

ABSTRACT
Elucidation of the molecular mechanisms underlying the human gut microbiota’s effects on health and disease has been complicated by difficulties in linking metabolic functions associated with the gut community as a whole to individual microorganisms and activities. Anaerobic microbial choline metabolism, a disease-associated metabolic pathway, exemplifies this challenge, as the specific human gut microorganisms responsible for this transformation have not yet been clearly identified. In this study, we established the link between a bacterial gene cluster, the choline utilization (cut) cluster, and anaerobic choline metabolism in human gut isolates by combining transcriptional, biochemical, bioinformatic, and cultivation-based approaches. Quantitative reverse transcription-PCR analysis and in vitro biochemical characterization of two cut gene products linked the entire cluster to growth on choline and supported a model for this pathway. Analyses of sequenced bacterial genomes revealed that the cut cluster is present in many human gut bacteria, is predictive of choline utilization in sequenced isolates, and is widely but discontinuously distributed across multiple bacterial phyla. Given that bacterial phylogeny is a poor marker for choline utilization, we were prompted to develop a degenerate PCR-based method for detecting the key functional gene choline TMA-lyase (cutC) in genomic and metagenomic DNA. Using this tool, we found that new choline-metabolizing gut isolates universally possessed cutC. We also demonstrated that this gene is widespread in stool metagenomic data sets. Overall, this work represents a crucial step toward understanding anaerobic choline metabolism in the human gut microbiota and underscores the importance of examining this microbial community from a function-oriented perspective.
IMPORTANCE
Anaerobic choline utilization is a bacterial metabolic activity that occurs in the human gut and is linked to multiple diseases. While bacterial genes responsible for choline fermentation (the cut gene cluster) have been recently identified, there has been no characterization of these genes in human gut isolates and microbial communities. In this work, we use multiple approaches to demonstrate that the pathway encoded by the cut genes is present and functional in a diverse range of human gut bacteria and is also widespread in stool metagenomes. We also developed a PCR-based strategy to detect a key functional gene (cutC) involved in this pathway and applied it to characterize newly isolated choline-utilizing strains. Both our analyses of the cut gene cluster and this molecular tool will aid efforts to further understand the role of choline metabolism in the human gut microbiota and its link to disease.
INTRODUCTION
The human gut is colonized by trillions of microbes that exert a profound influence on human health, in part by providing metabolic capabilities that extend beyond those of host cells (1, 2). There is growing evidence that biochemical functions associated with the gut microbiota affect human biology. In particular, metabolomics experiments have revealed that levels of human serum metabolites made or modified by gut microbes correlate strongly with both health and disease states (3–5). Recent advances in DNA sequencing technology have increased our understanding of the phylogenetic and functional complexity of the human gut microbiota (6–8). However, we still do not understand the vast majority of the molecular mechanisms underlying how these organisms affect host biology, a knowledge gap that currently limits our ability to intervene therapeutically in diseases involving the microbiota. A major reason for this deficiency is the difficulty of linking critical biochemical functions to specific organisms in this complex and dynamic microbial community.
Anaerobic choline metabolism is a disease-associated microbial metabolic activity that exemplifies the many challenges associated with understanding biochemical functions in the gut microbiota (Fig. 1A). This process, which occurs in the gastrointestinal tracts of humans (9–11) and other vertebrates (12–14), involves an initial C–N bond-cleaving step that generates trimethylamine (TMA) and acetaldehyde (15, 16). TMA, an exclusively microbial metabolite, is further oxidized in the liver to trimethylamine N-oxide (TMAO) by flavin-containing monooxygenase 3 (FMO3) (9, 17). Although bacteria can metabolize various substrates to TMA, studies in humans point toward choline, l-carnitine, and TMAO as the most significant precursors (9, 10, 18, 19). Multiple human diseases are associated with gut microbial TMA production. The inherited metabolic disorder trimethylaminuria (fish malodor syndrome) arises from mutations in the gene encoding FMO3 that result in an inability to completely oxidize TMA to TMAO and an associated malodor (20). Nonalcoholic fatty liver disease, a known consequence of choline deficiency, may be exacerbated by excess microbial choline metabolism (21). Most recently, metabolomics studies of humans have uncovered links between choline, TMAO, and susceptibility to both cardiovascular disease (22–24) and colon cancer (25). Despite the relevance of anaerobic choline metabolism to disease, the specific organisms responsible for this biochemical activity in the human gut remain unclear.
FIG 1 .
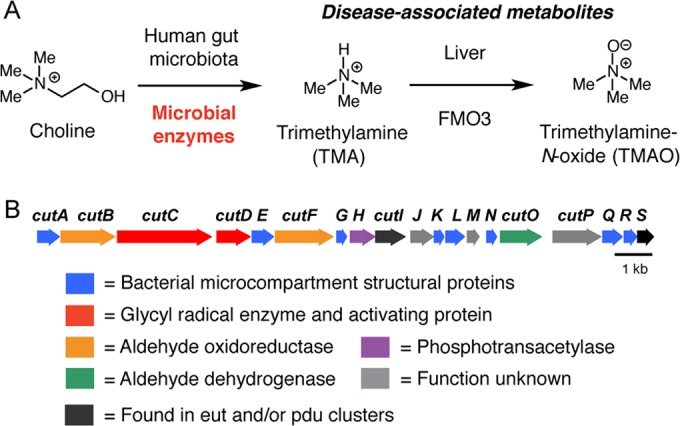
The choline utilization (cut) gene cluster encodes a pathway for anaerobic choline metabolism. (A) Anaerobic microbial metabolism in the human gut produces disease-associated metabolites. (B) Genetic organization of the Desulfovibrio desulfuricans ATCC 27774 cut gene cluster.
A major reason for this knowledge gap is the fact that the genetic and biochemical basis for anaerobic choline utilization remained uncharacterized until recently. We discovered the sole gene cluster known to mediate this process by mining the genomes of the sulfate-reducing bacteria Desulfovibrio desulfuricans and Desulfovibrio alaskensis (Fig. 1B) (26). Using genetics and heterologous expression, we demonstrated that two proteins encoded by the choline utilization (cut) gene cluster, previously unrecognized glycyl radical enzyme (GRE) choline TMA-lyase (CutC) and GRE activase (CutD), carry out the TMA-generating cleavage reaction. A preliminary bioinformatic analysis indicated that related gene clusters were present in sequenced human isolates from the gut, urogenital tract, airways, and oral cavity. Although our previous work established the relevance of the cut gene cluster to choline utilization, many important questions remained regarding its role in bacteria and its relevance to humans. In particular, it has been unclear whether the pathway encoded by the cut cluster is functional in members of the human gut microbiota, whether it is widely distributed among these organisms, and whether it represents a predominant route for choline degradation and TMA production in the gut environment.
Here we report the results of both functional and phylogenetic characterizations of the cut gene cluster in human gut bacteria. Using transcriptional analysis, biochemical analysis, and culture-based experiments, we further reinforce the connection between the cut genes and anaerobic choline metabolism in these organisms. Identification and analysis of cut gene clusters in additional sequenced bacterial genomes revealed an unexpectedly wide but discontinuous distribution for this pathway among bacterial phyla and evidence of acquisition via horizontal gene transfer. The observation that this function did not correlate with bacterial phylogeny led us to develop a PCR-based strategy for direct detection of the choline TMA-lyase gene (cutC). Using this method, cutC was universally found in newly isolated choline-fermenting bacteria from the human gut, suggesting that this pathway may be a predominate route for converting choline to TMA in this environment. The presence of the cutC gene in human stool metagenomes provided further support for this hypothesis. Overall, this study sheds new light on the distribution and importance of this metabolic pathway in the human gut and highlights the need to directly characterize the distribution and abundance of genes that give rise to biochemical functions associated with the gut microbiota.
RESULTS
Transcriptional and biochemical characterization of the cut gene cluster in Desulfovibrio desulfuricans.
The ~16-kb D. desulfuricans cut cluster comprises 19 open reading frames (ORFs) (Fig. 1B). In addition to the TMA-generating enzymes CutC and CutD, the cluster encodes other functions likely involved in choline metabolism. Many of the cut genes are closely related to components of the ethanolamine utilization (eut) and 1,2-propanediol utilization (pdu) pathways (27, 28), including eight genes encoding putative bacterial microcompartment (BMC) shell proteins (cutA, cutE, cutG, cutK, cutL, cutN, cutQ, and cutR), two predicted coenzyme A (CoA)-acylating aldehyde oxidoreductases (cutB and cutF), a phosphotransacetylase (cutH), a putative chaperonin (cutI), an alcohol dehydrogenase (cutO), and a Ras-like GTPase (cutS).
