-treated mDC (Fig. 6A and B).
FIG 6 .

Impact of FOH on ability of DC to act as professional antigen-presenting cells. FOH-treated mDC induced a >50%-reduced percentage of CFSElow T cells. (A) Representative histograms and dot plots of CFSE fluorescence intensity, gated on CD3+ T cells, are shown after 4 days of in vitro stimulation with untreated mDC and mDC pretreated with FOH (50 µM). As a negative control, CFSE-labeled unstimulated T cells are shown. In the histograms, each peak represents a round of cell division; percentages of cells that have undergone numbers of divisions are shown above the peaks. Representative dot plots show the percentages of CFSElow-gated cells. Similar results were obtained in three independent experiments. (B) Quantitative analysis of the percentage of CFSElow T cells by fluorescence-activated cell sorting analysis. Data are normalized to values for an untreated control (white bar) (**, P < 0.01).
FOH effects may be mediated in part by desensitizing monocytes to GM-CSF.
With regard to the changes in surface phenotype iDC generated in the presence of FOH seem to be trapped in a monocyte-like state. This could be due to a subpar response to the cytokines GM-CSF and IL-4 that induce differentiation. The transcriptomic data show that the gene encoding the GM-CSF receptor (CSF2R) is more than 3-fold down-regulated. Therefore, we checked the expression of the cytokine receptors for GM-CSF and IL-4 on the cell surface of iDC generated in the presence of FOH compared to mock-treated cells (Fig. 7). We observed a more than 50% decreased expression of GM-CSFR (44.5 ± 22.5; P < 0.001) on the cell surface after LPS stimulation. In agreement with the transcriptomic data, no difference in IL-4 receptor expression (data not shown) was detected.
FIG 7 .
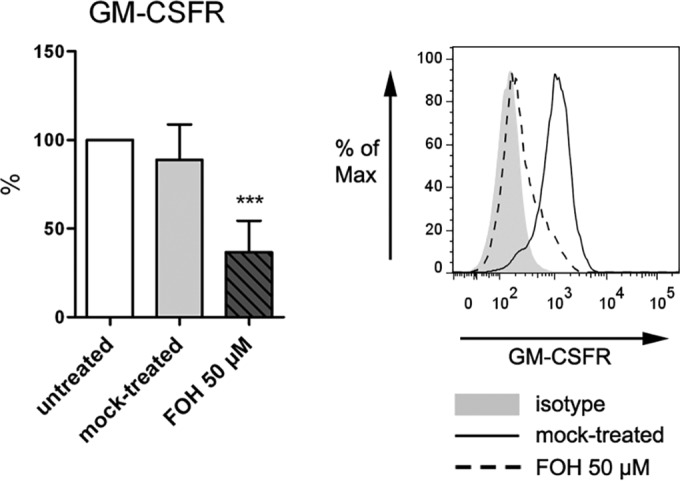
Effect of FOH on the GM-CSF receptor (GM-CSFR). GM-CSFR is reduced on the surface of DC generated in the presence of FOH. Surface receptor expression of GM-CSFR is shown after 24 h LPS stimulation of mock-treated mDC and mDC pretreated with FOH (50 µM). Data are means ± SD normalized to levels of untreated cells (white bar) (***, P < 0.001).
DISCUSSION
In the first study identifying FOH as a QS molecule of the pathogenic yeast C. albicans, Hornby et al. defined two competing hypotheses with FOH either being a possible therapeutic compound due to its ability to block the transition from yeast to filamentous form or acting as a virulence factor by triggering infection-promoting effects in host cells (4). Later, several publications indicated that FOH more likely acts as a virulence factor. C. albicans pretreated with subinhibitory concentrations of fluconazole showed increased extracellular, membrane-bound, and intracellular FOH levels associated with increased virulence and higher mortality rates in a murine infection model (28). In a more direct experiment, administration of FOH during murine C. albicans infection resulted in increased mortality and kidney fungal burden (29). During these experiments, it was also noted that FOH could be detected for several hours in the serum (29). Exogenous administration of FOH inhibited production of IL-12 in murine macrophages, thus preventing effective induction of protective immunity against C. albicans (30). Finally, treatment with pravastatin, which inhibits biosynthesis of FOH, or genetic deletion of DPP3, the phosphatase required for converting farnesyl pyrophosphate to FOH, resulted in reduced mortality and fungal burden (29, 31). In our study, we systematically analyzed effects of FOH on human innate immune cells. In line with previous studies, we showed that in contrast to the bacterial QS molecule homoserine lactone (HSL), proapoptotic activity of FOH can be observed only at very high concentrations in the presence of serum (17, 24, 32). For primary neutrophils and monocytes, we observed a low-grade activation characterized by an activated surface phenotype and induction of effector mechanisms like oxidative burst and secretion of cytokines, similar to what has been observed in previous experiments and also for HSL (33, 34). FOH-induced activation did not result in enhanced fungal uptake and killing. Major effects of FOH were observed for the differentiation of monocytes into iDC. iDC generated in the presence of FOH failed to undergo both phenotypic and functional maturation and retained monocyte specific markers like CD14. Functionally indispensable surface markers, e.g., the costimulatory molecules CD80 and CD86, and the maturation marker CD83 were significantly diminished on the cell surface (35).
