

ABSTRACT
Infection by alphaherpesviruses invariably results in invasion of the peripheral nervous system (PNS) and establishment of either a latent or productive infection. Infection begins with long-distance retrograde transport of viral capsids and tegument proteins in axons toward the neuronal nuclei. Initial steps of axonal entry, retrograde transport, and replication in neuronal nuclei are poorly understood. To better understand how the mode of infection in the PNS is determined, we utilized a compartmented neuron culturing system where distal axons of PNS neurons are physically separated from cell bodies. We infected isolated axons with fluorescent-protein-tagged pseudorabies virus (PRV) particles and monitored viral entry and transport in axons and replication in cell bodies during low and high multiplicities of infection (MOIs of 0.01 to 100). We found a threshold for efficient retrograde transport in axons between MOIs of 1 and 10 and a threshold for productive infection in the neuronal cell bodies between MOIs of 1 and 0.1. Below an MOI of 0.1, the viral genomes that moved to neuronal nuclei were silenced. These genomes can be reactivated after superinfection by a nonreplicating virus, but not by a replicating virus. We further showed that viral particles at high-MOI infections compete for axonal proteins and that this competition determines the number of viral particles reaching the nuclei. Using mass spectrometry, we identified axonal proteins that are differentially regulated by PRV infection. Our results demonstrate the impact of the multiplicity of infection and the axonal milieu on the establishment of neuronal infection initiated from axons.
IMPORTANCE
Alphaherpesvirus genomes may remain silent in peripheral nervous system (PNS) neurons for the lives of their hosts. These genomes occasionally reactivate to produce infectious virus that can reinfect peripheral tissues and spread to other hosts. Here, we use a neuronal culture system to investigate the outcome of axonal infection using different numbers of viral particles and coinfection assays. We found that the dynamics of viral entry, transport, and replication change dramatically depending on the number of virus particles that infect axons. We demonstrate that viral genomes are silenced when the infecting particle number is low and that these genomes can be reactivated by superinfection with UV-inactivated virus, but not with replicating virus. We further show that viral invasion rapidly changes the profiles of axonal proteins and that some of these axonal proteins are rate limiting for efficient infection. Our study provides new insights into the establishment of silent versus productive alphaherpesvirus infections in the PNS.
INTRODUCTION
Herpesviruses typically establish and maintain a quiescent (or latent) infection in specific cells in their immunocompetent hosts (1). This relatively silent state of infection is characterized by the expression of a limited number of transcripts and lack of active replication (2). The quiescent infection can be reactivated after various stimuli, and the new virions can spread to other hosts. Establishment, maintenance, and reactivation from the quiescent state require the concerted action of viral and cellular proteins (3, 4). The human alphaherpesviruses (e.g., herpes simplex virus 1 [HSV-1] and 2 [HSV-2] and varicella-zoster virus [VZV]) and animal alphaherpesviruses (e.g., pseudorabies virus [PRV], bovine herpesvirus 1 [BHV-1]) invade and establish reactivatable infections in neurons of the peripheral nervous system (PNS) ganglia, including the trigeminal ganglion (TG), dorsal root ganglion (DRG), and superior cervical ganglion (SCG) (2, 5–7).
Infection of PNS neurons requires the long-distance, retrograde axonal transport of invading viral particles to the neuronal nuclei in the ganglia (8). In humans, these PNS axons can be centimeters to meters in length. Alphaherpesvirus virions enter axons by receptor-mediated membrane fusion, which separates virion envelope, glycoproteins, and outer-tegument proteins from nucleocapsids and inner-tegument proteins (9, 10). Some virion components are transported separately in axons, which influences establishment of productive or quiescent infection in the cell bodies. It has been suggested that in axonal infections, required transcription activators carried in the virion tegument, such as VP16, may not reach the neuronal nucleus to facilitate productive infection (11, 12). VP16 is essential to initiate transcription of the HSV-1 immediate early (IE) genes: ICP4, ICP0, ICP22, ICP27, and ICP47 (2, 3). In the case of PRV, there is only one IE gene product, IE180, regulating the transcription of viral genes (13). If IE gene expression fails, the infection is not productive, and only the products of latency-associated transcript (LAT) and microRNAs can be expressed by the viral genome (4, 14–16). Consequently, viral DNA is covered with nucleosomes with histone modifications that repress HSV-1 promoters (17, 18). When these repressive nucleosomes are removed, the silenced viral genomes can be reactivated to replicate and produce progeny (2).
Our basic hypothesis was that the initial interaction of viral particles with axons determines whether the infection will be quiescent or productive in distant neuronal nuclei. This idea came from our previous finding that infection of isolated PNS axons by PRV virions induced synthesis of several proteins locally in axons, some of which were required for efficient transport of viral capsids to the cell bodies (19). In this report, we found that the number of particles infecting axons determines both the efficiency of axonal transport and the efficiency of replication in distant cell bodies. The entry and retrograde transport dynamics of viral particles in axons changed dramatically between multiplicities of infection (MOIs) of 1 and 10 but did not differ significantly between MOIs of 10 and 100. Moreover, we found that productive infection of neuronal cell bodies was efficient at MOIs of 10 and 100, was delayed at an MOI of 1, and was silenced below an MOI of 0.1. In a rescue experiment, high-MOI UV-inactivated virus was used to complement low-MOI replication-competent virus. Surprisingly, UV-inactivated virus particles did not complement infection by low-MOI infecting particles but actually blocked infection, most probably by competing for axonal proteins required for efficient retrograde transport. To identify axonal proteins that might limit viral infection, we used mass spectrometry analysis and found that the profile of axonal proteins changes rapidly after infection. Finally, we observed that low-MOI axonal infection, while inefficient for axonal transport, delivers genomes to cell bodies that are not expressed. However, these silenced genomes can be reactivated, even after 20 days, by infection with UV-inactivated virus particles, but not by replicating virus particles.
RESULTS
Postentry retrograde transport of PRV particles in axons is nonprocessive at and below an MOI of 1.
We infected neurite (N) compartment axons with 106, 105, 104, 103, and 102 PFU (Fig. 1A). We converted these absolute numbers of infectious virions to a relative MOI based on the number of cell bodies plated in the soma (S) compartment for ease of comparison and discussion—e.g., an MOI of 100 is 106 PFU, an MOI of 10 is 105 PFU, an MOI of 1 is 104 PFU, an MOI of 0.1 is 103 PFU, and an MOI of 0.01 is 102 PFU. To monitor infection by optical imaging, we used a PRV recombinant expressing an monomeric red fluorescent protein (mRFP)-VP26 fusion protein that efficiently labels capsids (PRV 180) (20). To quantify the efficiency of infection, we counted the number of red capsids in axons (Fig. 1B) and recorded the motility of these red capsids (see Movie S1 in the supplemental material). Figure 1C shows a maximum-intensity projection of the video frames in Movie S1; movement is recorded as lines of puncta in the projection.
FIG 1.