To better understand the connection between the entire D. desulfuricans cut gene cluster and anaerobic choline metabolism, we investigated its transcription under different growth conditions. Many bacterial metabolic pathways, including ethanolamine and propanediol utilization, are induced in the presence of substrate (29, 30). We used quantitative reverse transcription-PCR (qRT-PCR) to compare expression of the cut cluster and the genes immediately upstream (Ddes_1354) and downstream (Ddes_1374) during growth on lactate and choline (Fig. 2A). Transcript levels of all 19 cut genes increased dramatically during growth on choline relative to growth on lactate (49- to 1,971-fold), indicating that expression is induced in response to substrate. While the gene downstream of cutS (Ddes_1374) was expressed at the same level in both media, we detected a 32-fold induction of the gene upstream of cutA, the predicted transporter Ddes_1354, in the presence of choline. Interestingly, a gene encoding a putative transcriptional regulator (Ddes_1352) is located upstream of Ddes_1354. The close proximity of these two genes to the cut cluster suggests an involvement in regulation and choline transport, a proposal that was supported by the results of a recent, related transcriptional analysis of D. alaskensis G20 (31).
FIG 2 .
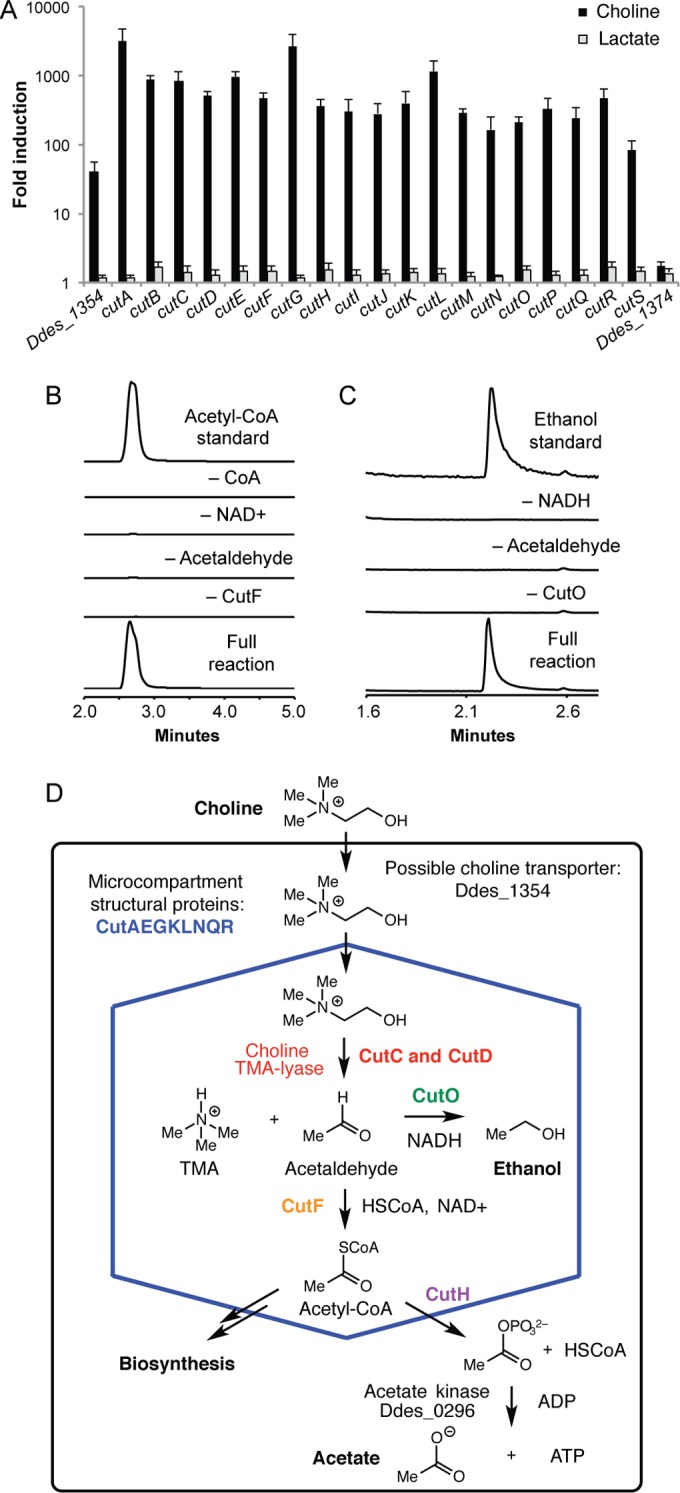
Transcription and biochemical analyses connect the entire cut gene cluster to choline metabolism. (A) qRT-PCR analysis results for cut genes during growth of D. desulfuricans in lactate versus choline fermentation media. Gene expression levels were calculated as the fold expression ratio (in choline versus lactate) after normalization to 16S rRNA gene expression levels. Values are presented as means ± standard deviations (SD) of three independent experiments. (B) Extracted ion chromatograms from LC-MS assays of CoA-acylating acetaldehyde oxidoreductase CutF. (C) Extracted ion chromatograms from GC-MS assays of acetaldehyde dehydrogenase CutO. (D) Proposed model for anaerobic choline catabolism.
To complement this analysis, we also examined the roles of additional enzymes encoded by the cut cluster. As genetic manipulations in Desulfovibrio are challenging, we used a biochemical approach to evaluate how specific gene products contribute to choline metabolism. We selected two D. alaskensis G20 enzymes for characterization: coenzyme A (CoA)-acylating aldehyde oxidoreductase CutF and alcohol dehydrogenase CutO. These enzymes are predicted to generate acetyl-CoA and ethanol, respectively, from acetaldehyde, metabolic functions that are consistent with the known end products of choline fermentation (ethanol and acetate) (15, 16, 32–35). Both Desulfovibrio cut clusters encoded two candidate acetaldehyde oxidoreductases (CutF and CutB). We chose to examine CutF because multiple-sequence alignment of these proteins with characterized homologs revealed CutB was missing an essential catalytic cysteine residue (see Fig. S1 in the supplemental material) and is likely nonfunctional. Recombinant CutF converted acetaldehyde to acetyl-CoA in the presence of both NAD+ and CoA (Fig. 2B) and displayed a 5-fold preference for NAD+ over NADP+, which is similar to the preference observed during choline fermentation (16). The amino acid sequence of CutO resembled the group III iron-containing alcohol dehydrogenases, including conserved active site residues (see Fig. S2 in the supplemental material) (36, 37). As these enzymes are oxygen sensitive, we expressed and purified CutO under strictly anaerobic conditions (38, 39). Recombinant CutO converted acetaldehyde to ethanol and selectively utilized NADH (Fig. 2C).
Bioinformatic analysis of the distribution of the cut gene cluster in sequenced bacterial genomes.
With an enhanced understanding of the connection between the cut cluster and choline metabolism, we assessed the distribution and diversity of homologous gene clusters in sequenced genomes. We used the D. desulfuricans CutC amino acid sequence as a search query and distinguished CutC sequences from other GREs based on insights from recent biochemical studies that identified essential CutC active site amino acids (Asp216, Phe395, Cys489, Glu491, and Gly821) (40). This analysis uncovered 459 CutC homologs (88% to 61% amino acid identity) distributed across four bacterial phyla (Proteobacteria, Firmicutes, Actinobacteria, and Fusobacteria). The majority of the hits were found in Proteobacteria (Gammaproteobacteria and Deltaproteobacteria) and Firmicutes (Clostridia and Bacilli), with less than 3% from Actinobacteria (Coriobacteridae) and Fusobacteria (Fusobacteriia). CutC was conspicuously absent from Bacteroidetes. All CutC-containing organisms were facultative or obligate anaerobes, which is consistent with the oxygen sensitivity of GREs. CutC-encoding strains originated from a range of microbial habitats, including the human body (50% of hits), with many of the human isolates obtained from the gut (35%). Consistent with our earlier findings, CutC was also found in strains isolated from the urogenital tract, oral cavity, and airways. We also identified CutC in three genomes assembled from human gut metagenomic DNA (Clostridium hathewayi CAG:224 [PRJNA221953], Collinsella sp. CAG:289 [PRJNA222147], and Klebsiella variicola CAG:634 [PRJNA221971]). CutC was also observed in gut isolates from other animals, including insects, fish, and mammals. Overall, we observed a discontinuous distribution of the cut gene cluster across most microbial species (Fig. 3A). For example, only 4% (98) of the 2,719 Escherichia coli genes and 15% (16) of the 101 Streptococcus suis genes encode a CutC homolog. Desulfosporosinus (6/6 genomes) and Proteus (17/17 genomes) are the only two bacterial genera that universally harbor the cut cluster. Other species that frequently harbor cutC include Clostridium botulinum (41/62 genomes; 66%) and Pectobacterium (18/29 genomes; 62%).
FIG 3 .

Analyses of the cut gene cluster and choline TMA-lyase in sequenced bacterial genomes. (A) The cut gene cluster is widely but unevenly distributed in sequenced human gut bacteria. The pie charts depict the proportions of bacterial genomes that contain the cut gene cluster (green) in selected bacterial genera that possess this pathway and have been isolated from the human gut. (B) Representative type I and type II cut gene cluster arrangements. (C) Maximum-likelihood phylogenetic trees showing the relationship between 16S rRNA genes (left) and the relationship between CutC amino acid sequences (right). Bootstrap confidence values of >50 are indicated by circles on the nodes. The CutC tree also contains the amino acid sequence of the glycyl radical enzyme glycerol dehydratase as an outgroup. Sequences from experimentally characterized choline-metabolizing strains are indicated with a red asterisk.
