

ABSTRACT
A 32-nucleotide (nt) RNA motif located at the 3′ end of the transmissible gastroenteritis coronavirus (TGEV) genome was found to specifically interact with the host proteins glutamyl-prolyl-tRNA synthetase (EPRS) and arginyl-tRNA synthetase (RRS). This RNA motif has high homology in sequence and secondary structure with the gamma interferon-activated inhibitor of translation (GAIT) element, which is located at the 3′ end of several mRNAs encoding proinflammatory proteins. The GAIT element is involved in the translation silencing of these mRNAs through its interaction with the GAIT complex (EPRS, heterogeneous nuclear ribonucleoprotein Q, ribosomal protein L13a, and glyceraldehyde 3-phosphate dehydrogenase) to favor the resolution of inflammation. Interestingly, we showed that the viral RNA motif bound the GAIT complex and inhibited the in vitro translation of a chimeric mRNA containing this RNA motif. To our knowledge, this is the first GAIT-like motif described in a positive RNA virus. To test the functional role of the GAIT-like RNA motif during TGEV infection, a recombinant coronavirus harboring mutations in this motif was engineered and characterized. Mutations of the GAIT-like RNA motif did not affect virus growth in cell cultures. However, an exacerbated innate immune response, mediated by the melanoma differentiation-associated gene 5 (MDA5) pathway, was observed in cells infected with the mutant virus compared with the response observed in cells infected with the parental virus. Furthermore, the mutant virus was more sensitive to beta interferon than the parental virus. All together, these data strongly suggested that the viral GAIT-like RNA motif modulates the host innate immune response.
IMPORTANCE
The innate immune response is the first line of antiviral defense that culminates with the synthesis of interferon and proinflammatory cytokines to limit virus replication. Coronaviruses encode several proteins that interfere with the innate immune response at different levels, but to date, no viral RNA counteracting antiviral response has been described. In this work, we have characterized a 32-nt RNA motif located at the 3′ end of the TGEV genome that specifically interacted with EPRS and RRS. This RNA motif presented high homology with the GAIT element, involved in the modulation of the inflammatory response. Moreover, the disruption of the viral GAIT-like RNA motif led to an exacerbated innate immune response triggered by MDA5, indicating that the GAIT-like RNA motif counteracts the host innate immune response. These novel findings may be of relevance for other coronaviruses and could serve as the basis for the development of novel antiviral strategies.
INTRODUCTION
Coronaviruses (CoVs) are enveloped, single-stranded, positive-sense RNA viruses that belong to the Coronaviridae family within the Nidovirales order (1). CoVs are vertebrate pathogens responsible mainly for respiratory and enteric infections in a wide range of animals and human (2). Among the high diversity of CoVs infecting animal species, transmissible gastroenteritis virus (TGEV) is of special relevance in pigs, causing a life-threatening disease with important economic losses (3). In humans, CoV infections have been historically associated with mild upper respiratory tract diseases (4). However, the identification of the severe acute respiratory syndrome CoV (SARS-CoV) in 2003 (5) and the recently emerged (April 2012) Middle East respiratory syndrome CoV (MERS-CoV) (6), both causing acute pneumonia and even death, redefined historic perceptions and potentiated the relevance of CoVs as important human pathogens. Therefore, understanding the molecular basis of CoV replication and pathogenesis will allow the development of effective strategies to prevent and control CoV infections.
CoVs contain the largest known RNA genome among RNA viruses, consisting of a plus-sense, 5′-capped, and polyadenylated RNA molecule of 27 to 31 kb in length (2, 7). The first two-thirds of the genome carries the replicase gene, which comprises two overlapping open reading frames (ORFs) named 1a and 1b. Both ORFs are directly translated from the viral genome, resulting in two large polyproteins (pp1a and pp1ab), which are autoproteolitically cleaved to release the replication-transcription complex components (8). The 3′ one-third of the genome includes the genes encoding the structural spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins, as well as the genus-specific proteins characteristic of each CoV, which are expressed from a nested set of 3′-coterminal subgenomic mRNAs (sgmRNAs) (2, 7, 8).
CoV replication and transcription are complex processes that require the specific recognition of RNA cis-acting elements located at the ends of the viral genome (9) and are mediated by a huge protein complex encoded by the viral replicase gene together with the participation of cellular proteins (2, 7, 9). To date, several cellular proteins implicated in CoV replication have been identified on the basis of their interaction with replicase proteins (10, 11) or with cis-acting elements required for viral genome replication (12, 13). Recently, we identified 10 cellular proteins interacting with the TGEV genome ends by RNA affinity chromatography and mass spectrometry analysis (13). These proteins included the polypyrimidine tract-binding protein (PTB), which preferentially interacted with the 5′ end of the genome, and a set of 9 proteins, including several heterogeneous nuclear ribonucleoproteins (hnRNPs) (A1, A2B1, A0, Q, and U), the translation factors glutamyl-prolyl-tRNA synthetase (EPRS), arginyl-tRNA synthetase (RRS), and poly(A)-binding protein (PABP), and the p100 transcriptional coactivator, which bound preferentially to the 3′ end of the genome. Silencing studies demonstrated a negative role in viral RNA synthesis for PTB (9) and a positive role for the 3′-end-interacting proteins hnRNP Q, EPRS, RRS, and PABP (13) (S. Marquez-Jurado, L. Enjuanes, and F. Almazán, unpublished data).
The innate immune response is the first line of antiviral defense. During infection, viral components are detected through a set of pathogen recognition receptors (PRRs) (14, 15), including the Toll-like receptors (TLRs), the retinoic acid-inducible gene I product (RIG-I), and the melanoma differentiation-associated gene 5 (MDA5) protein. This recognition triggers a signaling cascade that culminates with the synthesis of interferon (IFN) and proinflammatory cytokines to limit viral replication and spread (16). For RNA viruses, double-stranded RNAs (dsRNAs), which are present in the viral genome as stem-loop structures or are generated during viral replication, are the most important pathogen-associated molecular pattern (PAMP) recognized by cellular PRRs (15, 17). In the case of CoVs, MDA5 has been described as the main cytoplasmic PRR sensing the viral RNAs (18).
To evade the innate immune response, viruses have developed an extensive and highly diverse range of countermeasures to impair the antiviral response, including blocking different components of the innate immune pathways or hiding dsRNAs to make them inaccessible to sensors (14). In this sense, CoVs encode several proteins that interfere with type-I IFN and proinflammatory cytokine production at different levels (15). Similar to that of other positive-stranded RNA viruses, CoV replication takes place in the cytoplasm, leading to the generation of dsRNA molecules that trigger the host antiviral innate immune response. To overcome this response, CoVs have developed several strategies, including the induction of double-membrane vesicles (DMVs) (14), proposed to hide viral dsRNAs from innate sensors during genome replication. In addition to these dsRNA molecules, most likely, the viral genome and the sgmRNAs of CoVs could contain highly structured RNA domains that could be recognized by the cell guard sensors (19), as described for other viruses, like hepatitis C virus (20), reovirus (21), coxsackievirus (22), West Nile virus, and yellow fever virus (23). However, the nature of these highly structured RNA domains and the molecular mechanisms used by CoVs to avoid their recognition by the cellular sensors are still unknown. In addition, other viruses, such as adenoviruses, transcribe two adenovirus-associated RNA I (VAI) which fold into stem-loop structures that are involved in the blocking of some components of the innate immune pathway, such as the protein kinase R (PKR) (24). Whether RNA viruses, such as CoVs, contain similar RNAs interfering with the innate immune response remains to be determined.
In this work, a novel RNA domain located at the 3′ end of the TGEV genome that specifically interacted with the aminoacyl-tRNA synthetases EPRS and RRS has been identified and functionally characterized. This RNA motif presented high homology in sequence and secondary structure with the gamma interferon-activated inhibitor of translation (GAIT) element that is located at the 3′ end of several mRNAs encoding proinflammatory proteins. This element is involved in the translation silencing of these mRNAs to favor the resolution of inflammation. The functional role of the viral GAIT-like RNA motif during TGEV infection was further analyzed by mutational analysis using a TGEV infectious clone, demonstrating that the viral GAIT-like RNA motif influenced the innate immune response by affecting the MDA5 pathway. To our knowledge, this is the first time that a CoV RNA domain has been directly involved in the modulation of the innate immune response.
RESULTS
Relevance of the poly(A) tail in the binding of cellular proteins to the 3′ end of the TGEV genome.
In a previous study, nine cellular proteins were found to bind preferentially to the 3′ end of the TGEV genome, including several hnRNPs (A1, A2B1, A0, Q, and U), the translation factors EPRS, RRS, and PABP, and the p100 transcriptional coactivator (13). The specific RNA domain interacting with these cellular proteins was further analyzed. In a first approach, the role of the poly(A) tail in the binding of these proteins with the 3′ end of the genome was studied. To this end, three RNA constructs, one containing the first 504 nucleotides (nt) of the 5′ end of the genome and the others containing the last 493 nt of the 3′ end with or without the poly(A) tail, were generated by in vitro transcription, labeled with biotin, and used as baits for RNA affinity protein isolation (Fig. 1). The RNA baits were incubated with cytoplasmic cell extracts from infected human liver-derived Huh-7 cells, and the pulled-down proteins were detected by Western blotting by using specific antibodies. Huh-7 cells were used instead of swine testis (ST) cells, because Huh-7 cells are susceptible to TGEV and because of the availability of validated antibodies against human proteins (13). As expected, the PABP specifically bound to the 3′ end containing the poly(A) tail (Fig. 1). In contrast, all the remaining proteins interacted with the 3′ end both with and without the poly(A) tail (Fig. 1), indicating that EPRS, RRS, p100, and the hnRNPs Q, A1, U, and A0 did not require the poly(A) tail to interact with the genome 3′ end.