Effect of multiplicity of infection on PRV 180 capsid motility and infection efficiency during axonal infection. (A) The trichamber neuron culture is schematically represented at the top of the figure (soma [S], middle [M], and neurite [N] compartments). Low-magnification images of cell bodies in the S compartment and high-magnification images of axons in the N compartment are illustrated. (B) PRV 180 capsids (red fluorescent puncta) in infections with different MOIs (MOIs of 100, 10, and 1) in N-compartment axons are shown (0.5 to 1 h postinfection [hpi]). (a, c, and e) Capsid puncta (bars = 10 µm); (b, d, and f) merged images of capsid puncta and axons. In panels e and f, the white arrowhead points to a single capsid puncta. (C) Maximum-intensity projection of the video frames in Movie S1 in the supplemental material. Movement is recorded as lines of puncta. The black arrowhead in the projection for 1 MOI points to the short line corresponding to the back-and-forth movement of a single capsid (bars = 20 µm). (D) PRV 180 retrograde infection at an MOI of 1 in trichambers. Low-magnification images of cell bodies in the S compartment are shown. Virus inoculum was added to the N-compartment axons. The lipophilic dye DiO was added to the N-compartment medium at 3 hpi. Images of S compartments were taken at 24 and 48 hpi. Three independent experiments were performed. Bars = 1,000 µm.
Live-cell microscopy of red capsids in axons after a high-MOI infection of 100 or 10 revealed 325 ± 51.75 and 43 ± 16.8 capsids in axons in the imaging area (this should be corrected to ~1.65 × 10−2 mm2) (Table 1). Specifically, 44.5% ± 3.8% of these capsids were moving at an MOI of 100, and 46.5% ± 3.8% were moving at an MOI of 10 in axons during the first hour of infection (Fig. 1C; see Movie S1 in the supplemental material). However, at an MOI of 1, very few capsids were seen in axons, and the majority of these capsids were not moving during the imaging session. Capsid movement was observed to be erratic and nonprocessive at this MOI (Fig. 1C; Movie S1 shows an example of one capsid with this behavior). We concluded that there is a threshold of infecting PRV particles in axons above which capsids acquire properties of fast and processive retrograde transport toward distant neuronal nuclei.
TABLE 1.
Characterization of particle motility in axons and virus yield in cell bodies during high- and low-MOI retrograde PRV 180 infection
| MOI | No. of capsids/1.65 × 10−2 mm2 | Motility (% total particles)a,b | Virus yield (PFU/ml)a,c |
|
|---|---|---|---|---|
| 24 hpi | 48 hpi | |||
| 100 | 325 ± 51.8 | 44.5 ± 3.8 | 8.3 × 104 ± 3.4 × 104 | 1.1 × 106 ± 2.1 × 105 |
| 10 | 43.3 ± 16.8 | 46.5 ± 3.8 | 3.6 × 104 ± 1.2 × 104 | 5.5 × 105 ± 1.6 × 105 |
| 1 | 2.4 ± 2.5 | ND | <5 | 3.5 × 104 ± 6.7 × 104 |
| ≤0.1 | 0–1 | ND | <5 | <5 |
Values are means ± STDEV.
ND, not detected.
The virus yield detection limit is 5 PFU/ml.
The efficiency and mode of infection of neuronal cell bodies depend on the number of virus particles that infect the axons.
We determined the infectious virus yield from the cell bodies at 24 and 48 h after axonal infection at MOIs of 100, 10, and 1 (Table 1). We added green 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) to axons to label membranes of cell bodies that projected axons to the N compartment. Cell bodies that were primarily infected with PRV 180 appeared yellow. After 24 h, at an MOI of 10 or 100, almost all cell bodies in the S compartment were infected. The yield of infection was 8.3 × 104 ± 3.4 × 104 PFU/ml for the infection with an MOI of 100, and 3.6 × 104 ± 1.2 × 104 PFU/ml for the infection with an MOI of 10 (Table 1). Axons infected at an MOI of 1 showed no capsid accumulation in neuronal nuclei at 24 h postinfection (hpi) (Fig. 1D) with no production of infectious virions at this time. However, by 48 hpi, almost all the nuclei were filled with red capsids (Fig. 1D), and 3.5 × 104 ± 6.7 × 104 PFU/ml progeny were produced. At an MOI of 0.01, no red capsids were detected in neuronal nuclei even after 20 days. To show that the cell bodies could be infected even at a low MOI, infectious virus particles were produced when cell bodies in the S compartment were infected with all MOIs (MOIs from 0.01 to 100) (data not shown). These results, together with our previous work, indicate that efficient retrograde PRV infection is not a linear function of the number of particles added to axons. At an MOI of 100, about 10% of capsids are transported to the neuronal nuclei (19). However, as we show here, below an MOI of 1, capsid dynamics in axons are remarkably different, and infection in the cell bodies is delayed or undetectable, implicating a second bottleneck for productive infection in the neuronal cell bodies.
UV-inactivated PRV virions do not complement but further block infection with low-MOI replication-competent PRV.
Entry and directional movement of capsids are fast and processive after high-MOI axonal infections (MOIs of 10 or 100) but are inefficient after low-MOI infections (MOI below 1). We suspected that the low-MOI entry/transport deficiencies reflected an inability to stimulate downstream signaling in axons required for efficient infection. If this were so, the defects should be complemented by high-MOI coinfection with a nonreplicating virus that can enter and be transported in axons. We infected axons simultaneously with PRV 180 at an MOI of 1 and a UV-inactivated stock of wild-type PRV (Becker) at an MOI of 100. We monitored the cell bodies in the S compartments at 24 and 48 hpi. No PRV red capsid signal was detected in cell bodies by 24 hpi, and more importantly, no PRV red capsid signal was observed in cell bodies at 48 hpi (Fig. 2A). To confirm that UV-inactivated virions do not exclude infection by PRV 180, we coinfected neurons in the S compartment with UV-inactivated PRV Becker (UV-PRV Be or UV-Be) (MOI of 100) and PRV 180 (MOI of 1). There was no effect on the replication of low-MOI PRV 180 when both were added directly to the neuronal cell bodies (Fig. 2B). UV-inactivated PRV did not replicate, but the entry and transport of particles in axons were unaffected (see Fig. S1 in the supplemental material). We also showed that no inhibitory substances were in the media of UV-inactivated virus stocks (Fig. 2C). We further tested the effect of UV-Be complementation at an MOI of 100 on PRV 180 retrograde infection at an MOI of 10. At this ratio (10:1), we found a dramatic decrease both in the number of mRFP-positive neurons in the S chambers and infectious virus yield at 24 hpi (Fig. S2), indicating a stoichiometric interference. We conclude that the high-MOI UV-inactivated virions not only did not complement the low-MOI entry/transport defect but they also were effective inhibitors of low-MOI coinfecting PRV 180 virions.
FIG 2.

High-MOI UV-inactivated PRV Be and low-MOI PRV 180 coinfection. (A) N-compartment axons were infected with either PRV 180 at an MOI of 1 (control; top row of photographs) or together with UV-inactivated Be at an MOI of 100 (bottom row of photographs). The axons were also treated with DiO (green) to label cell bodies that project axons to the N compartment. Low-magnification images of cell bodies in the S compartment are shown in the photographs. (B) S-compartment neurons were either infected with 1 MOI of PRV 180 alone or together with UV-PRV Be at an MOI of 100. Images were taken at 48 hpi. Low-magnification images of cell bodies in the S compartment are shown in the photographs. (C) Infectious titers in S compartments at 48 hpi. Axons were infected with PRV 180 at an MOI of 1 alone (control) or together with UV-inactivated Be at an MOI of 100 in the N compartment (+UVBe N) or in the S compartment (+UVBe S) or with the filtered virus stock to test for inhibitors in the medium. (D) N-compartment axons were infected either with PRV 233 at an MOI of 1 alone or together with UV-PRV Be at an MOI of 100 in the N compartment (+UV-Be). Infectious titer values are means plus SEMs (error bars) from three independent experiments. Values that are significantly different by one-sample t test are indicated as follows: **, P < 0.01; ***, P < 0.001, using one sample t test.