We examined the genomic context of the cutC homologs and found that the additional elements of the D. desulfuricans cut gene cluster are largely conserved in other bacteria. The majority of the cutC sequences (~95%) clustered with homologs of 10 or more D. desulfuricans cut genes. A subset of these cut clusters (~10%) lacked genes encoding the acetaldehyde oxidoreductase, alcohol dehydrogenase, and/or microcompartment structural proteins, but in all cases homologs of the missing enzymes were identified elsewhere in the genome. cutC and cutD homologs failed to cluster with any other cut genes in only 3 of the 459 genomes, and only one strain possessed cutC but lacked cutD (Olsenella sp. oral taxon 809 F0356). We classified cut clusters into two groups (type I and type II), based on their gene content (Fig. 3B; see also Fig. S3 in the supplemental material). Type I clusters, found in Firmicutes, Actinobacteria, and Deltaproteobacteria, contained ~20 genes, including homologs of BMC-encoding genes (cutAEGKLNQR), cutB, cutC, cutD, cutF, cutHIJ, cutO, cutS, and in some cases cutP. Although gene content was consistent for these clusters, gene order and transcriptional orientation varied. In contrast to the type I clusters, type II cut clusters, found in Gammaproteobacteria, typically contained only 10 genes, including the central enzymes involved in choline fermentation (cutC, cutD, cutF, cutO, and cutH) and four BMC structural proteins. The overall arrangement of the type II cut clusters was identical in all species. While type I cut clusters were distributed among both facultative and obligate anaerobes, type II clusters were found exclusively in facultative anaerobes.
To deduce the phylogenetic relationship between choline TMA-lyases from different organisms, we constructed a maximum-likelihood phylogenetic tree of CutC amino acid sequences and compared it to the phylogeny of the 16S rRNA gene (Fig. 3C). We observed two distinct clades of CutC sequences, one consisting of sequences from Actinobacteria, Deltaproteobacteria, and Firmicutes (clade 1) and the other containing sequences from Gammaproteobacteria and Peptococcaceae (clade 2). Notably, this arrangement is incongruent with the 16S rRNA-based phylogeny, suggesting that cutC has been acquired through horizontal gene transfer. This hypothesis is also supported by the presence of putative mobile genetic elements in the vicinity of 35% of the cut clusters, including the uropathogenic E. coli strain 536, whose cluster is located within a known pathogenicity island (PAII) (41).
Confirmation that the cut gene cluster is a genetic marker for anaerobic choline metabolism.
To confirm that the cut gene cluster is a functional marker for anaerobic choline metabolism, we tested whether its presence in a genome was predictive of an organism’s ability to convert choline to TMA under anaerobic conditions. We obtained nine sequenced strains containing type I cut clusers (Clostridium citroniae WAL-17108, Streptococcus dysgalactiae subsp. equisimilis ATCC 12394, Olsenella sp. oral taxon 809 F0356, Olsenella uli DSM 7084) and type II cut clusters (Escherichia coli MS 69-1, Klebsiella sp. MS 92-3, Proteus mirabilis ATCC 29906, P. mirabilis HI4320, and P. mirabilis BB2000). These organisms represented each of the phyla identified in our bioinformatics search and, with the exception of S. dysgalactiae, were all human isolates. Each strain was grown anaerobically in rich medium supplemented with 1 mM (trimethyl-d9)-choline to approximate choline availability in the human gut (42). All organisms generated d9-TMA except for one of the Olsenella strains (Fig. 4A). This strain, Olsenella sp. oral taxon 809 F0356, was missing GRE activase cutD, whereas the other Olsenella strain, Olsenella uli DSM 7084, contained both cutC and cutD. The failure of the strain harboring an incomplete cut cluster to metabolize choline provided further evidence that activated CutC is required for this activity.
FIG 4 .

The cutC functional gene is diagnostic of anaerobic choline utilization and can be detected using degenerate PCR. (A) LC-MS quantification of d9-TMA produced by selected cutC-containing bacterial species grown in rich medium supplemented with d9-choline (1 mM); Olsenella sp. oral taxon 809 F0356 has cutC but is missing the gene encoding activase cutD. Each bar represents the mean ± standard deviation (SD) of d9-TMA from three cultures. (B) Agarose gel electrophoresis results for degenerate PCRs using cutC-specific degenerate primers and genomic DNA from representative cutC-containing, choline-utilizing strains and two sequenced organisms that lack cutC. (C) LC-MS quantification of d9-TMA produced ex vivo by a human fecal sample upon anaerobic incubation in BHI medium supplemented with (trimethyl-d9)-choline (1 mM) for 18 h at 37°C. Bar graphs represent the means ± SD of three independent incubations. (D) Abundances of translated cutC sequences in the 66-member clone library constructed from degenerate PCR with human gut metagenomic DNA and the percent identity to CutC proteins from sequenced isolates.
Development and validation of a PCR method for detection of choline TMA-lyase (cutC).
Understanding the role of anaerobic choline metabolism in the human gut and its connection to disease requires a means of identifying and quantifying this activity in individual gut communities. The discovery that bacterial phylogeny is not a good marker for choline utilization led us to explore methods for direct detection of the functional gene cutC. We began these efforts by developing a PCR method that could selectively amplify cutC in the presence of additional genes encoding functionally diverse GREs possessing similar sequences (43). Our primer design took advantage of our understanding of the relationship between CutC’s amino acid sequence and catalytic activity (40). By aligning 85 sequences, we identified conserved stretches of amino acids not found in other GREs and designed 20 potential pairs of degenerate PCR primers targeting these regions. We screened each of these candidate primer pairs for their ability to amplify cutC from genomic DNA isolated from a subset of sequenced strains with cut gene clusters. One of these primer pairs displayed particularly robust amplification across a range of strains and was chosen for further optimization. These primers, which target regions of the cutC gene that encode conserved, essential active site residues (Phe395, Cys489, and Glu491), amplify a 314-bp stretch of cutC that is readily distinguished from the corresponding regions of genes encoding unrelated GREs.
We validated the utility of this degenerate PCR approach by successfully amplifying cutC from the genomic DNA of 11 strains containing cut clusters. A product of the expected size was observed in all of the PCRs, with amplification efficiency varying moderately among the different isolates (Fig. 4B). We confirmed the identity of each PCR product through cloning and sequencing; in all cases, cutC was the only sequence obtained. No amplification was observed with DNA from a strain lacking a cut cluster but possessing other GREs (Roseburia inulinovorans A2-194). Because these PCR primers selectively amplified cutC from phylogenetically diverse isolates, they should be useful for surveying strains from different taxonomic groups. Finally, we performed serial dilutions to establish that our primers can detect as little as ~10 pg of cutC-containing DNA template, even in the presence of an excess of genomic DNA from an organism lacking this gene.
We also utilized our primers to amplify cutC sequences directly from human gut metagenomic DNA. We began by assessing whether biases in amplification efficiency might impact recovery of cutC sequences from a mixed DNA template by applying the primers to a mock community containing equal amounts of template DNA from four different isolates possessing the cut cluster, including a strain that had displayed lower levels of amplification in our original survey (S. dysgalactiae). Successful amplification of cutC sequences from 4 ng of community template DNA was followed by construction and sequencing of a 57-member clone library. We recovered all of the sequences present in the original sample, although in different proportions (see Fig. S3 in the supplemental material). This promising result prompted us to perform a proof-of-concept degenerate PCR with metagenomic DNA isolated from a human fecal sample that possessed the ability to convert choline to TMA ex vivo (Fig. 4C). We observed a band of the appropriate size in the PCR product from this complex sample and constructed a clone library of the amplified products. Sequencing of 66 clones revealed 33 unique cutC nucleotide sequences encoding 23 distinct amino acid sequences with high identity to CutC proteins from seven sequenced bacteria spanning three different genera (Fig. 4D). A phylogenetic analysis of the translated cutC sequence fragments further supported the distinct origins of these genes (see Fig. S4 in the supplemental material).
Isolation and characterization of new choline-utilizing human gut isolates.
With the ability to directly detect the cutC functional gene, we next tested the extent to which the pathway encoded by the cut gene cluster is a predominant route for TMA production from choline in the human gut by assessing the distribution of cutC in newly isolated choline-utilizing bacteria from this environment. We obtained isolates from two human fecal samples via enrichment culturing on six different choline-containing media. We included either ammonium, Casitone, or Trypticase peptone as a nitrogen source and also varied the electron acceptors present (nitrate, tetrathionate, fumarate, or none). Organisms were chosen for further characterization based on their ability to convert (trimethyl-d9)-choline to d9-TMA in either brain heart infusion (BHI) or enrichment medium (Fig. 5A and B). Using this approach, we accessed a total of nine distinct choline-utilizing strains from four genera (Clostridia, Klebsiella, Escherichia, and Enterococcus) as determined by 16S rRNA gene sequencing (see Table S3 in the supplemental material). We obtained cutC sequences from each of the isolates by using degenerate PCR (Fig. 5C), confirmed the identity of each PCR product by cloning and sequencing, and analyzed the phylogeny of each translated cutC sequence (see Fig. S4 in the supplemental material). This experiment revealed that our new choline-utilizing strains universally possess the cutC gene.