FIG 1 .
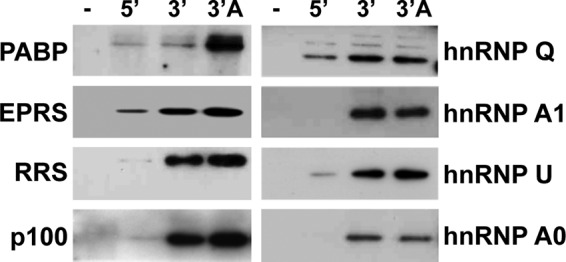
Relevance of the poly(A) tail in the binding of the 3′-end-interacting cellular proteins. Proteins (600 µg) from cytoplasmic extracts of TGEV-infected Huh-7 cells were pulled down without RNA (−) or with TGEV genome ends 5′ (5′) and 3′ without (3′) and with (3′ A) poly(A) tail, separated in SDS-PAGE gels, and analyzed by immunoblotting with specific antibodies against PABP, EPRS, RRS, p100, and the hnRNPs (Q, A1, U, and A0).
Mapping of TGEV 3′-end RNA domains interacting with cellular proteins.
To map the 3′-end RNA domains interacting with EPRS, RRS, p100, and the hnRNPs Q, A1, U, and A0, the last 493 nt of the TGEV genome were divided into six overlapping RNA fragments (F1 to F6) that were used as baits in pulldown assays (Fig. 2A). These RNA fragments were designed taking into consideration the predicted secondary structure of the 3′ end of the TGEV genome and the information available about the 3′-end cis-acting elements involved in CoV replication. Fragments F1 and F2, covering nt 28087 to 28207 and 28147 to 28277, respectively, contained four predicted stem-loops for which no function has been associated. Fragments F3, F4, and F5, spanning nt 28269 to 28388, 28358 to 28481, and 28289 to 28426, respectively, covered a region containing RNA structures similar to the bulged stem-loop (BSL) and the pseudoknot (PK), which have been described to be important for genome replication in mouse hepatitis virus (MHV) and bovine CoV (BCoV) (25–27). Finally, fragment F6, covering nt 28469 to 28580, contained the hypervariable region (HVR) and the highly conserved octanucleotide (OCT) (5′ GGA AGA GC 3′), originally described for MHV (27). The biotin-labeled RNA fragments, generated by in vitro transcription, were used as baits in pulldown assays, together with the full-length 5′ and 3′ ends of the genome as specificity controls. In addition to the expected preferential binding of all proteins to the 3′ end of the genome, a specific binding to F2 was observed for EPRS and RRS, and a preferential binding to F4 was found for hnRNP A1 (Fig. 2B). In the case of hnRNPs Q and A0, these proteins were preferentially pulled down with F2 and F6 and with F2 and F4, respectively (Fig. 2B). In contrast, no preferential binding for any fragment was detected in the case of hnRNP U and p100. Due to the specific interaction of EPRS and RRS with F2, and the positive role of these proteins in CoV RNA synthesis (13) (Marquez-Jurado et al., unpublished), this RNA fragment was selected to further delimitate the RNA domains interacting with both aminoacyl-tRNA synthetases.
FIG 2 .

Mapping of the RNA domains interacting with the 3′-end-interacting cellular proteins. (A) Predicted secondary structure of the 493 nt from the TGEV genome 3′ end, obtained using the Mfold 3.4 software (49). The sequence covered by the six overlapping fragments (F1 to F6) and the higher-order RNA structures similar to the bulge stem-loop (BSL), the pseudoknot (PK), the hypervariable region (HVR), and the conserved octanucleotide (OCT) are indicated. (B) Identification of the 3′-end domains interacting with the selected cellular proteins. Proteins (600 µg) from cytoplasmic extracts of TGEV-infected Huh-7 cells were pulled down with the six overlapping fragments (1 to 6) covering the last 493 nt of the TGEV genome or with TGEV genome ends 5′ and 3′ without poly(A) or without RNA (−) as specificity controls. The pulled down proteins were separated in SDS-PAGE gels together with 50 µg of the initial protein extract (I) and analyzed by immunoblotting with specific antibodies against EPRS, RRS, hnRNP Q, hnRNP A1, hnRNP A0, hnRNP U, and p100. To simplify the information of the figure, an empty lane was removed in the hnRNP A0 panel, in which the dashed line indicates the splicing site. The molecular masses in kDa of the analyzed proteins are indicated at the left.
Identification of the 3′-end RNA domains interacting with EPRS and RRS.
To delimitate the RNA domains within F2 interacting with EPRS and RRS, this fragment was divided into three smaller RNA fragments (F2.1, F2.2, and F2.3) according to its predicted secondary structure (Fig. 3A). The stability of the predicted harping loop structures present in these RNA fragments was equivalent to that of the corresponding harping loop structures presented in fragment F2 and in the full-length 3′ end of the genome. These fragments were chemically synthesized, labeled with biotin, and used as baits in pulldown assays, together with F2 and F3 as specificity controls. In addition to the expected binding to F2, a preferential binding to F2.2 was observed for both EPRS and RRS (Fig. 3A).
FIG 3 .

Mapping of the RNA domains interacting with EPRS and RRS. (A) Mapping of the region within F2 interacting with ERPS and RRS. F2 was divided into three fragments (F2.1, F2.2, and F2.3) that were used as bait in pulldown assays, together with fragments F2 and F3 or without RNA (−) as specificity controls. The presence of EPRS and RRS in the initial extract (I) and in the pulled-down fractions was analyzed by Western blotting. To simplify the information in the figure, an empty lane was removed in the EPRS panel, in which the dashed line indicates the splicing site. The molecular masses in kDa of EPRS and RRS are indicated at the left. (B) Identification of the F2.2 domain interacting with EPRS and RRS. F2.2 was divided into four fragments (F2.2L, F2.2R, F2.2U, F2.2D), and their interactions with EPRS and RRS were analyzed by pulldown assays and Western blotting. The same experiment was performed with fragment F2.2 or without RNA (−) as specificity controls. The molecular masses in kDa of EPRS and RRS are indicated at the left. (C) Sequence analysis of fragment F2.2L. The bar on the top represents the TGEV genome, in which the different genes (ORF 1a, ORF 1b, S, 3a, 3b, E, M, N, and 7 genes), the leader sequence (L), and the 3′ UTR are illustrated as boxes. The sequence alignment of the viral fragment F2.2L with the GAIT element of the ceruloplasmin (Cp GAIT) and their respective secondary structures, in which the A and U residues critical for the function of the GAIT element are outlined in green, are shown. Numbers above the sequence alignment and in the secondary structure indicate the genome position of fragment F2.2L.
The F2.2 RNA fragment was subsequently divided into four new fragments (F2.2L, F2.2R, F2.2U, and F2.2D), according to its sequence and predicted secondary structure that consists of a complex stem-loop with two internal bulges (Fig. 3B). F2.2L and F2.2R were comprised of the 5′ and 3′ halves of the fragment F2.2 sequence, respectively, while F2.2U contained the upper stem of the structure and F2.2D the two internal bulges. In addition to the expected binding to F2.2, both aminoacyl-tRNA synthetases preferentially bound to fragment F2.2L, which constitutes a 32-nt RNA domain located 410 nt upstream of the 3′ end of the viral genome, within the coding sequence of gene 7 (Fig. 3C). The sequence of this RNA domain showed high identity with the GAIT element, present in the 3′ untranslated region (UTR) of several mRNAs coding for proinflammatory proteins, such as the ceruloplasmin (Cp) (Fig. 3C) (28, 29). It has been described that IFN-γ induces the assembly of the GAIT complex, constituted by EPRS, hnRNP Q, the ribosomal protein L13a, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). This protein complex interacts with the GAIT element present in different mRNAs encoding proinflammatory proteins, inhibiting their translation and therefore the harmful accumulation of inflammatory products (29, 30). Interestingly, the secondary structure of the GAIT element, consisting of a stem-loop with an internal asymmetric bulge, was also predicted for the viral RNA domain, including the A and U residues located in the asymmetric internal bulge, which have been reported to be critical for the function of the cellular GAIT element (Fig. 3C) (28, 29). Overall, these data suggest that the viral RNA domain, expanding fragment F2.2L, could constitute a GAIT-like RNA motif.
Analysis of the interaction of EPRS and RRS with viral RNAs during infection.
The interaction of EPRS and RRS with viral RNAs in the context of TGEV infection was analyzed by RNA immunoprecipitation assays. To this end, cytoplasmic extracts of TGEV-infected ST cells were immunoprecipitated with EPRS- and RRS-specific antibodies, and the presence of the viral genome and sgmRNAs in the immunoprecipitated RNA-protein complexes was analyzed by reverse transcription-quantitative PCR (RT-qPCR) by using specific oligonucleotides. In contrast to the samples immunoprecipitated with the negative-control anti-green fluorescent protein (GFP) antibody, the viral genomic RNA and sgmRNAs were specifically detected in the RNA-protein complexes immunoprecipitated with anti-EPRS or anti-RRS antibodies (Fig. 4). These data indicated that both aminoacyl-tRNA synthetases interacted with the viral genome and the sgmRNAs during infection, as expected from the presence of the GAIT-like RNA motif in these viral RNAs.
FIG 4 .