Competition occurs in the primary infected neurons during the early stages of infection, not during viral replication or secondary spread.
The competition by UV-PRV Be could occur at entry or capsid transport or later during replication or spread of infection among cell bodies in the S compartment. To test this, we used PRV 233, a recombinant virus that encodes a diffusible green fluorescent protein (GFP) but lacks glycoprotein B (gB), a membrane protein absolutely required for virion entry and cell-cell spread (21–23). The PRV 233 stock was grown in gB-complementing cells, so that gB was incorporated in the virions. These gB-complemented gB null mutant virions can infect 1 cell; however, after replication, newly produced progeny will lack gB and cannot spread to the neighboring cells (21). Note that GFP expression does not depend on viral DNA replication and that accumulation of the fluorescent protein occurs faster than viral replication. We infected N-compartment axons with PRV 233 at an MOI of 1 to measure the number of GFP-positive cell bodies at 48 hpi. We also coinfected axons with PRV 233 (MOI of 1) and UV-PRV Be at an MOI of 100 and counted green cells at 48 hpi (Fig. 2D). The coinfecting UV-PRV Be virions inhibited infection by PRV 233 (from 37.7% ± 5.72% to 1.7% ± 0.45%), indicating that the competition likely occurred during entry, transport, or nuclear targeting, not during replication or spread in the neuronal cell bodies.
Entry-deficient PRV mutants do not interfere with low-MOI axonal infections.
To test whether interference happens during axonal attachment and entry, we employed entry-deficient PRV mutants in coinfection experiments. Concerted action of alphaherpesvirus envelope glycoproteins and their interaction with host cell membrane proteins are required for particle internalization via receptor-mediated membrane fusion. gB and gC interact with heparan sulfate proteoglycan (HSPG) moieties on the plasma membrane to facilitate interaction of gD with its receptor (e.g., nectin 1) (24). Upon gD binding to the receptor, gH/gL and gB fuse the plasma membrane with the viral envelope (10).
We used PRV mutants deleted for gB (PRV 233) or gD (PRV 357), both of which produce entry-deficient viral particles when grown in noncomplementing cell lines (see Materials and Methods). Equal titers of virus stocks were assayed by SDS-PAGE, and Western blot analysis confirmed the lack of gB and gD in the membranes of gB null and gD null virions, respectively (data not shown). Both gB and gD null virions should attach to axonal membranes, but neither will fuse the viral envelope with the plasma membrane and release capsids and tegument proteins into the axoplasm. Since PRV gB null virions contain gC and gD, which are required for HSPG and receptor (e.g., nectin 1) binding, they should engage these factors on the axonal membrane. On the other hand, PRV gD null virions should retain the capacity to bind to HSPG via gC or gB, but not to the nectin 1 receptor. When we coinfected axons with PRV 180 at an MOI of 1 together with PRV gB null or gD null virions, each at an MOI of 100, we did not detect any interference with entry, transport, or replication of PRV 180 particles (Fig. 3).
FIG 3.

Coinfection of low-MOI PRV 180 with high-MOI entry-deficient mutants. N-compartment axons were infected with PRV 180 (MOI of 1) and either simultaneously coinfected with PRV gB null or PRV gD null virus (MOI of 100) or incubated 1 h before infecting with UV-PRV Be (MOI of 100). The virus yield in cell bodies was determined 48 hpi. Values are means plus SEMs from three independent experiments.
We conclude that the competition by UV-inactivated wild-type virions does not occur at binding to axonal receptors. The competition may occur at engagement of entering capsids with motors or with other transport complexes. To confirm this hypothesis, we infected axons in the N compartment with PRV 180 at an MOI of 1 and waited 1 h before adding UV-Be virions at an MOI of 100 to give low-MOI PRV capsids time to internalize and bind retrograde transport machinery. When we monitored fluorescent capsid accumulation in the cell bodies and determined the virus yield 48 hpi, the infection efficiency was completely restored (Fig. 3).
Infection of axons rapidly changes the profile of axonal proteins.
The previous experiments suggest that one or more host axonal proteins required for efficient retrograde infection must be limiting. We know that high-MOI PRV infection induces new protein synthesis in axons and probably stimulates the degradation of others (19). Alternatively, after high-MOI infections, binding of capsid and tegument proteins to axonal retrograde transport complexes might deplete limiting components of this machinery. Therefore, we predicted that proteins that are decreased in axonal abundance after infection are critical factors in retrograde transport and would be prime candidates defining a rate-limiting step for high-MOI infections of axons. To investigate the changes in the axonal protein repertoire after high-MOI PRV infection, we analyzed the axonal proteome 1 h after infection with PRV Becker or UV-treated PRV Becker (Fig. 4A). Using mass spectrometry-based proteomics, we employed a label-free spectral counting approach to compare the axonal proteome under these conditions to that of mock-infected samples. Our trichamber system ensures physical and fluidic separation between neuronal cell bodies and axons by two barriers (25). After maturation, only axons that could penetrate these barriers are found in the N compartment (Fig. 4A). We combined 10 N compartments for each sample to average chamber variations.
FIG 4.

Mass spectrometry-based label-free analysis of PRV-infected axons. (A) N-compartment axons were infected with PRV Be or UV-Be at an MOI of 100. At 1 hpi, axons were washed twice with PBS and collected in protein lysis buffer. Equal volumes of protein lysates were resolved by one-dimensional (1D) SDS-PAGE, and gels were stained with Coomassie blue. Each gel lane was excised and cut into slices, and proteins were digested in the gel with trypsin. The resulting peptides were analyzed by nanoliquid chromatography-electrospray ionization-tandem mass spectrometry (nLC-MS/MS) using a LTQ Orbitrap XL spectrometer to characterize the axonal proteomes. (B) Total numbers of viral and host proteins detected in mock-, Be-, and UV-Be-infected samples. n is the number of biological replicates. (C) Viral proteins detected in Be- and UV-Be-infected axons. The total spectral counts (mean ± standard deviation [SD]) were normalized to the molecular mass (in kilodaltons) of the protein to calculate average relative viral protein abundance. (D) Correlation of Be-infected axonal proteome with mock- and UV-Be-infected samples. The x and y axes show the mean spectral counts of each protein detected in corresponding samples. (E) The graph shows the host proteins that have significantly increased or decreased spectral counts (corrected P value ≤0.05) in axons during the first hour of Be infection (blue bars) and UV-Be infection (red bars). Statistical significance was calculated by permutation testing and false discovery rate (FDR) correction by the Benjamini-Hochberg posttest as described in Supplemental Materials and Methods in Text S1 in the supplemental material (see also Table S1). M, mock infected.
In mock-infected axons, we identified 2,152 ± 147 host cell proteins, while in Be- and UV-Be-infected axons, 2,102 ± 30 and 2,061 ± 72 host proteins, respectively, and 12 ± 1 and 9 ± 1 PRV proteins were identified, respectively (Fig. 4B; see Table S1 in the supplemental material). The protein repertoires in mock-infected axons (n = 4), Be-infected axons (n = 3), and UV-Be-infected axons (n = 3) were similar, and the average protein spectral counts correlated well in all independent experiments. All of the identified viral proteins are known virion components and were expected to be delivered into the axoplasm upon entry (26). Spectral counts of 8 viral proteins were identified in all of the biological replicates in both Be- and UV-Be-infected samples (Fig. 4C). The tegument proteins VP22 and VP13/14 and the capsid proteins VP5 and VP23 comprised almost 80% of the viral protein abundance in PRV Be-infected axons, when spectral counts were normalized to the protein molecular mass (Fig. 4C).
