

Abstract
Objectives
Spinal muscular atrophy (SMA) is caused by reduced levels of SMN which results in motoneuron loss. Therapeutic strategies to increase SMN levels including drug compounds, antisense oligonucleotides or scAAV9 gene therapy have proved effective in mice. We wished to determine whether reduction of SMN in postnatal motoneurons resulted in SMA in a large animal model, whether SMA could be corrected after development of muscle weakness and the response of clinically relevant biomarkers.
Methods
Using intrathecal delivery of scAAV9 expressing a shRNA targeting pig SMN1, SMN was knocked down in motoneurons postnatally to SMA levels. This resulted in an SMA phenotype representing the first large animal model of SMA. Restoration of SMN was performed at different time points with scAAV9 expressing human SMN (scAAV9-SMN) and electrophysiology measures and pathology were performed.
Results
Knockdown of SMN in postnatal motoneurons results in overt proximal weakness, fibrillations on electromyography (EMG) indicating active denervation, and reduced compound muscle action potential (CMAP) and motor unit number estimates (MUNE), like human SMA. Neuropathology showed loss of motoneurons and motor axons. Pre-symptomatic delivery of scAAV9-SMN prevented SMA symptoms indicating all changes are SMN dependent. Delivery of scAAV9-SMN after symptom onset had a marked impact on phenotype, electrophysiological measures and pathology.
Interpretation
High SMN levels are critical in postnatal motoneurons and reduction of SMN results in a SMA phenotype which is SMN dependent. Importantly, clinically relevant biomarkers including CMAP and MUNE are responsive to SMN restoration and abrogation of phenotype can be achieved even after symptom onset.
Introduction
Spinal muscular atrophy (SMA) is a common genetic cause of infant death with an incidence of 1:10,000 live births 1–3. SMA is an autosomal recessive disorder caused by loss or mutation of the survival motor neuron 1 gene (SMN1) and retention of SMN2 resulting in low levels of the ubiquitous Survival Motor Neuron (SMN) protein 4–7. However, only motoneurons degenerate and this leads to proximal muscle weakness6, 8. SMA can be classified into 5 types based on the severity and onset of symptoms6, 9. In all cases, there is extensive evidence of motoneuron denervation based on muscle pathology and electromyography10, 11. Autopsies of type 1 SMA patients have indicated some classic neuropathological features that consist of a marked loss of motoneurons in the anterior horn of the spinal cord and loss of ventral roots axons12.
The SMN2 gene is only present in humans and other species have a single SMN gene, thus naturally occurring mutations in large animals are unlikely to occur as loss or mutation of SMN is embryonic lethal13, 14. In the Smn knockout mouse, the SMN2 gene was introduced to model the human condition, and these mice have been extensively used to develop treatments for SMA15–17. Antisense oligonucleotides (ASO) and orally available drug compounds can be used to block negative regulators of splicing increasing the amount of full-length SMN mRNA produced from SMN218–21. Remarkably, a single administration of ASO has a marked impact on survival of SMA mice21, 22. In a similar manner, self-complementary Adeno-Associated Virus containing SMN (scAAV9-SMN) delivered either via the blood stream or via the cerebrospinal fluid (CSF) has a major impact on survival of SMA mice23–27. Clinical trials for both ASOs and scAAV9-SMN in humans have started. There are a number of unresolved questions in these trials including whether correction can be obtained when symptoms are present, what is expected of electrophysiological biomarker measures and the role of peripheral SMN on other organs. To fully evaluate these factors a large animal model of SMA with correction is needed. Rodent models have a relative permeable blood brain barrier at early time points and distinct phenotypes such as tail necrosis and cardiac defects that are not associated with the human pathology and thus cannot give the required information 17, 28, 29. Models of SMA can be created by knockdown of SMN and in this case it is important to consider the degree of knockdown. Interestingly, in motoneurons or spinal cord of SMA mice the range of knockdown of full length SMN mRNA is 75–80% 30 and this compares to what is observed in human total spinal cord with approximately 78% knockdown of full length SMN31. Therefore we can assume that knockdown of ~80% of SMN would be necessary to generate a large animal model of SMA.
We show here that a large animal model of SMA in the pig is created by knockdown of SMN in postnatal motoneurons and that a marked correction of this phenotype occurs with reintroduction of SMN in the symptomatic phase. Furthermore, electrophysiological measures utilized in the clinic show improvement and give a reliable correlation to function. However the degree of recovery is tied to the severity of the animal and motor neuron number estimates do not fully recover, thus early symptomatic intervention is preferable in the design of clinical trials.
Material and Methods
Cells
Pig aorta-derived endothelial cells (PEDSV15) and Hela cells were maintained in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum, 1% L-glutamine and 1% penicillin/streptomycin. PEDSV15 cells were infected with a lentivirus expressing pig shRNA (shRNA1, 5′ ggaaataagccaaataaca 3′, shRNA2, 5′ gggaataatcagtttgaat 3′) under control of the H1 promoter. Cells were harvested 5 days post-infection and lysates analyzed for SMN levels by western blot.
Hela cells were transfected with plasmid pSUPuro containing the shRNAs being tested using Lipofectamine 2000. RNA were extracted 48 hours later using Trizol (Invitrogen) according to the manufacturer’s protocol and treated with RNase-free DNase (Ambion). cDNA was prepared using AMV Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions.
Vectors
shRNA1 sequence targeting pig SMN (or scrambled shRNA) under control of the H1 promoter was cloned into a self-complementary (sc) AAV9-based backbone plasmid along with reporter gene GFP under the CBA promoter. Human SMN cDNA under the CBA promoter was cloned into the same backbone plasmid as previously described24. ScAAV9-shRNA and -SMN vectors were produced by tri-transfection of HEK293 cells as previously described24. Vector titers were determined using Taqman PCR and expressed as viral genome (vg) per ml.
Intrathecal Injection
Farm-bred sows (Sus scrofa domestica) were obtained from a regional farm (Hartley Farm, OH). Five-day old (post-natal day 5, PND5) piglets were induced and maintained under anesthesia by mask inhalation of 5% isoflurane in oxygen. Body temperature, electrocardiogram and respiratory rate were monitored throughout the procedure. Piglets were injected with scAAV9-shRNA and/or scAAV9-SMN in the cisterna magna as previously described 32. Briefly, a 22-gauge needle was passed immediately caudal to the occipital bone and a flash of clear CSF confirmed entry into the cistern magna. A 1cc syringe was attached and Omnipaque™ administered to confirm delivery into the CSF. This syringe was removed while the needle was held in place and a 3 cc syringe containing a mixture of Omnipaque™ and scAAV9 virus was attached to the needle. The virus mixture was manually injected at a slow and constant rate. After delivery, approximately 0.25ml of Omnipaque™ was flushed through the spinal needle to ensure full delivery of the reagent. X-ray images were collected throughout the entire injection procedure using fluoroscopy. PND33-36 animals were injected following the same above procedure but anesthesia was induced with 0.15mg Telazol. All animals that were injected with scAAV9 vectors were treated with the immunosupressant Prograf (Tacrolimus) 0.1mg/kg and Cellcept 25mg/kg for a week after the initial injection or until the second injection at PND33 in the case of the treated symptomatic group.
