

Abstract
Disorders of the skeleton are one of the most common causes of chronic pain and long-term physical disability in the world. Chronic skeletal pain is caused by a remarkably diverse group of conditions including trauma-induced fracture, osteoarthritis, osteoporosis, low back pain, orthopedic procedures, celiac disease, sickle cell disease and bone cancer. While these disorders are diverse, what they share in common is that when chronic skeletal pain occurs in these disorders, there are currently few therapies that can fully control the pain without significant unwanted side effects. In this review we focus on recent advances in our knowledge concerning the unique population of primary afferent sensory nerve fibers that innervate the skeleton, the nociceptive and neuropathic mechanisms that are involved in driving skeletal pain, and the neurochemical and structural changes that can occur in sensory and sympathetic nerve fibers and the CNS in chronic skeletal pain. We also discuss therapies targeting nerve growth factor or sclerostin for treating skeletal pain. These therapies have provided unique insight into the factors that drive skeletal pain and the structural decline that occurs in the aging skeleton. We conclude by discussing how these advances have changed our understanding and potentially the therapeutic options for treating and/or preventing chronic pain in the injured, diseased and aged skeleton.
GRAPHICAL ABSTRACT: Skeletal pain is common and frequently difficult to fully control. Recent data suggests that the skeleton is innervated by a restricted set of nociceptors and that many skeletal pains have both a nociceptive and a neuropathic component. Significant progress has been made in defining the mechanisms that drive skeletal pain and the factors that control skeletal remodeling. These insights have the potential to fundamentally transform our understanding and ability to prevent and/or treat skeletal pain due to injury, disease, and aging.
Keywords: NGF, nociceptor, periosteum, marrow, age, cortical bone, articular cartilage, CRPS
Introduction
Painful skeletal conditions are prevalent and their impact is pervasive in both the developing and developed world (Lubeck, 2003; Woolf & Pfleger, 2003; Brooks, 2006; Kidd, 2006). A major reason skeletal pain occurs in such a diverse group of disorders (Fig. 1) is that the skeleton is required for structural support, movement, protection of the internal organs, mineral and growth factor storage and release, and the birth and maturation of blood cells. Importantly, if skeletal pain is not adequately controlled it oftentimes has secondary effects including loss of bone and muscle mass, cardiovascular function and cognitive health, all of which can significantly diminish the patient's functional status and quality of life.
Figure 1.

List of human disorders that are frequently accompanied by skeletal pain. A major reason that skeletal pain occurs in such a diverse group of disorders is that the skeleton participates in a variety of functions including structural support, ambulation, protection of internal organs, mineral and growth factor storage and release, and the birth and maturation of blood cells. As the skeleton is composed of tissues with very unique microenvironments, (such as articular cartilage, periosteum, mineralized bone, and bone marrow) injury, aging or disease in any of these compartments can result in skeletal pain. For an extensive list of diseases that are frequently accompanied by skeletal pain, see http://www.rightdiagnosis.com/symptoms/bone_pain/common.htm in children, http://www.nof.org/articles/5 and for a list of rare (orphan) bone diseases that may be accompanied by skeletal pain, http://www.usbji.org/projects/RBDPN_op.cfm?dirID=252.
Skeletal pain tends to increase with age as the mass, quality and strength of the human skeleton peaks at 25-30 years of age in both males and females (Heaney et al., 2000; Seeman, 2002) and then declines thereafter (Fig. 2A and B). As the lifespan of individuals in both the developing and developed world continues to increase (in many countries it is now >80 years old) and with the rise of lifestyle factors such as obesity and reduction in daily physical activity, both of which reduce skeletal health, the burden that skeletal pain will exact on individuals and society is expected to increase markedly in the coming decades (Peltonen et al., 2003; Stovitz et al., 2008).
Figure 2.
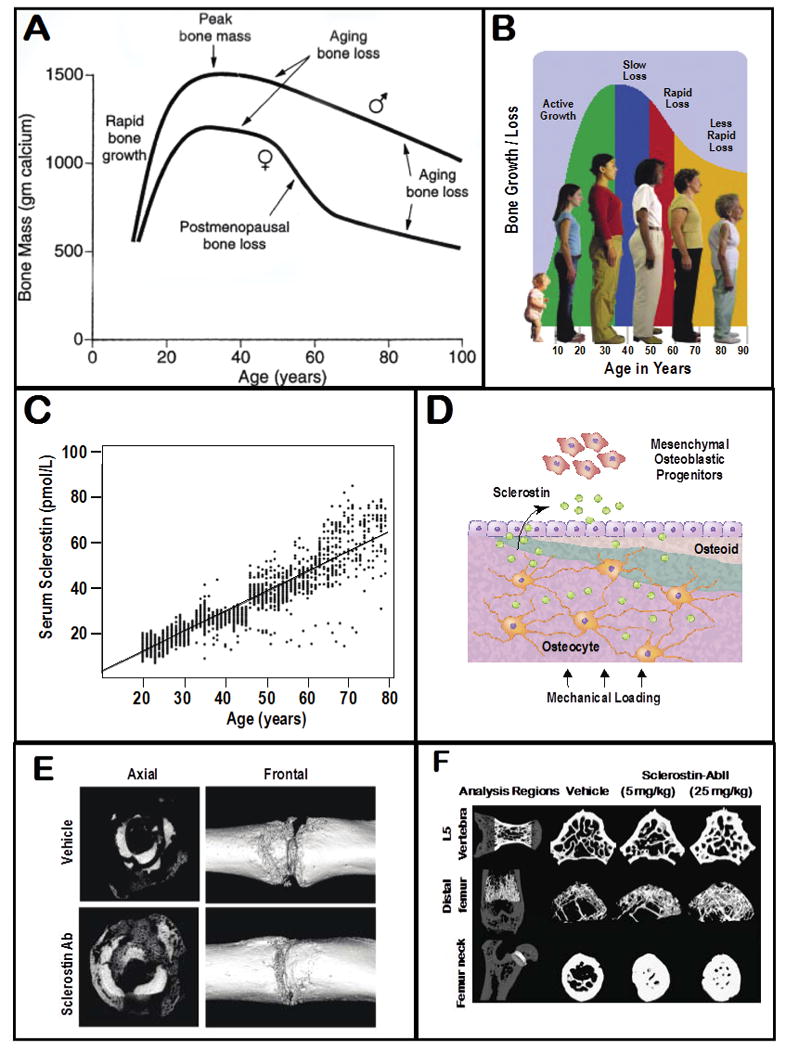
Changes in the skeleton and sclerostin levels with normal aging. (A) Human bone mass usually peaks at 25-30 years of age in both men and women and then generally declines thereafter, (B) resulting in stereotypic changes in posture and height. (C) One factor that appears to participate in driving age-related bone loss is sclerostin (here measured in plasma) which inhibits bone formation and increases linearly with age(D) The major cell type that expresses sclerostin in the adult is the osteocyte in bone, and sclerostin appears to exert its inhibitor effect by blocking bone progenitor cells and osteoblast function. (E) Administration of an antibody that sequesters sclerostin (Scl-Ab) has been shown to accelerate fracture healing in the primate tibia (reprinted with permission from Ominsky et al., 2011)(F) Administration of Scl-Ab to normal rats also shows a dose-related effect (as analyzed by μCT) in stimulating bone formation in the femoral neck of the femur, distal head of the femur and the L5 vertebrae (Figure reprinted with permission from Li et al., 2010).
In this review, we discuss the neurobiology of skeletal pain in terms of the unique repertoire of sensory nerve fibers that innervate the bone and joint, the sensory innervation in different compartments of the skeleton, the remarkable neurochemical and morphological plasticity that can occur in sensory nerves and their axons that innervate the skeleton following injury or disease, and how many forms of chronic skeletal pain appear to have both a nociceptive and neuropathic component. We conclude by discussing the role the central nervous system plays in maintaining and amplifying chronic skeletal pain and how current and emerging therapies have provided insight and potential therapeutic targets for treating / preventing chronic skeletal pain.
The types of sensory nerve fibers that innervate the skeleton
Previous studies have thoroughly and elegantly described the sensory nerve fiber innervation of the mammalian skin (Peters et al., 2002; Funfschilling et al., 2004; Zylka et al., 2005; Pare et al., 2007; Kwan et al., 2009; Albrecht & Rice, 2010; Kalliomaki et al., 2011). These studies have shown that both human and rodent skin are innervated by a rich variety of sensory nerve fibers. These include the thickly myelinated sensory nerve fibers (type II or A-beta), thinly myelinated sensory nerve fibers (type III or A-delta) and both major classes of unmyelinated nerve fibers (type IV or C-fibers): peptide-rich nerve fibers that express tropomysin receptor kinase A (TrkA) and calcitonin gene-related peptide (CGRP) as well as other neuropeptides, and the TrkA–, peptide-poor isolectin B4 nerve fibers which generally do not express CGRP (Karanth et al., 1991; Schulze et al., 1997; Zylka et al., 2005; Albrecht et al., 2006; Taylor et al., 2009). Each type of sensory nerve fiber that innervates the skin appears to have a unique conduction velocity, neurotransmitter and receptor expression profile, pattern of innervation, and function (Roosterman et al., 2006; Campero et al., 2009; Kwan et al., 2009; Smith & Lewin, 2009). Thickly myelinated A-beta fibers detect and signal non-noxious stimuli such as fine touch, light pressure, and brushing of the skin, do not express neuropeptide neurotransmitters, and have conduction velocities >30 m/sec. The thinly myelinated A-delta fibers are primarily involved in the rapid detection and signaling of nociceptive stimuli such as pin prick and noxious mechanical stimuli, such as pressure and mechanical distortion, express neuropeptides such as CGRP, and have conduction velocities between 2 and 30 m/s. Lastly, the largely unmyelinated C-fibers (both the peptide-rich and peptide-poor populations) have a slower conduction velocity (>2 m/s) than the A-fibers and are involved in detecting and signaling noxious heat, and mechanical and chemical stimuli.
In marked contrast to the skin, the adult skeleton (i.e. the bone and joint) is innervated largely by thinly myelinated, TrkA+ sensory nerve fibers (A-delta) and the peptide-rich CGRP, TrkA+ sensory nerve fibers (Fig 3), and receive little if any innervation by the larger more rapidly conducting A-beta fibers or the TrkA-, unmyelinated peptide-poor C-fibers (Zylka et al., 2005; Jimenez-Andrade et al., 2010). Recent work has shown that this difference in the population of sensory nerve fibers that innervates the adult skin vs. skeleton begins to be directed during perinatal and postnatal development. Thus, several transcription factors including Tlx3 and Runx1 establish the unique phenotype and cohort of sensory nerve fibers that innervate the skin vs. skeleton (Liu & Ma, 2011; Lopes et al., 2012). Functionally, the lack of innervation of bone and joint by A-beta sensory nerve fibers is probably related to the fact that fine touch, light pressure and brushing is not required in bone and joint as it is generally a deep structure so that most sensory nerve fibers that innervate the bone and joint are silent nociceptors, i.e. nerve fibers that are only activated by injury or damage to the bone or joint. Why bone does not receive significant innervation by the TrkA–, peptide-poor C-fibers (which do richly innervate the skin) is not completely clear, although it clearly suggests there is less nociceptor ‘redundancy’ in bone and joint than in skin (Fig. 3A and B).