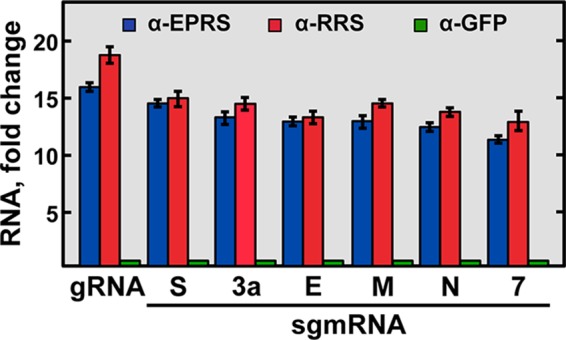
Analysis of the interaction of EPRS and RRS with viral RNAs by RNA immunoprecipitation assay. Cytoplasmic extracts of TGEV-infected ST cells were immunoprecipitated with specific antibodies for EPRS (α-EPRS), RRS (α-RRS), and GFP (α-GFP). The presence of viral genomic RNA (gRNA) and sgmRNAs (S, 3a, M, E, N, and 7 genes) in the immunoprecipitated RNA-protein complexes was quantified by RT-qPCR and expressed as the fold change of the RNA levels relative to the RNA levels in the samples immunoprecipitated with GFP antibody. Error bars represent the standard deviations from three independent experiments.
In vitro analysis of the interaction of EPRS and RRS with the viral GAIT-like RNA motif.
The pulldown assays did not discriminate whether the interaction of EPRS and RRS with the viral GAIT-like RNA motif was direct or mediated by other cellular or viral proteins. To clarify this issue, this interaction was analyzed by electrophoretic mobility shift assay (EMSA) by using purified proteins and the biotin-labeled GAIT-like RNA motif (Fig. 5). Purified GAPDH, a protein of the GAIT complex that does not directly interact with the GAIT element (30), and the DDK/Myc tag present in the purified EPRS were used as negative controls. A specific interaction of the TGEV GAIT-like RNA motif with the purified EPRS but not with the DDK/Myc tag or GADPH was observed (Fig. 5). Unfortunately, in the case of RRS, the purified protein did not enter into the gels used for the EMSA, and nonconclusive data were obtained using this technique (data not shown). These data indicated that, at the least, the EPRS interacted directly with the TGEV GAIT-like RNA motif in vitro, similar to what has been described for the cellular GAIT element (29, 31).
FIG 5 .
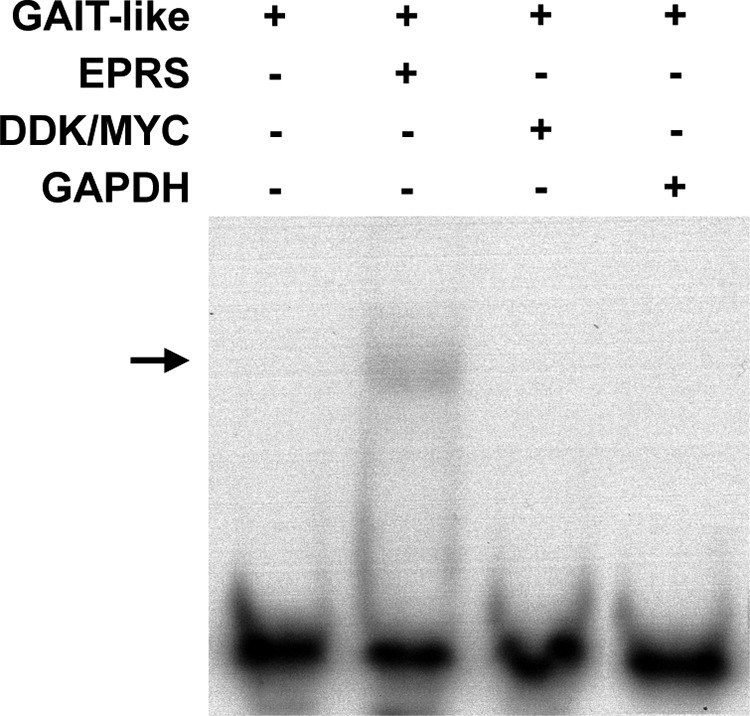
Study of the interaction between EPRS and the TGEV GAIT-like RNA motif by RNA EMSA. Purified EPRS, DDK/Myc tag, and GAPDH proteins were incubated with the 3′-biotinylated GAIT-like RNA motif riboprobe (GAIT-like). The RNA-protein complexes were resolved by electrophoresis on a nondenaturing 4% polyacrylamide gel, and the biotinylated riboprobe was detected using the BrightStar BioDetect kit (Ambion). The arrow indicates the shifted protein-RNA complex.
In vitro functional analysis of the viral GAIT-like RNA motif.
Based on the functional role of the cellular GAIT element on translation silencing (29) and the direct binding of EPRS to the viral GAIT-like RNA motif, the potential inhibitory effect of this viral RNA motif on translation was analyzed in vitro. To this end, three chimeric mRNAs expressing the luciferase protein, containing either the Cp GAIT element, the TGEV GAIT-like RNA motif, or no GAIT element in the 3′ UTR, were in vitro transcribed, and their translation efficiency was analyzed in the presence or in the absence of the GAIT complex by using a rabbit reticulocyte lysate system (Fig. 6). It has been described that the GAIT complex is assembled in monocytic cells after treatment with IFN-γ (30). Therefore, cytosolic extracts of human U937 monocytic cells untreated or treated with IFN-γ were used in the in vitro translation reactions as a source of the GAIT complex. As expected, translation of the luciferase gene was detected in all cases in the absence of the GAIT complex (Fig. 6). In contrast, a significant inhibition of luciferase translation was observed from the mRNA constructs containing either the cellular (75%) or the viral (50%) GAIT element in the presence of the GAIT complex (Fig. 6). Interestingly, when increasing amounts of either the cellular or the viral GAIT elements were added to the translation reaction mixtures as competitors, a reversion of the luciferase translation inhibition was observed (data not shown), indicating that the translation inhibition is due to the presence of the GAIT element. These data indicated that, similar to the cellular GAIT element, the viral GAIT-like RNA motif had an inhibitory effect on in vitro translation, probably by interaction with the GAIT complex.
FIG 6 .
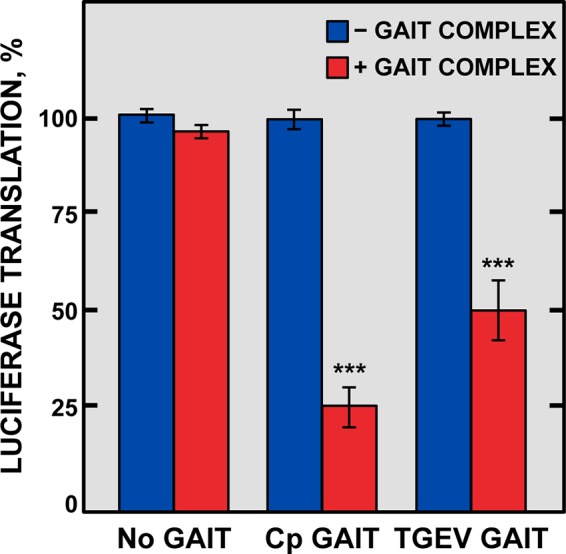
In vitro analysis of the inhibitory effect on translation of the TGEV GAIT-like RNA motif. Three chimeric luciferase reporter mRNA constructs, containing at the 3′ UTR no GAIT element (No GAIT), the Cp GAIT element (Cp GAIT), or the TGEV GAIT-like RNA motif (TGEV GAIT), were subjected to in vitro translation in a rabbit reticulocyte lysate in the absence (blue bars) or in the presence (red bars) of the GAIT complex. The translation of these chimeric constructs was quantitated by chemiluminescence and expressed as the percentage of translation of each construct in the presence of the GAIT complex compared to that in the absence of the GAIT complex. Error bars represent the standard deviations from three independent experiments. ***, P < 0.001.
Generation and characterization of a recombinant TGEV harboring mutations in the GAIT-like RNA motif.
Once the inhibitory effect on translation of the viral GAIT-like RNA motif was determined in vitro, the functional role of this RNA motif was studied by mutational analysis in the context of the TGEV infection. Based on previously reported functional data of the Cp GAIT element (28, 29), a TGEV recombinant virus harboring mutations in the GAIT-like RNA motif (rTGEV-mutG) was engineered by reverse genetics using a TGEV infectious clone (Fig. 7A) (32). The rTGEV-mutG virus contained the nucleotide substitutions A to G (genomic position 28178), U to C (genomic position 28181), and C to U (genomic position 28190), which were predicted to partially disrupt the stem structure and the internal asymmetric bulge. As the GAIT-like RNA motif is located within the coding region of gene 7, the mutations introduced were silent and maintained the amino acid sequence of protein 7. The rTGEV-mutG virus was successfully rescued and the mutations introduced were present in the recovered mutant virus, even after 20 serial passages in tissue culture of a plaque-purified virus, indicating that they were stably maintained in the rTGEV-mutG genome. In addition, no significant differences in the plaque size and the growth kinetics, at both low (0.05) and high (3) multiplicities of infection (MOI), were observed between the mutant and wild-type (WT) viruses (Fig. 7B), suggesting that the viral GAIT-like RNA motif was not essential for virus replication in cell cultures.
FIG 7 .

Characterization of rTGEV-mutG virus in cell culture. (A) Genetic structure of rTGEV-mutG virus. The bar on top represents the TGEV genome, in which the different genes (ORF 1a, ORF 1b, S, 3a, 3b, E, M, N, and 7 genes), the leader sequence (L), the 3′ UTR, and the GAIT-like RNA motif are indicated. The structures depicted below the genome show the sequence and predicted secondary structure of the GAIT-like RNA motif from the WT (rTGEV-WT) and mutant (rTGEV-mutG) viruses, where the mutations introduced in the mutant are indicated in red. (B) In vitro growth kinetics of the WT and mutant viruses. ST cells were infected at an MOI of 0.05 (left) and 3 (right) with rTGEV-WT (WT) and rTGEV-mutG (mutG) viruses, and at different times postinfection the viral titer was determined by plaque titration on ST cells. Error bars represent the standard deviations from three independent experiments.