The lipid antigen-presenting molecule CD1a, a functional marker for T-cell-activating DC, was minimally expressed on the surface of iDC generated in the presence of FOH (36, 37). In contrast to CD1a, CD1d levels increased on the surface of DC generated in the presence of FOH. CD1d presents lipid antigens to NKT cells that are known to be powerful modulators of immune response by producing Th1 and Th2 cytokines. However, the dominant expression of CD1d seems to be an alternative attempt of inducing an adequate immune response. Cohen et al. showed that CD1d-restricted T cells are important for anti-fungal immunity but pointed out that anti-fungal NKT cell cytokine secretion is dependent on IL-12 (38, 39). However, iDC generated in the presence of FOH showed an impaired release of IL-12 and an enhanced release of the anti-inflammatory cytokine IL-10. Our results, indicating a profound alteration of DC differentiation induced by FOH, were corroborated by analyses of transcriptome changes during the differentiation process, showing a profound rewiring of transcriptional regulation affecting major functional categories of both monocyte-to-DC differentiation and maturation of DC after LPS stimulation. DC generated in the presence of FOH displayed impaired migration and cell spreading and a delay in forming protrusions. Genes relevant for migration of mDC, such as MMP12, AMICA1, and MMP25, were not sufficiently up-regulated (40). In antifungal immunity, DC play a major role in triggering a protective Th1 response via IL-12 (41–45). However, iDC generated in the presence of FOH failed to induce an adequate T cell response. These results establish a novel function for FOH as an immune-modulatory compound. Interestingly, a comparable profile of monocyte-derived DC has been evaluated in patients with chronic mucocutaneous Candida infections (CMC). Ryan et al. (46) treated monocyte-derived DC from autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) and non-APECED CMC patients with C. albicans or LPS to assess cell surface maturation marker and cytokine production. Both APECED and non-APECED patients showed hyperresponsive cytokine expression profiles following LPS stimulation (e.g., hyperproduction of TNF-α) and impaired DC maturation following C. albicans stimulation (impaired up-regulation of CD83) (46). These data are consistent with our findings of suppressed DC maturation after FOH treatment. Those authors suggested that the impaired DC maturation in APECED patients might be the pathogenic mechanism behind the increased susceptibility to Candida infections. Our data suggest that the impaired maturation of DC may also be due to the chronic Candida infection. Compared to other QS molecules, similar effects have been observed for 3-oxo-HSL produced by Pseudomonas aeruginosa (34, 47, 48). Furthermore, our data provide a suitable explanation for the effects of FOH: in transcriptome analyses, we observed a significant down-regulation of the GM-CSF receptor, which could be confirmed by flow cytometry. GM-CSF is known to regulate multiple biological actions via the GM-CSF receptor (49) and is of central importance in promoting dendritic cell development from human monocytes (50–52). Overall, our results show that the fungal quorum-sensing molecule FOH modulates differentiation into iDC and shifts their response pattern toward a nonprotective and dysfunctional response that fails to activate T cells and lacks Th1-promoting cytokines. This establishes a new role of FOH as a virulence factor of C. albicans and strongly argues against the therapeutic use of FOH in systemic C. albicans infection.
MATERIALS AND METHODS
Ethics statement.
This study was conducted according to the principles expressed in the Declaration of Helsinki. All protocols were approved by the Ethics Committee of the University Hospital Jena (permit number 2734-12/09). Written informed consent was obtained from all blood donors.
Reagent preparation.
FOH was obtained as a 4 M stock solution (Sigma-Aldrich) and then diluted in 100% methanol. The FOH purity grade and molecular formula were assessed by HPLC, HPLC-HRESI-MS, and MALDI-MS analyses. Purity was found to be approximately 95%, and no contamination with LPS was detected by MALDI-MS analysis. Analytical HPLC was performed on a Shimadzu LC-10Avp series HPLC system consisting of an autosampler, high-pressure pumps, a column oven, and a photodiode array detector (Shimadzu, Japan). HPLC conditions were as follows: a C18 column (Eurospher 100-5, 250 by 4.6 mm) and a linear gradient from acetonitrile/0.1% (v/v) trifluoroacetic acid 0.5/99.5 to acetonitrile/0.1% (v/v) trifluoroacetic acid 100/0 in 30 min, followed by isocratic conditions of 100% acetonitrile for 10 min, at a flow rate of 1 ml min−1. LC-MS measurements were performed using a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer with an electrospray ion source coupled to an Accela HPLC system (Thermo, Fisher Scientific, Bremen, Germany). HPLC conditions were as follows: a C18 column (Accucore C18; 2.6 µm, 100 by 2.1 mm) and gradient elution (acetonitrile/H2O, each containing 0.1% [vol/vol] formic acid, at a ratio of 5/95, going up to 98/2 in 10 min, and then 98/2 for another 4 min), and a flow rate of 0.2 ml min−1. MALDI-TOF MS measurements were performed with a Bruker UltrafleXtreme mass spectrometer (Bruker Daltonics) in positive reflector mode using α-cyano-4-hydroxycinnamic acid or 2,5-dihydroxyacetophenone as a matrix and collecting data in the range of m/z 5 to 35 kDa. The working concentrations of FOH used ranged from 10 µM to 200 µM. Methanol was used as a solvent control (mock-treated). The biological activity of each lot of FOH was tested by a C. albicans filamentation inhibition assay, as previously described (24). N-3-Oxo-dodecanoyl-l-homoserine lactone (3-oxo-HSL) was purchased from Sigma-Aldrich, dissolved in DMSO, and used at concentrations up to 50 µM.