Figure 3.
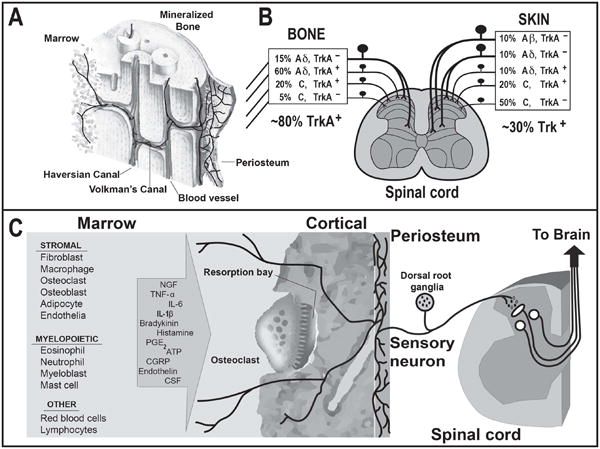
The types of cells in bone, algogenic factors that can be released from these cells, and the primary afferent sensory nerve fibers that innervate the bone. (A) Schematic diagram illustrating the general organization and pattern of sensory innervation of the skeleton. Note that the relative density of A-delta and C sensory fibers (nerve fibers per unit area) is greatest in the periosteum, followed by the bone marrow and cortical bone, with a ratio of 100: 2: 0.1, respectively. (B) Primary afferent neurons innervating the skeleton have their cell bodies in the dorsal root ganglia (DRG) and project to the spinal cord. The types of sensory neurons that innervate the bone are unmyelinated C fibers and thinly myelinated Aδ fibers. The great majority (>80%) of sensory nerve fibers that innervate the bone and articular cartilage express TrkA (which is the cognate receptor for NGF), whereas < 30% of the nerve fibers that innervate the skin express TrkA. (C) Bone contains a remarkably diverse population of stromal, myeloid and other cells which can release a wide variety of factors (some of which are listed in the arrow) that can activate and sensitize the primary afferent nerve fibers that innervate the bone.
The above immunohistochemical data concerning the innervation of the bone and articular cartilage are in close agreement with previous electron microscopy and electrophysiological studies that have examined the density and types of nerve fibers that innervate the bone and joint in animals and humans. For example, studies of the periosteum of the cat humerus (Ivanusic et al., 2006) and the bone marrow of the dog tibia (Seike, 1976) demonstrated that these bones are innervated primarily by unmyelinated and thinly myelinated nerve fibers, with few, if any, thickly myelinated nerve fibers. Similarly, electrophysiological recordings performed on nerve fibers entering the nutrient foramen of the humerus of the cat demonstrated that the conduction of nerve fibers separates into two categories, those with conduction velocities of < 2 m/s (presumably C-fibers) and those with conduction velocities between 2 and 30 m/s (presumably A-delta fibers), with no nerve fibers having conduction velocities >30 m/s, which would correspond to A-beta fibers (Mahns et al., 2006). Additionally, studies using either transgenic animals or retrograde tracers that were applied to different compartments of the skeleton also demonstrated the TrkA–, peptide-poor population of C-fibers, that richly innervate the skin, does not appear to innervate human (Ozawa et al., 2006) and rat intervertebral discs (Ozawa et al., 2003; Aoki et al., 2005; Ohtori et al., 2007), rat hip (Nakajima et al., 2008), rat wrist joint (Kuniyoshi et al., 2007), rat knee, humerus and bones of the skull (Kruger et al., 1989), or the mouse femur (Zylka et al., 2005; Jimenez-Andrade et al., 2010).
It should be noted that, while much of the recent work on the sensory and sympathetic innervation of bone has been done on the long bones of the body, several studies examining the bones of the skull and temporomandibular joint have found that, in general, bones that comprise the skull are generally innervated by the same subtypes and have an organization similar to that of the long bones of the body (Kruger et al., 1989; Silverman & Kruger, 1989; Hill & Elde, 1991; Hill et al., 1991; Takeuchi & Toda, 2003; Zylka et al., 2005; Kosaras et al., 2009). For example, in studies performed by the same authors on the same animals, it was noted the types, patterns and organization of sensory and sympathetic nerve fibers that innervate the rat calvaria and mandible were very similar to those that innervate a long bone such as the rat tibia (Hill & Elde, 1991; Hill et al., 1991).
The organization of sensory nerve fibers that innervate the skeleton
Although bone and joint (the two tissues which comprise the skeleton) appear to be innervated by the same sub-populations of nociceptive nerve fibers (A-delta and peptide-rich C-fibers), the density, pattern and morphology of nerve fibers in the two compartments of bone and joint are strikingly different (Mach et al., 2002; Kuniyoshi et al., 2007; Castañeda-Corral et al., 2011; Aso et al., 2013). The periosteum, which is the thin fibrous sheath that covers the entire surface of bone with the exception of the articular surface of the joint, receives the most dense sensory innervation (i.e. greatest number of sensory nerve fibers per unit area) of any compartment of the skeleton. The periosteum is composed of a thin outer ‘fibrous layer’ and an inner bone lining ‘cambium layer’ which contains the progenitor cells that are intimately involved and required for bone remodeling and fracture repair in adult bones (Hutmacher & Sittinger, 2003; Zhang et al., 2005; Bielby et al., 2007). In the periosteum, the A-delta and C-sensory nerve fibers are arranged in a fishnet-like pattern, which appears to be designed to act as a ‘neural net’ to detect mechanical injury or distortion of the underlying cortical bone (Martin et al., 2007). Thus, the initial sharp pain felt upon a kick to the shin bone (i.e. the tibia) or a fracture of any bone, is probably detected by mechanotransducers expressed by the A-delta and C-sensory fibers. Mechanotransducers are ion channels that detect mechanical stimuli such as stretching and pressure, although the exact set of mechanaotranducers that are expressed in each subset of mammalian sensory nerve fibers has yet to be fully elucidated (Delmas et al., 2011). Following bone fracture, movement before effective stabilization of the fractured bone can be remarkably painful. This pain is most likely attributable to the normally silent mechanosensitive nerve fibers in the periosteum that are discharging and signaling the pain. When the bone is stabilized with either an external cast or internal rod, significant relief of this sharp, stabbing and arresting pain occurs. In contrast, the dull aching pain following injury, bruising, or stabilization of the fractured bone is probably driven by the unmyelinated C-fibers present in the periosteum (Martin et al., 2007; Jimenez-Andrade et al., 2009).
The cortical bone and bone marrow are also innervated by the same population of A-delta and C-sensory nerve fibers that innervate the periosteum, although the relative density of sensory nerve fibers per unit area is markedly lower (Fig. 3A and B). The relative densities of A-delta and C-sensory nerve fibers in the periosteum, marrow and cortical bone are 100:2:0.1, respectively (Castañeda-Corral et al., 2011). In the cortical bone, both A-delta and C-fibers typically colocalize with blood vessels that run through the Haverisian and Volkmann canals although it is clear that the majority of blood vessels in Haversian or Volkmann canal in the mineralized bone are not innervated by sensory nerve fibers, which may be one reason why bone microfractures may not at least initially be perceived as painful. Likewise, the bone marrow is innervated by A-delta and C-sensory nerve fibers and, in most cases, these nerve fibers are associated with blood vessels.
In sharp contrast to periosteum, cortical bone and bone marrow, the normal articular cartilage of the knee and temporomandibular joint appears to completely lack any detectable innervation by sensory nerve fibers or vascularization by blood vessels (Archer et al., 2012), while the neighboring synovial membrane and the subchondral bone (i.e. bone immediately below the articular cartilage) in the normal joint is innervated by sensory nerve fibers and receives a significant vascular supply (Donaldson, 2009; Kelly et al., 2012) (Fig. 3A). In light of this lack of sensory nerve fibers in normal articular cartilage, the initial source of pain following acute injury of the joint in osteoarthritis (OA) (Campero et al., 2009) or temporomandibular joint disorder (TMD) must arise from adjacent structures such as the ligaments, synovium and muscle. As discussed below, nerve fibers may undergo sprouting and innervate the damaged articular cartilage following injury, inflammation or aging, and thus serve as a generator of chronic knee or TMD pain.
Sympathetic nerve fibers and skeleton pain
While primary afferent sensory nerve fibers are clearly involved in driving skeletal pain, the skeleton also receives a significant innervation by both adrenergic and cholinergic sympathetic nerve fibers. Previous studies have shown that sympathetic nerve fibers in bone can regulate bone destruction, bone formation, vasodilation, vasoconstriction, macrophage infiltration and bone progenitor cell function. As such, they may play a significant role in health and disease progression in both cartilage (i.e. rheumatoid arthritis) and bone (i.e. osteoporosis) and thus via modulating disease progression play a significant role in driving skeletal pain (Asmus et al., 2000; Elenkov et al., 2000; Bataille et al., 2012; Eimar et al., 2013).
It has also been shown that, following injury to the skeleton, sympathetic nerve fibers can modulate sensory nerve fiber function and this pathological interaction between sensory and sympathetic nerve fibers may play a role in OA and complex regional pain syndrome (Pepper et al., 2013). For example, in many cases of chronic skeletal pain, when significant sprouting of TrkA+ sensory nerve fibers occurs this is also accompanied by nearby sprouting on TrkA+, tyrosine hydroxylase sympathetic nerve fibers (Ghilardi et al., 2011). For example, in a rodent model of knee OA it was shown that sympathetic nerve sprouting occurred in the inflamed joint and that pharmacological suppression of sympathetic fiber function significantly decreased the OA pain-related behaviors (Longo et al., 2013). Previous data has suggested that noradrenaline released from these newly sprouted sympathetic fibers can bind to Beta-2 receptors expressed by nearby sensory nerve fibers, thus contributing to pain-related behavior associated with arthritis. Interestingly, peripheral noradrenaline that is mainly released by the sympathetic nervous system has little influence on healthy tissues. However, in injured or inflamed tissues it has varying effects, including aggravation of pain in neuropathy presumably due to injury-induced expression of novel noradrenergic receptors by nociceptors, sprouting of sympathetic nerve fibers, and pronociceptive changes in the ion channel properties on primary afferent nociceptors (Pertovaara, 2013). Developing a better understanding of the normal and pathological interactions of sensory and sympathetic nerve fibers in the skeleton may help understand how the nerve fibers may be involved in modulating both skeletal pain and disease progression.