The generation of rTGEV-mutG was based on the assumption that the introduced mutations abolish the binding of EPRS and RRS to the viral-GAIT-like RNA motif and, therefore, inactivate the putative function of this RNA motif. To confirm that this was the case, the binding of both aminoacyl-tRNA synthetases to either the native or the mutated GAIT-like RNA motif (see Table 2, F2.2L or F2.2L-mutG, respectively) was analyzed using pulldown assays (Fig. 8). As expected, the mutated viral GAIT-like RNA motif was not able to pull down both aminoacyl-tRNA synthetases, indicating that EPRS and RRS did not bind to the mutated viral GAIT-like RNA motif (Fig. 8).
TABLE 2 .
Biotin-labeled RNAs used in affinity chromatography experiments
| RNA | RNA bait sequence (5′→3′) |
|---|---|
| F2.1 | AGACUAUUACUUAAUCACUCUUUCAUACUAAAAGUGGUGCUUCGAGUAAU |
| F2.2 | AUCUUAAAACUGUCAAUGACUUUAAUAUCUUAUAUAGGAGUUUAGCAGAAACCAGAU |
| F2.3 | CUUUCUAGUCUUACUAGGAUUUUGCU |
| F2.2U | GACUUUAAUAUCUUAUAUAGGAGUU |
| F2.2D | AAUCUUAAAACUGUCAAUGUUAGCAGAAACCAGAUU |
| F2.2L | AAUCUUAAAACUGUCAAUGACUUUAAUAUCUU |
| F2.2L-mutG | AAUCUUAAGACCGUCAAUGAUUUUAAUAUCUU |
| F2.2R | AUAUAGGAGUUUAGCAGAAACCAGAUU |
FIG 8 .
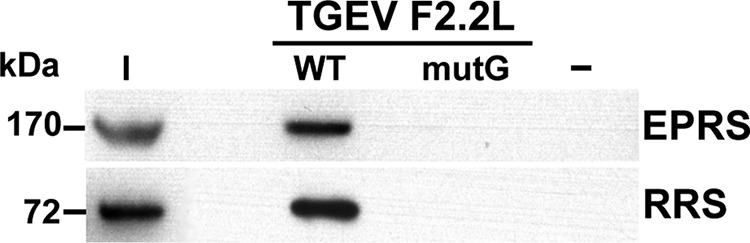
Analysis of the interaction of EPRS and RRS with the mutated TGEV GAIT-like RNA motif. Proteins (600 µg) from cytoplasmic extracts of TGEV-infected Huh-7 cells were pulled down with either the native (F2.2L-WT) or the mutated (F2.2L-mutG) TGEV GAIT-like RNA motif, together without RNA (−) as the specificity control. The presence of EPRS and RRS in the initial extract (I) and in the pulled-down fractions was further analyzed by Western blotting. The molecular masses in kDa of EPRS and RRS are indicated at the left.
All together, these data indicated that the viral GAIT-like RNA motif was dispensable for virus replication in cell culture.
Effect of GAIT-like mutations on the innate immune response.
Due to the dispensability of the GAIT-like RNA motif for virus replication in cell culture, and considering the reported role of the Cp GAIT element modulating the inflammatory response (30, 33), the potential role of the TGEV GAIT-like RNA motif in the modulation of the host inflammatory response was further studied. To this end, ST cells were infected at an MOI of 3 with the rTGEV-WT and rTGEV-mutG viruses, and the mRNA levels of a set of proinflammatory cytokines (tumor necrosis factor [TNF], CCL2, and CXCL9) were analyzed at different hours postinfection (hpi) by RT-qPCR using specific TaqMan assays. As a control of viral replication, the levels of genomic RNA and sgmRNA 7 were analyzed. In addition, to discard a general transcription up- or downregulation during virus infection that could affect the expression of all cellular genes, the mRNA levels of the transforming growth factor β (TGF-β) gene, which is involved in the immune response, were also evaluated as a control. As expected, all the analyzed genes increased their expression during the WT virus infection (Fig. 9). Interestingly, all of them were significantly upregulated (from 2- to 20-fold, depending on the gene) in cells infected with the rTGEV-mutG virus compared with in cells infected with the WT virus (Fig. 9). In contrast, no significant differences were detected in the levels of viral genomic RNA, sgmRNA 7, and TGF-β mRNA in cells infected with either the WT or mutant viruses (Fig. 9), discarding that proinflammatory RNA upregulation was due to a difference in virus replication or to a general transcription upregulation during rTGEV-mutG infection.
FIG 9 .

Accumulation kinetics of host genes involved in inflammation. ST cells were infected with either rTGEV-WT (blue) or rTGEV-mutG (red) viruses, and the expression of six genes involved in inflammation and innate immunity (TNF, CCL2, CXCL9, IFN-β, MDA5, and RIG-I) was analyzed at different hpi by RT-qPCR. In parallel, the expression of the TGF-β gene, whose expression is not affected by TGEV infection, and the levels of viral genomic RNA and sgmRNA 7 were also evaluated as controls. The β-glucuronidase (GUSB) mRNA levels were used as an endogenous mRNA control in all cases. The mRNA levels were relative to those in noninfected cells. The fold change of viral RNA and cellular mRNA levels in cells infected with rTGEV-mutG compared with cells infected with rTGEV-WT is also indicated. Error bars indicate the standard deviations from three independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
The host innate immune response led to both proinflammatory cytokines and type I IFN production. Therefore, IFN-β and IFN-stimulated genes (ISGs) (MDA5 and RIG-I) production was also analyzed. Similarly to the expression of proinflammatory cytokines, a significant increase in the IFN-β mRNA levels, and those of its downstream expressed MDA5 and RIG-I genes, was observed in cells infected with the rTGEV-mutG virus compared to those in cells infected with the WT virus (Fig. 9). These results indicated that the viral GAIT-like RNA motif interfered with the host innate immune response.
Signaling pathway affected by the viral GAIT-like RNA motif.
In nonimmune cells, the production of cytokines and IFN is mediated mainly by RIG-I and MDA5 cytoplasmic sensors. These proteins recognize dsRNAs, activating the transcription factors IRF3/7 and NF-κB, and the subsequent expression of proinflammatory cytokines and IFN (16, 34). In the case of CoVs, it has been described that the viral RNA is recognized by MDA5 (18), although a role for RIG-I was also proposed in several cell types (35). To analyze whether MDA5 or RIG-I signaling was affected by the GAIT-like RNA motif, both sensors were independently silenced in ST cells by using specific small interfering RNAs (siRNAs). The silenced cells were then infected with rTGEV-WT or rTGEV-mutG viruses, and the effect on TNF, IFN-β, and viral genomic RNA production was analyzed by RT-qPCR in comparison with the levels in cells transfected with a negative-control siRNA. Two siRNA transfection steps were needed to achieve a sustained silencing at the mRNA and protein levels of about 43% and 35% for RIG-I and MDA5, respectively (Fig. 10A). Despite the modest reduction of MDA5 expression, it was previously described that a similar reduction is enough to observe an effect on cytokine production (36). In infected MDA5-silenced cells but not in RIG-I-silenced cells, an increment in the viral genomic RNA levels of the rTGEV-WT and rTGEV-mutG viruses was observed compared with nonsilenced cells, being this increment higher in the case of the mutant virus (Fig. 10B, left). As previously observed (see above), in nonsilenced cells, infection by the rTGEV-mutG virus led to increased TNF and IFN-β mRNA levels compared with those caused by infection by the WT virus (Fig. 10B, right). Interestingly, in MDA5-silenced cells, the rTGEV-mutG virus showed the same pattern of cytokine reduction as the WT virus, while this did not occur in RIG-I-silenced cells (Fig. 10B, right). These results suggested that the GAIT-like RNA motif affects the MDA5 signaling pathway.
FIG 10 .

Effect of silencing the expression of RIG-I-like receptors on TNF and IFN-β production during rTGEV-WT and rTGEV-mutG infection. ST cells were transfected twice with a negative-control siRNA, an siRNA previously validated for porcine RIG-I, or an siRNA designed for porcine MDA5. After 24 h of the second transfection, cells were noninfected or infected with rTGEV-WT (WT) and rTGEV-mutG (mutG) viruses. Total RNA and proteins were extracted at 12 and 18 hpi (36 and 42 h after the second siRNA transfection, respectively) to evaluate the silencing, virus production, and mRNA levels of TNF and IFN-β. (A) Silencing of RIG-I and MDA5. Silencing at the mRNA level (left) was evaluated at 36 (blue) and 42 (red) h after the second transfection of the siRNAs by RT-qPCR and expressed as the percentage of RIG-I and MDA5 mRNA remaining after the silencing of target genes in comparison to reference levels from cells transfected with the negative-control siRNA. RIG-I and MDA5 silencing at the protein level (right) was evaluated by immunoblotting at 42 h after the second transfection of the RIG-I and MDA5 siRNAs (siRIG-I and siMDA5, respectively) in comparison to cells transfected with the negative-control siRNA (siNEG). The β-actin was used as a loading control. (B) Effect of RIG-I and MDA5 silencing on virus genomic RNA, IFN-β, and TNF production. Virus genomic RNA (gRNA) and IFN-β and TNF mRNA levels were quantified by RT-qPCR in cells transfected with the negative-control (siNEG; blue bars), RIG-I (siRIG-I; red bars), or MDA5 (siMDA5; green bars) siRNAs and infected with rTGEV-WT (WT) or rTGEV-mutG (mutG) viruses. Levels of virus gRNA are expressed as percentages in comparison with the gRNA levels in nonsilenced and infected cells (left). IFN-β and TNF mRNA levels are relative to those in nonsilenced and noninfected cells (right). The figure shows the effects observed at 18 hpi (42 h after the second siRNA transfection), when they were more evident. In all cases, GUSB mRNA levels were used as the endogenous control. Errors bars indicate standard deviations from three independent experiments. ***, P < 0.001.