Comparison of spectral counts for host proteins between Be and UV-Be infections were highly correlated (Fig. 4D, bottom), suggesting that both virus preparations had similar effects on the host axonal proteome. This finding contrasts with the comparison between Be infected and noninfected conditions, which showed less correlation (Fig. 4D, top), suggesting that the process of PRV infection affects axonal protein levels. Using spectral count differences as a measure of altered protein abundance, we identified a subset of proteins that changed significantly in comparison to the noninfected condition (P value of ≤0.05) (Fig. 4E). Specifically, 38 candidate host proteins showed decreases, while 34 were increased. For these candidates, the trend of the changes was the same in both Be- and UV-Be-infected samples (Fig. 4E; see Table S1 in the supplemental material), providing additional confidence that the majority of these changes result from bona fide virion-induced effects.
The levels of proteins involved in endocytosis (e.g., clathrin and dynamin), vesicle and protein transport (Snap91, Ap3d1, Kif5a, and Clip2), and nuclear translocation (e.g., exportin 1 and importin 5/importin beta 3) were all decreased in both Be- and UV-Be-infected axons (Fig. 5; see Table S1 for accession numbers). Proteins regulating ubiquitin-dependent proteasomal degradation, such as Nedd4, Cops2, and UBr4, were also reduced during the first hour of axonal infection. This observation is intriguing, since rapid decreases in the levels of proteins in axons most probably result from protein degradation, which might be induced by the process of viral entry.
FIG 5.

Tabulation of significantly changed proteins in axons 1 h after infection. Proteins were manually categorized based on their biological function or subcellular localization and color coded based on the significant decrease (blue) or increase (orange) in spectral counts following both PRV Be and UV-Be infections versus mock infection. Known direct or indirect functional relationships between proteins (obtained by STRING analysis) are indicated by solid lines between nodes. PT, permutation test.
A significant and consistent increase following infection was observed in the levels of vesicular transport regulators (e.g., Rabs 6a, 9b, and 11b and Ehd4), and signal transduction proteins (e.g., Ywhaz and Gng2). Various metabolic enzymes and regulators, cytoskeletal elements, and ion/small-molecule transporters were also affected after Be and UV-Be infection. Interestingly, the levels of cytoplasmic and mitochondrial glutamic-oxaloacetic transaminases, GOT1 and GOT2, respectively, increased rapidly upon viral infection of axons. In our previous study, local synthesis of mitochondrial GOT2 in axons was detected during the first 2 h of Be axonal infection (19). These enzymes regulate amino acid metabolism, glutamate levels in brain, and facilitate long-chain fatty acid uptake (27, 28).
Low-MOI infecting capsids reach the nucleus, but the DNA is silenced.
The efficiency of cell body infection is dramatically reduced after low-MOI infection of axons. This finding could simply reflect inefficient axonal transport, which we observed. However, as we show next, the cell bodies are indeed infected, but the viral genomes are silenced. To show this, we infected N-compartment axons with PRV 180 at an MOI of 0.01. We monitored cell bodies for 20 days, and during this entire period, no fluorescent capsid expression was observed in any cell bodies in the S compartment (Fig. 6A and C). At 20 days postinfection (dpi), we added UV-treated PRV 959 (at an MOI of 100) directly in the S compartment. Non-UV-treated PRV 959 expresses an mNeonGreen-VP26 fusion protein that labels capsids with a bright green fluorophore. This fusion protein is not expressed from the UV-treated PRV 959 genome. We detected mRFP-capsid accumulation in individual nuclei 4 days after UV-PRV 959 infection, indicating that the silenced PRV 180 genomes were reactivated. No expression of mNeonGreen VP26 was observed. Replicating PRV 180 particles subsequently spread through the entire S compartment in 6 days (Fig. 6B). This result shows that PRV 180 capsids after a very low-MOI infection of axons (MOI of ≤0.1) can reach neuronal nuclei but cannot complete a productive replication cycle. As further evidence that UV-treated PRV 959 indeed infected neurons in which the PRV 180 genome was present, in 1 of the 15 chambers we used in one experiment, mNeonGreen puncta were detected together with red capsids in the nuclei of a few neurons (Fig. 6E). These events most likely reflect recombination of the UV-inactivated PRV 959 genome with the replicating PRV 180 genome.
FIG 6.

Establishment and reactivation of low-MOI, quiescent PRV infection in neurons in trichambers. (A and C) N-compartment axons were infected with PRV 180 (red capsids) at an MOI of 0.01, and neuronal cell bodies in the corresponding S compartments were imaged 20 days postinfection (dpi). Low-magnification images of cell bodies in the S compartment together with RFP and GFP are shown. Bars, 1,000 µm. (B and D) After 20 days, the S compartments of chambers in panel A were superinfected with UV-inactivated PRV 959 (green capsids) at an MOI of 100 (B), and the S compartments of chambers in panel C were superinfected with non-UV-inactivated PRV 959 at an MOI of 0.1 (D). At 6 dpi, S compartments were imaged to detect fluorescent capsid accumulation (red from PRV 180 or green from PRV 959). (E) In one of the chambers shown in panel B, we observed expression of both mRFP and mNeonGreen capsid fusion proteins in cells most likely from a recombination event between reactivating PRV 180 and superinfecting UV-PRV 959. Panels a to d and panels e to h depict two different neuronal cell body clusters in the S compartment. The white arrows point to dual-color nuclei. A total of 20 to 24 chambers were used in 3 independent experiments. Bars = 100 µm.
Superinfection with a replicating virus does not allow reactivation of the silenced genomes.
When we repeated the reactivation experiment by adding a high or low MOI (MOI of 100 or 1, respectively) of non-UV-treated PRV 959 in the S compartments after the 20-day incubation period, no replicating PRV 180 (red capsids) was observed even after a week of incubation (Fig. 6D). All the cell bodies in the S compartment were infected with PRV 959 in 48 h. Importantly, superinfection with replication-competent virus did not allow derepression of silenced genomes, most probably due to the fast replication and reorganization of nuclear compartments during viral replication.
We conclude that not only the efficiency of axonal transport is regulated by the number of particles that infect axons but also the expression of genomes that reach the cell body nuclei is affected. Low-MOI axonal infections lead to silenced, but reactivatable viral genomes in the neuronal nucleus.
DISCUSSION
The life-long persistence of alphaherpesvirus genomes in the nervous systems of their hosts and the capacity to reactivate these silent genomes to produce infectious particles are remarkable for the precision of their control. The establishment of this quiescent infection in the neurons of peripheral ganglia depends on many parameters, including the efficiency of virus replication in epithelial cells, successful virion entry into axon terminals, subsequent retrograde transport of nucleocapsids to the neuronal cell body, as well as effective control of intrinsic and innate antiviral responses in the infected neuron and ganglia. Several studies have suggested that virus infection in the peripheral ganglia is inherently biased toward quiescent or latent infection (2, 11, 29). This bias is more obvious in homologous infections of human viruses with human neurons or swine viruses in swine neurons. When alphaherpesvirus virions enter axons, some tegument proteins are transported toward the cell body separately from the capsid (8). The bias toward the latent infection presumably occurs because a productive infection may require that both tegument proteins and capsids containing the viral genome arrive at the nucleus at the same time (11, 30). Cytokine and interferon (IFN) production at the site of infection also influence the outcome of neuronal infection (31).