Clinically relevant models of skeletal pain
Recent research into defining the mechanisms that drive skeletal pain have significantly benefited from the development of new preclinical models of skeletal pain (Honore & Mantyh, 2000; Halvorson et al., 2005; Ballas et al., 2012; Pepper et al., 2013; Vincent et al., 2013). The development and usefulness of these models is predicated on three insights.
First, as noted above, the skeleton is innervated by a very different repertoire of sensory nerve fibers from the skin and thus models of skin pain and skeletal pain cannot serve as surrogates of each other.
Second, even when modeling skeletal pain, it is very useful to attempt to have the model closely mirror a specific type of human skeletal pain, as the various bone microenvironments (marrow, mineralized bone, articular cartilage and periosteum) involved in driving the pain can be remarkably heterogeneous (Ballas et al., 2012; Mantyh, 2013). Thus, while there may be similarities in the mechanisms that drive skeletal pain in different diseases, the mechanisms that drive sickle cell pain due to a vascular occlusion episode in the marrow may be quite different from the pain following mechanical injury to articular cartilage or periosteum.
Third, models of skeletal pain that closely mirror the underlying skeletal disease process have the advantage of being able to simultaneously determine not only whether the therapy is an effective analgesic but whether it also has an impact the disease process itself. Examples of therapies having an analgesic and disease modifying effect in skeletal disorders are bisphosphonates and Denosumab in bone cancer, NSAIDS and COX-2 inhibitors in bone fracture, and tumor necrosis factor (TNF)-alpha in rheumatoid arthritis(Gaston, 2007; Brown-Glaberman & Stopeck, 2013; Sakthiswary & Das, 2013).
Nociceptive component of skeletal pain
Until recently, the major factors thought to drive acute and chronic skeletal pain were that following overuse, injury, inflammation, disease or aging of the bone or cartilage, the sensory nerve fibers that innervate the skeleton were activated and/or sensitized by either direct mechanical injury or the release of algogenic substances from the bone or joint (Fig. 3C).
A good example of mechanical distortion of sensory nerve fibers inducing significant skeletal pain is the immediate pain that is perceived following bone fracture. Initial fracture of the bone is thought to be detected by the mechanotransducers expressed by nociceptors that densely innervate the periosteum, with this initial sharp, stabbing and arresting pain being transmitted by the thinly myelinated A-delta nerve fibers. Upon stabilization of the fracture, the initial sharp, stabbing pain usually subsides and is replaced by a lesser dull, aching pain that is most likely conveyed by the C-fibers in the periosteum, bone and marrow. This dull aching pain is probably due to activation and sensitization of both the A-delta and C-fibers by algogenic factors released by the fractured bone cells and the invading inflammatory and immune cells. These algogenic factors include bradykinin, colony stimulating factors, nerve growth factor (NGF), prostaglandin E2, serotonin, protease-activated receptor 2, and TNF-alpha (Fig. 3C) (Inglis et al., 2005; Schweizerhof et al., 2009; Gold & Gebhart, 2010; Kurejova et al., 2010; Ye et al., 2011; Lam et al., 2012). If mechanical distortion of the periosteum of the fractured bone recurs, the now sensitized A-delta and C-fibers avidly discharge, and the sharp, stabbing pain immediately returns.
A similar duality of sharp stabbing pain, and a lesser dull aching pain, appears to occur following injury to the articular cartilage in OA or TMD. With either trauma or degeneration of the articular cartilage of the joint, A-delta and C-fibers in the synovium and subchondral bone are sensitized by algogenic substances so that normally non-noxious loading and movement of the joint are perceived as noxious stimuli. As the normal articular cartilage completely lacks any detectable innervation by sensory nerve fibers, the exact location(s) of nerve fibers that drive OA pain are as yet unknown. Thus, while OA of TMD pain may arise from direct mechanical stimulation of sensitized nerve fibers that are present in the adjacent bone (Kidd, 2006; Hunter et al., 2008), if this were the only mechanism driving arthritic joint pain, a clear correlation should appear between joint deterioration or destruction and joint pain. However, in both trauma and age-related OA and TMD, there is a poor correlation between the extent of joint destruction and the frequency and severity of joint pain (Felson et al., 1987; Hannan et al., 2000; Bedson & Croft, 2008).
Decades of research have demonstrated that NGF directly activates and sensitizes TrkA-expressing sensory neurons and that NGF activation of TrkA could play a key role in the sensitization of TrkA-expressing nociceptors to mechanical, chemical and thermal stimuli (Levi-Montalcini, 1987; Lewin & Mendell, 1993; Petruska & Mendell, 2004). It had been shown that NGF induces a rapid phosphorylation and sensitization of transient receptor potential cation channel subfamily V member 1 (TrpV1) channel (which detects acid and heat), as well as nociceptor mechanotransducers (Mendell et al., 1999; Zhang et al., 2005). Additionally, retrograde transport of the NGF–TrkA complex to the neuronal cell body of nociceptors induces increased synthesis of the neurotransmitters substance P and CGRP, and receptors (bradykinin), channels (P2X3, TRPV1, ASIC-3 and sodium channels), transcription factors (ATF-3) and structural molecules (neurofilaments and the sodium channel-anchoring molecule p11) (Julius & Basbaum, 2001; Patapoutian & Reichardt, 2001). However, sequestration of NGF showed little efficacy in a rat skin incision model of pain, suggesting that targeting of NGF–TrkA may not be efficacious in relieving chronic pain in skin (Zahn et al., 2004; Banik et al., 2005).
Although anti-NGF did not appear to have a major effect on reducing incisional skin pain, a major question was that, given that most sensory nerve fibers that innervated the skeleton were TrkA+, could blockage of NFG–TrkA significantly reduce severe skeletal pain?. To test this hypothesis, blockade of NGF–TrkA was tested in three separate mouse models of bone cancer pain using sarcoma, prostate and breast cancer cell implantation, all of which generated a significant skeletal pain that was accompanied by bone remodeling similar to that seen in human patients with bone cancer. Surprisingly, administration of anti-NGF attenuated skeletal pain by 50% in all three mouse models of bone cancer pain, regardless of whether the injected cancer cells expressed NGF (Mantyh et al., 2010; Jimenez-Andrade et al., 2011). These data demonstrated that NGF appeared to play a significant role in driving severe bone cancer pain, and that the relief of bone cancer pain did not depend on the type of tumor or whether the cancer cells expressed NGF (Halvorson et al., 2005). These data suggested not only that blockade of NGF–TrkA could significantly attenuate skeletal pain but that blockade of NGF–TrkA might also relieve non-malignant skeletal pain as the major source of NGF was not from the cancer cells themselves but from bone stromal, immune and/or inflammatory cells that are present in both non-malignant and malignant skeletal conditions.
The fact that the efficacy of anti-NGF therapy was not limited to malignant mouse models was then demonstrated in rodent models of knee arthritis pain in the rat (Albrecht et al., 2006; Ashraf et al., 2013) and mouse (Ghilardi et al., 2012), and bone fracture pain in the mouse (Jimenez-Andrade et al., 2007; Koewler et al., 2007; Ghilardi et al., 2011) and the rat (Sabsovich et al., 2008). In most of these studies similar efficacy (an ∼50% reduction in pain-related behaviors) was observed as in the malignant models of skeletal pain (Fig. 4). Furthermore, subsequent human clinical trials demonstrated that anti-NGF therapy could relieve OA pain by 50% and human low back pain by 40% (Fig. 4) whereas in human visceral pain (interstitial cystitis) and human neuropathic pain (sciatica), anti-NGF showed less efficacy (Nickel et al., 2012).
Figure 4.
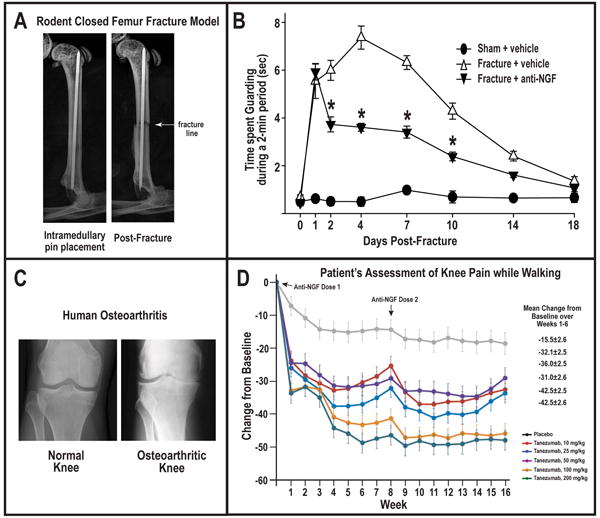
The efficacy of anti-NGF in blocking skeletal pain in pre-clinical and human models. (A) Radiograph of the normal mouse bone with intramedullary pin, and following a closed femoral fracture that generates fracture pain. (B) Sustained anti-NGF therapy (commenced immediately after fracture) attenuated fracture-induced skeletal pain-related guarding behaviors by 40-50%. Figure reprinted with permission from Koewler et al. (2007). (C) Radiograph of the normal and osteoarthritic human knee. (D) The assessment of the patient's knee pain while walking showed a significant reduction (40-50%) with human anti-NGF therapy (Tanezumab). A decrease in the change from baseline indicates an improvement (i.e., less pain). Figure reprinted with permission from Lane et al. (2010).
Neuropathic component of skeletal pain
While most preclinical and clinical scientists who study skeletal pain would agree that a significant nociceptive component in most chronic skeletal pains (OA, TMD, bone cancer, bone fracture and sickle cell), recent data has suggested there may also be a neuropathic component in many types of skeletal pain. For example, in both the sarcoma, prostate and breast mouse models of bone cancer pain, as tumor cells invade the normal tissue the tumor appears to first come into contact then injure and destroy the very distal processes of sensory fibers. This tumor-induced injury and destruction of the distal ends of the sensory nerve fibers that innervate the skeleton is accompanied by an increase in ongoing and movement-evoked pain behaviors. This data, along with data suggesting sensory nerve fibers are injured in OA and low back pain, suggests a component of malignant and non-malignant skeletal pain can be neuropathic in origin (Ohtori et al., 2012; Schaible, 2012). This, as well as the fact that several skeletal pains can be attenuated by gabapentin (Rosenberg et al., 1997; Wetzel&Connelly), which is approved for the treatment of neuropathic pain, suggests that injury to sensory neurons may play a role in several types of skeletal pain (Donovan-Rodriguez et al., 2005; Kurejova et al., 2010).