Cell isolation and stimulation.
Human polymorphonuclear neutrophilic granulocytes (PMN) were isolated and purified from peripheral venous blood of healthy donors by density gradient centrifugation using the Polymorphprep system (Progen Biotechnik GmbH) as described elsewhere (53). Human monocytes were isolated from buffy coats of healthy volunteers. First, primary human peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation using Biocoll (Biochrom AG). Monocytes were separated from PBMC by negative magnetic bead selection via a magnetic cell sorting system (MACS) using isolation kit II (Miltenyi Biotech) or, when used for dendritic cell generation, via CD14+ microbeads (Miltenyi Biotech) according to the manufacturer’s instructions. Freshly isolated monocytes were either used immediately for stimulation assay or differentiated to monocyte-derived dendritic cells (iDC) over 6 days in the presence of GM-CSF (800 U/ml) and IL-4 (1,000 U/ml; Miltenyi Biotech). Culture in LPS (10 ng/ml; Sigma-Aldrich) was used for the final maturation step to mature DC (mDC). All stimulation assays were performed in RPMI 1640 (Biochrom AG) with 10 mM l-glutamine containing 5% heat-inactivated fetal bovine serum (FBS) for PMN and 10% FBS for monocytes and DC. Cells were stimulated with methanol as a solvent control (mock-treated) and FOH at different concentrations (25 µM to 200 µM) at 37°C for various times. For experiments concerning the impact of FOH on the differentiation process to iDC, freshly isolated monocytes were exposed to cytokines for differentiation and in addition to FOH in concentrations ranging from 10 µM to 100 µM or the solvent control. To ensure that FOH was present during the entire differentiation process, FOH was included in the medium, and therefore the concentration was constant even after medium replenishment. Supernatants were obtained by centrifugation and stored at −80°C until further analysis.
Antibodies and flow cytometry.
Phenotypic characterization of PMN, monocytes, and dendritic cells was performed by flow cytometry using fluorochrome-conjugated antibodies. For this purpose, 100 µl of cell suspension was stained with the following antibodies: mouse anti-human CD16-APC, CD66b-FITC, CD62L-FITC, and CD11b-PE (for PMN), HLA-DR-PerCp, CD14-FITC, CD1d-APC, and CD116-PE (for monocytes and DC), and CD80-PE, CD1a-PE, CD83-APC, and CD86-V450 (for DC). All antibodies were purchased from BioLegend except CD86-V450, which was obtained from BD. In parallel, staining with the appropriate isotype control (mouse IgG) was performed as a binding specificity control. After 15 min staining at 4°C, cells were washed and harvested in CellWash (BD Bioscience). For determination of cell death, cells were harvested and stained using annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit II (BD Biosciences) according to the manufacturer’s instructions. The percentage of viable (annexin V-FITC-negative and propidium iodide [PI]-negative) cells was quantified, and the distribution of cell death was displayed in a dot plot. Annexin V-positive and PI-negative cells were considered apoptotic, whereas annexin V- and PI-positive cells represent either late apoptotic cells or necrotic cells. All flow cytometry analyses were performed on a FACSCanto II analyzer (BD Bioscience), and the data were analyzed with FlowJo software 7.6.4.
Quantification of oxidative burst.
The oxidative burst of PMN was measured using the Bursttest from Orpegen Pharma. After 30 min of incubation with FOH, 100 µl of cell suspension was treated with the fluorogenic substrate DHR-123. After incubation for 10 min at 37°C, formation of reactive oxidant species was monitored by oxidation of DHR-123 to R-123. After analysis with FlowJo software 7.6.4, results were presented as median fluorescence intensity.
Measurement of secreted proteins.
Following stimulation with FOH (50 µM), secretion of cytokines and other proteins (MMP12) by monocytes, dendritic cells, and antimicrobial peptides (lactoferrin, elastase 2, and myeloperoxidase) released by PMN was assessed in cell-free supernatants using the MilliplexMap kits from Millipore and the ProcartaPlex immunoassay from eBioscience according to the manufacturer’s instructions. Soluble CD14 was measured in cell-free supernatants according to the instructions of the manufacturer of the Quantikine ELISA kit from R&D Systems.