Nerve conduction study and EMG
Electrophysiological measurements were performed by a single blinded investigator (W. D. A.). The animals were induced with Telazol and maintained under anesthesia using inhaled isoflurane. Animals were placed in a side-lying position and oxygen flow and isoflurane were adjusted to maintain adequate sedation. Surface temperature was maintained between 34–37°C. Hair was removed from the lateral and posterior portions of the left hind limb with clippers. Sciatic motor nerve conduction studies and needle electromyography (EMG) were performed using a portable clinical electrodiagnostic system (Synergy; Natus Neurology, Middleton, WI). Sciatic compound muscle action potential (CMAP) and motor unit number estimation (MUNE) were recorded from the left hind limb while stimulating the sciatic nerve. The electrode placement for the CMAP and MUNE recordings is pictured in Fig 4B. The low pass filter was set at 20 Hz and the high-pass filter at 10 kHz. A pair of insulated 28 gauge monopolar needles (Teca, Oxford Instruments Medical, NY) were used as the cathode and anode to stimulate the sciatic nerve at the proximal hind limb. The cathode was inserted at the region of the proximal hind limb and the anode was inserted more proximally in the subcutaneous tissue overlying the lumbar paraspinal muscles. A pair of 10 mm disc electrodes was used for recording (Teca, Oxford Instruments Medical, NY). The active (E1) disc electrode was placed on the skin overlying the mid-belly region of the biceps femoris muscle and the reference (E2) disc electrode was placed on the skin over the lateral malleolus. In order to reduce impedance, the disk electrodes were coated with electrode gel (Spectra 360 by Parker laboratories, Fairfield, NJ). A disposable disc electrode (Carefusion, Middleton, WI) was placed on the left hind limb between the cathode and active electrode. To obtain the CMAP response, a supramaximal stimulation was delivered to the sciatic nerve using single square-wave pulses of 0.1–0.2 ms duration and a stimulus intensity ranging from 10–100 mA. The CMAP amplitudes of the responses were measured baseline-to-peak and peak-to-peak and stored.
MUNE was performed using an incremental technique maintaining the placement of the stimulating, recording, and ground electrodes utilized for the sciatic CMAP 33–35. Submaximal stimulation at 0.1 ms duration was delivered to obtain a minimal all-or-none response. Then 9 additional incremental responses were obtained by gradually increasing the stimulus intensity. Incremental responses were required to be stable and without fractionation, established by observing three duplicate responses. Additionally each response was required to increase in voltage in both the negative and positive directions on comparison to the prior response to ensure the bulk of the response was originating from beneath active electrode and not volume conduction from a more distant sciatic-innervated muscle. The amplitude of each response was calculated peak-to-peak by subtracting the peak-to-peak amplitude of the prior response. The minimum amplitude for each incremental response allowed was 25 μV. The 10 incremental values were then averaged to give an estimation of the average single motor unit potential (SMUP) amplitude. MUNE was calculated by dividing the maximum CMAP amplitude (peak-to-peak) by the average SMUP amplitude (peak-to-peak).
Needle electromyography (EMG) was performed to assess for abnormal spontaneous activity. A 30 gauge concentric needle electrode (Natus, San Carlos, CA) was inserted into the left biceps femoris, gluteus medius, lumbar paraspinal, and trapezius muscles. A disposable disc electrode (Carefusion, Middleton, WI) was placed on the left hind limb. Settings for the high and low frequency filters were set at 20 Hz to 10 kHz, respectively. Needle electrode insertions into four quadrants of each muscle were used to trigger the occurrence of fibrillation potentials. Fibrillation potentials were judged to be absent or present in each muscle as determined by the identification of spontaneous potentials with characteristics of a single muscle fiber action potential source (having either a morphology of a fibrillation or positive sharp wave) and regular firing rate (Fig 4C) 36.
Figure 4. scAAV9-shSMN leads to SMA-like clinical symptoms in piglets.

(A) Time course of symptoms and progression of phenotype. (B) Recording set up for the measurement of the sciatic CMAP and MUNE responses. 1, recording electrodes E1 and E2; 2, anode and 3, cathode stimulating electrodes; 4, ground electrode. (C) Representative EMG recording from a control and a scAAV9-shSMN injected animal showing fibrillations with positive wave morphology. (D) CMAP and MUNE at PND54 were significantly reduced in scAAV9-shSMN animals (N=4) compared to the non-injected control group (N=6). The CMAP and MUNE values were preserved in the treated pre-symptomatic group that received scAAV9-shSMN and scAAV9-SMN vector 24hrs apart and the values were not significantly different from those of the unaffected controls. ** P<0.01
Perfusion and tissue-processing
Piglets received an intramuscular injection of 50 mg Telazol and were maintained under deep anesthesia by mask inhalation of 5% isoflurane in oxygen. Animals were perfused with 1 to 1.5L of 0.1M phosphate buffer (pH 7.4). Spinal cord was dissected out and alternating segments were flash frozen in LN2 or post-fixed for 48 hours in 4% paraformaldehyde (PFA). Flash frozen spinal cord samples were stored at −80°C until further processing. Following fixation in 4% PFA, samples were transferred in 0.1M phosphate buffer and serial 40μm spinal cord sections were collected using a Leica VT1200 vibrating blade microtome for histology analysis and stored in 0.1M phosphate buffer at 4°C.
For the biodistribution of AAV vector genome, biopsy samples from various organs including the brain were flash frozen in LN2 and genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instruction.
Western blot
PEDSV.15 cells were detached using Trypsin/EDTA, washed once with PBS 1x and lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and completed with protease inhibitor). Protein lysates were loaded onto a 10% polyacrylamide gel and the separated protein were transferred to a PVDF-FL membrane (Millipore) using a semi dry transfer. The membrane was probed with the following primary antibodies prepared in Licor blocking buffer: anti-human SMN mouse monoclonal antibody (7B10 from immunoGlobe) 1:1000, Lamin A/C (636) (sc-7292, Santa cruz) 1:1000.
Frozen sections from spinal cord L4 segment of 3 scAAV9-shSMN and 3 control animals were obtained using a cryostat and protein extracted using SDS blending buffer (5% SDS, 62.5 mM Tris pH 6.8, 5 mM EDTA). 25 μg of protein was loaded onto a 12% SDS polyacrylamide gel. The separated proteins were transferred to a nitrocellulose membrane and blocked for 1 hour in Odyssey Blocking buffer (LI-COR Biosciences). The membrane was then incubated overnight with primary antibodies prepared in Odyssey Blocking buffer: mouse anti-SMN (MANSMA) 1:100 and monoclonal mouse anti-β-actin (A5441, Sigma) 1:50,000. All blots were incubated with a goat anti-mouse IRDye 800CW antibody (926-32210, LI-COR), 1:10,000. For all quantitative western blots, detection was performed using the LI-COR Odyssey Imaging System (Biosciences) and quantification was determined using Odyssey Infrared Imaging System Application Software (Biosciences).
cDNA preparation from LCM-collected material
14 μm sections from flash frozen lumbar spinal cord were collected onto PEN membrane slides (Zeiss), fix for 1 minute in methanol, stained with 1% cresyl violet in methanol for 1 minute 30 second, quickly rinsed in methanol and immediately stored at −80°C for no more than a week. Laser capture microdissection (LCM) was performed with the Palm Microbeam IV (Carl Zeiss MicroImaging) under 10x magnification. Motoneurons were identified based on their localization in the ventral horn and morphology (large cell body). Approximately 2,000,000 μm2 of motoneuron tissue was collected per sample. In addition, 2,000,000 μm2 of dorsal horn tissue was collected from the same sections. RNA was isolated using the RNaqueous Micro Kit (Ambion) according to the manufacturer’s instructions and cDNA was obtained using AMV-RT (Invitrogen) as described by manufacturer.