Another intriguing, but largely unexplored, mechanism by which neuropathic skeletal pain may be generated is not by injury, but rather by an active and pathological sprouting and neuroma formation by sensory and sympathetic nerve fibers that innervate the skeleton. To explore this possibility, active nerve sprouting and formation of neuroma-like structures was examined using mouse models of sarcoma, breast and prostate bone cancer cells growing in the bone marrow. Using these models it was noted that there was a remarkable and dramatic sprouting of sensory and sympathetic nerve fibers, and that these newly sprouted nerve fibers (which can be observed in the periosteum, mineralized bond and marrow) have a unique morphology, organization and high density that is never observed in the normal bone (Fig. 5). What is impressive is how exuberant this sprouting can be: in the bone cancer prostate model (where the prostate cancer cells do not express NGF) the number of nerve fibers per unit area increased to 10-70× what is observed in the normal bone marrow. This sprouting appears to require NGF, as sustained administration of anti-NGF or Pan-Trk inhibitor largely blocked the pathological sprouting of sensory nerve fibers and the formation of neuroma-like structures, and significantly inhibited the generation of pain (Mantyh et al., 2010; Ghilardi et al., 2012).
Figure 5.
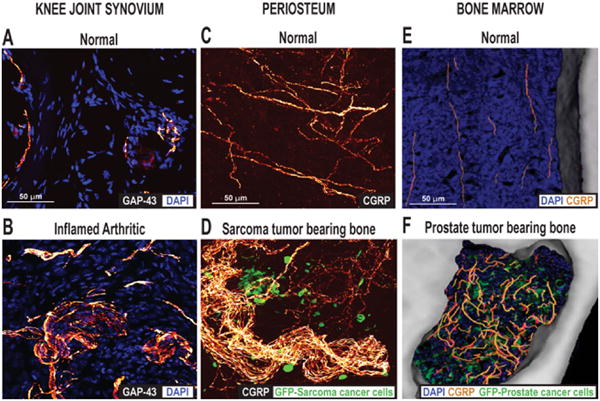
Ectopic sprouting of primary afferent sensory nerve fibers occurs in a variety of skeletal pain conditions. (A) Confocal images of sections of (A) the normal and (B) the inflamed knee joint that have been immunostained for DAPI, which stains nuclei, and growth-associated protein (GAP-43), which stains sprouting nerve fibers. Twenty-eight days after the initial injection of Complete Freund's Adjuvant (CFA) into the rat knee-joint, a significant number of GAP-43+ nerve fibers in the synovial knee joint had sprouted and had a disorganized appearance, as compared with vehicle-injected mice. Reprinted with permission from Jimenez-Andrade & Mantyh (2012). Confocal images of periosteal whole mounts of (C) normal and (D) sarcoma tumor-bearing bone immunostained for calcitonin gene-related peptide (CGRP+) and green fluorescent protein (GFP)-labeled sarcoma. As tumor cells invade the periosteum of the bone, ectopic sprouting of CGRP+ sensory fibers occurs and neuroma-like structures form. Reprinted with permission from Mantyh et al. (2010). Confocal images of bone marrow of (E) normal and (F) prostate tumor-bearing bone marrow, immunostained for DAPI, CGRP and GFP-expressing prostate cancer cells. Note that in the normal mice, CGRP+ nerve fibers present in the marrow space of normal mice appear as single nerve fibers with a highly linear morphology. As GFP+ prostate tumor cells proliferate and form tumor colonies, the CGRP+ sensory nerve fibers undergo marked sprouting which produces a highly branched, disorganized and dense meshwork of sensory nerve fibers that is never observed in normal marrow. Reprinted with permission from Jimenez-Andrade et al. (2010).
The above studies suggest ectopic sprouting of nerve fibers may occur in several types of bone cancer pain, whereas previous studies have reported that ectopic sprouting of sensory nerve fibers has been observed in several types of non-malignant skeletal pain. For example, in human chronic vertebral discogenic pain it has been shown that there is growth of CGRP sensory nerves fibers into normally aneural and avascular areas of the human intervertebral disc (Freemont et al., 2002); there is also sprouting of CGRP nerve fibers into the callus that forms around the bone fracture in rat, and in the synovoium of arthritic joints of humans and animals (Buma et al., 1992; Wu et al., 2002; Suri et al., 2007). Similar to what is observed in models of bone cancer, it has been suggested that the sprouting of sensory nerve fibers in non-malignant skeletal pain is driven by NGF released by endogenous stromal, immune and/or inflammatory cells (Ehrhard et al., 1993; Skaper et al., 2001; Artico et al., 2008).
Given the above observations on ectopic sprouting in the injured or diseased skeleton, a major question is to what extent does this pathological sprouting drive skeletal pain? Previous studies in animals and humans have demonstrated that inappropriate sprouting and/or neuroma formation in non-skeletal conditions is often is accompanied by a remarkable change in the phenotype and hypersensitivity to normally non-noxious stimuli (Kryger et al., 2001). Given the exuberant nature of this observed sprouting in the injured skeleton, and that in adult humans newly sprouted sensory nerve fibers (such as that which follows nerve transection of injury) frequently exhibit both a spontaneous and movement-evoked ectopic discharges, it will be important to determine the role that ectopic nerve sprouting plays in the generation and maintenance of a chronic skeletal pain.
Central sensitization and skeletal pain
Studies in both animals and humans have demonstrated that, following injury to the skin, muscle and joint, neurons responsible for pain processing in the spinal cord and brain can also undergo sensitization (i.e. ‘central sensitization’) that amplifies the perception and severity of pain (Woolf, 1983; Woolf & Salter, 2000). Central sensitization is thought to occur when the chemical, electrophysiological and pharmacological systems that transmit and modulate pain are altered in both the spinal cord and higher centers of the brain. These changes cause an exaggerated perception of painful stimuli so that normally mild painful stimuli are perceived as highly painful (hyperalgesia) and normal non-painful stimuli such as normal loading or use of joint or bone is now perceived as a painful event (allodynia). Additionally, central sensitization also contributes to the phenomenon of referred pain, where areas adjacent to the initial injury become hypersensitive. A common example of this is in whiplash injuries to the neck: following injury to one cervical vertebra, nearby areas such as the shoulder, arm and back also become hypersensitive (Graven-Nielsen & Arendt-Nielsen, 2010).
Currently, we know remarkably little as to the specific mechanisms that drive central sensitization in chronic skeletal pain. Changes that have been described include upregulation of dynorphin, CGRP and substance P in the spinal cord, astrocyte hypertrophy and microglial activation (Voscopoulos&Lema,2010). Other reports have demonstrated that increases in skeletal pain-related behaviors are accompanied by increased expression of NR2B (an NMDA receptor subunit), and interleukin-1β (released from glial cells) and enhanced phosphorylation of NMDA receptor NR-1 subunits (Ren&Dubner,2008).Understanding the specific cascade of changes that generate and maintain central sensitization would significantly expand our ability to design targeted therapies to treat the central sensitization that occurs following many types of skeletal pain. These neurochemical and electrophysiological changes appear to occur in the ascending and descending pain pathways of the spinal cord, brainstem, thalamus and cerebral cortex, and all of these changes may contribute to the amplified perception of skeletal pain (Basbaum&Fields,1978; Gebhart, 2004; Robinson et al., 2004; Hunt, 2009; Latremoliere&Woolf, 2009; Woolf, 2011; Yoshida et al., 2013).
While we do not yet know the specific mechanisms that generate central sensitization, what we do know is that injury to the skeleton seems to be much more effective at inducing central sensitization as compared to injury to skin or muscle (WoolfWall, 1986). For example as noted by Woolf and Wall "..a twisted ankle invokes relatively little destruction of tissue and elicits an abrupt localized stabbing pain that dies down quickly but is followed by a prolonged period of spreading, poorly localized deep pain, and tenderness that affects reflexes and gait. In contrast, localized skin damage produces an acute burst of pain that gradually dies down over minutes but is associated with a spatially restricted response of flair, wheal and surrounding tenderness”(Woolf&Wall,1986). These authors also noted that small skin lesions produce comparatively less widespread and prolonged disturbances to sensation and reflex patterns than skeletal injuries. They then postulated that the varied pattern of post-injury pain hypersensitivities resulting from injury to different tissues may be the consequence of the activation of distinct primary afferent neurons with differing central actions, or that the unique subtypes of sensory neurons that innervate the skeleton might be uniquely effective at inducing central sensitization in the spinal cord and higher centers of the brain.
Anabolic therapies and skeletal pain
Analgesics with increased efficacy and fewer side effects are clearly needed to improve control of skeletal pain. However, another complimentary approach would be therapies that induce bone or cartilage formation and/or promote more rapid healing following injury, disease or aging of the skeleton. Currently, injury and age-related loss of bone injury to the skeleton, and the frequently slow healing of these injures that comes with aging, have enormous consequences in terms of recovery rates and the functional status of the affected individual (Wang & Seeman, 2008; Ominsky et al.; Woolf et al., 2012). Thus, most fractures in people over age 50 are the result of age-related bone loss and the resulting fragility of bone, and these fractures result in high rates of hospitalization, frequently a permanent decrease in function status, and mortality (Woolf & Pfleger, 2003).
Currently, there are two main classes of drugs available to treat age-related bone loss. Antiresorptives (e.g., bisphosphonates, denosumab) work to slow bone loss by inhibiting the activity of osteoclasts. However, with long-term use there is a decrease in the rate of bone formation because the cyclic activity of osteoclasts and osteoblasts is disrupted. The disturbance slows bone remodeling (Baron & Hesse, 2012) and leads to a state of low bone turnover, which over time can cause the accumulation of microcracks and compromise the strength and integrity of the bone.
The other relevant class of drugs is osteoanabolic: true bone-building agents. The first osteoanabolic to be put into clinical use was intermittent parathyroid hormone (PTH), which exerts its effects by preferentially stimulating osteoblasts over osteoclasts. However, bone density seems to plateau after 18-24 months of PTH therapy, and the treatment may also increase the risk of osteosarcoma (Baron & Hesse, 2012). Bone morphogenic proteins represent another anabolic option, though these are limited by their high cost (Lane et al., 2010) and the difficulties of administration (Albrecht et al., 2006; Lane & Silverman, 2010). Thus, the niche for a safe and effective osteoanabolic drug to treat age-related bone loss remains largely unfilled.