Candida culture.
C. albicans wild-type SC5314 or the GFP-expressing mutant strain was maintained on YPD agar. For experiments, colonies were transferred to YPD medium (2% d-glucose, 1% peptone, 0.5% yeast extract in water) and cultured at 30°C to stationary phase. Germ tubes were induced by culturing in RPMI 1640 medium for 1 h at 37°C. For confrontation with PMN or monocytes, germ tubes were washed in phosphate-buffered saline (PBS) and used at a multiplicity of infection of 0.5.
Phagocytosis assay.
Phagocytosis of C. albicans by PMN and monocytes was measured via flow cytometry after 1 h of coincubation. To distinguish between intra- and extracellular fungal cells, a GFP-expressing C. albicans strain was used, and extracellular fungal cells were stained with a specific anti-Candida antibody after coincubation with immune cells. Phagocytosis rate was calculated as a percentage of all Candida cells associated with either PMN or monocytes.
Metabolic activity assay.
Following confrontation with PMN or monocytes the metabolic activity of Candida wild-type was determined via the XTT {2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)-carbonyl]-2H-tetrazolium hydroxide} (Sigma) assay as described previously (53). In a Tecan Infinite 200 plate reader, absorbance of supernatants was measured at 450 nm. Fungal metabolic activity was determined as percentage of the metabolic activity of Candida in medium.
RNA isolation and quantification and Illumina expression array.
Cells treated with FOH or solvent control were harvested after various times, and the cell pellet was resuspended in RNAprotect cell reagent (Qiagen) and stored at −20°C until further use for RNA isolation. RNA isolation was performed using the RNeasy Plus Mini Kit (Qiagen) corresponding with manufacturer’s instruction. The Illumina TotalPrep RNA amplification kit (Ambion) was used for RNA amplification and cRNA transcription. RNA concentrations and quality were assessed with a NanoDrop 1000 spectrophotometer (Thermo Scientific) and an Agilent 2100 Bioanalyzer (Agilent Technologies). Expression levels of RNA samples were analyzed with an Illumina HumanHT-12 v4 Expression BeadChip kit following the manufacturer’s protocol.
Preprocessing of Illumina HT-12 chips.
Raw microarray data were preprocessed using functions of the R package lumi. Intensity values were log2 transformed and quantile normalized to allow further statistical analysis of the samples. Detection calls were used to select probes with detectable intensity values in at least three samples. Differentially expressed genes were identified using the Limma package. A linear model was generated using provided functions, followed by the application of specific contrasts to obtain significantly differentially expressed probes. Selection criteria required an adjusted P value of <0.05 and a minimum 2-fold change. Probes were linked using the Bioconductor annotation package illuminaHumanv4.db. Enrichment of Gene Ontology categories and KEGG pathways within the set of differentially expressed genes was carried out to associate genes with known cellular functions. Annotation lists and manually downloaded KEGG pathways were tested for statistical enrichment based on a background gene set, which included all annotated genes on the microarray. Significant categories with P values of <0.05 were selected. Pearson product measure correlation (PPMC) was used to calculate correlation. Correlation network figures were visualized using Cytoscape software (http://www.cytoscape.org) version 3.1.1. Significant correlations of ≥0.85 are displayed, with nodes representing genes and edges representing correlation.
Allogeneic mixed lymphocyte reaction.
Dendritic cells were generated from peripheral blood monocytes as described before. At day 5 iDC were stimulated with LPS (10 ng/ml; Sigma-Aldrich) and pulsed with tetanus toxoid (10 μg/ml; Sigma-Aldrich). On day 6, when DC were harvested, fresh CD3+ lymphocytes were obtained from buffy coats by density gradient centrifugation and separation via AutoMACS device (Miltenyi Biotech) using CD3+ microbeads according to the manufacturer’s instructions. Purified CD3+ T cells were suspended in 0.5 µM CFSE (Invitrogen) diluted in RPMI 1640, and incubated at 37°C in the dark for 10 min. An equal volume of RPMI 1640 containing 10% human serum (type AB, PAA Laboratories GmbH) was added to inactivate the extracellular CFSE. The cells were washed three times in PBS and then resuspended in the proliferation medium consisting of RPMI 1640 medium enriched with 10% human serum, IL-2 (10 U/ml; Immunotools), IL-4 (145 U/ml; Miltenyi Biotech), and GM-CSF (140 U/ml). Mature dendritic cells (mDC) were harvested at day 6, washed with PBS, and resuspended in proliferation medium as well. CFSE CD3+ T cells were mixed at a ratio of 10:1 (105 T cells to 104 dendritic cells per well) in a volume of 1 ml. CFSE-labeled T cells alone were used as a negative control. After day 4, cells were harvested, and CFSE fluorescence was analyzed via flow cytometry.
Cell spreading.