Droplet digital PCR
For pig and human SMN mRNA analysis, the following primers and probe were used: pig SMN Fd 5′-ggcggcagcggtgtt-3′, pig SMN Rv 5′-gaatcatcactctggcctgca-3′, pig SMN MGB probe 5′-ctgaggcggaggact-3,. human SMN Fd 5′-gtttcagacaaaatcaaaaagaagga-3′, human SMN Rv 5′-tctataacgcttcacattccagatct-3′, human SMN MGB probe 5′-atgccagcatttctccttaatttaagg-3′, pig GAPDH fd 5′-ccccaacgtgtcggttgt-3′, pig GAPDH Rv 5′-cctgcttcaccaccttcttga-3′, pig GAPDH MGB probe 5′-agaaacctgcaaaata-3′.
For biodistribution study using gDNA, the following primers and probes were used: GFP Fd 5′-cactacctgagcacccagtc-3′, GFP Rv 5′-tccagcaggaccatgtgatc-3′, GFP MGB probe 5′-tgagcaaagaccccaacgagaagcg-3′, GAPDH Fd 5′-ccccaacgtgtcggttgt-3′, GAPDH Rv 5′-caaagctgttccaacccaaac-3′, GAPDH MGB probe 5′-atctgacctgccgcctg-3′ and the human SMN primers/probe set described above.
Reactions were done in triplicate as a multiplex with 2 μl of cDNA, 1.6 μl of GFP, human or pig SMN (800 nM final) forward and reverse primers, 1.6 μl of GAPDH forward and reverse primers, 0.5 μl of each appropriate probe (250nM final) and 10 μl of 2x ddPCR supermix in 20 μl final volume. Samples were converted into droplets with the QX100 droplet generator (Bio-Rad) and run on a classic thermal cycler under standard conditions: 95°C for 10 min followed by 40 cycles of 94°C for 30 sec, 60°C for 1 min and a final step of 98°C for 10 min. After PCR, the plate was loaded onto the QX100 droplet digital reader (Bio-Rad) and results analyzed using the QuantaSoft software (Bio-Rad).
Histology
For immunofluorescence labeling, 40 μm spinal cord sections were stained as floating sections. Dorsal roots (DR) and ventral roots (VR) were dissected from postfixed spinal cord segments, incubated in 15% sucrose for 1 week and embedded in O.C.T. compound frozen in LN2-cooled isopentane. Samples were cut with a cryostat at 8μm thickness. Sections from spinal cord, DR and VR were blocked for 1 hour in blocking solution (Tris-Buffer Saline, TBS, containing 10% donkey serum and 1% triton X-100) at room temperature. Primary antibodies were diluted in blocking solution as follows: rabbit anti-GFP 1:500 (A6455, Invitrogen), goat anti-ChAT 1:100 (AB144P, Millipore), mouse monoclonal anti-SMN 1:10 (SMN-KH,19), mouse anti-neurofilament 1:5,000 (MAB5254, Millipore). The SMN-KH antibody preferentially detects human SMN. In our hands SMN-KH was found to react weakly with pig SMN and not at all with mouse SMN. Sections were incubated with primary antibodies for 24 to 72 hours at 4°C and then washed 3 times over 10 min with TBS. Sections were then incubated with appropriate secondary antibodies in blocking solution as follows: donkey anti-rabbit A488 1:1000 (Invitrogen), donkey anti-mouse A555 1:1000 (Invitrogen), donkey anti goat A555 1:1000 (Invitrogen), donkey anti-goat A647 1:1000 (Invitrogen). Sections were washed with TBS 3 times over 10 min and mounted with Fluoromount G. All images were captured on a Zeiss laser-scanning microscope or Olympus BX61 fluorescence microscope equipped with a Hamamatsu ORCA-ER digital camera.
For cresyl violet staining, 40 μm floating sections were mounted onto slides and air dried for 24 hours before staining. Slides were submerged in increasing dilutions of ethanol to remove fat, rehydrated for staining with 0.5% cresyl violet acetate, followed by dehydratation in successive graded dilutions of ethanol and cleared in Histo-Clear. Sections were mounted with Permount and images capture with a Zeiss Axioscop light microscope.
Dorsal roots and ventral roots thick section (1 μm) were obtained from the Campus Microscopy and Imaging facility at OSU. Briefly, samples were dissected out of the lumbar 4 segment of the spinal cord and cut into small pieces of ~2 mm cubed, fixed in 4% PFA for 24 hours and transferred to 0.1M phosphate buffer. Tissues were post-fixed in 1% osmium tetroxide, dehydrated, infiltrated with resin, embedded and sectioned. Thick sections were stained with Toluidine blue and images captured with an Olympus microscope.
Motoneuron and motor axon quantification
For cresyl violet motoneuron counts, 5 to 6 serial 40 μm thick sections from lumbar 4 (L4) spinal cord segments separated by 800μm were stained as described above. Spinal cord sections of 4 or 5 animals per group were examined. Images of each ventral horn region were captured with an Axioscop light microscope equipped with an Olympus camera DP71 and motoneurons were manually counted by a blinded operator. Motoneurons were identified based on their location in the ventral horn, morphology as well as presence of Nissl-positive staining in the cytoplasm. For immunostained motoneurons counting, 10 to 12 serial sections stained as described above were used and captured with an Olympus BX61 fluorescence microscope equipped with a Hamamatsu ORCA-ER digital camera. Images were processed with the free NIH imageJ software.
For axonal count from Toluidine stain section, the whole ventral root area was captured with a Zeiss Axioscop light microscope equipped with an Olympus camera DP71 and images analyzed with the ImageJ software. GFP-positive axon counts were performed on double labeled ventral and dorsal roots sections as described above. Three representative fields were captured at 20x with a Zeiss laser-scanning confocal microscope and the total number of axons and GFP-positive axons analyzed with the ImageJ software.
Statistics
Statistical analyses were performed using Sigma Plot 12.0. Values are shown as the mean ± s.e.m. and statistical significance difference was set at P<0.05. We used a one way ANOVA for motoneuron, axon counts and student t-test for RNA analysis. The motoneuron and axon counts were recorded by an investigator (Xiaohui Li) blinded to the treatment group of the animal. We used power analysis to calculate the sample size of animals to use in this study. Power calculations were performed for the fibrillation contingency table (Table S1) data using GraphPad and the consideration that fibrillations do not occur in controls and will be uncommon (1) in treated animals. The power was 90% with 3 animals in each group. In the case of ANOVA analysis of CMAP and MUNE data, the power was determined in SYSTAT13. The CMAP deviation was set as 4 and CMAP mean of 20 for controls and 7 for knockdown animals with restoration in treated (pre-symptomatic and symptomatic) animals at 17. For MUNE data, the standard deviation was set at 70 and the MUNE for controls was 380 and 133 for knockdown animals, treated animals (pre-symptomatic and symptomatic) were considered to return to close to control at 224. Under these conditions, 4 animals in each group gave 99% power for MUNE and 100% power for CMAP. The differences in mean motoneuron and axon counts between either control and knockdown or knockdown and treated was analyzed using a student t-test and had powers above 90% in all instances. As the control and treated animals have very similar numbers of motoneurons and motor axons, there is not sufficient difference in the means to reliably indicate a difference between these groups. The RNA analysis was also performed with a student t-test and indicated a power above 99%. The recorder of CMAP and MUNE data was blinded to the pig treatment group at all intervals (W. D. A.). The animals were assigned to treatment groups at the time of injection by Sandra Duque.