Several new therapeutic targets for building bone have profoundly changed our understanding as to why age-related bone loss occurs. Two inhibitory proteins, known as sclerostin (Burgers & Williams) and Dickkopf-1 (DKK1), have been identified and both proteins interact with the Wnt coreceptors LRP5/6 to inhibit the canonical Wnt/beta-catenin signaling pathway, causing a decrease in bone formation (Moester et al., 2010; Palaniswamy et al., 2010; Paszty et al., 2010; Lewiecki, 2011; Costa & Bilezikian, 2012; Ke et al., 2012; Lim & Clarke, 2012; Ohlsson, 2013). DKK1 appears to have a greater role in developing animals than in adults (Ke et al., 2012). In contrast, sclerostin which is a small (24 Da) secreted glycoprotein that is expressed by osteocytes in bone (Moester et al., 2010; Costa & Bilezikian, 2012) and appears to play a greater role in the adult than in the the developing skeleton. In adults, the expression and release of sclerostin by osteocytes is modulated by local mechanical loading of the bone (Costa & Bilezikian, 2012)(Fig. 2D). Aside from its normal physiological role, sclerostin has been directly implicated as a factor in skeletal disease, as studies in humans have shown a relatively linear increase in plasma levels of sclerostin with age (Modder et al., 2011) (Fig. 2C) and older women with high expression of sclerostin have greater risk of hip fracture (Albrecht et al., 2006). An antibody that sequesters sclerostin has been developed and administration of this therapy increased bone growth and accelerated fracture healing in preclinical trials in osteoporotic rats and monkeys (Li et al., 2009; Ominsky et al., 2010) (Fig. 2E). In a Phase I study, a single dose of anti-sclerostin antibody increased bone density in the hip and spine in healthy men and postmenopausal women (Padhi et al., 2011)(Fig. 2F). The administration of the drug for one year was well tolerated, and in a Phase II trial, for osteoporotic women, it increased bone density (Reid, 2012; Burgers & Williams, 2013).
In many older patients with bone fractures due to low-impact falls, appropriate healing of the skeleton never occurs and many patients develop chronic pain at the site of non-healed injury. Given the exuberant sensory nerve sprouting that has been shown following other skeletal injury and diseases, one possibility is that ectopic sensory nerve sprouting occurs at the non-healed skeletal injury site and this then drives chronic skeletal pain. However, if ectopic nerve sprouting and skeletal pain could be attenuated by blockade of NGF–TrkA, and fracture healing accelerated by blocking sclerostin, this could potentially transform how we treat fracture healing in the aged or diseased skeleton.
Analgesics and their potential effects on the skeleton
Although it may seem counterintuitive, one question that needs to be addressed when considering therapies to relieve skeletal pain is how much relief of skeletal pain is desirable? Obviously, one would probably want to eliminate all skeletal pain due to bone cancer, leukemia, fibrous dysplasia, Gaucher's disease and sickle-cell disease, as these skeletal pains appear to serve little or no protective purpose. However, in patients with skeletal pain due to OA, TMD, bone fracture or aging, eliminating all skeletal pain could potentially lead to inappropriate loading and/or overuse of the skeleton that could drive more rapid deterioration of the diseased, injured or aged skeleton. For example, this issue as to the extent of relief of skeletal pain occurred in a phase III clinical trial with anti-NGF therapy in patients with knee and hip OA. Here it was noted that a small subset of patients being treated simultaneously with anti-NGF and an NSAID developed rapidly progressing OA although it was unclear whether this deterioration was due to patients overusing their arthritic joints or a direct effect of anti-NGF and NSAIDS on the skeleton itself (Lane & Silverman, 2010).
A second major issue is whether any analgesics used to treat skeletal pain may have direct negative effects on skeletal health and/or healing of the injured skeleton. Here the answer is probably yes, as the administration of Ibuprofen or COX-2 inhibitors have been shown to markedly slow down fracture healing in rodent models of bone fracture (O'Connor et al., 2009; Barry, 2010). While the mechanism(s) by which these side effects occurs is not completely clear, prostaglandins do appear to play a significant role in Wnt signaling (Genetos et al., 2011), a key pathway required for bone formation in both the normal and fractured bone. Thus, inhibition of prostaglandin synthesis might be expected to inhibit bone formation. Which and how much NSAIDS and COX-2 inhibitors reduce normal bone formation and fracture healing in humans remains to be determined (Su & O'Connor, 2013). However, what is clear is that it is probably wise to include bone formation and skeletal healing as key endpoints in assessing novel skeletal analgesics in both preclinical studies and human clinical trials.
Lastly, if reducing skeletal pain by any mechanism has the potential to induce overuse of the injured, diseased or aged skeleton by the patient, is there an approach that ameliorates or prevents potential injury do to overuse? Obviously, more effective monitoring and feedback to the patient concerning desired activity and movement could prevent overuse. However, another possibility would be to simultaneously block NGF to attenuate skeletal pain and block sclerostin to promote bone formation and fracture healing. Whether this combined therapy would simultaneously reduce skeletal pain, increase bone formation and healing, and improve the patient's functional status and ability to participate in physical therapy has not yet been determined.
Future perspectives
Although skeletal pain is one of the most common causes of chronic pain and long-term physical disability, we currently have relatively few pharmacological therapies that can fully manage the pain, stimulate skeletal repair following injury, or reverse the bone and cartilage loss that occurs with aging. However, in the last decade significant progress has been made in developing more clinically relevant models of skeletal pain, understanding some of the mechanisms that drive skeletal pain, and identifying key molecules involved in the regulation of skeletal repair and the decline in the skeleton that occurs with aging.
In terms of skeletal pain, we now know that both the bone and joint are innervated by a limited repertoire of sensory nerve fibers and that many forms of skeletal pain have both a nociceptive and a neuropathic component. Researchers have identified the restricted repertoire of primary afferent sensory nerve fibers that innervate the skeleton, the majority of which express TrkA and thus respond to NGF released by bone stromal cells that invade the skeleton following injury or in disease. Sensory nerve fibers in an injured or diseased skeleton also display a remarkable neurochemical and morphological plasticity by upregulating neurotransmitters and receptors and undergoing a level of nerve sprouting that is never observed in the normal skeleton. In agreement with these observations, human clinical trials have demonstrated that sequestration of NGF results in significant attenuation of OA and low back pain.
Progress has also been made in beginning to understand the molecules that contribute to the deterioration and lack of effective skeletal repair that frequently occur with aging. For example, sclerostin has bee identified as a protein that is expressed and released by osteocytes when the bone is mechanically unloaded, inhibits bone formation, and whose plasma levels increase dramatically with age. As the incidence of low-trauma fractures increases and the rate of rapid and effective bone fracture healing decreases with age, inhibiting sclerostin may reduce the likelihood of fragility fractures, stimulate more rapid and effective fracture healing, and thereby reduce the incidence and duration of skeletal pain that frequently accompanies failed healing of bone fractures. Indeed, data from preclinical and human studies suggests that inhibition of endogenous sclerostin can both build bone and promote fracture healing in both young and aged bone (Lane & Silverman, 2010; Moester et al., 2010).
Whether therapies targeting the NGF or sclerostin will ultimately receive approval for broad use in humans will depend on their safety and side-effect profile. However, what is clear is that NGF plays a major role in driving skeletal pain and sclerostin plays a major role in driving the age-related decline in normal bone remodeling and fracture healing. Increasing our understanding of the mechanisms that drive skeletal pain and defining the factors that control bone remodeling in the young and old have the potential to fundamentally transform our understanding and ability to prevent and/or treat skeletal pain due to injury, disease, and aging.
Acknowledgments
This work was supported by the National Institutes of Health grant (NS023970, CA154550, CA157449). P.W.M. received anti-NGF as a research tool and gift from Rinat Laboratories, Pfizer Inc. (South San Francisco, CA, USA) and has served as a consultant and received research grants from Abbott (Abbott Park, IL, USA), Adolor (Exton, PA, USA), Array Biopharma (Boulder, CO, USA), Johnson and Johnson (New Brunswick, NJ, USA), Pfizer (New York, NY, USA), Plexxikon (Berkeley, CA, USA) and Roche (South San Francisco, CA, USA). The author thanks Stephane Chartier, Michelle Fealk, Lisa Majuta, Gwen McCaffrey and Michelle L. Thompson for editing and reviewing the manuscript.