In a complementary approach, ST cells were treated with swine IFN-β for 16 h to induce the overexpression of RIG-I and MDA5 (Fig. 11, left). Cells were then infected with the rTGEV-WT and rTGEV-mutG viruses, and the genomic RNA was quantified at 8 hpi by RT-qPCR. A significant reduction in the genomic RNA levels of the rTGEV-WT virus was observed in cells treated with IFN-β compared with those in untreated cells. Interestingly, this reduction was higher in IFN-β-treated cells infected with the rTGEV-mutG virus (Fig. 11, right) than in cells infected with the rTGEV-WT virus, confirming that the GAIT-like RNA motif modulates cytoplasmic sensor signaling.
FIG 11 .
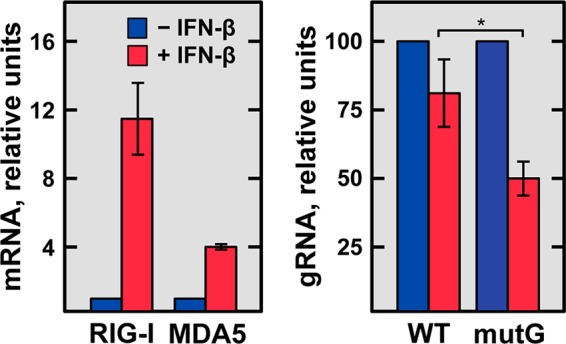
IFN-β sensitivity of rTGEV-WT and rTGEV-mutG viruses. Confluent ST cells were untreated (blue) or treated (red) with 4 ng/106 cells of porcine IFN-β (Kingfisher Biotech) for 16 h and then noninfected or infected with either rTGEV-WT (WT) or rTGEV-mutG (mutG) at an MOI of 3. At 8 hpi, total RNA was extracted, and the levels of viral genomic RNA (gRNA) were quantified by RT-qPCR and expressed relative to those in IFN-β-untreated and infected cells (right). As a control of the IFN-β treatment, the MDA5 and RIG-I mRNA levels were quantified by RT-qPCR in noninfected cells, untreated or treated with IFN-β (left). GUSB mRNA levels were used as an endogenous control in all cases. Error bars indicate the standard deviations from three independent experiments. *, P < 0.05.
DISCUSSION
The identification of host factors and the elucidation of their functions in viral replication are major areas in current virology research that will allow the development of novel antiviral strategies to control viral diseases. In this sense, we previously identified nine cellular proteins that interact with the 3′ end of the TGEV genome, from which a functional role in virus replication was described for hnRNP Q, PABP, and the aminoacyl-tRNA synthetases EPRS and RRS (13) (Marquez-Jurado et al., unpublished). In this study, we have identified a novel viral RNA motif, consisting of a 32-nt domain located 410 nt from the 3′ end of the TGEV genome, which directly interacted with the cellular proteins EPRS and RRS. This RNA motif shared high homology in sequence and secondary structure with the cellular GAIT element that is involved in the modulation of the inflammatory response (29). Similar to the cellular GAIT element, we have shown that the viral RNA motif inhibited the in vitro translation of a chimeric mRNA containing this motif, indicating that in vitro it behaves as a GAIT-like RNA motif. Finally, a novel finding of this study was that the GAIT-like RNA motif counteracted the host innate immune response triggered by MDA5, at least in cell cultures. Further studies will be needed to confirm this effect in vivo and to analyze the role of the GAIT-like RNA motif in pathogenesis.
In addition to the canonical function of aminoacyl-tRNA synthetases in translation, higher eukaryotic aminoacyl-tRNA synthetases have acquired a variety of noncanonical roles, including signaling, stress response, and transcriptional regulation (37, 38). Based in part on this versatility, these enzymes have been recruited by viruses to perform essential functions (39, 40). In this respect, EPRS and RRS could be involved in different essential processes of the TGEV life cycle, through their interaction with the 3′ end of the genome.
A key observation of this study was the high homology in sequences and secondary structures between the viral motif interacting with EPRS and RRS and the cellular GAIT element present at the 3′ UTR of several mRNAs encoding proinflammatory proteins (29). Monocytes and macrophages promote the synthesis of inflammatory cytokines and chemokines to counteract invading microorganisms. However, an unregulated synthesis of these inflammatory products can have detrimental effects in the host. Therefore, the host must have different mechanisms to regulate cytokine production and allow the resolution of inflammation. One of these mechanisms is provided by the GAIT system in monocytic cells (29). In response to IFN-γ, the GAIT complex (constituted by EPRS, hnRNP Q, L13a, and GAPDH) is assembled and, by interaction with the GAIT element present in the 3′ UTR of several mRNAs encoding proinflammatory proteins, represses their translation. In this study, we have shown that the viral RNA motif is a GAIT-like RNA motif, based on its capacity to bind the GAIT complex and to inhibit the in vitro translation of a chimeric mRNA containing this motif. In this sense, EPRS and/or RRS could be components of a protein complex that regulate viral translation by its interaction with the viral GAIT-like RNA motif. Nevertheless, the characterization of an rTGEV-mutG virus, harboring mutations that disrupt the GAIT-like RNA motif, indicated that the GAIT-like RNA motif was not critical in CoV translation and replication in cell cultures, suggesting that during infection the viral RNA motif could have a function different to that described for the cellular GAIT element.
After confirming the dispensability of the GAIT-like motif for virus replication in cell cultures, its potential role modulating the host inflammatory response and more specifically the innate immune response was further analyzed in cell cultures. The innate immune response is the first line of antiviral defense that triggers the synthesis of proinflammatory cytokines and type I IFN to prevent viral infections (15). In this study, an exacerbated expression of both proinflammatory cytokines and IFN-β, together with its downstream ISGs (MDA5 and RIG-I), has been observed during infection with the rTGEV-mutG virus, harboring mutations in the GAIT-like RNA motif, compared with that in the rTGEV-WT virus infection. The level of the effect observed on the expression of proinflammatory cytokines and IFN-β was similar or even higher than that reported for CoV and other RNA viruses (14). In addition, we have identified that the MDA5 pathway was the innate immune pathway significantly affected by the GAIT-like RNA motif. In contrast to other RNA viruses, in which structured RNA domains present in their genome are recognized by the cell sensor triggering the innate immune response (20–23, 41), the data presented in this study suggested that the TGEV GAIT-like RNA motif interferes with the innate immune response, at least in cell cultures. Regarding the mechanism of action of the GAIT-like RNA motif, it could be postulated that this motif may act directly, preventing the activation of MDA5. Alternatively, the interaction with EPRS and RRS could mask the GAIT-like RNA motif to make it inaccessible to cellular sensors, such as MDA5, thus avoiding the triggering of the innate immune response. In this sense, mutations in the viral GAIT-like RNA motif could facilitate its sensing by MDA5, eliciting a faster and stronger antiviral response, such as the one observed during the infection with the rTGEV-mutG virus. Similar mechanisms have been described in negative-stranded RNA viruses, such as influenza and Ebola viruses, in which a viral protein instead of a cellular protein binds a dsRNA signature in their genomes, hiding them from recognition by the cellular sensors (14). To our knowledge, this is the first report of a cellular protein proposed to mask a viral PAMP RNA domain to subvert the host antiviral response.
The identification of the TGEV GAIT-like motif and the clarification of its role in the modulation of the antiviral response may be of relevance for other coronaviruses, such as the highly pathogenic SARS-CoV and MERS-CoV. In fact, several GAIT-like RNA motif candidates have been found within the 3′ UTR of SARS-CoV and MERS-CoV by bioinformatics analysis (unpublished results), although complementary approaches will be needed to clarify whether these putative GAIT-like RNA motifs could act as such.
MATERIALS AND METHODS
Cells and viruses.
Swine testis (ST) cells (ATCC, CRL-1746) (42) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Baby hamster kidney cells, stably transformed with the porcine aminopeptidase N gene (BHK-pAPN; kindly provided by H. Laude, Unité de Virologie Immunologie Moléculaires, France) to make them susceptible to TGEV, were grown in DMEM supplemented with 5% FBS and Geneticin (1.5 mg/ml) as the selection agent. Human liver-derived Huh-7 cells (kindly provided by R. Bartenschlager, University of Heidelberg, Germany) were grown in DMEM supplemented with 10% heat-inactivated FBS. Human monocytic U937 cells (kindly provided by M. Fresno, Centro de Biología Molecular Severo Ochoa, CBMSO, CSIC-UAM, Spain) were grown in RPMI 1640 medium supplemented with 10% FBS.
The TGEV PUR46-MAD strain (43) was used to infect ST cells, and the TGEV PUR46-C11 strain (44) was used to infect Huh-7 cells. In all cases, virus titration was performed on ST cell monolayers as previously described (45).
DNA constructs and RNA transcripts used for RNA affinity chromatography.
The DNA templates for in vitro transcription of the precise TGEV 5′ end, TGEV 3′ end without or with a poly(A) tail, and fragments F1 to F6 were generated by PCR by using specific oligonucleotides (Table 1) and the pML33L-637G plasmid (46) as the template. To amplify the first 504 nt of the TGEV 5′ end, PCR was performed with the oligonucleotides XmaI-SP6-5′-VS, which includes the SP6 promoter, and 5′TGEV-504-RS. To amplify the last 493 nt of the genome without or with a 25-nt poly(A) tail, the PCRs were performed with the oligonucleotides XmaI-T7-VS (including the T7 promoter) and either TGEV-3′-RS to generate the 3′ end without poly(A) or TGEV-3′-pA-RS to amplify the 3′ end with the poly(A) tail. Finally, to amplify the 3′-end fragments F1 to F6, the PCRs were performed with the oligonucleotides XmaI-T7-VS and F1-RS for fragment F1, F2-VS and F2-RS for fragment F2, F3-VS and F3-RS for fragment F3, F4-VS and F4-RS for fragment F4, F5-VS and F5-RS for fragment F5, and F6-VS and TGEV-3′-RS for fragment F6. In all cases, the virus sense primer (VS) included the T7 promoter to allow the in vitro transcription. All PCRs were performed with the platinum Pfx DNA polymerase (Invitrogen) by following the manufacturer’s recommended conditions. The resulting amplicons were purified with the QIAquick gel extraction kit (Qiagen) and used as the templates for in vitro transcription.