Study approval
All animals procedures performed were in accordance to The Ohio State University Institutional Animal Care and Use Committees (IACUC).
Results
scAAV9-shSMN efficiently knockdowns pig SMN1
We first determined the efficiency of two shRNAs to knockdown pig SMN in vitro and western blot analysis showed that SMN was reduced by > 75% (Fig 1A). The SMN:shRNA1 (referred to as shSMN hereafter) was the most consistent in knocking down SMN over multiple transductions of PEDSV15 cells and was selected. Given the strong sequence homology of pig and human SMN (Fig 1B), we tested the specificity of shSMN for pig SMN in Hela cells. Human SMN mRNA levels were not affected by pig shSMN (Fig 1C). Based on these results, the shSMN construct was cloned into a self-complementary (sc) AAV plasmid backbone containing the reporter gene GFP under control of the CBA promoter, and scAAV9-shSMN vectors were produced (Fig 1D).
Figure 1. Design of the shRNA construct targeting pig SMN.
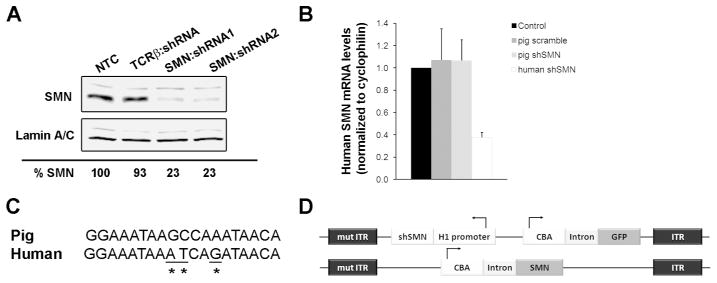
(A) Western blot from lysates of pig aorta-derived endothelial (PEDSV.15) cells collected 5 days after lentiviral infection showed effective knockdown of pig SMN with construct shRNA1 and shRNA2, targeting exon 4 and 8 respectively. TCRβ:shRNA encodes a shRNA targeting T cell receptor beta chain. (B) Sequence of the shRNA1 (hereafter referred to as shSMN) binding site in exon 4 of pig SMN aligned to the human sequence (C) Quantification of human SMN mRNA levels (hSMN) from transiently transfected Hela cells showing that pig shSMN does not affect human SMN. A shRNA targeting human SMN (human shRNA) was used as a positive control and shows significant decrease of human SMN levels (P=0.015). (D) Maps of the scAAV9-shSMN and scAAV9-SMN vectors used in this study. The shSMN is transcribed from the human promoter H1 and was cloned into a scAAV vector along with the GFP reporter gene under the chicken β-actin promoter. ITR, Inverted Terminal Repeats; mutITR, mutated ITR; NTC, non-transfected cells.
We and others previously demonstrated the potential of the scAAV9 vector to efficiently transduce motoneurons in piglets after a single intrathecal injection 32, 37. We administered scAAV9-shSMN or a scAAV9 vector expressing a scrambled shRNA into the cisterna magna of five-day old piglets (Fig 2A and Table 1). As an additional control group, some scAAV9-shSMN injected animals received a second intrathecal injection 24 hours later with the rescue vector scAAV9-SMN (Treated pre-symptomatic group, Table 1). Six to ten weeks following the injection, we performed immunofluorescence on lumbar spinal cord sections and observed robust transduction of the motoneurons (Fig 2B)32. Quantification of GFP-positive axons from the corresponding lumbar ventral root allowed us to estimate the number of motoneurons transduced. Up to 78% of the motor axons were found to be GFP-positive. (Fig 2B, C). Sensory neurons in the dorsal horn and dorsal root ganglia were also highly transduced (Fig 2C). However motoneurons, especially in the lumbar region, were the main GFP-expressing cells in all injected piglets.
Figure 2. scAAV9-shSMN efficiently knockdown pig SMN in vivo.
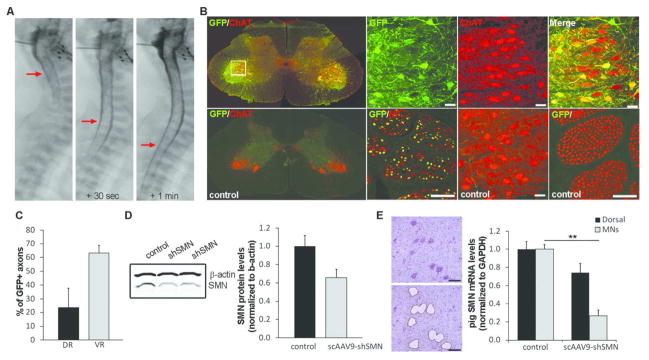
(A) X-ray images taken during the intrathecal injection procedure. AAV vector was mixed with Omnipaque™ and this mixture injected in the cisterna magna of a 5-day old piglet (left panel). Within seconds, the dye is observed in the lower segment of the spinal cord (middle and right panels, red arrows) indicating proper delivery in the CSF. (B) Distribution of the scAAV9-shSMN vector in the lumbar region (L4) of the spinal cord was analyzed using GFP and ChAT (Choline Acetyl Transferase, motoneuron marker) staining. As previously published, scAAV9 mediates robust transduction of motoneurons in the pig32. Lumbar ventral roots stained for GFP and NF (neurofilament) confirmed high transduction of the motor axons. (C) Axon counts from lumbar dorsal root (DR) and ventral root (VR) of scAAV9-shSMN injected animals was performed on sections stained for GFP and NF (N=5) and show an average of 63% of GFP-positive motor axons. Sensory axons were also transduced but at a lower efficiency (24%) (D) Pig SMN protein levels in lumbar spinal cord of scAAV9-shSMN injected animals and non-injected controls were analyzed by western blot (N=3). Although western blot quantification indicates a decrease in SMN protein levels in shSMN-injected animals, no statistical difference was observed between the 2 groups. (E) Lumbar motoneurons were laser-capture microdissected and pig mRNA levels from scAAV9-shSMN injected animals and non-injected controls analyzed using ddPCR. ScAAV9-shSMN injected animals showed a 73±6% (** P<0.001 with standard T-test and P=0.008 with Mann-Whitney Rank Sum Test) and 26±10% reduction in pig SMN mRNA in the motoneurons and dorsal horn respectively compared to controls. Scale bar: 100μm
Table 1.