Cells were seeded in an 8-well ibidi plate coated with fibronectin (10 µg/ml; Sigma-Aldrich) at a density of 2 × 104 cells per well. Following 40 min of incubation, cell spreading was monitored via microscopy. Nonspread cells were defined as small and round with few or no membrane protrusions, while spread cells were defined as larger cells with visible lamellipodia. Bright field images were obtained with a Zeiss LSM 780 confocal microscope, and the percentage of spread cells was scored.
Statistics.
Statistical significance was determined by a two-tailed unpaired Student’s t test. Data were analyzed using GraphPad Prism v5.0.
Microarray raw data.
Microarray raw data are available at ArrayExpress (https://www.ebi.ac.uk/arrayexpress/arrays/) under accession no. E-MTAB-2813.
SUPPLEMENTAL MATERIAL
Impact of FOH on viability of innate immune cells. (A to C) PMN (A), monocytes (B), and iDC (C) were incubated with different concentrations of FOH for 30 min (PMN) or 24 h (monocytes and DC), and cell death was quantified via flow-cytometric analysis. Representative scatter plots show annexin V and PI staining for mock-treated and FOH-treated cells. Bars show the quantitative analysis of the percentage of viable cells (annexin V-negative and PI-negative cells). All PMN experiments were performed in medium with 5% serum. Viability of monocytes and DC was compared in media with both 5% and 10% serum. (D and E) Quantification of viability of monocytes (D) and DC (E) treated with 3-oxo-HSL (HSL) at different concentrations. Data shown in all diagrams are the means ± SD from at least four independent experiments normalized to the basal level of the untreated control (white bars) (**, P < 0.01; ***, P < 0.001). Download
Activated surface pattern of FOH-treated PMN and monocytes. (A) Representative histogram profiles (left) and arithmetic means (right) show changes in the surface expression levels of activation markers CD66b, CD11b, CD16, and CD62L on primary PMN treated with FOH (dashed line) compared to mock-treated PMN (solid line) following a 30-min treatment. FOH activates monocytes by enhancing surface expression of CD86 and HLA-DR. (B) Representative histogram profiles (left) and quantification of surface levels (right) of activation markers CD86 and HLA-DR on primary monocytes that were either mock treated (solid line, light grey bar) or treated with FOH (dashed line, striped bar) for 24 h. Data are means ± SD normalized to the values for an untreated control (white bar) (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Download
ACKNOWLEDGMENTS
We gratefully acknowledge all volunteers who donated blood for this study. We thank Seána Duggan, Jessica Voigt, and Michael Böhringer for helpful discussion and Cindy Reichmann for technical assistance. In addition, we acknowledge the help of Maria Poetsch with MALDI measurements.
This work was supported by the German Ministry for Education and Science in the program Unternehmen Region (grant number BMBF 03Z2JN21) and by the Deutsche Forschungsgemeinschaft (DFG) within the Collaborative Research Center CRC124 FungiNet (projects A2 to J.L. and C3 to O.K.).
We have no conflicts of interest to declare.
The funding bodies had no role in the design of the study and no input on the decision to publish this work.
Footnotes
Citation Leonhardt I, Spielberg S, Weber M, Albrecht-Eckardt D, Bläss M, Claus R, Barz D, Scherlach K, Hertweck C, Löffler J, Hünniger K, Kurzai O. 2015. The fungal quorum-sensing molecule farnesol activates innate immune cells but suppresses cellular adaptive immunity. mBio 6(2):e00143-15. doi:10.1128/mBio.00143-15.
Contributor Information
Mihai G. Netea, Radboud University Nijmegen Medical Center.
Arturo Zychlinsky, Max Planck Institute for Infection Biology.