TABLE 1 .
Oligonucleotides used for PCR amplifications
| Oligonucleotidea | Oligonucleotide sequence (5′→3′)b |
|---|---|
| XmaI-sp6-5′-VS | CGCGCCCGGGATTTAGGTGACACTATA(G)ACTTTTAAAGTAAAGTGAGTGTAGC |
| 5′TGEV-504-RS | CACCAATGACGTAGTGATCCTTACG |
| XmaI-T7-VS | CGCGCCCGGGTAATACGACTCACTATA(G)GGATGCTGTATTTATTACAGTTTTAATC |
| TGEV-3′-RS | GTATCACTATCAAAAGGAAAATTTTCAAACAATTTTAC |
| TGEV-3′-pA-RS | TTTTTTTTTTTTTTTTTTTTTTTTGTATCACTATCAAAAGG |
| F1-RS | CTATATAAGATATTAAAGTCATTGACAGTTTTAAGATTG |
| F2-VS | CGCGCCCGGGTAATACGACTCACTATA(G)GGACTATTACTTAATCACTCTTTCAAT |
| F2-RS | GCAAAATCCTAGTAAGACTAGAAAGATTACTCGAAGC |
| F3-VS | CGCGCCCGGGTAATACGACTCACTATA(G)GGATTTTGCTGCTACAGATTGTTAG |
| F3-RS | GTGCTTACCATTCTGTACAAGAGTAGACAGCG |
| F4-VS | CGCGCCCGGGTAATACGACTCACTATA(G)GGCTGTCTACTCTTGTACAGAATGGTA |
| F4-RS | CTTTAAACTTCTCTAAATTACTAAATCTAGCATTGC |
| F5-VS | CGCGCCCGGGTAATACGACTCACTATA(G)GGTTAGTCACATTAATGTAAGGCAAC |
| F5-RS | CTAATATGCAATAGGGTTGCTTGTACCTCCTATTAC |
| F6-VS | CGCGCCCGGGTAATACGACTCACTATA(G)GGAGAAGTTTAAAGATCCGCTACGA |
VS, virus sense; RS, reverse sense.
Transcription promoters are in bold. Transcription initiation sites are in parentheses. The template sequence corresponding to poly(A) is underlined
In vitro transcription reactions were performed from 600 ng of each PCR amplicon using the SP6/T7 transcription kit (Roche). To label the transcript with biotin, biotin-14-CTP (Invitrogen) was added to the transcription reaction at a final concentration of 0.16 mM in a 1:6.25 ratio to unlabeled CTP. The transcription reaction mixtures were incubated for 2 h at 37°C and treated with 10 U of DNase I (Roche) for 30 min at 37°C. The resulting transcripts were purified with the RNeasy minikit (Qiagen) by following the RNA cleanup protocol, analyzed by denaturing electrophoresis in 2.5% (wt/vol) agarose gels, and spectrophotometrically quantified.
The RNA fragments F2.1, F2.2, F2.3, F2.2U, F2.2D, F2.2L, F2.2L-mutG, and F2.2R (Table 2), labeled at the 3′ end with biotin, were chemically synthesized (Sigma Aldrich).
RNA affinity chromatography.
Huh-7 cells were grown to confluence in 15-cm-diameter dishes and infected at an MOI of 5 with TGEV PUR46-C11. At 16 hpi, cytoplasmic cell extracts were prepared as described previously (13), and total protein concentration was determined with the BCA protein assay kit (Thermo Scientific). To perform the RNA affinity chromatography, 600 µg of cytoplasmic cell extract was diluted 1:1 in binding buffer (50 mM HEPES [pH 7.9], 150 mM KCl, 5% glycerol, 0.01% NP-40) and precleared three times by incubation with 20 µl of streptavidin Sepharose beads (streptavidin Sepharose high performance; Amersham) for 4 h at 4°C. Biotinylated RNA (between 5 and 10 µg) was diluted in 200 µl of RNA-binding buffer (5 mM HEPES [pH 7.9], 0.5 mM EDTA, 1 M KCl) and incubated with 20 µl of fresh streptavidin Sepharose beads at 4°C for 1 h. The immobilized RNA was washed three times for 15 min with 200 µl of binding buffer and then incubated with the precleared protein extract overnight at 4°C. The RNA-protein complexes were washed three times for 15 min with 200 µl of binding buffer, pulled down, and resuspended in 30 µl of NuPAGE sample buffer (Invitrogen). After incubation for 10 min at 65°C, the beads were centrifuged and the supernatant containing RNA-interacting proteins was recovered. All incubations were performed at 4°C in an orbital shaker, and the beads were collected by centrifugation at 500 × g.
Western blot analysis.
Cell lysates or pulled down RNA-binding proteins were resolved by denaturing electrophoresis in NuPAGE 4 to 12% bis-Tris gels with morpholinepropanesulfonic acid (MOPS) SDS running buffer (Invitrogen) and transferred electrophoretically to nitrocellulose membranes (Hybond-C extra nitrocellulose; Amersham Biosciences) by following the manufacturer’s recommended conditions. Membranes were blocked for 1 h at room temperature with 5% dried skimmed milk in TBS (20 mM Tris-HCl [pH 7.5], 150 mM NaCl) and then probed with specific antibodies diluted in TBS and 0.1% Tween 20 (TTBS) containing 3% dried skimmed milk. Primary antibodies for hnRNP A1 (rabbit polyclonal E17), hnRNP Q (mouse monoclonal antibody [MAb] I8E4:sc-56703), and hnRNP A0 (goat polyclonal G-17:sc-16509) were purchased from Santa Cruz Biotechnology. Antibodies for hnRNP U (rabbit polyclonal ab20666), EPRS (rabbit polyclonal ab31531), RRS (rabbit polyclonal ab31537), and β-actin (mouse MAb ab8226) were purchased from Abcam. The p100 transcriptional coactivator guinea pig polyclonal antiserum GP25 was purchased from Progen Biotechnik. The MDA5 rabbit monoclonal antibody (D74E4) and the RIG-I rabbit polyclonal antiserum were purchased from Cell Signaling and AnaSpec, respectively. The PABP rabbit polyclonal antiserum was kindly provided by Amelia Nieto (Centro Nacional de Biotecnología, CNB-CSIC, Spain). The blots were then incubated with horseradish peroxidase-conjugated secondary antibodies diluted in TTBS-3% dried skimmed milk for 1 h at room temperature, and the immune complexes were detected using the Immobilon Western chemiluminescent substrate (Millipore) according to the manufacturer’s instructions.
RNA EMSA.
RNA-protein binding reactions were performed by incubating 100 pmol of the 3′ biotinylated TGEV GAIT-like RNA motif (Table 2, F2.2L) with 1 pmol of recombinant purified EPRS (OriGene), GAPDH (Abcam), and DDK/Myc tag (NH2-EQKLISEEDLAANDILDYKDDDDKV-OH) in binding buffer (12% glycerol, 20 mM Tris-HCl [pH 7.4], 50 mM KCl, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol [DTT]) containing 0.2 U/µl SUPERase inhibitor (Ambion) for 30 min at 25°C. The RNA-protein complexes were resolved in a 4% nondenaturing PAGE gel using 0.5× TBE as the running buffer. After electrophoresis, the gel was blotted onto positively charged nylon membranes (BrightStar-Plus; Ambion), and the biotinylated RNA was detected using the BrightStar BioDetect kit (Ambion), following the manufacturer’s instructions.
RNA immunoprecipitation assay.
ST cells were grown in 15-cm-diameter dishes to confluence and infected at an MOI of 5 with TGEV PUR46-MAD. At 8 hpi, the cells were harvested and centrifuged at 2,000 × g for 5 min at 4°C, and the cell pellets were resuspended in RIP lysis buffer (MagnaRIP; Millipore) containing protease inhibitor cocktail (Roche) and 1 U/µl RNasin RNase inhibitor (Promega). Then, the cell suspension was incubated at 4°C for 5 min and centrifuged at 3,000 × g for 2 min at 4°C, and the supernatant was collected to perform the RNA immunoprecipitation assay using the EZ-Magna RIP kit (Millipore) by following the manufacturer’s instructions. Briefly, cytoplasmic supernatant, corresponding to 1 × 107 cells, was immunoprecipitated with anti-EPRS, anti-RRS, or anti-GFP (Roche) antibodies (attached to magnetic beads). Immunoprecipitated RNA-protein complexes were incubated with proteinase K for 30 min at 55°C, and the RNA was isolated using the RNeasy minikit (Qiagen), according to the manufacturer’s recommendations. Finally, the purified RNA was subjected to RT-qPCR for the detection of EPRS-, RRS-, or GFP-associated viral genomic RNA and sgmRNAs by using specific oligonucleotides (Table 3) and SYBR green-based detection.
TABLE 3 .