Summary of the experimental treatment groups used in this study.
| Group | # of animal | scAA9-shSMN vector | scAAV9-SMN vector | ||
|---|---|---|---|---|---|
| Injected at | Dose | Injected at | Dose | ||
| Control | 6 | - | - | - | - |
| scAAV9-scrambled | 6 | PND5 | 6.5×1012 vg/kg | - | - |
| scAAV9-shSMN | 5 | PND5 | 6.5×1012 vg/kg | - | - |
| Treated Pre-symptomatic | 4 | PND5 | 6.5×1012 vg/kg | PND6 | 8×1012 vg/kg |
| Treated Symptomatic | 5 | PND5 | 6.5×1012 vg/kg | PND33-36 | 2×1013 vg |
We next determined the level of knockdown achieved in vivo with our scAAV9-shSMN vector. Western blot of lumbar spinal cord lysates obtained from scAAV9-shSMN injected animals showed a 30% reduction in SMN protein compared to non-injected controls (Fig 2D). Since protein analysis was performed on whole lumbar spinal cord extracts, we next performed laser capture micro-dissection (LCM) of motoneurons followed by reverse transcriptase ddPCR to more accurately assess pig SMN mRNA expression in motoneurons. Quantification with primers specific for pig SMN revealed an effective 73±6% knockdown of SMN in the motoneurons and a 26±10% knockdown in the dorsal horn compared to samples from control littermates (Fig 2E), thus indicating successful reduction of pig SMN.
Biodistribution of the vector throughout the central nervous system (CNS) and major peripheral organs was assessed by droplet digital PCR (ddPCR) (Fig 3). A high number of vector genomes were found at all levels of the spinal cord and in the brain. Consistent with previous reports, vector genomes were also found in organs outside the CNS, although the levels were more variable38, 39.
Figure 3. Biodistribution of scAAV9-shSMN and scAAV9-SMN vectors in the CNS and major peripheral organs after intracisternal injection.

Total DNA was purified from various regions of the (A) brain, (B) spinal cord and (C) peripheral organs. Vector genome (vg) copy numbers for each vector were determined using ddPCR with primers specific to GFP or human SMN (and normalized to pig GAPDH).
Knockdown of pig SMN leads to the development of SMA-like symptoms
Three to four weeks following intrathecal injection of scAAV9-shSMN, piglets developed progressive muscle weakness, particularly in the hind limbs. ScAAV shows rapid expression with high expression in the first week and reaches maximum at 2 weeks after injection40. Because of the stability of SMN protein41 as well as time needed for the reduction in the critical RNP particles, the three to four weeks for onset of phenotype is reasonable. Initial clinical signs consisted of a wide-based stance and abnormal gait with difficulty standing for prolonged periods (Fig 4A). Weakness progressed rapidly to complete posterior paresis and difficulty in ascending a carpeted incline (Supplemental Movie 1). The piglets then lost the ability to stand independently and developed weakness in their front legs (Supplemental Movie 2). Hind limb muscle atrophy and fasciculations were noted to develop in concert with hind limb weakness. The piglets otherwise behaved normally and there were no apparent alterations in alertness, appetite, ability to swallow or ability to breathe. scAAV9-scrambled injected piglets did not develop any noticeable clinical symptoms.
To assess the function of the motor unit, we evaluated the electrophysiological biomarkers normally reduced in SMA including compound muscle action potential (CMAP), motor unit number estimate (MUNE), and electromyography (EMG) for presence of fibrillation potentials (Fig 4B). At PND54, no fibrillation potentials were observed for the control or scAAV9-scrambled animals (Fig 4C and Table 2). However, fibrillation potentials were observed in each muscle tested (gluteus medius, paraspinal, biceps femori and trapezius) from scAAV9-shSMN injected animals (Table 2). Consistent with SMA patient measurements, a significant decrease in CMAP and MUNE was observed in shSMN injected animals compared to controls (P<0.001 and P=0.003) (Fig 4D, E). Longitudinal measurements of CMAP and MUNE did not indicate any electrophysiological changes before the onset of symptoms (Fig 5). Interestingly, a recovery in the CMAP amplitude was observed at later time points while the MUNE values remained low.
Table 2.
2×2 contingency table showing the presence or absence of fibrillations.
| Group | Fibrillation | No fibrillation | Total | P-value vs control | P-value vs scAAV9-shSMN |
|---|---|---|---|---|---|
| Control | 0 | 6 | 6 | ||
| scAAV9-shSMN | 5 | 0 | 5 | 0.002 | |
| Treated Pre-symptomatic | 1 | 3 | 4 | 0.400 | 0.048 |
| Treated Symptomatic | 3 | 2 | 5 | 0.061 | 0.222 |
A one-tail Fisher’s exact test for the expected distribution was performed and showed significant difference between the control group and scAAV9-shSMN injected animals. 100% scAAV9-shSMN injected animals showed fibrillations in all tested muscles (gluteus medius, biceps femori, gastrocnemius and trapezius) against 0% for non-injected controls. Significant differences were also observed between the control and scAAV9-shSMN groups as well as the scAAV9-shSMN and treated pre-symptomatic groups.
Figure 5. Longitudinal sciatic CMAP and sciatic MUNE measurements in scAAV9-shSMN, treated pre-symptomatic, treated symptomatic and control piglets.
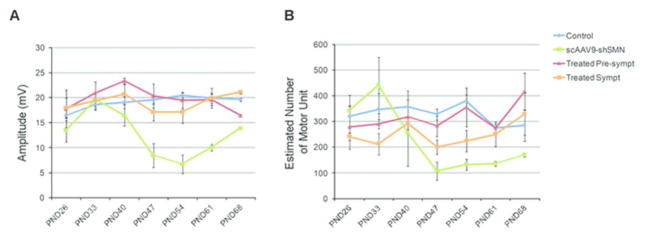
(A) SMA-induced animals show a decrease in their CMAP amplitudes at PND40 that reaches statistical significance by PND47. The apparent recovery at PND68 is due to the number of animals analyzed at this time point (only 2 remaining). Treated pre-symptomatic and treated symptomatic animals show a recovery in their CMAP. (B) scAAV9-shSMN animals show a significant reduction in MUNE by PND47. The sciatic MUNE of treated pre-symptomatic animals is identical to control animals throughout the study. PND: Postnatal Day.
Prevention of SMA-like phenotype and electrophysiological changes with human SMN administration
We then evaluated the impact of scAAV9-SMN intracisternal administration in scAAV9-shSMN injected piglets to determine SMN dependence on the phenotype. Twenty-four hours following the injection of scAAV9-shSMN vectors, piglets received a dose of 8×1012 vg/kg scAAV9-SMN and we investigated these animals both phenotypically and electrophysiologically. Since the rescue vector was administered 24 hours after the scAAV-shSMN, this group of piglet was identified as pre-symptomatic. Animals treated pre-symptomatically did not develop a clinical phenotype or severe proximal weakness (one piglet in the treated group developed mild weakness but did not show electrophysiological change) (Supplemental Movie 3). The CMAP amplitude of the pre-symptomatically treated animals was significantly preserved compared to the affected animals as well as the MUNE values (P<0.01) (Fig 4D, E). Another piglet showed transient fibrillations (PND54 and PND61) in the gluteus medius and paraspinal muscles but appeared phenotypically normal and no other electrophysiological changes were observed (Table 2). Overall, electrophysiological examination of the treated pre-symptomatically animals demonstrated that the reduction in CMAP amplitude and MUNE values were prevented and that EMG evidence of denervation was minimal. Together, these results indicate that in the pig there is an SMN dependence for these electrophysiological biomarkers of SMA and muscle weakness.