Our study provides four important observations on the roles of local axonal responses that influence the overall infection of PNS neurons (Fig. 7). First, the efficiency of retrograde transport in axons depends on the number of virus particles that infect axons. There is a threshold above which particle movement is fast and efficient and below which the entry and transport of particles are delayed and inefficient. When the MOI was less than 1, those few capsids that entered axons exhibited nonprocessive motion, suggesting that the particles could not efficiently engage the transport machinery. Second, we showed that UV-inactivated PRV virions do not complement the low-MOI retrograde infection deficiencies. In fact, we show that UV-inactivated PRV virions actually compete with low-MOI coinfecting particles. Third, we showed that infection of axons rapidly changes the profile of axonal proteins. Fourth, we demonstrated that despite inefficient entry and trafficking, low-MOI infecting capsids do reach the nucleus, but the viral DNA is silenced, indicating a productive infection threshold between MOIs of 1 and 0.1.
FIG 7.
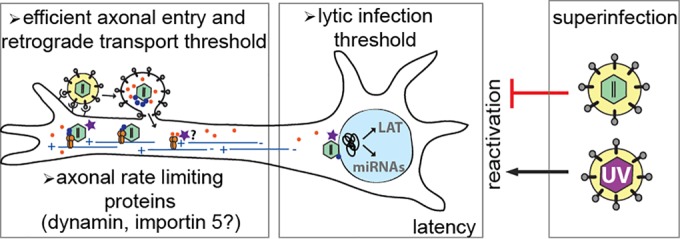
Model of alphaherpesvirus spread into the peripheral nervous system. Upon entry into axons via receptor-mediated membrane fusion, viral nucleocapsids with inner-tegument proteins (e.g., UL36/UL37 [blue dots]) and outer-tegument proteins (e.g., VP16 [orange dots]) are separately transported toward the distant neuronal nuclei by dynein motors on microtubules. Fast and efficient viral entry and transport require high-MOI infection. However, proteins (e.g., dynamin and importin, shown as a purple star) are rate limiting and restrict the number of genomes that may be delivered to the nucleus. In the nucleus, when a low number of viral genomes enter, they are silenced, and a quiescent/latent infection is established. During such an infection, viral gene expression is shut off, and only a few transcripts are expressed (e.g., LAT, microRNAs [miRNAs]). This silent state can be reversed by superinfecting these neurons with UV-inactivated virus. However, reactivation cannot be detected after superinfection with replicating virus.
The reason for the inefficiency of low-MOI infections might be that a limited number of entering virions (or virion components) cannot trigger a signaling cascade required for efficient recruitment of retrograde transport complexes. The coinfection experiments with UV-inactivated wild-type PRV particles prove that the mechanism is not that simple. Even though high-MOI UV-inactivated virus particles are able to stimulate fast transport machinery, they do not complement the low-MOI retrograde infection defect of PRV 180 particles. Importantly, this competition is only for factors in the axons and not in the cell bodies, as there was no competition when cell bodies were directly infected with UV-inactivated PRV and PRV 180. When we infected axons at a low MOI with PRV 180 and then coinfected the axons with UV-PRV at a high MOI after 1 h, there was no competition, and replication of the low-MOI PRV 180 was restored. The observed competition must be for a protein that associates with capsid or tegument proteins during or early after axonal entry. Interestingly, we saw no competition with high-MOI infection of either gB null or gD null virions, indicating that the entry receptors or mediators are not the major rate-limiting factors in axons. These data suggest that one or more host cell protein(s) in the axoplasm must be rate limiting, restricting the number of retrogradely trafficking capsids or tegument proteins that move toward the neuronal nucleus to start transcription and replication. Dynein motor subunits or their regulators, transcription, or nuclear import proteins that associate with the transport complex immediately after entry are likely to be rate limiting in axons.
To investigate how axonal protein levels are affected in the first hour of high-MOI PRV infection, we analyzed axons infected with both wild-type PRV and UV-inactivated PRV by mass spectrometry and compared the results to that of mock-infected axons. Proteomic analyses showed that the axonal proteome changes rapidly and similarly after infection by wild-type or UV-inactivated PRV. We identified several candidates for the limiting protein(s) in axons required for efficient retrograde infection.
First, we noted that dynamin levels drop significantly after infection. The entry of HSV-1 virions into keratinocytes is dependent on dynamin (32), and dynamin and endocytosis machinery are required for cell-to-cell fusion (33). Therefore, dynamin might play a role in viral entry into axons and be a local rate-limiting factor for axonal herpesvirus infection. The idea that competition for limiting proteins happens after receptor attachment and might be at the membrane fusion level is supported by the fact that entry-deficient gD and gB null mutants did not compete with low-MOI PRV 180 infection. Both of these recombinant viruses are defective in membrane fusion. The endocytosis regulatory proteins that decrease after axonal infection are not likely to be candidates for rate-limiting proteins because PRV does not enter axons primarily by endocytosis (unpublished observations).
Importin 5 (Ipo5, i.e., karyopherin beta 3) is a strong candidate for the infection-limiting protein. We know that docking of HSV-1 nucleocapsids at nuclear pores requires importin beta interaction with capsids (34). Efficient localization of viral nucleocapsids to the nuclear pore may require long-distance axonal cotransport of importin and capsids. Moreover, previous reports indicate that newly synthesized importin beta in axons directs molecules containing nuclear localization signal (NLS) to the nucleus in the case of axonal injury (35). Our data and those of others suggest that upon entry into axoplasm, herpesvirus nucleocapsids interact with Ipo5 through the NLS of the inner-tegument protein UL36 (VP1/2) and form a complex with dynein motors (36, 37). These nucleus-targeted complexes move long distances in axons on microtubules toward the minus end in the cell body. If Ipo5 is limited in axons, after high-MOI infections, only a fraction of nucleocapsids will efficiently move to and dock at the nuclear pores for genome delivery. Taken together, both membrane fusion proteins, such as dynamin, and nuclear import factors, such as importin 5, may be rate limiting in axons after high-MOI infection. These interactions might provide new targets that could interfere with the establishment of latent herpesvirus infections in the PNS.
The final observation of our studies is the identification of a threshold for establishing a quiescent or productive infection that is set by the number of infecting particles in axons. Below this threshold (below an MOI of 0.1 in our studies), infecting genomes are delivered to the neuronal nucleus and are silenced. Previous publications have shown that at least two HSV-1 proteins, VP16 and ICP0 play major roles in the establishment of productive infections (3, 38–40). If VP16 does not arrive at the nucleus at the same time as nucleocapsids, and as a result, ICP0 is not expressed quickly, viral genomes are covered with histones by the host cell machinery and effectively silenced (3, 11, 29). An alternative hypothesis is that neuronal innate immunity might be able to keep low-MOI infecting genomes silenced, which may be overcome by high-MOI infecting particles. On the other hand, extensive membrane fusion events resulting from high-MOI virus infection may trigger a local antiviral response in axons as suggested by Noyce et al. (41). However, we do not see a reduction in particle transport in axons or in the efficiency of infection in cell bodies when we compare infections at MOIs of 10 and 100. In our previous studies, PRV infections of rodent neurons were always productive when we used high-MOI infections. De Regge et al. showed that both HSV-1 and PRV infections can be silenced in two-chamber porcine trigeminal ganglion (TG) cultures, when these neurons were treated with IFN alpha before and during infection (42). Our in vitro studies show that in the absence of interferon or any replication inhibitor, low-MOI infection of axons by PRV also results in a silent, but reactivatable, infection in neurons cultured in trichambers (43).