FIG 5 .
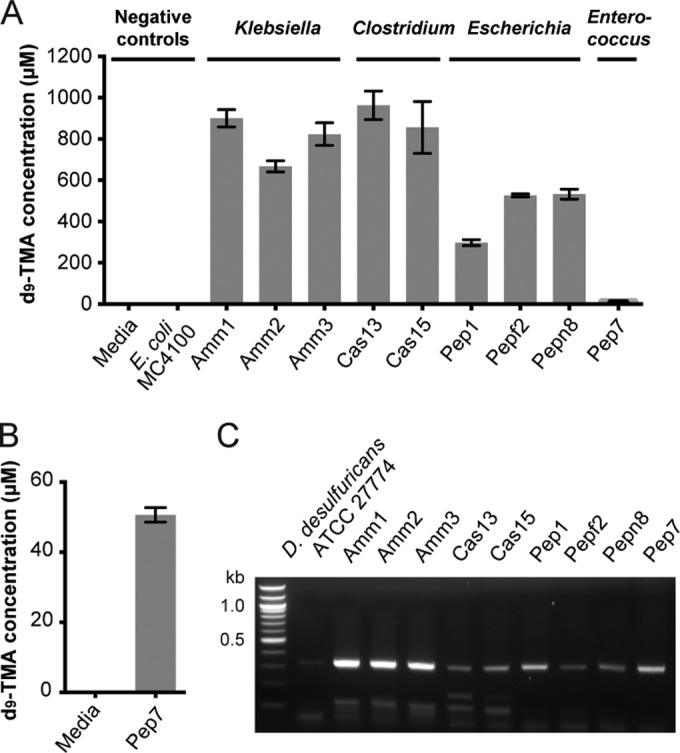
Choline-utilizing isolates from the human gut universally possess cutC. (A) LC-MS quantification of d9-TMA produced by human gut isolates grown anaerobically in BHI medium supplemented with d9-choline (1 mM) for 20 h at 37°C. Bar graphs represent the means ± standard deviations (SD) of three independent cultures. (B) LC-MS quantification of d9-TMA produced by isolate Pep7 grown anaerobically in enrichment medium supplemented with d9-choline (1 mM) for 20 h at 37°C. Bar graphs represent the means ± SD of three independent cultures. (C) Agarose gel electrophoresis results for PCRs with cutC-specific degenerate primers and genomic DNA isolated from choline-utilizing human isolates.
Identification of cutC sequences in human stool metagenomes.
In addition to examining the distribution of cutC in culturable members of the human gut microbiota, we also assessed whether this gene was present in the stool metagenomes of healthy individuals; these samples were obtained as part of the Human Microbiome Project (HMP) (6). We built an independent BLAST database for each assembled stool metagenome and separately searched for the CutC sequence from D. alaskensis G20 within each database, resulting in a total of 12,534 full and partial cutC homologs in all 148 samples. However, as glycyl radical enzymes are widespread in the microbiome and display varied functions despite high sequence conservation (43), simply using BLAST to search for cutC in assembled metagenomes is likely to generate many false positives. To better identify cutC sequences, we built a separate multiple-sequence alignment with each of the 1,000 most similar cutC homologs and a set of nine cutC sequences selected for high sequence diversity. We then assessed each hit for the presence of conserved, essential active site residues. Of the 1,000 most similar hits, 445 sequences representing 143 different stool samples (96.6% of individuals) contained the three conserved active site residues (Phe395, Cys489, and Glu491) that were targeted by our degenerate PCR and that have been biochemically verified as essential for CutC catalysis (40).
DISCUSSION
This study used a multidisciplinary approach to explore the connection between cut genes and choline metabolism in human gut microbes. Prior to this work, there was no experimental evidence that this cluster was functional in human gut microbes, and no molecular tools were available to investigate this activity. Our studies of the transcription and biochemical functions of the Desulfovibrio cut cluster provide both further support for the connection between the entire gene cluster and anaerobic choline utilization and also additional information about the reactions involved in this pathway that enhance our understanding of this metabolic activity. Overall, the qRT-PCR analysis demonstrated that transcription of the cut gene cluster is strongly induced during growth on choline, suggesting that several, if not all, of the proteins encoded by the cluster are involved in anaerobic choline metabolism. Interestingly, the genes with the highest induction levels (cutA, cutE, cutG, and cutL) encode the shell proteins of BMCs, which are large, intracellular polyhedral structures that encapsulate metabolic enzymes (44, 45). The dramatic upregulation of these genes supports the involvement of a BMC in anaerobic choline degradation, as similar expression levels have been previously reported for other BMC-utilizing pathways (46, 47). The choline utilization-associated BMC may encapsulate the acetaldehyde generated by choline TMA-lyase to limit its toxicity or prevent its escape from the cell, as has been proposed for the BMC that is involved in ethanolamine metabolism (48).
During this analysis, we also identified a candidate regulatory protein encoded adjacent to the cut gene cluster (Ddes_1352). This protein belongs to the MerR family of transcriptional regulators and was recently demonstrated to be an activator of the cut genes in D. alaskensis during growth on choline (32). Our transcriptional analysis confirmed that the presence of choline induces the expression of the cut cluster. Interestingly, this regulator is present in almost all type I clusters but it is absent in type II clusters, which have a different predicted regulatory protein, a TetR family transcriptional regulator. This finding suggests that the cut genes are controlled in different organisms by at least two distinct regulatory systems. Elucidating the factors that regulate the expression of both types of cut gene clusters is an important area for future research, as this knowledge could guide the development of strategies (dietary interventions, probiotics, etc.) for reducing TMA formation in the gut.
The biochemical characterization of CutF and CutO confirmed that these enzymes possess activities consistent with their proposed roles in choline fermentation. These data, together with the cluster annotation and transcription experiments, allowed us to propose a model for anaerobic choline metabolism in Desulfovibrio (Fig. 2D). After transport of choline into the cell, choline cleavage and TMA generation likely occur inside a BMC (CutAEGKLNQR). CutF then converts the acetaldehyde generated in this step to acetyl-CoA. Phosphotransacetylase CutH could react with acetyl-CoA phosphate to give acetyl phosphate, which could then be converted to ATP by an acetate kinase homolog encoded elsewhere in the genome (Ddes_0296). To maintain redox balance and/or regenerate NAD+ inside the BMC, CutO would reduce acetaldehyde to ethanol. Disproportionation of acetaldehyde through this pathway would generate the observed fermentation end products and one molecule of ATP for every two molecules of choline. Further experiments are needed to elucidate the roles of the remaining cut genes in choline degradation and to characterize the regulatory elements that control gene cluster expression.
Our bioinformatic analysis of cut clusters in sequenced bacterial genomes and the comparison of their gene content and arrangement provides the first information regarding the prevalence of anaerobic choline metabolism across a wide range of diverse bacteria. Overall, this search revealed cut gene clusters in bacterial species from three phyla, indicating that anaerobic choline utilization is a widely distributed microbial metabolic pathway. This metabolic activity is present in strains isolated from the human gut and other body sites. The cut cluster is also found in many environmental isolates and in bacteria isolated from both plants and animals. This observation suggests that anaerobic choline utilization may be important for survival in many microbial habitats. The conservation of cutC, cutD, cutF, cutH, cutO, and BMC-encoding genes in cut clusters from diverse phylogenetic groups indicates the central importance of these genes for choline metabolism and provides further support for our model for pathway function. Additionally, we observed two distinct subtypes (types I and II) among the cut clusters that differed in the number of BMC structural genes and the presence or absence of an additional N-terminal domain on CutC. These arrangements may reflect structural and potentially functional differences among BMCs from various choline-metabolizing organisms. The observation that the type II cluster arrangement is present only in facultative anaerobes suggests the possibility that these differences have arisen in response to various exposures of choline-metabolizing organisms to molecular oxygen.
With the exception of the extra N-terminal domain found in CutC homologs from type II clusters, we found that the amino acid sequence of this key TMA-generating enzyme is highly conserved across organisms possessing the cut cluster. The phylogenetic reconstruction of the CutC protein sequence was incongruous with the phylogeny based on 16S rRNA sequencing, suggesting that horizontal gene transfer events have mediated acquisition of these clusters in multiple organisms. This proposal is also supported by the discontinuous distribution of this metabolic pathway across the majority of bacterial species possessing cut clusters and the presence of mobile genetic elements near many of these gene clusters. We hypothesize that acquisition of cut genes could occur in habitats in which choline is an abundant growth substrate, as it would confer a competitive advantage in such environments. Recent work has suggested that horizontal gene transfer occurs more frequently in the human gut than in other habitats, potentially due to the high population densities and increased metabolic activity in this setting (49). While exchange of antibiotic resistance genes among gut microbes has received particular attention (50), there is growing evidence for transfer of genes encoding metabolic pathways (51–53). The cut gene cluster therefore represents an additional example of a bacterial metabolic function that may have been exchanged in the gut environment.