Knockdown of pig SMN in vivo leads to neuropathological changes
We evaluated the effects of scAAV9-shSMN intracisternal administration on the central and peripheral nervous systems from end stage scAAV9-shSMN animals (PND46-69). Nissl staining of the lumbar tissue revealed dramatic pathological changes within the ventral horn of the spinal cord, with numerous motoneurons presenting swelling of the perikarya and chromatolysis (Fig 6A). Motoneuron counts confirmed a significant loss of motor cell bodies with a 74% decrease compared to control animals (Fig 6B). Histopathology of ventral root tissues indicated a major loss of motor axons (Fig 6C, D). No morphological changes were observed in other regions of the spinal cord or in the dorsal root ganglia. Importantly, re-introduction of SMN with scAAV9-SMN corrected the morphological changes observed in SMA-like piglets (Fig 6). These results indicate that CSF delivery of scAAV9-shSMN in neonatal pigs leads to electrophysiological and pathological changes similar to those observed in SMA patients. These changes are SMN-dependent. Scrambled shRNA delivered by scAAV9 resulted in no pathological or electrophysiological changes.
Figure 6. scAAV9-shSMN induces neuropathological changes in the central and peripheral nervous system.

(A) Representative ventral horn motoneurons from lumbar spinal cord sections stained with cresyl violet. Motoneuron morphology was drastically changed in scAAV9-shSMN injected animals with numerous cells showing signs of central chromatolysis and swelling of the perikarya (red star). Motoneuron morphology was preserved in treated pre-symptomatic animals and chomatolytic neurons were observed only on rare occasions. (B) Motoneuron counts showing significant cell loss in scAAV9-shSMN injected animals (11.3±2.8, N=5) compare to non-injected control (43.6±5.5, N=6). Motoneuron loss was reduced in treated pre-symptomatic animals (31.7±4.6, N=4). (C) Semi-thick lumbar ventral root sections stained with toluidine blue showing axonal loss in end stage scAAV9-shSMN animals. Middle panel and corresponding high magnification showing Wallerian degeneration, only a few myelinated axons are visible and myelin is forming globules in Schwann cells and macrophages. (D) The number of myelinated axons per 1,000 μm2 in the lumbar ventral root was also significantly reduced in scAAV9-shSMN animals compared to controls (3.2±0.4, N=5 versus 7±0.3, N=4) and preserved in the treated pre-symptomatic group (6.2±0.4, N=4). Scale bars: 50μm in (A) and 100μm in (C). ** P<0.01
Delivery of human SMN at onset of symptoms has a major influence on disease outcome
We evaluated the effect of scAAV9-SMN intracisternal administration in scAAV9-shSMN piglets at onset of symptoms. Onset of symptoms was determined based on clinical examination. In particular piglets showed a splayed gait (wide stance of legs) as well as a spread gait while either moving in their pens or on a ramp used to evaluate the pigs (ability to mount an inclined ramp to receive a reward). They also showed difficulty standing for prolonged periods. This was defined as symptomatic onset and indication of proximal weakness. A dose of 2×1013 vg was administered when signs of hind limb weakness and abnormal gait were observed (Table 1). Longitudinal electrophysiological studies for CMAP, MUNE and EMG examination were performed as before and no electrophysiological changes were observed at the time of the rescue vector injection (Fig 5). Remarkably, CMAP values at PND54 of the treated symptomatic group were significantly increased compared to affected animals (17.2±2.3 versus 6.8±1.9, P=0.005) and were similar to values obtained in the control and treated pre-symptomatic group (Fig 7C and Fig 5). Interestingly, MUNE was only partially corrected with a value higher than in affected animals (225±41 versus 133±21, P=0.397), but not similar to control animals (380±25, P=0.058) (Fig 7C). We observed variability in the response of some animals. In 3 out of 5 piglets, hind limb weakness was still present until the time of euthanasia but symptoms never evolved to severe weakness (Supplemental Movie 4). In the other 2 piglets, the hind limb weakness progressed to severe paresis but did not develop to complete paralysis. In these 2 animals, CMAP and MUNE values at PND54 were markedly reduced. One of these piglets had to be euthanized due to a rectal prolapse and therefore we do not have electrophysiological measurements for this piglet after PND54. The MUNE values of the other piglet remained low (~100) from PND54 to PND68 but the CMAP showed recovery (13.1mV at PND54 and 21.8 at PND68). EMG assessment of hind limb muscles at PND54 demonstrated fibrillation potentials in 3 of 5 treated symptomatic piglets but not in all the muscles tested (Table 2).
Figure 7. scAAV9-SMN treatment at onset of symptoms partially corrects the electrophysiological and histopathological changes observed in SMA affected animals.

(A) Immunofluorescence analysis of lumbar spinal cord sections shows robust GFP (green) and human SMN (red) expression in ChAT-positive cells (blue) of treated pre-symptomatic and treated symptomatic animals with numerous cells expressing both transgenes. (B) LCM-collected lumbar motoneurons were analyzed for pig or human SMN mRNA levels by using ddPCR. Motoneurons from treated pre-symptomatic and treated symptomatic animals have an 87±4% (N=4) and 69±7% (N=5) reduction in pig SMN levels, respectively. Human SMN mRNA levels relative to pig SMN are significantly increased in motoneurons from treated pre-symptomatic and treated symptomatic animals (44±13% and 72±32%). (C) CMAP responses obtained at PND54 in treated symptomatic animals are significantly improved (17.2±2.3) compared\ to SMA-like affected animals (6.8±1.9). MUNE responses were only partially rescued in the treated symptomatic group (225±41 versus 380±25 in control animals) (D) Histopathology performed on the lumbar spinal cord and ventral roots from treated symptomatic animals show scattered chromatolytic motoneurons and axonal degeneration (E) Motoneuron and motor axon counts show a moderate loss of ventral motoneurons and motor axons in treated symptomatic animals indicating partial rescue of the motor unit in these animals. * P<0.05, ** P<0.01. Scale bars: 40μm (A) and 25μm (D).
We evaluated the degree of pig SMN knockdown and human SMN expression achieved in these animals. GFP staining showed strong transduction of the motoneurons indicating that efficient knockdown of pig SMN was achieved in the motoneurons of these animals (Fig 7A). SMN staining using an antibody specific for human SMN showed robust expression of the scAAV9-SMN vector in motoneurons (Fig 7A and Fig 8). Quantitative mRNA analysis using primer sets specific for pig SMN or human SMN was performed on motoneurons and dorsal horn elements collected by LCM (Fig 7B). As expected, we observed a robust decrease in pig SMN levels in the motoneurons from the groups treated pre-symptomatically (87% decrease) and treated symptomatically (69% decrease). This level of knockdown is sufficient to induce SMA-like changes in the piglets. In contrast, high levels of human SMN mRNA were detected in motoneurons in animals treated pre-symptomatically or symptomatically (Fig 7B).
Figure 8. Motoneuron transduction efficiency of scAAV9-shSMN and scAAV9-SMN in treated pre-symptomatic and treated symptomatic animals.