REFERENCES
- 1.Atkinson S, Williams P. 2009. Quorum sensing and social networking in the microbial world. J R Soc Interface 6:959–978. doi: 10.1098/rsif.2009.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nealson KH, Platt T, Hastings JW. 1970. Cellular control of the synthesis and activity of the bacterial luminescent system. J Bacteriol 104:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waters CM, Bassler BL. 2005. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol 21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 4.Hornby JM, Jensen EC, Lisec AD, Tasto JJ, Jahnke B, Shoemaker R, Dussault P, Nickerson KW. 2001. Quorum sensing in the dimorphic fungus Candida albicans is mediated by farnesol. Appl Environ Microbiol 67:2982–2992. doi: 10.1128/AEM.67.7.2982-2992.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albuquerque P, Casadevall A. 2012. Quorum sensing in fungi—a review. Med Mycol 50:337–345. doi: 10.3109/13693786.2011.652201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh KB, Miyazawa H, Naito T, Matsuoka H. 2001. Purification and characterization of an autoregulatory substance capable of regulating the morphological transition in Candida albicans. Proc Natl Acad Sci U S A 98:4664–4668. doi: 10.1073/pnas.071404698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Fujita M, Feng Q, Clardy J, Fink GR. 2004. Tyrosol is a quorum-sensing molecule in Candida albicans. Proc Natl Acad Sci U S A 101:5048–5052. doi: 10.1073/pnas.0401416101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber K, Sohr R, Schulz B, Fleischhacker M, Ruhnke M. 2008. Secretion of E,E-farnesol and biofilm formation in eight different Candida species. Antimicrob Agents Chemother 52:1859–1861. doi: 10.1128/AAC.01646-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber K, Schulz B, Ruhnke M. 2010. The quorum-sensing molecule E,E-farnesol—its variable secretion and its impact on the growth and metabolism of Candida species. Yeast 27:727–739. doi: 10.1002/yea.1769. [DOI] [PubMed] [Google Scholar]
- 10.Ramage G, Saville SP, Wickes BL, López-Ribot JL. 2002. Inhibition of Candida albicans biofilm formation by farnesol, a quorum-sensing molecule. Appl Environ Microbiol 68:5459–5463. doi: 10.1128/AEM.68.11.5459-5463.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martins M, Henriques M, Azeredo J, Rocha SM, Coimbra MA, Oliveira R. 2007. Morphogenesis control in Candida albicans and Candida dubliniensis through signaling molecules produced by planktonic and biofilm cells. Eukaryot Cell 6:2429–2436. doi: 10.1128/EC.00252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westwater C, Balish E, Schofield DA. 2005. Candida albicans-conditioned medium protects yeast cells from oxidative stress: a possible link between quorum sensing and oxidative stress resistance. Eukaryot Cell 4:1654–1661. doi: 10.1128/EC.4.10.1654-1661.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cugini C, Calfee MW, Farrow JM III, Morales DK, Pesci EC, Hogan DA. 2007. Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol Microbiol 65:896–906. doi: 10.1111/j.1365-2958.2007.05840.x. [DOI] [PubMed] [Google Scholar]
- 14.Brehm-Stecher BF, Johnson EA. 2003. Sensitization of Staphylococcus aureus and Escherichia coli to antibiotics by the sesquiterpenoids nerolidol, farnesol, bisabolol, and apritone. Antimicrob Agents Chemother 47:3357–3360. doi: 10.1128/AAC.47.10.3357-3360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semighini CP, Hornby JM, Dumitru R, Nickerson KW, Harris SD. 2006. Farnesol-induced apoptosis in Aspergillus nidulans reveals a possible mechanism for antagonistic interactions between fungi. Mol Microbiol 59:753–764. doi: 10.1111/j.1365-2958.2005.04976.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Wang Y, Zhou Y, Wei X. 2014. Farnesol induces apoptosis-like cell death in the pathogenic fungus Aspergillus flavus. Mycologia 106:881–888. doi: 10.3852/13-292. [DOI] [PubMed] [Google Scholar]
- 17.Scheper MA, Shirtliff ME, Meiller TF, Peters BM, Jabra-Rizk MA. 2008. Farnesol, a fungal quorum-sensing molecule triggers apoptosis in human oral squamous carcinoma cells. Neoplasia 10:954–963. doi: 10.1593/neo.8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joo JH, Liao G, Collins JB, Grissom SF, Jetten AM. 2007. Farnesol-induced apoptosis in human lung carcinoma cells is coupled to the endoplasmic reticulum stress response. Cancer Res 67:7929–7936. doi: 10.1158/0008-5472.CAN-07-0931. [DOI] [PubMed] [Google Scholar]
- 19.Saidi S, Luitaud C, Rouabhia M. 2006. In vitro synergistic effect of farnesol and human gingival cells against Candida albicans. Yeast 23:673–687. doi: 10.1002/yea.1389. [DOI] [PubMed] [Google Scholar]
- 20.Rennemeier C, Frambach T, Hennicke F, Dietl J, Staib P. 2009. Microbial quorum-sensing molecules induce acrosome loss and cell death in human spermatozoa. Infect Immun 77:4990–4997. doi: 10.1128/IAI.00586-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abe S, Tsunashima R, Iijima R, Yamada T, Maruyama N, Hisajima T, Abe Y, Oshima H, Yamazaki M. 2009. Suppression of anti-Candida activity of macrophages by a quorum-sensing molecule, farnesol, through induction of oxidative stress. Microbiol Immunol 53:323–330. doi: 10.1111/j.1348-0421.2009.00128.x. [DOI] [PubMed] [Google Scholar]