Abbreviations
- CGRP
calcitonin gene-related peptide
- NGF
nerve growth factor
- OA
osteoarthritis
- TMD
temporomandibular joint disorder
- TrkA
tropomysin receptor kinase A
References
- Albrecht PJ, Hines S, Eisenberg E, Pud D, Finlay DR, Connolly MK, Pare M, Davar G, Rice FL. Pathologic alterations of cutaneous innervation and vasculature in affected limbs from patients with complex regional pain syndrome. Pain. 2006;120:244–266. doi: 10.1016/j.pain.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Albrecht PJ, Rice FL. Role of small-fiber afferents in pain mechanisms with implications on diagnosis and treatment. Current pain and headache reports. 2010;14:179–188. doi: 10.1007/s11916-010-0105-y. [DOI] [PubMed] [Google Scholar]
- Aoki Y, Ohtori S, Takahashi K, Ino H, Douya H, Ozawa T, Saito T, Moriya H. Expression and co-expression of VR1, CGRP, and IB4-binding glycoprotein in dorsal root ganglion neurons in rats: differences between the disc afferents and the cutaneous afferents. Spine. 2005;30:1496–1500. doi: 10.1097/01.brs.0000167532.96540.31. [DOI] [PubMed] [Google Scholar]
- Archer CW, Williams R, Nelson L, Khan IM. Articular Cartilage-Derived Stem Cells: Identification, Characterisation and their Role in Spontaneous Repair. Rheumatology: Current Research 2012 [Google Scholar]
- Artico M, Bronzetti E, Felici LM, Alicino V, Ionta B, Bronzetti B, Magliulo G, Grande C, Zamai L, Pasquantonio G, De Vincentiis M. Neurotrophins and their receptors in human lingual tonsil: an immunohistochemical analysis. Oncology reports. 2008;20:1201–1206. [PubMed] [Google Scholar]
- Ashraf S, Mapp PI, Burston J, Bennett AJ, Chapman V, Walsh DA. Augmented pain behavioural responses to intra-articular injection of nerve growth factor in two animal models of osteoarthritis. Annals of the rheumatic diseases. 2013:1–9. doi: 10.1136/annrheumdis-2013-203416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmus SE, Parsons S, Landis SC. Developmental changes in the transmitter properties of sympathetic neurons that innervate the periosteum. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:1495–1504. doi: 10.1523/JNEUROSCI.20-04-01495.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso K, Ikeuchi M, Izumi M, Sugimura N, Kato T, Ushida T, Tani T. Nociceptive phenotype of dorsal root ganglia neurons innervating the subchondral bone in rat knee joints. European journal of pain (London, England) 2013:1532–2149. doi: 10.1002/j.1532-2149.2013.00360.x. [DOI] [PubMed] [Google Scholar]
- Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120:3647–3656. doi: 10.1182/blood-2012-04-383430. [DOI] [PubMed] [Google Scholar]
- Banik RK, Subieta AR, Wu C, Brennan TJ. Increased nerve growth factor after rat plantar incision contributes to guarding behavior and heat hyperalgesia. Pain. 2005;117:68–76. doi: 10.1016/j.pain.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Baron R, Hesse E. Update on bone anabolics in osteoporosis treatment: rationale, current status, and perspectives. The Journal of clinical endocrinology and metabolism. 2012;97:311–325. doi: 10.1210/jc.2011-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry S. Non-steroidal anti-inflammatory drugs inhibit bone healing: a review. Veterinary and comparative orthopaedics and traumatology : V.C.O. T. 2010;23:385–392. doi: 10.3415/VCOT-10-01-0017. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. Endogenous pain control mechanisms: review and hypothesis. Annals of neurology. 1978;4:451–462. doi: 10.1002/ana.410040511. [DOI] [PubMed] [Google Scholar]
- Bataille C, Mauprivez C, Hay E, Baroukh B, Brun A, Chaussain C, Marie PJ, Saffar JL, Cherruau M. Different sympathetic pathways control the metabolism of distinct bone envelopes. Bone. 2012;50:1162–1172. doi: 10.1016/j.bone.2012.01.023. [DOI] [PubMed] [Google Scholar]
- Bedson J, Croft PR. The discordance between clinical and radiographic knee osteoarthritis: a systematic search and summary of the literature. BMC musculoskeletal disorders. 2008;9:116. doi: 10.1186/1471-2474-9-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielby R, Jones E, McGonagle D. The role of mesenchymal stem cells in maintenance and repair of bone. Injury, 38 Suppl. 2007;1:S26–32. doi: 10.1016/j.injury.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Brooks PM. The burden of musculoskeletal disease--a global perspective. Clinical rheumatology. 2006;25:778–781. doi: 10.1007/s10067-006-0240-3. [DOI] [PubMed] [Google Scholar]
- Brown-Glaberman U, Stopeck AT. Impact of denosumab on bone mass in cancer patients. Clinical pharmacology : advances and applications. 2013;5:117–129. doi: 10.2147/CPAA.S30330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buma P, Verschuren C, Versleyen D, Van der Kraan P, Oestreicher AB. Calcitonin gene-related peptide, substance P and GAP-43/B-50 immunoreactivity in the normal and arthrotic knee joint of the mouse. Histochemistry. 1992;98:327–339. doi: 10.1007/BF00270017. [DOI] [PubMed] [Google Scholar]
- Burgers TA, Williams BO. Regulation of Wnt/beta-catenin signaling within and from osteocytes. Bone. 2013;54:244–249. doi: 10.1016/j.bone.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campero M, Baumann TK, Bostock H, Ochoa JL. Human cutaneous C fibres activated by cooling, heating and menthol. The Journal of physiology. 2009;587:5633–5652. doi: 10.1113/jphysiol.2009.176040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda-Corral G, Jimenez-Andrade JM, Bloom AP, Taylor RN, Mantyh WG, Kaczmarska MJ, Ghilardi JR, Mantyh PW. The majority of myelinated and unmyelinated sensory nerve fibers that innervate bone express the tropomyosin receptor kinase A. Neuroscience. 2011;178:196–207. doi: 10.1016/j.neuroscience.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AG, Bilezikian JP. Sclerostin: therapeutic horizons based upon its actions. Current osteoporosis reports. 2012;10:64–72. doi: 10.1007/s11914-011-0089-5. [DOI] [PubMed] [Google Scholar]
- Delmas P, Hao J, Rodat-Despoix L. Molecular mechanisms of mechanotransduction in mammalian sensory neurons. Nature reviews. Neuroscience. 2011;12:139–153. doi: 10.1038/nrn2993. [DOI] [PubMed] [Google Scholar]
- Donaldson LF. Neurogenic Mechanisms in Arthritis. Neurogenic Inflammation in Health and Disease. 2009;8:211–241. [Google Scholar]
- Donovan-Rodriguez T, Dickenson AH, Urch CE. Gabapentin normalizes spinal neuronal responses that correlate with behavior in a rat model of cancer-induced bone pain. Anesthesiology. 2005;102:132–140. doi: 10.1097/00000542-200501000-00022. [DOI] [PubMed] [Google Scholar]
- Ehrhard PB, Erb P, Graumann U, Otten U. Expression of nerve growth factor and nerve growth factor receptor tyrosine kinase Trk in activated CD4-positive T-cell clones. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:10984–10988. doi: 10.1073/pnas.90.23.10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimar H, Tamimi I, Murshed M, Tamimi F. Cholinergic regulation of bone. Journal of musculoskeletal & neuronal interactions. 2013;13:124–132. [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacological reviews. 2000;52:595–638. [PubMed] [Google Scholar]
- Felson DT, Naimark A, Anderson J, Kazis L, Castelli W, Meenan RF. The prevalence of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis and rheumatism. 1987;30:914–918. doi: 10.1002/art.1780300811. [DOI] [PubMed] [Google Scholar]
- Freemont AJ, Watkins A, Le Maitre C, Baird P, Jeziorska M, Knight MT, Ross ER, O'Brien JP, Hoyland JA. Nerve growth factor expression and innervation of the painful intervertebral disc. The Journal of pathology. 2002;197:286–292. doi: 10.1002/path.1108. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Ng YG, Zang K, Miyazaki J, Reichardt LF, Rice FL. TrkC kinase expression in distinct subsets of cutaneous trigeminal innervation and nonneuronal cells. The Journal of comparative neurology. 2004;480:392–414. doi: 10.1002/cne.20359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston MS, Simpson AHRW. Inhibition of fracture healing. The Journal of Bone and Joint Surgery (Br) 2007;89B:1553–1560. doi: 10.1302/0301-620X.89B12.19671. [DOI] [PubMed] [Google Scholar]
- Gebhart GF. Descending modulation of pain. Neuroscience and biobehavioral reviews. 2004;27:729–737. doi: 10.1016/j.neubiorev.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Genetos DC, Yellowley CE, Loots GG. Prostaglandin E2 signals through PTGER2 to regulate sclerostin expression. PloS one. 2011;6:e17772. doi: 10.1371/journal.pone.0017772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi JR, Freeman KT, Jimenez-Andrade JM, Coughlin KA, Kaczmarska MJ, Castaneda-Corral G, Bloom AP, Kuskowski MA, Mantyh PW. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis and rheumatism. 2012;64:2223–2232. doi: 10.1002/art.34385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi JR, Freeman KT, Jimenez-Andrade JM, Mantyh WG, Bloom AP, Bouhana KS, Trollinger D, Winkler J, Lee P, Andrews SW, Kuskowski MA, Mantyh PW. Sustained blockade of neurotrophin receptors TrkA, TrkB and TrkC reduces non-malignant skeletal pain but not the maintenance of sensory and sympathetic nerve fibers. Bone. 2011;48:389–398. doi: 10.1016/j.bone.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nature medicine. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graven-Nielsen T, Arendt-Nielsen L. Assessment of mechanisms in localized and widespread musculoskeletal pain. Nature reviews. Rheumatology. 2010;6:599–606. doi: 10.1038/nrrheum.2010.107. [DOI] [PubMed] [Google Scholar]
- Halvorson KG, Kubota K, Sevcik MA, Lindsay TH, Sotillo JE, Ghilardi JR, Rosol TJ, Boustany L, Shelton DL, Mantyh PW. A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer research. 2005;65:9426–9435. doi: 10.1158/0008-5472.CAN-05-0826. [DOI] [PubMed] [Google Scholar]
- Hannan MT, Felson DT, Pincus T. Analysis of the discordance between radiographic changes and knee pain in osteoarthritis of the knee. The Journal of rheumatology. 2000;27:1513–1517. [PubMed] [Google Scholar]
- Heaney RP, Abrams S, Dawson-Hughes B, Looker A, Marcus R, Matkovic V, Weaver C. Peak bone mass. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2000;11:985–1009. doi: 10.1007/s001980070020. [DOI] [PubMed] [Google Scholar]
- Hill EL, Elde R. Distribution of CGRP-, VIP-, D beta H-, SP-, and NPY-immunoreactive nerves in the periosteum of the rat. Cell and tissue research. 1991;264:469–480. doi: 10.1007/BF00319037. [DOI] [PubMed] [Google Scholar]
- Hill EL, Turner R, Elde R. Effects of neonatal sympathectomy and capsaicin treatment on bone remodeling in rats. Neuroscience. 1991;44:747–755. doi: 10.1016/0306-4522(91)90094-5. [DOI] [PubMed] [Google Scholar]
- Honore P, Mantyh PW. Bone cancer pain: from mechanism to model to therapy. Pain medicine. 2000;1:303–309. doi: 10.1046/j.1526-4637.2000.00047.x. [DOI] [PubMed] [Google Scholar]
- Hunt SP. Genes and the dynamics of pain control. Functional neurology. 2009;24:9–15. [PubMed] [Google Scholar]