To confirm that the cut genes are indeed associated with anaerobic choline utilization across a broad range of organisms, we examined the metabolic activities of strains identified in our bioinformatic search. Culturing a phylogenetically diverse set of sequenced isolates possessing cut clusters in the presence of labeled choline confirmed that the presence of these genes is indicative of an organism’s ability to convert choline to TMA. Interestingly, all of the strains metabolized choline in rich media containing alternate carbon and energy sources. This observation suggests that choline is a preferred substrate or that this pathway contributes to cellular processes distinct from growth. Deciphering the specific advantages associated with this mode of metabolism will require further experiments, but we hypothesize that preferential use of choline is indicative of resource partitioning, providing bacteria harboring this cluster with a unique metabolic niche and allowing them to avoid competition for more energy-rich growth substrates available in the gut environment (54).
Altogether, this work demonstrates that the presence of the cut genes in a bacterial genome is diagnostic of anaerobic choline utilization, making this cluster a useful molecular marker for this biochemical function. Several of the cut clusters revealed in our search had been identified in previous bioinformatics analyses but were annotated as mediating either pyruvate or diol metabolism (55, 56). Our work clarifies the biochemical function encoded by these genes. Confirming that the cut cluster predicts choline metabolism across a variety of bacterial phyla has important implications for efforts to understand choline metabolism in natural communities. The wide but discontinuous distribution of this pathway in bacteria indicates that the phylogenetic composition of the microbiota is likely a poor predictor of anaerobic choline metabolism. Indeed, attempts to identify TMA-generating microbes by correlating either disease phenotypes or metabolite levels with changes in gut community composition have been unsuccessful (18, 57). This finding highlights the hazards of inferring biochemical functions from phylogenetic information, as pathways of interest may not be universally present in a given species or phylogenetic group nor limited to a narrow subset of microbial diversity.
The lack of a correlation between phylogeny and the presence of cutC in sequenced bacteria prompted us to develop a method for the direct detection of this gene. We used our understanding of the CutC sequence-activity relationship to design degenerate PCR primers based on regions of sequence encoding conserved amino acid residues. These primers amplified cutC sequences from template DNA derived from a phylogenetically diverse set of isolates with generally good efficiency. Further optimization of the primers or PCR conditions may still be required to access the broadest possible range of sequences present in a given community, but these primers did successfully amplify multiple cutC sequences from stool metagenomic DNA. This experiment provided a first glimpse into the diversity of cutC sequences present in an individual human gut microbial community, revealing that multiple types of bacteria have the capacity to perform anaerobic choline metabolism in this environment. We note that while the translated cutC sequences obtained in this experiment matched CutC protein sequences from sequenced bacteria, many of these sequences did not show significant similarity to known cutC nucleotide sequences, suggesting we have accessed sequences from organisms that are not yet represented in genome sequence databases. Though this analysis demonstrates the presence of cutC in multiple community members, it does not reveal which of these organisms may be responsible for generating TMA from choline in vivo.
Studies of particular metabolic pathways in microbial habitats are greatly facilitated by the use of molecular approaches that target functional genes, particularly when the activities of interest have a broad phylogenetic distribution. Previous efforts to characterize gut microbial metabolism using function-oriented strategies have largely focused on pathways and activities that are well understood in comparison to anaerobic choline metabolism, including bile acid metabolism, short-chain fatty acid synthesis, and β-glucuronidase activity (53, 58–61). These studies have provided important insights into the distribution and abundance of these core functions in gut microbial communities, including information about how activities and pathways vary between individuals and how functional gene abundance changes with diet. Our work illustrates the benefits of applying such approaches, which are greatly enabled by the wealth of available bacterial genome sequences, to study newly characterized functions.
An important unanswered question regarding the pathway encoded by the cut gene cluster is the extent to which it represents a predominant route for choline utilization and TMA generation in the human gut. We have begun to explore this problem using two complementary approaches: screening new choline-metabolizing isolates for the presence of the cutC functional gene and searching for cutC genes in human stool metagenomes. By employing a range of different growth media, we obtained nine new choline-utilizing organisms. To our knowledge, these experiments represent the first targeted isolation of choline-utilizing bacteria from the human gut. Importantly, we were able to correlate anaerobic choline metabolism in each isolate with the presence of the cutC gene. These results further establish the validity of the cutC gene as a molecular marker for anaerobic choline metabolism and demonstrate the utility of directly detecting this biochemical function. Most significantly, the observation that these gut isolates universally possess cutC confirms that choline-utilizing organisms are present in human gut microbial communities and suggests that the pathway encoded by the cut cluster could be a predominant mechanism for choline degradation and TMA production within this environment.
This hypothesis is also supported by the high prevalence of CutC homologs in assembled HMP stool metagenomes (96.6% of healthy individuals). Analysis of metagenomic data enables the identification of functional genes directly in communities containing organisms that are difficult to culture, complementing culture-based methods. However, a consistent challenge in searching metagenomes has been the accurate identification of homologs of important genes from partial sequences without resorting to arbitrary similarity score thresholds. Our search strategy represents a step toward solving this problem: by using higher-quality multiple-sequence alignments instead of pairwise alignments and assessing sequences for the presence of conserved, essential active site residues, we can be more confident that the metagenomic hits we have identified are CutC homologs. Overall, the high prevalence of CutC homologs in the HMP stool metagenomes further supports the hypothesis that the pathway encoded by the cut gene cluster has a large impact on choline degradation and TMA production within the gut microbiome.
Together, these experimental and computational findings suggest that the pathway encoded by the cut gene cluster may be a major mechanism for the direct conversion of choline to TMA in gut bacteria. However, many central questions remain regarding the factors influencing TMA production by the human gut microbiota and TMAO levels in humans. Choline may be utilized via alternate pathways that do not yield TMA, such as oxidation to betaine (62) and demethylation to produce dimethylaminoethanol (63). The factors that influence levels of choline and other potential TMA precursors in the gut, including diet, availability of other bacterial metabolic activities such as lipid metabolism, and other environmental factors, are not well established. TMA and TMAO levels may also be influenced by the presence of TMA-utilizing archaea (64). We expect that the ability to directly detect choline TMA-lyase in gut microbial communities, along with other “omics”-based approaches, will prove broadly useful in future efforts to correlate this metabolic pathway with metabolite levels, to identify alternate routes for choline utilization, and to understand the connection between this bacterial activity and human disease. More generally, our findings illuminate the importance of employing methods that directly detect the microbial functional genes responsible for activities of interest. We anticipate that implementation of similar function-oriented approaches will help to reveal gut microbial processes that are the bases for disease and can potentially guide therapeutic development.
MATERIALS AND METHODS
General procedures.
Standard molecular biology techniques, protein purification methods, liquid chromatography-tandem mass spectrometry (LC-MS/MS) and gas chromatography-MS (GC-MS) analyses and chemical sources are described in the supplemental material. All anaerobic experiments were conducted in an MBraun glove box (Stratham, NH) under an atmosphere consisting of 99.997% N2 with less than 5 ppm O2 or in an anaerobic chamber (Coy Laboratory Products, Grass Lake Charter Township, MI) under an atmosphere of 98% nitrogen and 2% hydrogen.
RNA isolation and quantitative RT-PCR.
D. desulfuricans ATCC 27774 cultures were grown in six 18- by 160-mm modified Hungate tubes containing 10 ml of lactate sulfate (LS) medium and six tubes containing choline sulfate (CS) medium with argon in the headspace, as previously described (26). RNAprotect bacteria reagent (Qiagen) and RNase-free DNase were used with the RNeasy Protect bacteria kit (Qiagen) to isolate the RNA, which was converted to cDNA by using SuperScript II reverse transcriptase (Invitrogen). Specific primers for qRT-PCR, listed in Table S1 in the supplemental material, were designed using Primer3 (65). The cutC gene was amplified by PCR from D. desulfuricans ATCC 27774 genomic DNA and gel purified. Transcription levels of the genes analyzed were normalized to 16S rRNA levels. The relative fold change of RNA transcription (choline/lactate) was determined via the 2−ΔΔCt method (66). Additional details are in provided in the supplemental material.
Cloning, expression, and purification of CutF and CutO.
The cutF and cutO genes were PCR amplified from Desulfovibrio alaskensis G20 genomic DNA using the primers listed in Table S2 in the supplemental material. Restriction digests were purified directly by using agarose gel electrophoresis and then ligated into a linearized expression vector (pET-29b for cutF and pET-28a for cutO) using T4 DNA ligase (New England Biolabs). These constructs were transformed into chemically competent E. coli BL21(DE3) cells (Invitrogen) and stored at −80°C as frozen LB/glycerol stocks. Protein overexpression and purification afforded yields of 0.22 mg/liter of C-His6-tagged CutF (stock solution concentration, 49 µM) and 0.15 mg/liter for N-His6-tagged CutO (stock solution concentration, 1.5 µM). Additional details are provided in the supplemental material.