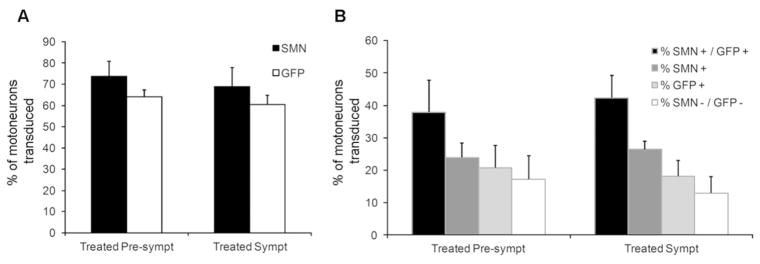
Lumbar spinal cords were stained for GFP, SMN and ChAT. ChAT-positive motoneurons were considered for SMN and GFP expression (n=4 in each group). (A) Total percentage of GFP-positive and SMN-positive motoneurons in treated pre-symptomatic and symptomatic animals. (B) Distribution of SMN- and GFP-positive cells within the ChAT-positive motoneuron population. 46% of the treated pre-symptomatic and 42% of the treated symptomatic motoneurons expressed both transgenes.
Interestingly, neuropathology of lumbar spinal cord and corresponding ventral root tissues of animals treated at the onset of symptoms showed marked improvement with fewer chromatolytic motoneurons and degenerative motor axons observed (Fig 7D). Indeed, a 39% increase in motoneuron count was observed in the onset treated group compare to the non-treated animals (28±8 versus 11±3, P=0.167) as well as an increase in the number of motor axons per x1,000 μm2 (4.8± .5 versus 3.2±0.4, P = 0.009) (Fig 7E).
Discussion
Here we report the creation of the first large SMA model in the domestic pig. We showed that reduction of SMN mRNA levels by 73% in motoneurons postnatally was sufficient to induce a SMA-like phenotype. Importantly, this animal model recapitulates characteristic electrophysiological and histological changes associated with SMA.
Animal models of SMA have been created in species from C.elegans to mice and now pig. In invertebrate organisms, there is contribution of maternal SMN from the yolk. This maternal contribution allows deletion or nonfunctional missense mutations in the homozygous state to survive to various stages of development8. In Drosophila and C. elegans, this produce larvae with motility and neuromuscular junction defects42, 43. However maternal SMN becomes depleted and this results ultimately in larval lethality. Knockdown of Smn in Zebrafish using morpholin ASO results in axonal defects44 that are not present in zebrafish smn mutants. Furthermore when motor axons are examined in SMA mice through various stages of development no axonal patterning defects are found45.
In spinal cord material from SMA patients, full-length SMN mRNA is reduced to 20–25% of normal levels31. Similar levels of full-length SMN mRNA have been found in laser microdissected motoneurons from SMA mice containing two copies of SMN230. In addition, a marked reduction of SMN protein level is found in SMA type 1 with fetal spinal cords showing 22–23% SMN levels46 and even more marked reduction in autopsy samples from type 1 patients47. Together, those reports suggest that knockdown of ~80% of SMN would be sufficient to produce an SMA-like phenotype. In the current study we obtained 73% knockdown of SMN on average in the lumbar spinal cord motoneurons which is a level consistent with the phenotype we observed. While some motoneurons could take up more scAAV9 and thus have further knockdown of SMN, symptomatic-treated piglets still respond and benefit from scAAV9-SMN thus indicating they have not reached the point of no return and further validates the efficacy of gene replacement.
In mice some evidence indicates that correction of SMN levels in the periphery has an impact on the survival of SMA mice22. Other studies indicate the importance of delivery to the CNS at least in SMA mice21, 27. One issue with all these studies is the relative permeability of the blood brain barrier in the neonatal stages of mouse development. In addition the question can be asked as to whether the peripheral features found in the mouse are present in humans. Necrosis is observed in mice even with higher copy of SMN2 or after treatments but only very rarely in humans 17, 24, 25, 27, 48. In addition cardiac defects clearly present in SMA mice are not often present in humans29, 49. One possible reason is that the human SMN2 regulatory regions may not be responsive in the mouse resulting in lower SMN levels in the periphery. Alternatively there could be a species dependence of high SMN levels as regards splicing of particular genes as the intron structure changes between species. Regardless in mouse it is clear that dependence on just 2 copies of SMN2 in the neurons of the CNS is not sufficient for normal function50. Furthermore SMA types other than type 1 maybe even less dependent on peripheral SMN levels49. Another interesting aspect of motor neuron disorders is a role for non-motoneuron cells of the CNS in the development of the phenotype51, 52. In SMA, cells such as astrocytes, schwann cells and sensory neurons have been evoked53–55. However whether changes seen contribute to the pathology of SMA or are a result of motoneuron distress in response to low SMN levels remain unclear. In our study, immunostaining of pig spinal cord reveals a strong tropism of AAV9 for the motoneuron with glial cells transduced only on rare occasions although we cannot eliminate low transduction of these cells contributing to the phenotype. Sensory neurons within the dorsal horn of the spinal cord were found to be transduced relatively efficiently thus they could contribute to the phenotype observed. However, no neuropathological changes were noted. In addition, although leakage of virus from the CNS does occur into the periphery in our study, the number of vg found in those tissues is unlikely to cause sufficient knockdown to induce a phenotype. Thus we have shown in this large animal model the critical nature of high SMN levels in motoneurons, which is consistent with SMA pathology in humans.
A powerful advantage of our animal model is that the CMAP and MUNE measures correlate with the progression of the phenotype in the SMA-like piglets similarly to the progression of SMA in patients. In humans, electrophysiological studies suggest that CMAP and MUNE values are preserved until the clinical manifestation of the disease is observed. However, the drop in CMAP and MUNE occurs within the first 2 years of life in both SMA types 1 and 2 patients56, 57. In our model, the first clinical symptoms were observed at ~PND24-34 while reductions in CMAP and MUNE were observed between PND40-47 which correlates with clinical data from human patients. Interestingly, a recovery in the CMAP value was observed at later time points and is consistent with collateral reinnervation by the remaining motor units. A similar situation is observed in patients with milder forms of SMA56.
Studies in mice using inducible transgenes indicate that early delivery is the most effective means of treatment of SMA24, 58, 59. We have delivered SMN when piglets had their first symptoms of weakness to more closely mimic the timing of treatment in human patients. Children with type 1 SMA are often normal at birth and then develop symptoms rapidly. Indeed CMAP and MUNE values in many pre-symptomatic SMA patients are normal at early time points but quickly drop upon symptom appearance56, 57. Treatment prior to loss of CMAP amplitude can be predicted to delay or prevent the phenotype of SMA. However the critical question in a clinical trial will be whether an impact of treatment occurs when administered upon development of SMA symptoms. The studies presented here indicate that a therapeutic benefit will be obtained, provided administration occurs early after symptom onset. In milder cases there is a possibility to encourage and enhance the remaining motoneurons to sprout and re-innervate the muscles6. Remarkably, when we administered treatment at onset of symptoms in piglets, but before any changes in CMAP and MUNE, we were able to alter the time course and progression of the symptoms. Indeed, based on SMN protein turnover and well-known capability of scAAV9 vector for quick transgene expression, SMN knockdown should reach its maximum 2 to 3 weeks after vector injection40, 41. Another consideration is the biological activity of SMN in particular its role in the assembly of snRNP particles which are critical in splicing of genes. Given that there are excessive amount of snRNPs, their depletion can take a relative long time to induce a specific phenotype like SMA. Indeed this appears to be the case since pigs do not show clinical symptoms until 3 to 4 weeks post-injection. We also induced reduction of SMN in the neonatal period when SMN appears to be critical to be at high levels from mice studies59, 60. The rescue scAAV9-hSMN vector was delivered at 4 weeks post-knockdown and it improved the neuropathology which indicates that the degeneration process induced by low SMN levels in the motoneurons can be reversed after symptoms appear. Also, the biomarkers CMAP and MUNE as well as EMG (fibrillations) clearly respond to SMN restoration indicating that positive outcomes can be obtained later in the disease process.