- 22.Décanis N, Savignac K, Rouabhia M. 2009. Farnesol promotes epithelial cell defense against Candida albicans through Toll-like receptor 2 expression, interleukin-6 and human beta-defensin 2 production. Cytokine 45:132–140. doi: 10.1016/j.cyto.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Shirtliff ME, Krom BP, Meijering RA, Peters BM, Zhu J, Scheper MA, Harris ML, Jabra-Rizk MA. 2009. Farnesol-induced apoptosis in Candida albicans. Antimicrob Agents Chemother 53:2392–2401. doi: 10.1128/AAC.01551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosel DD, Dumitru R, Hornby JM, Atkin AL, Nickerson KW. 2005. Farnesol concentrations required to block germ tube formation in Candida albicans in the presence and absence of serum. Appl Environ Microbiol 71:4938–4940. doi: 10.1128/AEM.71.8.4938-4940.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao X, Sugita M, Van Der Wel N, Lai J, Rogers RA, Peters PJ, Brenner MB. 2002. CD1 molecules efficiently present antigen in immature dendritic cells and traffic independently of MHC class II during dendritic cell maturation. J Immunol 169:4770–4777. doi: 10.4049/jimmunol.169.9.4770. [DOI] [PubMed] [Google Scholar]
- 26.Kariyawasam HH, Robinson DS. 2006. The eosinophil: the cell and its weapons, the cytokines, its locations. Semin Respir Crit Care Med 27:117–127. doi: 10.1055/s-2006-939514. [DOI] [PubMed] [Google Scholar]
- 27.Al-Alwan MM, Rowden G, Lee TD, West KA. 2001. The dendritic cell cytoskeleton is critical for the formation of the immunological synapse. J Immunol 166:1452–1456. doi: 10.4049/jimmunol.166.3.1452. [DOI] [PubMed] [Google Scholar]
- 28.Navarathna DH, Hornby JM, Hoerrmann N, Parkhurst AM, Duhamel GE, Nickerson KW. 2005. Enhanced pathogenicity of Candida albicans pre-treated with subinhibitory concentrations of fluconazole in a mouse model of disseminated candidiasis. J Antimicrob Chemother 56:1156–1159. doi: 10.1093/jac/dki383. [DOI] [PubMed] [Google Scholar]
- 29.Navarathna DH, Hornby JM, Krishnan N, Parkhurst A, Duhamel GE, Nickerson KW. 2007. Effect of farnesol on a mouse model of systemic candidiasis, determined by use of a DPP3 knockout mutant of Candida albicans. Infect Immun 75:1609–1618. doi: 10.1128/IAI.01182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Navarathna DH, Nickerson KW, Duhamel GE, Jerrels TR, Petro TM. 2007. Exogenous farnesol interferes with the normal progression of cytokine expression during candidiasis in a mouse model. Infect Immun 75:4006–4011. doi: 10.1128/IAI.00397-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tashiro M, Kimura S, Tateda K, Saga T, Ohno A, Ishii Y, Izumikawa K, Tashiro T, Kohno S, Yamaguchi K. 2012. Pravastatin inhibits farnesol production in Candida albicans and improves survival in a mouse model of systemic candidiasis. Med Mycol 50:353–360. doi: 10.3109/13693786.2011.610037. [DOI] [PubMed] [Google Scholar]
- 32.Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, Standiford TJ, Ishiguro M, Yamaguchi K. 2003. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun 71:5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner C, Zimmermann S, Brenner-Weiss G, Hug F, Prior B, Obst U, Hänsch GM. 2007. The quorum-sensing molecule N-3-oxododecanoyl homoserine lactone (3OC12-HSL) enhances the host defence by activating human polymorphonuclear neutrophils (PMN). Anal Bioanal Chem 387:481–487. doi: 10.1007/s00216-006-0698-5. [DOI] [PubMed] [Google Scholar]
- 34.Glucksam-Galnoy Y, Sananes R, Silberstein N, Krief P, Kravchenko VV, Meijler MM, Zor T. 2013. The bacterial quorum-sensing signal molecule N-3-oxo-dodecanoyl-L-homoserine lactone reciprocally modulates pro- and anti-inflammatory cytokines in activated macrophages. J Immunol 191:337–344. doi: 10.4049/jimmunol.1300368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kruse M, Rosorius O, Krätzer F, Stelz G, Kuhnt C, Schuler G, Hauber J, Steinkasserer A. 2000. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J Virol 74:7127–7136. doi: 10.1128/JVI.74.15.7127-7136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang CC, Wright A, Punnonen J. 2000. Monocyte-derived CD1a+ and CD1a− dendritic cell subsets differ in their cytokine production profiles, susceptibilities to transfection, and capacities to direct Th cell differentiation. J Immunol 165:3584–3591. doi: 10.4049/jimmunol.165.7.3584. [DOI] [PubMed] [Google Scholar]
- 37.Cernadas M, Lu J, Watts G, Brenner MB. 2009. CD1a expression defines an interleukin-12 producing population of human dendritic cells. Clin Exp Immunol 155:523–533. doi: 10.1111/j.1365-2249.2008.03853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spada FM, Grant EP, Peters PJ, Sugita M, Melián A, Leslie DS, Lee HK, van Donselaar E, Hanson DA, Krensky AM, Majdic O, Porcelli SA, Morita CT, Brenner MB. 2000. Self-recognition of CD1 by gamma/delta T cells: implications for innate immunity. J Exp Med 191:937–948. doi: 10.1084/jem.191.6.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen NR, Tatituri RV, Rivera A, Watts GF, Kim EY, Chiba A, Fuchs BB, Mylonakis E, Besra GS, Levitz SM, Brigl M, Brenner MB. 2011. Innate recognition of cell wall beta-glucans drives invariant natural killer T cell responses against fungi. Cell Host Microbe 10:437–450. doi: 10.1016/j.chom.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hijova E. 2005. Matrix metalloproteinases: their biological functions and clinical implications. Bratisl Lek Listy 106:127–132. [PubMed] [Google Scholar]