Biochemical characterization of CutF and CutO.
CutF assay mixtures (100 µl) contained 50 mM Tris (pH 8.0), 0.4 mM NAD+, 0.1 mM CoA, and 5 µM C-His6-tagged CutF (final concentration). Reactions were initiated by the addition of acetaldehyde to a final concentration of 10 mM. Assays were incubated at room temperature for 1 h, quenched with ice-cold methanol (200 µl), and centrifuged (16,100 × g for 15 min). A volume of 100 µl was removed from the supernatant and added to 900 µl of methanol for analysis by LC-MS as described in the general procedures provided in the supplemental material.
CutO assays were performed under anaerobic conditions. Reaction mixtures (250 µl) contained 100 mM NaH2PO4 (pH 7.0), 5 mM NADH, and 1 µM N-His6-tagged CutO (final concentration). The reactions were initiated by the addition of acetaldehyde to a final concentration of 150 mM. After incubation at 22°C for 12 h, each reaction mixture was transferred to a 10-ml-headspace vial (Sigma-Aldrich) containing 2.25 ml of water and 1.8 g of NaCl. Vials were capped tightly, and samples were analyzed using GC-MS as detailed in the supplemental material.
Identification and analysis of cut gene clusters in sequenced bacterial genomes.
The choline TMA-lyase (CutC) protein sequence from D. desulfuricans ATCC 27774 (EMBL accession number ACL49259.1) was used as a query for BLASTP searches against the nonredundant protein sequences (nr) database from NCBI (performed on 27 November 2014) and all genomes in the IMG database (performed on 29 November 2014). BLAST hits having an identity between 88 and 61% to CutC from D. desulfuricans were aligned using ClustalW2 (67) to confirm the presence of conserved active site residues. Strains containing a cutC homolog were classified according to the source of the isolate based on the information available at the Genome Online Database, the IMG database, or bibliographic searches. The content and arrangement of each of the cut clusters identified in the initial BLAST search were determined by the examination of the genetic context of the cutC gene. Putative functions were assigned to neighboring genes based on the genome annotations and similarities to the cut genes from D. desulfuricans.
Phylogenetic analysis of 16S rRNA and CutC protein sequences.
Nucleotide and protein sequences were aligned with ClustalW2 using the default parameters. For sequences identified in bacterial genomes, alignments were manually curated to a final length of 794 amino acids (aa) for CutC and 948 nt for 16S rRNA. Translated CutC nucleotide sequences from isolates obtained via enrichment culturing and metagenomic clone libraries were aligned with CutC sequences from bacterial genomes. These alignments were manually edited to a final length of 104 aa. Maximum-likelihood trees were constructed in MEGA 5 (68) using the Jones-Taylor-Thorton (JTT) method of substitution for CutC and the Tamura-Nei method for 16S rRNA. Statistical support for both trees was obtained by bootstrapping 100 iterations. Trees were visualized with iTOL (69).
Quantitation of TMA production from choline.
To confirm and quantify the TMA formed during microbial choline utilization, reference strains were grown anaerobically on BHI medium containing 1 mM (trimethyl-d9)-choline. Three 4-ml replicates were inoculated with 40 µl of an overnight starter culture and grown for 20 h (10 days for Olsenella strains) at 37°C. The concentration of d9-TMA in culture medium was determined using LC-MS after derivatization with ethyl bromoacetate using a published procedure (26). Additional details are provided in the supplemental material.
Detection of cutC via degenerate PCR.
To amplify an internal fragment of cutC, degenerate primers were designed based on an alignment from 85 CutC sequences from different species. These primers amplify a 314-bp conserved portion of the cutC gene. Primer sequences were 5′-TTYGCIGGITAYCARCCNTT-3′ (Fw cutC-dPCR-389 aa-A) and 5′-TGNGGRTCIACYCAICCCAT-3′ (Rv cutC-dPCR-492 aa-B). This primer set was used to amplify cutC from genomic DNA isolated from both reference strains and isolates obtained from enrichment culturing. PCRs were analyzed by agarose gel electrophoresis with ethidium bromide staining. Bands of the expected length (314 bp) were gel purified and cloned into the pCR4-TOPO vector using the TOPO TA cloning kit for sequencing (Invitrogen). The identity of each clone was confirmed by sequencing. To assess the amplification bias of the degenerate primers, a “mock” community containing equal amounts of genomic DNA from E. coli MS 69-1, Klebsiella sp. MS 92-3, C. citroniae WAL-17108, and S. dysgalactiae subsp. equisimilis ATCC 12394 was prepared. PCR-amplified fragments were gel purified and cloned as described above, and 60 clones were randomly chosen for analysis. Plasmid purification and sequencing were performed by Beckman Coulter Genomics (Danvers, MA). Low-quality, unsuccessful, and no-insert reaction mixtures were discarded, reducing the number of clones to 57. Clones were identified by aligning cutC sequences from the corresponding organisms with the obtained nucleotide sequences. Additional details are provided in the supplemental material.
Clone library construction and sequence analysis.
A clone library specific for cutC was constructed by amplification of human fecal DNA with the degenerate primers described above. The PCR mixture and cycling parameters tested were the same as those used for the amplification of cutC from genomic DNA, except for the template DNA concentration, which was changed to 300 or 50 ng. PCR products were analyzed by agarose gel electrophoresis with ethidium bromide staining. Amplified PCR products from different template DNA concentrations were pooled, gel purified, and cloned into the pCR4-TOPO vector using the TOPO TA cloning kit for sequencing (Invitrogen) according to instructions from the manufacturer. A total of 96 clones were randomly selected for sequencing. Plasmid purification and sequencing were performed by Beckman Coulter Genomics (Danvers, MA). Low-quality, unsuccessful, and no-insert reaction mixtures were discarded, which reduced the size of the library to 66 clones. Nucleotide sequences were subjected to BLASTn searches against the NCBI and IMG databases and translated to protein sequences to perform a BLASTp search against the NCBI database. All translated sequences had 90 to 98% amino acid identity to CutC sequences in the database.
Ex vivo incubation.
A frozen fecal sample from a healthy individual was diluted 1:10 (wt/vol) in sterile prereduced PBS. The dilution was vortexed for 1 min and allowed to settle for 15 min at room temperature. A 40-µl aliquot of the resulting supernatant was transferred to a capped 5-ml serum vial (Supelco, Bellefonte, PA) containing 4 ml of anaerobic BHI medium supplemented with 1 mM (trimethyl-d9)-choline. Samples were incubated in triplicate for 18 h at 37°C. The concentration of d9-TMA in culture medium was determined using LC-MS after derivatization with ethyl bromoacetate according to a published procedure (26). Medium blanks consisted of 4 ml uninoculated medium containing 1 mM (trimethyl-d9)-choline.
Enrichment culturing.
All experiments were performed under the guidance of the Harvard Committee on the Use of Human Subjects in Research. Two healthy volunteers provided an intact fecal sample. Six types of medium were used for enrichment culturing. Individual strains were isolated after two passages in medium containing choline as the sole carbon source and streaking for isolation on agar plates. To verify that the isolates were converting choline into TMA, each strain was grown on BHI or enrichment medium supplemented with choline-d9, and TMA-d9 was quantified by LC-MS/MS (26). Additional details are provided in the supplemental material.
16S rRNA sequencing.
PCR amplification of 16S rRNA genes was performed on genomic DNA isolated from all of the new choline-utilizing strains with previously reported primers fD1 (5′-TCCTACGGGAGGCAGCAGT-3′) and rP2 (5′-GGACTACCAGGGTATCTAATCCTGTT-3′) (70). PCR products were gel purified, cloned, and sequenced to confirm the identity and purity of the strains. DNA sequences were analyzed with a BLASTn search against the nucleotide collection (nr/nt). 16S rRNA gene sequences with >98% identity were classified as the same species.
Identifying cutC homologs in human stool metagenomes.
The 148 stool metagenome assemblies made available by the HMP were downloaded and made into separate local BLAST databases with the BLAST+ command line applications (71). The tBLASTn algorithm was then used to search for a known CutC sequence (D. alaskensis G20; accession number ABB40076) against each individual BLAST database using all six reading frames. Hits were collated and ranked by BLAST expectation score. Clustal Omega (72) was then subsequently used to create a multiple-sequence alignment with each of the 1,000 hits with the lowest BLAST expectation scores and a set of nine known CutC variants selected for high sequence diversity (Desulfovibrio alaskensis G20, ABB40076; Clostridium ljungdahlii DSM 13528, ADK17007; Collinsella tanakaei YIT 12063, EGX67047; Anaeroccocus vaginalis ATCC 51170, EEU12078; Streptococcus dysgalactiae ATCC 12394, ADX25417; Desultifobacterium hafniense DP7, EHL05181; Klebsiella variicola At-22, ADC60394; Escherichia coli MS 200-1, EFJ62362; Proteus mirabilis ATCC 29906, EEI47333). Alignments were then assessed for the presence of the conserved active site residues Phe395, Cys489, and Glu491, and the number of hits with each combination of residues was determined (see Table S4 in the supplemental material).