Oligonucleotides used for RT-qPCR of viral gRNA and sgmRNA
| Amplicon | Oligonucleotidea | Oligonucleotide sequence (5′→3′)b | Methodc |
|---|---|---|---|
| gRNA | Rep-VS | TTCTTTTGACAAAACATACGGTGAA | SYBR |
| Rep-RS | CTAGGCAACTGGTTTGTAACATCTTT | SYBR | |
| sgmRNA S | LCS1-VS | CCAACTCGAACTAAACTTTGGTAACC | SYBR |
| LCS1-RS | TCAATGGCATTACGACCAAAAC | SYBR | |
| sgmRNA 3a | rt3a-VS2 | CGGACACCAACTCGAACTAAACTTAC | SYBR |
| rt3a-RS | ATCAAGTTCGTCAAGTACAGCATCTAC | SYBR | |
| sgmRNA E | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | SYBR |
| mRNAE-RS | CAATGCCCTAGGAAACGTCATAG | SYBR | |
| sgmRNA M | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | SYBR |
| mRNAM-RS | GCATGCAATCACACACGCTAA | SYBR | |
| sgmRNA N | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | SYBR |
| N82−RS | TCTTCCGACCACGGGAATT | SYBR | |
| sgmRNA 7 | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | SYBR |
| 7 (38)-RS | AAAACTGTAATAAATACAGCATGGAGGAA | SYBR | |
| gRNA | Rep-VS | TTCTTTTGACAAAACATACGGTGAA | TaqMan |
| Rep-RS | CTAGGCAACTGGTTTGTAACATCTTT | TaqMan | |
| Rep-MGB | FAM-AGGGCACCGTTGTCA-MGB | TaqMan | |
| sgmRNA 7 | Ldrt-VS | CGTGGCTATATCTCTTCTTTTACTTTAACTAG | TaqMan |
| 7 (38)-RS | AAAACTGTAATAAATACAGCATGGAGGAA | TaqMan | |
| mRNA7-MGB | FAM-CGAACTAAACGAGATGCT-MGB | TaqMan |
VS, virus sense; RS, reverse sense; MGB, minor groove binder group.
FAM, 6-carboxyfluorescein.
SYBR, SYBR green-based detection chemistry for qPCR; TaqMan, TaqMan-based detection for qPCR.
Generation of plasmids and RNA constructs for in vitro translation.
To analyze the effect on translation of the TGEV GAIT-like RNA motif, the plasmids pTNT-Luc-Cp, pTNT-Luc-TGEV, and pTNT-Luc were generated. These plasmids expressed (under the control of the T7 promoter) three chimeric mRNAs carrying the luciferase gene, which contained at the 3′ UTR the Cp GAIT element, the TGEV GAIT-like RNA motif, or no GAIT element. The pTNT-Luc-Cp plasmid was generated by cloning into the pTNT vector (Promega) digested with MluI and NotI a chemically synthesized (GeneArt; Life Technologies) DNA fragment (1,761 bp), which contained the restriction site MluI, the luciferase gene, the first 48 nt of the luciferase mRNA 3′ UTR, a 15-nt polylinker, and the 29-nt Cp GAIT element (28), flanked by the restriction sites SalI and NotI. To generate the pTNT-Luc-TGEV plasmid, the SalI-NotI DNA fragment from pTNT-Luc-Cp was exchanged with a chemically synthesized (GeneArt; Life Technologies) SalI-NotI DNA fragment containing the 32-nt TGEV GAIT-like RNA motif. Finally, the plasmid pTNT-Luc, which did not contain any GAIT element, was generated by religation of pTNT-Luc-Cp digested with SalI and NotI and treated with Klenow polymerase.
To generate the capped mRNA construct for in vitro translation, the pTNT-Luc-Cp, pTNT-Luc-TGEV, and pTNT-Luc plasmids were linearized with BamHI and in vitro transcribed with T7 polymerase by using the mMESSAGE mMACHINE T7 kit (Ambion), by following the manufacturer’s recommendations. The resulting transcripts were purified with the RNeasy minikit (Qiagen), analyzed by denaturing electrophoresis in 2.5% (wt/vol) agarose gels, and spectrophotometrically quantified.
Preparation of cytosolic extracts from U937 cells.
To prepare cytosolic extracts of U937 cells, 1 × 107 cells grown in 10-cm-diameter dishes were incubated in 10 ml of RPMI 1640 containing 0.5% FBS for 1 h at 37°C and then treated or not treated with 500 U/ml of recombinant human IFN-γ (R&D Systems) for 24 h at 37°C. After IFN treatment, cytosolic extracts were prepared using the PhosphoSafe extraction reagent (Novagen) containing protease inhibitor cocktail (Roche), by following the manufacturer’s recommendations, and the protein concentration was determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific).
In vitro translation of chimeric luciferase mRNAs.
In vitro transcribed chimeric luciferase mRNAs, containing the Cp GAIT element, the TGEV GAIT-like RNA motif, or no GAIT element at the 3′ UTR, were in vitro translated in the absence or in the presence of the GAIT complex. To this end, 200 ng of capped chimeric transcripts were in vitro translated with 35 µl of rabbit reticulocyte lysate (Promega) in the presence of 4 µg of cytosolic extracts from U937 monocytic cells untreated or treated with IFN-γ, 1 µM amino acid mixture, and 40 U of RNasin RNase inhibitor (Promega). The translation reactions were performed at 30°C for 90 min in a final volume of 50 µl. Finally, the translation efficiency was analyzed by luciferase activity quantification using the luciferase assay system (Promega) and an Orion microplate luminometer (Berthold Detection System).
Construction of a TGEV cDNA clone harboring mutations in the GAIT-like RNA motif.
The TGEV infectious clone harboring mutations in the GAIT-like RNA motif (pBAC-TGEV-mutG) was generated from a TGEV cDNA clone (pBAC-TGEVFL-SPTV) containing the S gene of the TGEV Purdue-type virus isolate (32, 44, 47). To this end, a DNA fragment (mutG) expanding the last 727 nt of the viral genome was generated by chemical synthesis (GeneArt; Life Technologies). This fragment was flanked by the restriction sites KpnI and BamHI and contained three point mutations, A to G, U to C, and C to U, at genomic positions 28178, 28181, and 28190, respectively. The DNA fragment was digested with KpnI and BamHI and cloned into the intermediate plasmid pSL-3EMN7 (containing the TGEV genome nt 20372 to 28087) digested with the same restriction sites, leading to plasmid pSL-3EMN7-mutG. To generate pBAC-TGEV-mutG, the MluI-BamHI DNA fragment from the infectious clone pBAC-TGEVFL-SPTV was exchanged with the MluI-BamHI DNA fragment from plasmid pSL-3EMN7-mutG. All cloning steps were checked by restriction analysis and sequencing.
Recovery of the recombinant virus from the cDNA clone.
To recover the rTGEV-mutG recombinant virus, BHK-pAPN cells were grown to 95% confluence on 35-mm-diameter plates and transfected with 4 µg of infectious cDNA clone pBAC-TGEV-mutG using 12 µl of Lipofectamine 2000 (Invitrogen), according to the manufacturer’s specifications. After 6 h of incubation at 37°C, cells were trypsinized, plated over confluent ST monolayers grown on a 35-mm-diameter plate, and incubated at 37°C for 72 h. The cell supernatants were harvested, and the recovered virus was cloned by three rounds of plaque purification. Recombinant virus was grown and titrated as previously described (45).
Analysis of cellular gene expression and viral RNA synthesis.
Cellular gene expression and viral RNA synthesis were quantified by RT-qPCR. Total RNA from uninfected or infected ST cells was prepared using the RNeasy minikit (Qiagen), and total cDNA was synthesized with random hexamers from 100 ng of total RNA, using the high-capacity cDNA transcription kit (Life Technologies), by following the manufacturers’ instructions. Cellular gene expression was analyzed using TaqMan gene expression assays (Life Technologies) specific for porcine TNF (Ss03391318_g1), CCL2 (Ss03394377_m1), CCL4 (Ss03381395_u1), CXCL9 (Ss03390033_m1), CXCL11 (Ss03648935_g1), IFN-β (Ss03378485_u1), MDA5 (Ss03386373_u1), RIG-I (Ss03381552_u1), or TFG-β (Ss03382325_u1) genes. To analyze viral RNA levels, custom TaqMan assays (Life Technologies) specific for the TGEV genomic RNA and sgmRNA 7 (Table 3) were used. The β-glucuronidase (GUSB) gene (TaqMan code Ss03387751_u1) was used as a reference housekeeping gene, since its expression remains constant in infected cells compared to that in uninfected cells. Data were acquired with a 7500 real-time PCR system (Applied Biosystems) and analyzed with 7500 software v2.0.6. All experiments and data analysis were MIQE compliant (48).
siRNA transfection and silencing analysis.
ST cells were transfected twice with specific Silencer Select siRNAs (Ambion-Life Technologies) by following a reverse transfection protocol. Briefly, for each well of a 6-well plate, 8.4 × 105 cells were transfected in suspension with 25 nM of each siRNA using 8 µl of siPORT amine (Ambion-Life Technologies) diluted in 100 µl of Opti-MEM medium (Gibco BRL-Life Technologies), by following the manufacturer’s instructions. For the porcine MDA5 gene, the siRNA used in the assay (sense sequence of 5′ CCC AGA AUA UGA AAA UGA Att 3′ and antisense sequence of 5′ UUC AUU UUC AUA UUC UGG Gtt 3′ [lowercase indicates nucleotides protruding at the 3′ ends]) was selected from three designed siRNAs, as it provided the highest silencing efficiency (around 40%) in previous MDA5-silencing experiments (data not shown). To silence the RIG-I gene, an siRNA (sense sequence of 5′ CAG UCA GAG ACA ACU UGG Att 3′ and antisense sequence of 5′ UCC AAG UUG UCU CUG ACU Gtt 3′ [lowercase indicates nucleotides protruding at the 3′ ends]) previously validated in porcine cells was used (36). As a negative control, an irrelevant siRNA (sense sequence of 5′ CUGCCCCAGCGAUAUCCAGtt 3′ and antisense sequence of 5′ CUGGAUAUCGCUGGGGCAGtt 3′ [lowercase indicates nucleotides protruding at the 3′ ends]) previously described for porcine cells (36) was used. After transfection, cells were plated onto each well using DMEM with 10% FBS without antibiotic and incubated at 37°C for 48 h. Then, cells were trypsinized, retransfected in suspension with 25 nM of each siRNA, seeded in 24-well plates at a confluence of 2 × 105 cells per well, and incubated at 37°C for 24 h. Transfection medium was then discarded, and cells were infected with rTGEV-WT and rTGEV-mutG viruses at an MOI of 3. Total RNA and protein extract were collected at 12 and 18 hpi (36 and 42 h after the second siRNA transfection, respectively) for further analysis.