- 41.Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O’Garra A. 1995. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol 154:5071–5079. [PubMed] [Google Scholar]
- 42.Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman RM, Romani N, Schuler G. 1996. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur J Immunol 26:659–668. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 43.Romani L. 1997. The T cell response against fungal infections. Curr Opin Immunol 9:484–490. doi: 10.1016/S0952-7915(97)80099-4. [DOI] [PubMed] [Google Scholar]
- 44.Romani L, Puccetti P. 2008. Immune regulation and tolerance to fungi in the lungs and skin. Chem Immunol Allergy 94:124–137. doi: 10.1159/000154957. [DOI] [PubMed] [Google Scholar]
- 45.Romani L. 2011. Immunity to fungal infections. Nat Rev Immunol 11:275–288. doi: 10.1038/nri2939. [DOI] [PubMed] [Google Scholar]
- 46.Ryan KR, Hong M, Arkwright PD, Gennery AR, Costigan C, Dominguez M, Denning D, McConnell V, Cant AJ, Abinun M, Spickett GP, Lilic D. 2008. Impaired dendritic cell maturation and cytokine production in patients with chronic mucocutanous candidiasis with or without APECED. Clin Exp Immunol 154:406–414. doi: 10.1111/j.1365-2249.2008.03778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boontham P, Robins A, Chandran P, Pritchard D, Cámara M, Williams P, Chuthapisith S, McKechnie A, Rowlands BJ, Eremin O. 2008. Significant immunomodulatory effects of Pseudomonas aeruginosa quorum-sensing signal molecules: possible link in human sepsis. Clin Sci (Lond) 115:343–351. doi: 10.1042/CS20080018. [DOI] [PubMed] [Google Scholar]
- 48.Skindersoe ME, Zeuthen LH, Brix S, Fink LN, Lazenby J, Whittall C, Williams P, Diggle SP, Froekiaer H, Cooley M, Givskov M. 2009. Pseudomonas aeruginosa quorum-sensing signal molecules interfere with dendritic cell-induced T-cell proliferation. FEMS Immunol Med Microbiol 55:335–345. doi: 10.1111/j.1574-695X.2008.00533.x. [DOI] [PubMed] [Google Scholar]
- 49.Van de Laar L, Coffer PJ, Woltman AM. 2012. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 119:3383–3393. doi: 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- 50.Sallusto F, Lanzavecchia A. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med 179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Liu YJ. 1999. Reciprocal control of T helper cell and dendritic cell differentiation. Science 283:1183–1186. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 52.Conti L, Gessani S. 2008. GM-CSF in the generation of dendritic cells from human blood monocyte precursors: recent advances. Immunobiology 213:859–870. doi: 10.1016/j.imbio.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 53.Wozniok I, Hornbach A, Schmitt C, Frosch M, Einsele H, Hube B, Löffler J, Kurzai O. 2008. Induction of ERK-kinase signalling triggers morphotype-specific killing of Candida albicans filaments by human neutrophils. Cell Microbiol 10:807–820. doi: 10.1111/j.1462-5822.2007.01086.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Impact of FOH on viability of innate immune cells. (A to C) PMN (A), monocytes (B), and iDC (C) were incubated with different concentrations of FOH for 30 min (PMN) or 24 h (monocytes and DC), and cell death was quantified via flow-cytometric analysis. Representative scatter plots show annexin V and PI staining for mock-treated and FOH-treated cells. Bars show the quantitative analysis of the percentage of viable cells (annexin V-negative and PI-negative cells). All PMN experiments were performed in medium with 5% serum. Viability of monocytes and DC was compared in media with both 5% and 10% serum. (D and E) Quantification of viability of monocytes (D) and DC (E) treated with 3-oxo-HSL (HSL) at different concentrations. Data shown in all diagrams are the means ± SD from at least four independent experiments normalized to the basal level of the untreated control (white bars) (**, P < 0.01; ***, P < 0.001). Download
Activated surface pattern of FOH-treated PMN and monocytes. (A) Representative histogram profiles (left) and arithmetic means (right) show changes in the surface expression levels of activation markers CD66b, CD11b, CD16, and CD62L on primary PMN treated with FOH (dashed line) compared to mock-treated PMN (solid line) following a 30-min treatment. FOH activates monocytes by enhancing surface expression of CD86 and HLA-DR. (B) Representative histogram profiles (left) and quantification of surface levels (right) of activation markers CD86 and HLA-DR on primary monocytes that were either mock treated (solid line, light grey bar) or treated with FOH (dashed line, striped bar) for 24 h. Data are means ± SD normalized to the values for an untreated control (white bar) (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Download