Nucleotide sequence accession numbers.
The 16S rRNA and CutC sequences obtained in this study were deposited in GenBank under accession numbers KR029063-KR029080.
SUPPLEMENTAL MATERIAL
Materials and methods Download
Multiple-sequence alignment of the N-terminal region of AdhE, the bifunctional aldehyde-alcohol dehydrogenase (P0A9Q7) from E. coli (AdhEEc); acetaldehyde dehydrogenases (YP_389772 and YP_389768) from D. alaskensis (CutFDa and CutBDa); acetaldehyde dehydrogenase (P41793) from S. enterica (EutESe); and propionaldehyde dehydrogenase (NP_460996) from S. enterica (PduPSe). The catalytic cysteine residue is indicated with an asterisk. Download
Multiple-sequence alignment of alcohol dehydrogenase (ACK56133) from Thermococcus sp. ES1 (AdhTher); 1,2-propanediol oxidoreductase (AAC75841) from E. coli (FucOEc); alcohol dehydrogenase II (YP_163331) from Zymomonas mobilis (AdhIIZm); 1,3-propanediol oxidoreductase (AAP97875) from Klebsiella pneunomiae (DhaTKp); alcohol dehydrogenase (NP_461396) from S. enterica (EutGSe); 1-propanol dehydrogenase (AAD39016) from S. enterica (PduQSe); and alcohol dehydrogenase from D. alaskensis (CutODa). Black asterisks indicate residues that coordinate the Fe2+ ion. The red asterisk shows an essential histidine residue which putatively interacts with the substrate. Download
Comparison of the composition and organization of cut gene clusters in sequenced bacteria revealed two cluster architectures. Experimentally characterized strains are indicated in red text. Download
Maximum-likelihood phylogenetic tree of translated cutC sequences amplified from new choline-utilizing isolates and human gut metagenomic DNA. Download
Primers used for qRT-PCR analysis.
Primers used for cloning. Restriction sites are underlined.
Molecular characterization of choline-utilizing human isolates.
Relative number of CutC hits with conserved/essential active site residues in Human Microbiome Project stool metagenomes.
Putative cut gene clusters in sequenced bacterial genomes. Download
ACKNOWLEDGMENTS
We received assistance from Kelly Chatman and Jennifer Wang with MS experiments (Harvard University Small Molecule Mass Spectrometry Core Facility). We also acknowledge Carina Chittim for help with enrichment culturing, and we thank Karine A. Gibbs (Harvard University) and the Stanhope group (Cornell University) for providing P. mirabilis and S. dysgalactiae strains.
Financial support was provided by the Smith Family Award for Excellence in Biomedical Research, the Packard Fellowship for Science and Engineering, the Milton Fund, the Corning Foundation, the Eli Lilly New Faculty Award, and Amgen. S. Bodea acknowledges a graduate fellowship from the Howard Hughes Medical Institute.
Footnotes
Citation Martínez-del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, Balskus EP. 2015. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 6(2):e00042-15. doi:10.1128/mBio.00042-15.
REFERENCES
- 1.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. 2012. Host-gut microbiota metabolic interactions. Science 336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 2.Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, Mazmanian SK. 2014. Specialized metabolites from the microbiome in health and disease. Cell Metab 20:719–730. doi: 10.1016/j.cmet.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. 2011. Understanding the role of microbiome-host metabolic signal disruption in health and disease. Trends Microbiol 19:349–359. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Martin FP, Collino S, Rezzi S, Kochhar S. 2012. Metabolomic applications to decipher gut microbial metabolic influence in health and disease. Front Physiol 3:113. doi: 10.3389/fphys.2012.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schicho R, Shaykhutdinov R, Ngo J, Nazyrova A, Schneider C, Panaccione R, Kaplan GG, Vogel HJ, Storr M. 2012. Quantitative metabolomic profiling of serum, plasma, and urine by 1H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J Proteome Res 11:3344–3357. doi: 10.1021/pr300139q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Human Microbiome Project Consortium 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. 2012. Human gut microbiome viewed across age and geography. Nature 486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sichertiz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, MetaHIT Consortium, Bork P, Ehrlich SD, Wang J. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeisel SH, Wishnok JS, Blusztajn JK. 1983. Formation of methylamines from ingested choline and lecithin. J Pharmacol Exp Ther 225:320–324. [PubMed] [Google Scholar]
- 10.Zhang AQ, Mitchell SC, Smith RL. 1999. Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol 37:515–520. doi: 10.1016/S0278-6915(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 11.Bain MA, Fornasini G, Evans AM. 2005. Trimethylamine: metabolic, pharmacokinetic and safety aspects. Curr Drug Metab 6:227–240. doi: 10.2174/1389200054021807. [DOI] [PubMed] [Google Scholar]
- 12.Neill AR, Grime DW, Dawson RM. 1978. Conversion of choline methyl groups through trimethylamine into methane in the rumen. Biochem J 170:529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Waiz M, Mikov M, Mitchell SC, Smith RL. 1992. The exogenous origin of trimethylamine in the mouse. Metabolism 41:135–136. doi: 10.1016/0026-0495(92)90140-6. [DOI] [PubMed] [Google Scholar]
- 14.Niizeki N, Daikoku T, Hirata T, El-Shourbagy I, Song X, Sakaguchi M. 2002. Mechanism of biosynthesis of trimethylamine oxide from choline in the teleost tilapia, Oreochromis niloticus, under freshwater conditions. Comp Biochem Physiol B Biochem Mol Biol 131:371–386. doi: 10.1016/S1096-4959(01)00508-5. [DOI] [PubMed] [Google Scholar]
- 15.Hayward HR, Stadtman TC. 1959. Anaerobic degradation of choline. I. Fermentation of choline by an anaerobic, cytochrome-producing bacterium, Vibrio cholinicus n. sp. J Bacteriol 78:557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradbeer C. 1965. The clostridial fermentations of choline and ethanolamine. 1. Preparation and properties of cell-free extracts. J Biol Chem 240:4669–4674. [PubMed] [Google Scholar]
- 17.Krueger SK, Williams DE. 2005. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther 106:357–387. doi: 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WHW, Bushman FD, Lusis AJ, Hazen SL. 2013. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller CA, Corbin KD, da Costa KA, Zhang S, Zhao X, Galanko JA, Blevins T, Bennett BJ, O’Connor A, Zeisel SH. 2014. Effect of egg ingestion on trimethylamine-N-oxide production in humans: a randomized, controlled, dose-response study. Am J Clin Nutr 100:778–786. doi: 10.3945/ajcn.114.087692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christodoulou J. 2012. Trimethylaminuria: an under-recognised and socially debilitating metabolic disorder. J Paediatr Child Health 48:E153–E155. doi: 10.1111/j.1440-1754.2010.01978.x. [DOI] [PubMed] [Google Scholar]
- 21.Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK. 2006. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 103:12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. 2011. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. 2013. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. 2014. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J 35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae S, Ulrich CM, Neuhouser ML, Malysheva O, Bailey LB, Xiao L, Brown EC, Cushing-Haugen KL, Zheng Y, Cheng TY, Miller JW, Green R, Lane DS, Beresford SA, Caudill MA. 2014. Plasma choline metabolites and colorectal cancer risk in the Woman’s Health Initiative Observational Study. Cancer Res 74:7442–7452 doi: 10.1158/0008-5472.CAN-14-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Craciun S, Balskus EP. 2012. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A 109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bobik TA, Havemann GD, Busch RJ, Williams DS, Aldrich HC. 1999. The propanediol utilization (pdu) operon of Salmonella enterica serovar Typhimurium LT2 includes genes necessary for formation of polyhedral organelles involved in coenzyme B12-dependent 1,2-propanediol degradation. J Bacteriol 181:5967–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kofoid E, Rappleye C, Stojiljkovic I, Roth J. 1999. The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins. J Bacteriol 181:5317–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roof DM, Roth JR. 1988. Ethanolamine utilization in Salmonella typhimurium. J Bacteriol 170:3855–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bobik TA, Ailion M, Roth JR. 1992. A single regulatory gene integrates control of vitamin B12 synthesis and propanediol degradation. J Bacteriol 174:2253–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuehl JV, Price MN, Ray J, Wetmore KM, Esquivel Z, Kazakov AE, Nguyen M, Kuehn R, Davis RW, Hazen TC, Arkin AP, Deutschbauer A. 2014. Functional genomics with a comprehensive library of transposon mutants for the sulfate-reducing bacterium Desulfovibrio alaskensis G20. mBio 5(3):e01041-14. doi: 10.1128/mBio.01041-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayward HR. 1960. Anaerobic degradation of choline. III. Acetaldehyde as an intermediate in the fermentation of choline by extracts of Vibrio cholinicus. J Biol Chem 235:3592–3596. [PubMed] [Google Scholar]