We do not know how many neurons sustain quiescent infection after low-MOI infection or how many genomes are present in a single nucleus during quiescence, but superinfection of this neuron population with a UV-inactivated virus was sufficient to reactivate the genomes even after 20 days of quiescence. The quiescently infected neurons do not exhibit superinfection exclusion. The fact that UV-inactivated virions reactivate the silent genomes suggests that either viral/cellular proteins in the virion itself stimulate reactivation or that cellular responses produced against entering virus particles induce activation of viral promoters. We know that individual expression of HSV-1 proteins ICP0, ICP4, and VP16 is sufficient to reactivate latent HSV-1 genomes in TG neurons (44). Therefore, we anticipate PRV homologues early protein zero (EPO) and VP16 in the UV-inactivated virions to act similarly in derepressing silent genomes. We further showed that UV-irradiated viral genomes can be delivered into the neuronal nuclei. Foreign DNA in the nucleus bearing pyrimidine dimers may activate DNA damage response and apoptotic signaling that was previously shown to stimulate HSV-1 reactivation and gene expression (45, 46). Interestingly, we observed that the silent genome was available for recombination with the superinfecting nonreplicating genome in the nucleus, confirming the colocalization of both genomes in the neuronal nucleus.
An important observation was that a replicating virus (either at high or low MOI) could not fully reactivate the silent genomes. We believe that PRV replication is so rapid that the silenced genomes are unable to compete. This effect may also reflect the rapid reorganization of nuclear architecture and segregation of nuclear factors in favor of replicating viral DNA. As previously published, during HSV-1 infection, nuclear factors such as nuclear domain 10 (ND10) components and DNA repair proteins are redistributed and cellular chromatin is marginalized (47–49). Moreover, beginning 2 hpi, replicating virus starts to exclude infection by a secondary virus, resulting in complete inhibition of superinfection by 8 hpi (13). If this hypothesis is true, even if the viral proteins in the tegument of superinfecting virus (e.g., EPO and VP16) are able to induce derepression of silent genomes, spatial segregation of replication compartments and cellular domains (e.g., ND10) by the replication-competent virus precedes the complete transactivation of silenced promoters. After approximately 8 h, superinfection exclusion may further block replication of the reactivated genome. This hypothesis is currently under study. We observed that reactivation does not happen before replication of the superinfecting virus initiates and infection spreads.
Our work raises a number of other questions. What is the mechanism that initiates fast retrograde transport immediately after axonal infection? Why are low-MOI infecting particles much less efficient at initiating this process? How do tegument proteins (e.g., VP16) engage fast axonal transport machinery? Is the productive infection threshold defined only by the number of capsids that are transported or by the ratio of transported capsids and VP16 that arrive in the nucleus?
MATERIALS AND METHODS
Cell lines and virus strains.
The porcine kidney epithelial cell line (PK15) was purchased from the American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin and streptomycin. Viral titers were determined on PK15 cells by plaque assays. PRV Becker is a wild-type laboratory strain (50). PRV 180 encodes mRFP1-VP26 in a PRV Becker background (20) (see Supplemental Materials and Methods in Text S1 in the supplemental material for further details).
Neuronal cultures.
Superior cervical ganglia (SCG) were isolated from day 17 Sprague-Dawley rat embryos (Hilltop Labs, Inc., Scottsdale, PA), and neurons were cultured in trichambers as described previously (25). Briefly, the SCG were incubated in 250 µg/ml of trypsin (Worthington Biochemicals) for 15 min. Trypsin inhibitor (1 mg/ml; Sigma Aldrich) was added to neutralize the trypsin for 5 min and then removed and replaced with neuron culture medium (see also Supplemental Materials and Methods in Text S1 in the supplemental material). All animal work was done in accordance with the Institutional Animal Care and Use Committee of the Princeton University Research Board under protocol 1947-13.
Virus infection in compartmented neuronal cultures.
For the analysis of retrograde spread in the soma (S) compartment, 1% Methocel including neuronal medium was placed in the middle (M) compartment 30 min before the infection to block possible diffusion of viral particles through the grooves. PRV inoculum was then added to the neurite (N) compartment. Depending on the assay, infected axons or soma were imaged either immediately or at indicated time (hours) postinfection. For titer assays, the contents of the S compartments were collected by scraping the dish with a pipette tip. All titers were determined on PK15 cells and are expressed in PFU/ml.
Antibodies and reagents.
The mouse monoclonal anti-beta-actin antibody (Sigma) was used at a dilution of 1:5,000. Mouse monoclonal anti-VP5 antibody was used at a 1:2,000 dilution (gift of H. Rziha). Monoclonal antibody M2 against PRV gB was used at a 1:800 dilution (51). A polyclonal antiserum against PRV gD was kindly provided by Krystyna Bienkowska-Szewczyk (University of Gdansk). The horseradish peroxidase-coupled secondary mouse and rabbit antibodies (KPL) were used at 1:10,000 dilution. The lipophilic tracer DiO (Invitrogen) was used at a 2.5-µg/ml concentration.
Live-cell imaging.
Live-cell imaging was performed on an Ti-Eclipse inverted epifluorescence microscope (Nikon) equipped with a Cool Snap ES2 camera (Photometrics). Neuron cultures were kept in a humidified stage-top incubator (Live Cell Instrument) at 37°C with 5% CO2 during imaging. The percentage of moving virus particles was calculated manually using NIS-Elements imaging software. Movies were processed using ImageJ Stacks tools.
Western blot analysis.
Total cell extracts were prepared in radioimmunoprecipitation assay (RIPA) buffer without sodium deoxycholate supplemented with 1 mM dithiothreitol (DTT) and protease inhibitor cocktail (Roche). Lysates were kept 30 min on ice and sonicated to shear genomic DNA. Cell lysates were centrifuged at 11,000 rpm at 4°C. Supernatants were then transferred into new tubes, and their protein concentrations were measured by Bradford assay (BioRad) (see also Supplemental Materials and Methods in Text S1 in the supplemental material).
Mass spectrometry-based proteomic sample preparation and data analysis.
N-compartment axons were either mock infected or infected with PRV Be or UV-Be at an MOI of 100 (10 chambers were used for each sample). At 1 h postinfection, axons were washed twice with phosphate-buffered saline (PBS) and collected in 1× NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Life Technologies). Samples were first incubated with 100 mM DTT at 70°C for 10 min and then with 100 mM iodoacetamide (IAA) at room temperature for 30 min protected from light. Part (20%) of each sample was loaded onto 4 to 12% NuPAGE BisTris gels (Invitrogen) and stained with Coomassie blue. GeLC-MS/MS (LC stands for liquid chromatography, and MS/MS stands for tandem mass spectrometry) analysis was performed as previously described with some modifications (52, 53) (see also Supplemental Materials and Methods in Text S1 in the supplemental material).
Q-RT-PCR.
Quantitative reverse transcription-PCR (Q-RT-PCR) was performed with an Eppendorf Realplex Mastercycler. Reaction mixture was prepared using Kapa Sybr Fast quantitative PCR (qPCR) kit, and three replicate samples were prepared. Each experiment was done in duplicate. Ninety microliters of virus stock was first digested with 100 U of DNase I (Invitrogen) to remove contaminating DNA before proteinase K treatment. Capsids were digested with proteinase K (New England Biolabs) for 50 min at 55°C followed by 5 min at 95°C. Viral DNA released from capsids was quantified by using UL54-specific primers as published previously (54). PRV Becker nucleocapsid DNA and PRV Becker virus stock (1 × 108 PFU/ml) were used as standards to determine the amount of viral DNA corresponding to 1 PFU.