- Hunter DJ, McDougall JJ, Keefe FJ. The symptoms of osteoarthritis and the genesis of pain. Rheumatic diseases clinics of North America. 2008;34:623–643. doi: 10.1016/j.rdc.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutmacher DW, Sittinger M. Periosteal cells in bone tissue engineering. Tissue engineering, 9 Suppl. 2003;1:S45–64. doi: 10.1089/10763270360696978. [DOI] [PubMed] [Google Scholar]
- Inglis JJ, Nissim A, Lees DM, Hunt SP, Chernajovsky Y, Kidd BL. The differential contribution of tumour necrosis factor to thermal and mechanical hyperalgesia during chronic inflammation. Arthritis research & therapy. 2005;7:R807–816. doi: 10.1186/ar1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanusic JJ, Mahns DA, Sahai V, Rowe MJ. Absence of large-diameter sensory fibres in a nerve to the cat humerus. Journal of anatomy. 2006;208:251–255. doi: 10.1111/j.1469-7580.2006.00519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Bloom AP, Mantyh WG, Koewler NJ, Freeman KT, Delong D, Ghilardi JR, Kuskowski MA, Mantyh PW. Capsaicin-sensitive sensory nerve fibers contribute to the generation and maintenance of skeletal fracture pain. Neuroscience. 2009;162:1244–1254. doi: 10.1016/j.neuroscience.2009.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Ghilardi JR, Castaneda-Corral G, Kuskowski MA, Mantyh PW. Preventive or late administration of anti-NGF therapy attenuates tumor-induced nerve sprouting, neuroma formation, and cancer pain. Pain. 2011;152:2564–2574. doi: 10.1016/j.pain.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Mantyh WG, Bloom AP, Xu H, Ferng AS, Dussor G, Vanderah TW, Mantyh PW. A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone. 2010;46:306–313. doi: 10.1016/j.bone.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Martin CD, Koewler NJ, Freeman KT, Sullivan LJ, Halvorson KG, Barthold CM, Peters CM, Buus RJ, Ghilardi JR, Lewis JL, Kuskowski MA, Mantyh PW. Nerve growth factor sequestering therapy attenuates non-malignant skeletal pain following fracture. Pain. 2007;133:183–196. doi: 10.1016/j.pain.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Kalliomaki M, Kieseritzky JV, Schmidt R, Hagglof B, Karlsten R, Sjogren N, Albrecht P, Gee L, Rice F, Wiig M, Schmelz M, Gordh T. Structural and functional differences between neuropathy with and without pain? Experimental neurology. 2011;231:199–206. doi: 10.1016/j.expneurol.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Karanth SS, Springall DR, Kuhn DM, Levene MM, Polak JM. An immunocytochemical study of cutaneous innervation and the distribution of neuropeptides and protein gene product 9.5 in man and commonly employed laboratory animals. The American journal of anatomy. 1991;191:369–383. doi: 10.1002/aja.1001910404. [DOI] [PubMed] [Google Scholar]
- Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocrine reviews. 2012;33:747–783. doi: 10.1210/er.2011-1060. [DOI] [PubMed] [Google Scholar]
- Kelly S, Dunham JP, Murray F, Read S, Donaldson LF, Lawson SN. Spontaneous firing in C-fibers and increased mechanical sensitivity in A-fibers of knee joint-associated mechanoreceptive primary afferent neurones during MIA-induced osteoarthritis in the rat. Osteoarthritis and cartilage/OARS, Osteoarthritis Research Society. 2012;20:305–313. doi: 10.1016/j.joca.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Kidd BL. Osteoarthritis and joint pain. Pain. 2006;123:6–9. doi: 10.1016/j.pain.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Koewler NJ, Freeman KT, Buus RJ, Herrera MB, Jimenez-Andrade JM, Ghilardi JR, Peters CM, Sullivan LJ, Kuskowski MA, Lewis JL, Mantyh PW. Effects of a monoclonal antibody raised against nerve growth factor on skeletal pain and bone healing after fracture of the C57BL/6J mouse femur. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2007;22:1732–1742. doi: 10.1359/jbmr.070711. [DOI] [PubMed] [Google Scholar]
- Kosaras B, Jakubowski M, Kainz V, Burstein R. Sensory innervation of the calvarial bones of the mouse. The Journal of comparative neurology. 2009;515:331–348. doi: 10.1002/cne.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger L, Silverman JD, Mantyh PW, Sternini C, Brecha NC. Peripheral patterns of calcitonin-gene-related peptide general somatic sensory innervation: cutaneous and deep terminations. The Journal of comparative neurology. 1989;280:291–302. doi: 10.1002/cne.902800210. [DOI] [PubMed] [Google Scholar]
- Kryger GS, Kryger Z, Zhang F, Shelton DL, Lineaweaver WC, Buncke HJ. Nerve growth factor inhibition prevents traumatic neuroma formation in the rat. The Journal of hand surgery. 2001;26:635–644. doi: 10.1053/jhsu.2001.26035. [DOI] [PubMed] [Google Scholar]
- Kuniyoshi K, Ohtori S, Ochiai N, Murata R, Matsudo T, Yamada T, Ochiai SS, Moriya H, Takahashi K. Characteristics of sensory DRG neurons innervating the wrist joint in rats. European journal of pain (London, England) 2007;11:323–328. doi: 10.1016/j.ejpain.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Kurejova M, Nattenmuller U, Hildebrandt U, Selvaraj D, Stosser S, Kuner R. An improved behavioural assay demonstrates that ultrasound vocalizations constitute a reliable indicator of chronic cancer pain and neuropathic pain. Molecular pain. 2010;6:18. doi: 10.1186/1744-8069-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Glazer JM, Corey DP, Rice FL, Stucky CL. TRPA1 modulates mechanotransduction in cutaneous sensory neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:4808–4819. doi: 10.1523/JNEUROSCI.5380-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DK, Dang D, Zhang J, Dolan JC, Schmidt BL. Novel animal models of acute and chronic cancer pain: a pivotal role for PAR2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:14178–14183. doi: 10.1523/JNEUROSCI.2399-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane NE, Schnitzer TJ, Birbara CA, Mokhtarani M, Shelton DL, Smith MD, Brown MT. Tanezumab for the treatment of pain from osteoarthritis of the knee. The New England journal of medicine. 2010;363:1521–1531. doi: 10.1056/NEJMoa0901510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane NE, Silverman SL. Anabolic therapies. Current osteoporosis reports. 2010;8:23–27. doi: 10.1007/s11914-010-0005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. The journal of pain : official journal of the American Pain Society. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Lewiecki EM. Sclerostin: a novel target for intervention in the treatment of osteoporosis. Discovery medicine. 2011;12:263–273. [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Nerve growth factor and nociception. Trends in neurosciences. 1993;16:353–359. doi: 10.1016/0166-2236(93)90092-z. [DOI] [PubMed] [Google Scholar]
- Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2009;24:578–588. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- Lim V, Clarke BL. New therapeutic targets for osteoporosis: beyond denosumab. Maturitas. 2012;73:269–272. doi: 10.1016/j.maturitas.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ma Q. Generation of somatic sensory neuron diversity and implications on sensory coding. Current opinion in neurobiology. 2011;21:52–60. doi: 10.1016/j.conb.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo G, Osikowicz M, Ribeiro-da-Silva A. Sympathetic fiber sprouting in inflamed joints and adjacent skin contributes to pain-related behavior in arthritis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:10066–10074. doi: 10.1523/JNEUROSCI.5784-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes C, Liu Z, Xu Y, Ma Q. Tlx3 and Runx1 act in combination to coordinate the development of a cohort of nociceptors, thermoceptors, and pruriceptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:9706–9715. doi: 10.1523/JNEUROSCI.1109-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck DP. The costs of musculoskeletal disease: health needs assessment and health economics. Best practice & research. Clinical rheumatology. 2003;17:529–539. doi: 10.1016/s1521-6942(03)00023-8. [DOI] [PubMed] [Google Scholar]
- Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, O'Leary P, Mantyh PW. Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience. 2002;113:115–166. doi: 10.1016/s0306-4522(02)00165-3. [DOI] [PubMed] [Google Scholar]
- Mahns DA, Ivanusic JJ, Sahai V, Rowe MJ. An intact peripheral nerve preparation for monitoring the activity of single, periosteal afferent nerve fibres. Journal of neuroscience methods. 2006;156:140–144. doi: 10.1016/j.jneumeth.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Mantyh P. Bone cancer pain: Causes, consequences, and therapeutic opportunities. Pain. 2013 doi: 10.1016/j.pain.2013.07.044. [DOI] [PubMed] [Google Scholar]
- Mantyh WG, Jimenez-Andrade JM, Stake JI, Bloom AP, Kaczmarska MJ, Taylor RN, Freeman KT, Ghilardi JR, Kuskowski MA, Mantyh PW. Blockade of nerve sprouting and neuroma formation markedly attenuates the development of late stage cancer pain. Neuroscience. 2010;171:588–598. doi: 10.1016/j.neuroscience.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CD, Jimenez-Andrade JM, Ghilardi JR, Mantyh PW. Organization of a unique net-like meshwork of CGRP+ sensory fibers in the mouse periosteum: Implications for the generation and maintenance of bone fracture pain. Neuroscience letters. 2007;427:148–152. doi: 10.1016/j.neulet.2007.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell LM, Albers KM, Davis BM. Neurotrophins, nociceptors, and pain. Microscopy research and technique. 1999;45:252–261. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<252::AID-JEMT9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Modder UI, Hoey KA, Amin S, McCready LK, Achenbach SJ, Riggs BL, Melton LJ, 3rd &, Khosla S. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26:373–379. doi: 10.1002/jbmr.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcified tissue international. 2010;87:99–107. doi: 10.1007/s00223-010-9372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Ohtori S, Yamamoto S, Takahashi K, Harada Y. Differences in innervation and innervated neurons between hip and inguinal skin. Clinical orthopaedics and related research. 2008;466:2527–2532. doi: 10.1007/s11999-008-0432-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel JC, Atkinson G, Krieger JN, Mills IW, Pontari M, Shoskes DA, Crook TJ. Preliminary assessment of safety and efficacy in proof-of-concept, randomized clinical trial of tanezumab for chronic prostatitis/chronic pelvic pain syndrome. Urology. 2012;80:1105–1110. doi: 10.1016/j.urology.2012.07.035. [DOI] [PubMed] [Google Scholar]
- O'Connor JP, Capo JT, Tan V, Cottrell JA, Manigrasso MB, Bontempo N, Parsons JR. A comparison of the effects of ibuprofen and rofecoxib on rabbit fibula osteotomy healing. Acta orthopaedica. 2009;80:597–605. doi: 10.3109/17453670903316769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson C. Bone metabolism in 2012: Novel osteoporosis targets. Nature reviews. Endocrinology. 2013;9:72–74. doi: 10.1038/nrendo.2012.252. [DOI] [PubMed] [Google Scholar]