- 33.Hayward HR, Stadtman TC. 1960. Anaerobic degradation of choline. II. Preparation and properties of cell-free extracts of Vibrio cholinicus. J Biol Chem 235:538–543. [PubMed] [Google Scholar]
- 34.Sandhu SS, Chase T. 1986. Aerobic degradation of choline by Proteus mirabilis: enzymatic requirements and pathway. Can J Microbiol 32:743–750. doi: 10.1139/m86-135. [DOI] [PubMed] [Google Scholar]
- 35.Chao CK, Zeisel SH. 1990. Formation of trimethylamine from dietary choline by Streptococcus sanguis I, which colonizes the mouth. J Nutr Biochem 1:89–97. doi: 10.1016/0955-2863(90)90055-P. [DOI] [PubMed] [Google Scholar]
- 36.Reid MF, Fewson CA. 1994. Molecular characterization of microbial alcohol dehydrogenases. Crit Rev Microbiol 20:13–56. doi: 10.3109/10408419409113545. [DOI] [PubMed] [Google Scholar]
- 37.Montella C, Bellsolell L, Pérez-Luque R, Badía J, Baldoma L, Coll M, Aguilar J. 2005. Crystal structure of an iron-dependent group III dehydrogenase that interconverts l-lactaldehyde and L-1,2-propanediol in Escherichia coli. J Bacteriol 187:4957–4966. doi: 10.1128/JB.187.14.4957-4966.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cabiscol E, Aguilar J, Ros J. 1994. Metal-catalyzed oxidation of Fe2+ dehydrogenases. Consensus target sequence between propanediol oxidoreductase of Escherichia coli and alcohol dehydrogenase II of Zymomonas mobilis. J Biol Chem 269:6592–6597. [PubMed] [Google Scholar]
- 39.Cheng S, Fan C, Sinha S, Bobik TA. 2012. The PduQ enzyme is an alcohol dehydrogenase used to recycle NAD+ internally within the Pdu microcompartment of Salmonella enterica. PLoS One 7:e47144. doi: 10.1371/journal.pone.0047144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Craciun S, Marks JA, Balskus EP. 2014. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol 9:1408–1413. doi: 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- 41.Dobrindt U, Blum-Oehler G, Nagy G, Schneider G, Johann A, Gottschalk G, Hacker J. 2002. Genetic structure and distribution of four pathogenicity islands (PAI I536 to PAI IV536) of uropathogenic Escherichia coli strain 536. Infect Immun 70:6365–6372. doi: 10.1128/IAI.70.11.6365-6372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeisel SH, daCosta KA, Youssef M, Hensey S. 1989. Conversion of dietary choline to trimethylamine and dimethylamine in rats: dose-response relationship. J Nutr 119:800–804. [DOI] [PubMed] [Google Scholar]
- 43.Selmer T, Pierik AJ, Heider J. 2005. New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol Chem 386:981–988. doi: 10.1515/BC.2005.114. [DOI] [PubMed] [Google Scholar]
- 44.Yeates TO, Kerfeld CA, Heinhorst S, Cannon GC, Shively JM. 2008. Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol 6:681–691. doi: 10.1038/nrmicro1913. [DOI] [PubMed] [Google Scholar]
- 45.Kerfeld CA, Heinhorst S, Cannon GC. 2010. Bacterial microcompartments. Annu Rev Microbiol 64:391–408. doi: 10.1146/annurev.micro.112408.134211. [DOI] [PubMed] [Google Scholar]
- 46.Cai F, Heinhorst S, Shively JM, Cannon GC. 2008. Transcript analysis of the Halothiobacillus neapolitanus cso operon. Arch Microbiol 189:141–150. doi: 10.1007/s00203-007-0305-y. [DOI] [PubMed] [Google Scholar]
- 47.Bertin Y, Girardeau JP, Chaucheyras-Durand F, Lyan B, Pujos-Guillot E, Harel J, Martin C. 2011. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ Microbiol 13:365–377. doi: 10.1111/j.1462-2920.2010.02334.x. [DOI] [PubMed] [Google Scholar]
- 48.Penrod JT, Roth JR. 2006. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol 188:2865–2874. doi: 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 50.Sommer MO, Dantas G, Church GM. 2009. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325:1128–1131. doi: 10.1126/science.1176950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. 2010. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464:908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- 52.Louis P, McCrae SI, Charrier C, Flint HJ. 2007. Organization of butyrate synthetic genes in human colonic bacteria: phylogenetic conservation and horizontal gene transfer. FEMS Microbiol Lett 269:240–247. doi: 10.1111/j.1574-6968.2006.00629.x. [DOI] [PubMed] [Google Scholar]
- 53.Vital M, Howe AC, Tiedje JM. 2014. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio 5(2):e00889-14. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown SA, Palmer KL, Whiteley M. 2008. Revisiting the host as a growth medium. Nat Rev Microbiol 6:657–666. doi: 10.1038/nrmicro1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wackett LP, Frias JA, Seffernick JL, Sukovich DJ, Cameron SM. 2007. Genomic and biochemical studies demonstrating the absence of an alkane-producing phenotype in Vibrio furnissii M1. Appl Environ Microbiol 73:7192–7198. doi: 10.1128/AEM.01785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jorda J, Lopez D, Wheatley NM, Yeates TO. 2013. Using comparative genomics to uncover new kinds of protein-based metabolic organelles in bacteria. Protein Sci 22:179–195. doi: 10.1002/pro.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. 2012. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat J Commun 3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. 2008. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A 105:13580–13585. doi: 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dabek M, McCrae SI, Stevens VJ, Duncan SH, Louis P. 2008. Distribution of β-glucosidase and β-glucuronidase activity and of β-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol Ecol 66:487–495. doi: 10.1111/j.1574-6941.2008.00520.x. [DOI] [PubMed] [Google Scholar]
- 60.Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P. 2014. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J 8:1323–1335. doi: 10.1038/ismej.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McIntosh FM, Maison N, Holtrop G, Young P, Stevens VJ, Ince J, Johnstone AM, Lobley GE, Flint HJ, Louis P. 2012. Phylogenetic distribution of genes encoding β-glucuronidase activity in human colonic bacteria and the impact of diet on faecal glycosidase activities. Environ Microbiol 14:1876–1887. doi: 10.1111/j.1462-2920.2012.02711.x. [DOI] [PubMed] [Google Scholar]
- 62.Wargo MJ. 2013. Homeostasis and catabolism of choline and glycine betaine: lessons from Pseudomonas aeruginosa. Appl Environ Microbiol 79:2112–2120. doi: 10.1128/AEM.03565-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Müller E, Fahlbusch K, Walther R, Gottschalk G. 1981. Formation of N,N-dimethylglycine, acetic acid, and butyric acid from betaine by Eubacterium limosum. Appl Environ Microbiol 42:439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brugère JF, Borrel G, Gaci N, Tottey W, O’Toole PW, Malpuech-Brugère C. 2014. Proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes 5:5–10. doi: 10.4161/gmic.26749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rozen S, Skaletsky H. 1999. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386. [DOI] [PubMed] [Google Scholar]
- 66.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 67.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 68.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Letunic I, Bork P. 2011. Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nadkarni MA, Martin FE, Jacques NA, Hunter N. 2002. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148:257–266. [DOI] [PubMed] [Google Scholar]
- 71.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods Download
Multiple-sequence alignment of the N-terminal region of AdhE, the bifunctional aldehyde-alcohol dehydrogenase (P0A9Q7) from E. coli (AdhEEc); acetaldehyde dehydrogenases (YP_389772 and YP_389768) from D. alaskensis (CutFDa and CutBDa); acetaldehyde dehydrogenase (P41793) from S. enterica (EutESe); and propionaldehyde dehydrogenase (NP_460996) from S. enterica (PduPSe). The catalytic cysteine residue is indicated with an asterisk. Download
Multiple-sequence alignment of alcohol dehydrogenase (ACK56133) from Thermococcus sp. ES1 (AdhTher); 1,2-propanediol oxidoreductase (AAC75841) from E. coli (FucOEc); alcohol dehydrogenase II (YP_163331) from Zymomonas mobilis (AdhIIZm); 1,3-propanediol oxidoreductase (AAP97875) from Klebsiella pneunomiae (DhaTKp); alcohol dehydrogenase (NP_461396) from S. enterica (EutGSe); 1-propanol dehydrogenase (AAD39016) from S. enterica (PduQSe); and alcohol dehydrogenase from D. alaskensis (CutODa). Black asterisks indicate residues that coordinate the Fe2+ ion. The red asterisk shows an essential histidine residue which putatively interacts with the substrate. Download
Comparison of the composition and organization of cut gene clusters in sequenced bacteria revealed two cluster architectures. Experimentally characterized strains are indicated in red text. Download
Maximum-likelihood phylogenetic tree of translated cutC sequences amplified from new choline-utilizing isolates and human gut metagenomic DNA. Download
Primers used for qRT-PCR analysis.
Primers used for cloning. Restriction sites are underlined.
Molecular characterization of choline-utilizing human isolates.
Relative number of CutC hits with conserved/essential active site residues in Human Microbiome Project stool metagenomes.
Putative cut gene clusters in sequenced bacterial genomes. Download