Statistical analysis.
One-way analysis of variance (ANOVA) with Tukey’s posttest or Student’s t test were performed using GraphPad Prism 5.0d for Max OS X (GraphPad Software). Values in the text, bar graphs, and figure legends throughout the manuscript are means ± standard errors of the means (SEMs).
SUPPLEMENTAL MATERIAL
Supplemental text including extended results and supplemental Materials and Methods. Download
UV inactivation of PRV particles. (A) PRV Be aliquots that were treated with increasing amounts of UV light were graphed against their virus yield (n = 3). (B) PK15 cells were either infected with PRV Be or UV-PRV Be (MOI of 10) or mock infected. At 24 hpi, cells were harvested and lysed, and Western blot assay was performed. Membranes were probed with antibodies against the major capsid protein VP5 and envelope proteins gB and gD. (C) N-compartment axons were infected with PRV 180 and UV-PRV 180, and capsid motility was assayed by live-cell imaging in the M-compartment axons. (D) N-compartment axons were infected with PRV 426, PRV 926, UV-PRV 426, and UV-PRV 926 at an MOI of 10. Neuronal cell bodies in the corresponding S compartments were imaged at 48 hpi. Low-magnification images of cell bodies in the S compartment are shown. RFP and GFP channels are shown in the figure. Values are means ± SEMs. Bars = 1,000 µm. Download
Coinfection with UV-inactivated PRV Be (MOI of 100) and PRV 180 (MOI of 10). (A) N-compartment axons were infected with either PRV 180 at an MOI of 10 (control) (top) or together with UV-inactivated Be at an MOI of 100 (bottom). The axons were also treated with DiO (green) to label cell bodies that project axons to the N compartment. Low-magnification images of cell bodies in the S compartment are shown. (B) Infectious titers in S compartments at 24 hpi. Axons were infected with PRV 180 at an MOI of 10 alone (control) or together with UV-inactivated Be at an MOI of 100 in the N compartment (+UV Be). Download
Proteins identified in axons. Axons were mock infected (M) and infected with PRV Becker (Be) or UV-inactivated PRV Becker (UV-Be). Protein name, description, UniProt accession number, and normalized spectral counts (see Supplemental Materials and Methods in Text S1 in the supplemental material) at the protein cluster/group level are reported for each biological replicate (n = 4 for M; n = 3 for Be and UV). Averages of replicates were calculated, and spectral counts of each protein in Be- and UV-Be-infected samples were compared to that of the mock-infected sample after addition of a value of one (plus 1) to obtain the fold change value. On the second worksheet, differential abundance calculations are reported separately for Be versus mock and UV Be versus mock, using a permutation test with a Benjamini-Hochberg posttest (P < 0.05) to correct FDR for multiple comparisons. For this comparison, normalized spectral counts of individual proteins in a cluster/group were used.
Time-lapse microscopy recording of PRV 180 capsids in SCG axons during infections at MOIs of 1, 10, and 100. Capsid motility was recorded 10 to 50 min after infection at a rate of approximately 2 frames/s. The playback speed is 10 frames/s. (Top) A single capsid (arrow) infected at an MOI of 1 is shown. Capsids infected at an MOI of 10 (middle) or 100 (bottom) are shown. Download
ACKNOWLEDGMENTS
We thank all the members of the Enquist lab, especially Jens B. Bosse, Julian Scherer, Ian Hogue, Esteban Engel, Karen Lancaster, and Emre Koyuncu for sharing reagents and critical reading of the manuscript.
L.W.E. was supported by U.S. National Institutes of Health grants RO1 NS33506 and RO1 NS060699. I.M.C. was supported by grants from NIDA A (DP1DA026192) and NIAID (R21AI102187), and T.M.G. was supported by an NJCCR postdoctoral fellowship.
Footnotes
Citation Koyuncu OO, Song R, Greco TM, Cristea IM, Enquist LW. 2015. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. mBio 6(2):e00276-15. doi:10.1128/mBio.00276-15.
REFERENCES
- 1.Speck SH, Ganem D. 2010. Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8:100–115. doi: 10.1016/j.chom.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67:355–374. doi: 10.1146/annurev-micro-092412-155654. [DOI] [PubMed] [Google Scholar]
- 3.Preston CM, Efstathiou S. 2007. Molecular basis of HSV latency and reactivation, chapter 33 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 4.Wagner EK, Bloom DC. 1997. Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10:419–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung AK. 1989. Detection of pseudorabies virus transcripts in trigeminal ganglia of latently infected swine. J Virol 63:2908–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilden D, Mahalingam R, Nagel MA, Pugazhenthi S, Cohrs RJ. 2011. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol 37:441–463. doi: 10.1111/j.1365-2990.2011.01167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Microbiol Rev 16:79–95. doi: 10.1128/CMR.16.1.79-95.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hafezi W, Lorentzen EU, Eing BR, Müller M, King NJ, Klupp B, Mettenleiter TC, Kühn JE. 2012. Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog 8:e1002679. doi: 10.1371/journal.ppat.1002679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu BW, Engel EA, Enquist LW. 2014. Characterization of a replication-incompetent pseudorabies virus mutant lacking the sole immediate early gene IE180. mBio 5(6):e01850. doi: 10.1128/mBio.01850-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kubat NJ, Amelio AL, Giordani NV, Bloom DC. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol 78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloom DC. 2004. HSV LAT and neuronal survival. Int Rev Immunol 23:187–198. doi: 10.1080/08830180490265592. [DOI] [PubMed] [Google Scholar]
- 16.Jarman RG, Wagner EK, Bloom DC. 1999. LAT expression during an acute HSV infection in the mouse. Virology 262:384–397. doi: 10.1006/viro.1999.9861. [DOI] [PubMed] [Google Scholar]
- 17.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol 78:1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koyuncu OO, Perlman DH, Enquist LW. 2013. Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host Microbe 13:54–66. doi: 10.1016/j.chom.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Del Rio T, Ch’ng TH, Flood EA, Gross SP, Enquist LW. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J Virol 79:3903–3919. doi: 10.1128/JVI.79.7.3903-3919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curanovic D, Enquist LW. 2009. Virion-incorporated glycoprotein B mediates transneuronal spread of pseudorabies virus. J Virol 83:7796–7804. doi: 10.1128/JVI.00745-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babic N, Mettenleiter TC, Flamand A, Ugolini G. 1993. Role of essential glycoproteins gII and gp50 in transneuronal transfer of pseudorabies virus from the hypoglossal nerves of mice. J Virol 67:4421–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu WW, Goodhouse J, Jeon NL, Enquist LW. 2008. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS One 3:e2382. doi: 10.1371/journal.pone.0002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grünewald K, Miller RJ, Osten P, Spear PG. 2009. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A 106:17916–17920. doi: 10.1073/pnas.0908892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ch’ng TH, Enquist LW. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J Virol 79:10875–10889. doi: 10.1128/JVI.79.17.10875-10889.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramer T, Greco TM, Enquist LW, Cristea IM. 2011. Proteomic characterization of pseudorabies virus extracellular virions. J Virol 85:6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guidetti P, Amori L, Sapko MT, Okuno E, Schwarcz R. 2007. Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. J Neurochem 102:103–111. doi: 10.1111/j.1471-4159.2007.04556.x. [DOI] [PubMed] [Google Scholar]