- Ohtori S, Inoue G, Koshi T, Ito T, Yamashita M, Yamauchi K, Suzuki M, Doya H, Moriya H, Takahashi Y, Takahashi K. Characteristics of sensory dorsal root ganglia neurons innervating the lumbar vertebral body in rats. The journal of pain : official journal of the American Pain Society. 2007;8:483–488. doi: 10.1016/j.jpain.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Ohtori S, Orita S, Yamashita M, Ishikawa T, Ito T, Shigemura T, Nishiyama H, Konno S, Ohta H, Takaso M, Inoue G, Eguchi Y, Ochiai N, Kishida S, Kuniyoshi K, Aoki Y, Arai G, Miyagi M, Kamoda H, Suzkuki M, Nakamura J, Furuya T, Kubota G, Sakuma Y, Oikawa Y, Suzuki M, Sasho T, Nakagawa K, Toyone T, Takahashi K. Existence of a neuropathic pain component in patients with osteoarthritis of the knee. Yonsei medical journal. 2012;53:801–805. doi: 10.3349/ymj.2012.53.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, Gong J, Gao Y, Cao J, Graham K, Tipton B, Cai J, Deshpande R, Zhou L, Hale MD, Lightwood DJ, Henry AJ, Popplewell AG, Moore AR, Robinson MK, Lacey DL, Simonet WS, Paszty C. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25:948–959. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Aoki Y, Ohtori S, Takahashi K, Chiba T, Ino H, Moriya H. The dorsal portion of the lumbar intervertebral disc is innervated primarily by small peptide-containing dorsal root ganglion neurons in rats. Neuroscience letters. 2003;344:65–67. doi: 10.1016/s0304-3940(03)00416-6. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Ohtori S, Inoue G, Aoki Y, Moriya H, Takahashi K. The degenerated lumbar intervertebral disc is innervated primarily by peptide-containing sensory nerve fibers in humans. Spine. 2006;31:2418–2422. doi: 10.1097/01.brs.0000239159.74211.9c. [DOI] [PubMed] [Google Scholar]
- Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- Palaniswamy C, Selvaraj DR, Rao V, Patel U. Newer therapies for osteoporosis. American journal of therapeutics. 2010;17:197–200. doi: 10.1097/MJT.0b013e3181a6ee1c. [DOI] [PubMed] [Google Scholar]
- Pare M, Albrecht PJ, Noto CJ, Bodkin NL, Pittenger GL, Schreyer DJ, Tigno XT, Hansen BC, Rice FL. Differential hypertrophy and atrophy among all types of cutaneous innervation in the glabrous skin of the monkey hand during aging and naturally occurring type 2 diabetes. The Journal of comparative neurology. 2007;501:543–567. doi: 10.1002/cne.21262. [DOI] [PubMed] [Google Scholar]
- Paszty C, Turner CH, Robinson MK. Sclerostin: a gem from the genome leads to bone-building antibodies. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2010;25:1897–1904. doi: 10.1002/jbmr.161. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Current opinion in neurobiology. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Peltonen M, Lindroos AK, Torgerson JS. Musculoskeletal pain in the obese: a comparison with a general population and long-term changes after conventional and surgical obesity treatment. Pain. 2003;104:549–557. doi: 10.1016/S0304-3959(03)00091-5. [DOI] [PubMed] [Google Scholar]
- Pepper A, Li W, Kingery WS, Angst MS, Curtin CM, Clark JD. Changes resembling complex regional pain syndrome following surgery and immobilization. The journal of pain : official journal of the American Pain Society. 2013;14:516–524. doi: 10.1016/j.jpain.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara A. The noradrenergic pain regulation system: a potential target for pain therapy. European journal of pharmacology. 2013;716:2–7. doi: 10.1016/j.ejphar.2013.01.067. [DOI] [PubMed] [Google Scholar]
- Peters EM, Botchkarev VA, Muller-Rover S, Moll I, Rice FL, Paus R. Developmental timing of hair follicle and dorsal skin innervation in mice. The Journal of comparative neurology. 2002;448:28–52. doi: 10.1002/cne.10212. [DOI] [PubMed] [Google Scholar]
- Petruska JC, Mendell LM. The many functions of nerve growth factor: multiple actions on nociceptors. Neuroscience letters. 2004;361:168–171. doi: 10.1016/j.neulet.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Reid IR. Osteoporosis treatment at ASBMR 2012. IBMS BoneKEy. 2012;9 [Google Scholar]
- Ren K, Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Current opinion in anaesthesiology. 2008;21:570–579. doi: 10.1097/ACO.0b013e32830edbdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DA, Calejesan AA, Wei F, Gebhart GF, Zhuo M. Endogenous facilitation: from molecular mechanisms to persistent pain. Current neurovascular research. 2004;1:11–20. doi: 10.2174/1567202043480189. [DOI] [PubMed] [Google Scholar]
- Roosterman D, Goerge T, Schneider SW, Bunnett NW, Steinhoff M. Neuronal control of skin function: the skin as a neuroimmunoendocrine organ. Physiological reviews. 2006;86:1309–1379. doi: 10.1152/physrev.00026.2005. [DOI] [PubMed] [Google Scholar]
- Rosenberg JM, Harrell C, Ristic H, Werner RA, de Rosayro AM. The effect of gabapentin on neuropathic pain. The Clinical journal of pain. 1997;13:251–255. doi: 10.1097/00002508-199709000-00011. [DOI] [PubMed] [Google Scholar]
- Sabsovich I, Wei T, Guo TZ, Zhao R, Shi X, Li X, Yeomans DC, Klyukinov M, Kingery WS, Clark JD. Effect of anti-NGF antibodies in a rat tibia fracture model of complex regional pain syndrome type I. Pain. 2008;138:47–60. doi: 10.1016/j.pain.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakthiswary R, Das S. The Effects of TNFalpha Antagonist Therapy on Bone Metabolism in Rheumatoid Arthritis: A Systematic Review. Current drug targets. 2013 doi: 10.2174/13894501113149990176. [DOI] [PubMed] [Google Scholar]
- Schaible HG. Mechanisms of chronic pain in osteoarthritis. Current rheumatology reports. 2012;14:549–556. doi: 10.1007/s11926-012-0279-x. [DOI] [PubMed] [Google Scholar]
- Schulze E, Witt M, Fink T, Hofer A, Funk RH. Immunohistochemical detection of human skin nerve fibers. Acta histochemica. 1997;99:301–309. doi: 10.1016/S0065-1281(97)80024-4. [DOI] [PubMed] [Google Scholar]
- Schweizerhof M, Stosser S, Kurejova M, Njoo C, Gangadharan V, Agarwal N, Schmelz M, Bali KK, Michalski CW, Brugger S, Dickenson A, Simone DA, Kuner R. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nature medicine. 2009;15:802–807. doi: 10.1038/nm.1976. [DOI] [PubMed] [Google Scholar]
- Seeman E. Pathogenesis of bone fragility in women and men. Lancet. 2002;359:1841–1850. doi: 10.1016/S0140-6736(02)08706-8. [DOI] [PubMed] [Google Scholar]
- Seike W. Electrophysiological and histological studies on the sensibility of the bone marrow nerve terminal. Yonago acta medica. 1976;20:192–211. [PubMed] [Google Scholar]
- Silverman JD, Kruger L. Calcitonin-gene-related-peptide-immunoreactive innervation of the rat head with emphasis on specialized sensory structures. The Journal of comparative neurology. 1989;280:303–330. doi: 10.1002/cne.902800211. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Pollock M, Facci L. Mast cells differentially express and release active high molecular weight neurotrophins. Brain research. Molecular brain research. 2001;97:177–185. doi: 10.1016/s0169-328x(01)00314-x. [DOI] [PubMed] [Google Scholar]
- Smith ES, Lewin GR. Nociceptors: a phylogenetic view. Journal of comparative physiology. A, Neuroethology, sensory, neural, and behavioral physiology. 2009;195:1089–1106. doi: 10.1007/s00359-009-0482-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stovitz SD, Pardee PE, Vazquez G, Duval S, Schwimmer JB. Musculoskeletal pain in obese children and adolescents. Acta paediatrica. 2008;97:489–493. doi: 10.1111/j.1651-2227.2008.00724.x. [DOI] [PubMed] [Google Scholar]
- Su B, O'Connor JP. NSAID Therapy Effects on Healing of Bone, Tendon, and the Enthesis. Journal of applied physiology. 2013 doi: 10.1152/japplphysiol.00053.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri S, Gill SE, Massena de Camin S, Wilson D, McWilliams DF, Walsh DA. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Annals of the rheumatic diseases. 2007;66:1423–1428. doi: 10.1136/ard.2006.063354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Toda K. Subtypes of nociceptive units in the rat temporomandibular joint. Brain research bulletin. 2003;61:603–608. doi: 10.1016/s0361-9230(03)00219-3. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Peleshok JC, Ribeiro-da-Silva A. Distribution of P2X(3)-immunoreactive fibers in hairy and glabrous skin of the rat. The Journal of comparative neurology. 2009;514:555–566. doi: 10.1002/cne.22048. [DOI] [PubMed] [Google Scholar]
- Vincent L, Vang D, Nguyen J, Gupta M, Luk K, Ericson ME, Simone DA, Gupta K. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. 2013;122:1853–1862. doi: 10.1182/blood-2013-04-498105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voscopoulos C, Lema M. When does acute pain become chronic? British journal of anaesthesia, 105 Suppl. 2010;1:i69–85. doi: 10.1093/bja/aeq323. [DOI] [PubMed] [Google Scholar]
- Wang Q, Seeman E. Skeletal growth and peak bone strength. Best practice & research. Clinical endocrinology & metabolism. 2008;22:687–700. doi: 10.1016/j.beem.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Wetzel CH, Connelly JF. Use of gabapentin in pain management. The Annals of pharmacotherapy. 1997;31:1082–1083. doi: 10.1177/106002809703100920. [DOI] [PubMed] [Google Scholar]
- Woolf AD, Erwin J, March L. The need to address the burden of musculoskeletal conditions. Best practice & research. Clinical rheumatology. 2012;26:183–224. doi: 10.1016/j.berh.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Woolf AD, Pfleger B. Burden of major musculoskeletal conditions. Bulletin of the World Health Organization. 2003;81:646–656. [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152:S2–15. doi: 10.1016/j.pain.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Wall PD. Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1986;6:1433–1442. doi: 10.1523/JNEUROSCI.06-05-01433.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Nagata K, Iijima T. Involvement of sensory nerves and immune cells in osteophyte formation in the ankle joint of adjuvant arthritic rats. Histochemistry and cell biology. 2002;118:213–220. doi: 10.1007/s00418-002-0443-x. [DOI] [PubMed] [Google Scholar]
- Ye Y, Dang D, Zhang J, Viet CT, Lam DK, Dolan JC, Gibbs JL, Schmidt BL. Nerve growth factor links oral cancer progression, pain, and cachexia. Molecular cancer therapeutics. 2011;10:1667–1676. doi: 10.1158/1535-7163.MCT-11-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida W, Seymour B, Koltzenburg M, Dolan RJ. Uncertainty increases pain: evidence for a novel mechanism of pain modulation involving the periaqueductal gray. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:5638–5646. doi: 10.1523/JNEUROSCI.4984-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn PK, Subieta A, Park SS, Brennan TJ. Effect of blockade of nerve growth factor and tumor necrosis factor on pain behaviors after plantar incision. The journal of pain : official journal of the American Pain Society. 2004;5:157–163. doi: 10.1016/j.jpain.2004.02.538. [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. The EMBO journal. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Rice FL, Anderson DJ. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron. 2005;45:17–25. doi: 10.1016/j.neuron.2004.12.015. [DOI] [PubMed] [Google Scholar]