Statistical analysis.
A two-tailed, unpaired Student t test was used to analyze differences in mean values between groups. All results were expressed as means ± standard deviations of the means. P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank M. Fresno for kindly providing U937 cells and for his advice on the preparation of U937 cytoplasmic extracts containing the GAIT complex.
This work was supported by grants from the Ministry of Science and Innovation of Spain (MCINN) (BIO2010-16705), the European Community’s Seventh Framework Programme (FP7/2007-2013) under the project “EMPERIE” (HEALTH-F3-2009-223498), and the National Institutes of Health (NIH) of The United States (2P01AI060699-06A1). S.M.-J. received a predoctoral fellowship from the National Institutes of Health (ISCIII) of Spain and a contract from the National Institutes of Health (NIH) of The United States. A.N. and S.Z. received contracts from EU and NIH.
Footnotes
Citation Marquez-Jurado S, Nogales A, Zuñiga S, Enjuanes L, Almazán F. 2015. Identification of a gamma interferon-activated inhibitor of translation-like RNA motif at the 3′ end of the transmissible gastroenteritis coronavirus genome modulating innate immune response. mBio 6(2):e00105-15. doi:10.1128/mBio.00105-15.
REFERENCES
- 1.De Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2012. Coronaviridae, p 774–796. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 2.Masters PS. 2006. The molecular biology of coronaviruses. Adv Virus Res 66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enjuanes L, Van der Zeijst B. 1995. Molecular basis of transmissible gastroenteritis coronavirus epidemiology, p 337–376. In Siddell SG (ed), The Coronaviridae. Plenum Press, New York, NY. [Google Scholar]
- 4.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Peñaranda S, Bankamp B, Maher K, Chen M-H, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rassmussen M, Fouchier R, Gunther S, Osterhaus ADME, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 6.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 7.Enjuanes L, Almazán F, Sola I, Zuñiga S. 2006. Biochemical aspects of coronavirus replication and virus-host interaction. Annu Rev Microbiol 60:211–230. doi: 10.1146/annurev.micro.60.080805.142157. [DOI] [PubMed] [Google Scholar]
- 8.Ziebuhr J. 2005. The coronavirus replicase, p 57–94. In Enjuanes L (ed), Coronavirus replication and reverse genetics, vol 287 Springer-Verlag, Berlin, Germany. [Google Scholar]
- 9.Sola I, Mateos-Gomez PA, Almazan F, Zuñiga S, Enjuanes L. 2011. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol 8:237–248. doi: 10.4161/rna.8.2.14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JY, Chen WN, Poon KM, Zheng BJ, Lin X, Wang YX, Wen YM. 2009. Interaction between SARS-CoV helicase and a multifunctional cellular protein (Ddx5) revealed by yeast and mammalian cell two-hybrid systems. Arch Virol 154:507–512. doi: 10.1007/s00705-009-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu L, Khadijah S, Fang S, Wang L, Tay FP, Liu DX. 2010. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J Virol 84:8571–8583. doi: 10.1128/JVI.00392-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi ST, Lai MM. 2005. Viral and cellular proteins involved in coronavirus replication. Curr Top Microbiol Immunol 287:95–131. doi: 10.1007/3-540-26765-4_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galán C, Sola I, Nogales A, Thomas B, Akoulitchev A, Enjuanes L, Almazán F. 2009. Host cell proteins interacting with the 3′ end of TGEV coronavirus genome influence virus replication. Virology 391:304–314. doi: 10.1016/j.virol.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zinzula L, Tramontano E. 2013. Strategies of highly pathogenic RNA viruses to block dsRNA detection by RIG-I-like receptors: hide, mask, hit. Antiviral Res 100:615–635. doi: 10.1016/j.antiviral.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Totura AL, Baric RS. 2012. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen S, Thomsen AR. 2012. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol 86:2900–2910. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 18.Züst R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. 2009. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr.. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Q, Langereis MA, Olagnier D, Chiang C, van de Winkel R, van Essen P, Zoll J, Hiscott J, van Kuppeveld FJ. 2014. Coxsackievirus cloverleaf RNA containing a 5′ triphosphate triggers an antiviral response via RIG-I activation. PLoS One 9:e95927. doi: 10.1371/journal.pone.0095927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uzri D, Gehrke L. 2009. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol 83:4174–4184. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Launer-Felty K, Cole JL. 2014. Domain interactions in adenovirus VAI RNA mediate high-affinity PKR binding. J Mol Biol 426:1285–1295. doi: 10.1016/j.jmb.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsue B, Masters PS. 1997. A bulged stem-loop structure in the 3′ untranslated region of the genome of the coronavirus mouse hepatitis virus is essential for replication. J Virol 71:7567–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams GD, Chang R-Y, Brian DA. 1999. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J Virol 73:8349–8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Züst R, Miller TB, Goebel SJ, Thiel V, Masters PS. 2008. Genetic interactions between an essential 3′ cis-acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J Virol 82:1214–1228. doi: 10.1128/JVI.01690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sampath P, Mazumder B, Seshadri V, Fox PL. 2003. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol 23:1509–1519. doi: 10.1128/MCB.23.5.1509-1519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay R, Jia J, Arif A, Ray PS, Fox PL. 2009. The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci 34:324–331. doi: 10.1016/j.tibs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, Ting SM, Dignam JD, Kim S, Driscoll DM, Fox PL. 2004. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell 119:195–208. doi: 10.1016/j.cell.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 31.Jia J, Arif A, Ray PS, Fox PL. 2008. WHEP domains direct noncanonical function of glutamyl-prolyl tRNA synthetase in translational control of gene expression. Mol Cell 29:679–690. doi: 10.1016/j.molcel.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Almazán F, González JM, Pénzes Z, Izeta A, Calvo E, Plana-Durán J, Enjuanes L. 2000. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc Natl Acad Sci U S A 97:5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray PS, Fox PL. 2007. A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J 26:3360–3372. doi: 10.1038/sj.emboj.7601774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gantier MP, Williams BR. 2007. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev 18:363–371. doi: 10.1016/j.cytogfr.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Liu Y, Zhang X. 2010. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol 84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hüsser L, Alves MP, Ruggli N, Summerfield A. 2011. Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus Res 159:9–16. doi: 10.1016/j.virusres.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Ibba M, Soll D. 2000. Aminoacyl-tRNA synthesis. Annu Rev Biochem 69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 38.Guo M, Schimmel P. 2013. Essential nontranslational functions of tRNA synthetases. Nat Chem Biol 9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dewan V, Reader J, Forsyth KM. 2014. Role of aminoacyl-tRNA synthetases in infectious diseases and targets for therapeutic development. Top Curr Chem 344:293–329. doi: 10.1007/128_2013_425. [DOI] [PubMed] [Google Scholar]
- 40.Dreher TW. 2009. Role of tRNA-like structures in controlling plant virus replication. Virus Res 139:217–229. doi: 10.1016/j.virusres.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodríguez Pulido MR, Sobrino F, Borrego B, Sáiz M. 2013. Use of RNA domains in the viral genome as innate immunity inducers for antiviral strategies and vaccine improvement, p 193–212. In Romanowski V (ed), Current issues in molecular virology—viral genetics and biotechnological applications. InTech, Rijeka, Croatia. doi: 10.5772/56099. [DOI] [Google Scholar]
- 42.McClurkin AW, Norman JO. 1966. Studies on transmissible gastroenteritis of swine. II. Selected characteristics of a cytopathogenic virus common to five isolates from transmissible gastroenteritis. Can J Comp Med Vet Sci 30:190–198. [PMC free article] [PubMed] [Google Scholar]
- 43.Sánchez CM, Jiménez G, Laviada MD, Correa I, Suñe C, Bullido Mj, Gebauer F, Smerdou C, Callebaut P, Escribano JM, Enjuanes L. 1990. Antigenic homology among coronaviruses related to transmissible gastroenteritis virus. Virology 174:410–417. doi: 10.1016/0042-6822(90)90094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sánchez CM, Izeta A, Sánchez-Morgado JM, Alonso S, Sola I, Balasch M, Plana-Durán J, Enjuanes L. 1999. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J Virol 73:7607–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiménez G, Correa I, Melgosa MP, Bullido MJ, Enjuanes L. 1986. Critical epitopes in transmissible gastroenteritis virus neutralization. J Virol 60:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galán C, Enjuanes L, Almazán F. 2005. A point mutation within the replicase gene differentially affects coronavirus genome versus minigenome replication. J Virol 79:15016–15026. doi: 10.1128/JVI.79.24.15016-15026.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruz JL, Sola I, Becares M, Alberca B, Plana J, Enjuanes L, Zuñiga S. 2011. Coronavirus gene 7 counteracts host defenses and modulates virus virulence. PLoS Pathog 7:e1002090. doi: 10.1371/journal.ppat.1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 49.Zuker M. 2003. Mfold Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]