- 28.Panteghini M. 1990. Aspartate aminotransferase isoenzymes. Clin Biochem 23:311–319. doi: 10.1016/0009-9120(90)80062-N. [DOI] [PubMed] [Google Scholar]
- 29.Roizman B, Sears AE. 1987. An inquiry into the mechanisms of herpes simplex virus latency. Annu Rev Microbiol 41:543–571. doi: 10.1146/annurev.mi.41.100187.002551. [DOI] [PubMed] [Google Scholar]
- 30.Kristie TM, Roizman B. 1988. Differentiation and DNA contact points of host proteins binding at the cis site for virion-mediated induction of alpha genes of herpes simplex virus 1. J Virol 62:1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikloska Z, Cunningham AL. 2001. Alpha and gamma interferons inhibit herpes simplex virus type 1 infection and spread in epidermal cells after axonal transmission. J Virol 75:11821–11826. doi: 10.1128/JVI.75.23.11821-11826.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahn E, Petermann P, Hsu MJ, Rixon FJ, Knebel-Mörsdorf D. 2011. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS One 6:e25464. doi: 10.1371/journal.pone.0025464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin NY, Choi H, Neff L, Wu Y, Saito H, Ferguson SM, De Camilli P, Baron R. 2014. Dynamin and endocytosis are required for the fusion of osteoclasts and myoblasts. J Cell Biol 207:73–89. doi: 10.1083/jcb.201401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol 20:4922–4931. doi: 10.1128/MCB.20.13.4922-4931.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perry RB, Doron-Mandel E, Iavnilovitch E, Rishal I, Dagan SY, Tsoory M, Coppola G, McDonald MK, Gomes C, Geschwind DH, Twiss JL, Yaron A, Fainzilber M. 2012. Subcellular knockout of importin beta1 perturbs axonal retrograde signaling. Neuron 75:294–305. doi: 10.1016/j.neuron.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O’Hare P. 2012. A nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J Virol 86:8998–9014. doi: 10.1128/JVI.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wysocka J, Herr W. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci 28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 39.Wilson AC, LaMarco K, Peterson MG, Herr W. 1993. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell 74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 40.Xiao P, Capone JP. 1990. A cellular factor binds to the herpes simplex virus type 1 transactivator Vmw65 and is required for Vmw65-dependent protein-DNA complex assembly with Oct-1. Mol Cell Biol 10:4974–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noyce RS, Taylor K, Ciechonska M, Collins SE, Duncan R, Mossman KL. 2011. Membrane perturbation elicits an IRF3-dependent, interferon-independent antiviral response. J Virol 85:10926–10931. doi: 10.1128/JVI.00862-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Regge N, Van Opdenbosch N, Nauwynck HJ, Efstathiou S, Favoreel HW. 2010. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS One 5:e13076. doi: 10.1371/journal.pone.0013076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halford WP, Kemp CD, Isler JA, Davido DJ, Schaffer PA. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J Virol 75:6143–6153. doi: 10.1128/JVI.75.13.6143-6153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A 109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Volcy K, Fraser NW. 2013. DNA damage promotes herpes simplex virus-1 protein expression in a neuroblastoma cell line. J Neurovirol 19:57–64. doi: 10.1007/s13365-012-0140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol 75:1223–1233. [DOI] [PubMed] [Google Scholar]
- 48.Monier K, Armas JC, Etteldorf S, Ghazal P, Sullivan KF. 2000. Annexation of the interchromosomal space during viral infection. Nat Cell Biol 2:661–665. doi: 10.1038/35023615. [DOI] [PubMed] [Google Scholar]
- 49.Weller SK. 2010. Herpes simplex virus reorganizes the cellular DNA repair and protein quality control machinery. PLoS Pathog 6:e1001105. doi: 10.1371/journal.ppat.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platt KB, Maré CJ, Hinz PN. 1979. Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky’s disease) virus: thermal sensitivity and rabbit virulence markers. Arch Virol 60:13–23. doi: 10.1007/BF01318093. [DOI] [PubMed] [Google Scholar]
- 51.Hampl H, Ben-Porat T, Ehrlicher L, Habermehl KO, Kaplan AS. 1984. Characterization of the envelope proteins of pseudorabies virus. J Virol 52:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kramer T, Greco TM, Taylor MP, Ambrosini AE, Cristea IM, Enquist LW. 2012. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 12:806–814. doi: 10.1016/j.chom.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai YC, Greco TM, Cristea IM. 2014. Sirtuin 7 plays a role in ribosome biogenesis and protein synthesis. Mol Cell Proteomics 13:73–83. doi: 10.1074/mcp.M113.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tombácz D, Tóth JS, Petrovszki P, Boldogkoi Z. 2009. Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative RT-PCR assay. BMC Genomics 10:491. doi: 10.1186/1471-2164-10-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental text including extended results and supplemental Materials and Methods. Download
UV inactivation of PRV particles. (A) PRV Be aliquots that were treated with increasing amounts of UV light were graphed against their virus yield (n = 3). (B) PK15 cells were either infected with PRV Be or UV-PRV Be (MOI of 10) or mock infected. At 24 hpi, cells were harvested and lysed, and Western blot assay was performed. Membranes were probed with antibodies against the major capsid protein VP5 and envelope proteins gB and gD. (C) N-compartment axons were infected with PRV 180 and UV-PRV 180, and capsid motility was assayed by live-cell imaging in the M-compartment axons. (D) N-compartment axons were infected with PRV 426, PRV 926, UV-PRV 426, and UV-PRV 926 at an MOI of 10. Neuronal cell bodies in the corresponding S compartments were imaged at 48 hpi. Low-magnification images of cell bodies in the S compartment are shown. RFP and GFP channels are shown in the figure. Values are means ± SEMs. Bars = 1,000 µm. Download
Coinfection with UV-inactivated PRV Be (MOI of 100) and PRV 180 (MOI of 10). (A) N-compartment axons were infected with either PRV 180 at an MOI of 10 (control) (top) or together with UV-inactivated Be at an MOI of 100 (bottom). The axons were also treated with DiO (green) to label cell bodies that project axons to the N compartment. Low-magnification images of cell bodies in the S compartment are shown. (B) Infectious titers in S compartments at 24 hpi. Axons were infected with PRV 180 at an MOI of 10 alone (control) or together with UV-inactivated Be at an MOI of 100 in the N compartment (+UV Be). Download
Proteins identified in axons. Axons were mock infected (M) and infected with PRV Becker (Be) or UV-inactivated PRV Becker (UV-Be). Protein name, description, UniProt accession number, and normalized spectral counts (see Supplemental Materials and Methods in Text S1 in the supplemental material) at the protein cluster/group level are reported for each biological replicate (n = 4 for M; n = 3 for Be and UV). Averages of replicates were calculated, and spectral counts of each protein in Be- and UV-Be-infected samples were compared to that of the mock-infected sample after addition of a value of one (plus 1) to obtain the fold change value. On the second worksheet, differential abundance calculations are reported separately for Be versus mock and UV Be versus mock, using a permutation test with a Benjamini-Hochberg posttest (P < 0.05) to correct FDR for multiple comparisons. For this comparison, normalized spectral counts of individual proteins in a cluster/group were used.
Time-lapse microscopy recording of PRV 180 capsids in SCG axons during infections at MOIs of 1, 10, and 100. Capsid motility was recorded 10 to 50 min after infection at a rate of approximately 2 frames/s. The playback speed is 10 frames/s. (Top) A single capsid (arrow) infected at an MOI of 1 is shown. Capsids infected at an MOI of 10 (middle) or 100 (bottom) are shown. Download