In conclusion we show that postnatal reduction of SMN in motoneurons results in SMA. This has resulted in the creation of the first large animal model of SMA. Furthermore, we show the ability to correct the SMA phenotype when symptoms are present in this large animal model. Lastly, we demonstrated that CMAP, MUNE and EMG may be used to predict the response to SMN therapy in SMA. Thus for clinical trial design early symptomatic restoration of SMN should result in an improved phenotype that does not progress and recovery of CMAP indicating better function of the available motoneurons.
Supplementary Material
Supplementary Video 1. Loss of motor function in scAAV9-shSMN injected animals
Supplementary Video 2. End stage scAAV9-shSMN injected piglet (PND40).
Supplementary Video 3. Motor function is preserved in treated pre-symptomatic animal.
Supplementary Video 4. Moderate motor function loss in treated symptomatic piglet.
Acknowledgments
We thank Dr. D.J. Coble, L. Mattox and C.L. Sims from the ULAR Wiseman Hall Surgery Services at OSU for excellent technical help with the pigs, Dr. M. Joseph for technical assistance with fluoroscopy. We also thanks N. Madabusi, C. Iyer, A. Blatnik, S. Renusch and K. Marhenke for technical help and Dr. V.L. McGovern for critical reading of the manuscript. This work was funded by the Sofia’s Cure Foundation, SMA Europe and NINDS grant RC2NS069476 and R21NS083804 (to A.H.M.B). S.I.D was supported by a postdoctoral fellowship from SMA Europe. S.J.K received funding from NINDS (K08NS067282 and U01NS079163). W.D.A. received funding from NIH-NICHD (5K12HD001097-17).
Footnotes
Author contributions. S.I.D. designed most of the experiments, performed intrathecal injection, behavioral experiments, performed necropsy and tissue collection, analyzed histological experiments, performed, analyzed mRNA and protein levels in tissues, wrote the manuscript. X.L. helped with necropsy, tissue collection, performed histological staining and motoneuron quantification. L.S. contributed to plasmid production for AAV stocks and performed tissue sectioning. K.M. provided technical assistance with the piglets. W.D.A. performed intrathecal injection and designed, conducted and analyzed all the electrophysiological studies, performed necropsy and tissue collection and contributed to the manuscript writing. P.N.P. performed intrathecal injection. S.J.K. performed intrathecal injections, contributed to the study design, clinical evaluation of the piglets and manuscript writing. P.O and D.S. designed the shRNA constructs and performed testing in vitro. A.H.M.B contributed to perform necropsy and tissue collection. B.K.K. and A.H.M.B. conceived the study, analyzed data and wrote the manuscript.
Conflict of Interest. Dr. Kaspar has patents, AAV9 and blood brain barrier/CSF barrier, licensed to AveXis Inc., and a patent, AAV9-SMN, licensed to AveXis Inc. Dr Burghes serves on the SAB of AveXis.
References and Notes
- 1.Pearn J. Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet. 1978;156:409–413. doi: 10.1136/jmg.15.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 2010;152A7:1608–1616. doi: 10.1002/ajmg.a.33474. [DOI] [PubMed] [Google Scholar]
- 3.Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;201:27–32. doi: 10.1038/ejhg.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;87:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 5.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;801:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 6.Arnold WD, Burghes AH. Spinal muscular atrophy: development and implementation of potential treatments. Annals of neurology. 2013;743:348–362. doi: 10.1002/ana.23995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;9611:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;108:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prior TW, Russman BS. Spinal Muscular Atrophy. 1993. [Google Scholar]
- 10.Hausmanowa-Petrusewicz I, Karwanska A. Electromyographic findings in different forms of infantile and juvenile proximal spinal muscular atrophy. Muscle Nerve. 1986;91:37–46. doi: 10.1002/mus.880090106. [DOI] [PubMed] [Google Scholar]
- 11.Buchthal F, Olsen PZ. Electromyography and muscle biopsy in infantile spinal muscular atrophy. Brain. 1970;931:15–30. doi: 10.1093/brain/93.1.15. [DOI] [PubMed] [Google Scholar]
- 12.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;32:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 13.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;9418:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Workman E, Saieva L, Carrel TL, et al. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Human molecular genetics. 2009;1812:2215–2229. doi: 10.1093/hmg/ddp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le TT, Pham LT, Butchbach ME, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;146:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 16.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;93:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 17.Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nature genetics. 2000;241:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 18.Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;3456197:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- 19.Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes & development. 2010;2415:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;372:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porensky PN, Mitrpant C, McGovern VL, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;217:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;4787367:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valori CF, Ning K, Wyles M, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;235:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 24.Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nature biotechnology. 2010;283:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Human molecular genetics. 2011;204:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 26.Glascock JJ, Shababi M, Wetz MJ, et al. Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochemical and biophysical research communications. 2012;4171:376–381. doi: 10.1016/j.bbrc.2011.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Passini MA, Bu J, Roskelley EM, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. The Journal of clinical investigation. 2010;1204:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palladino A, Passamano L, Taglia A, et al. Cardiac involvement in patients with spinal muscular atrophies. Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology / edited by the Gaetano Conte Academy for the study of striated muscle diseases. 2011;303:175–178. [PMC free article] [PubMed] [Google Scholar]
- 29.Bevan AK, Hutchinson KR, Foust KD, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Human molecular genetics. 2010;1920:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruggiu M, McGovern VL, Lotti F, et al. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Molecular and cellular biology. 2012;321:126–138. doi: 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tisdale S, Lotti F, Saieva L, et al. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell reports. 2013;55:1187–1195. doi: 10.1016/j.celrep.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bevan AK, Duque S, Foust KD, et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Molecular therapy : the journal of the American Society of Gene Therapy. 2011;1911:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnold WD, Porensky PN, McGovern VL, et al. Electrophysiological Biomarkers in Spinal Muscular Atrophy: Preclinical Proof of Concept. Annals of clinical and translational neurology. 2014;11:34–44. doi: 10.1002/acn3.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McComas AJ, Fawcett PR, Campbell MJ, et al. Electrophysiological estimation of the number of motor units within a human muscle. Journal of neurology, neurosurgery, and psychiatry. 1971;342:121–131. doi: 10.1136/jnnp.34.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shefner JM, Cudkowicz ME, Brown RH., Jr Comparison of incremental with multipoint MUNE methods in transgenic ALS mice. Muscle & nerve. 2002;251:39–42. doi: 10.1002/mus.10000. [DOI] [PubMed] [Google Scholar]
- 36.Willmott AD, White C, Dukelow SP. Fibrillation potential onset in peripheral nerve injury. Muscle & nerve. 2012;463:332–340. doi: 10.1002/mus.23310. [DOI] [PubMed] [Google Scholar]
- 37.Federici T, Taub JS, Baum GR, et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2012;198:852–859. doi: 10.1038/gt.2011.130. [DOI] [PubMed] [Google Scholar]
- 38.Gray SJ, Nagabhushan Kalburgi S, McCown TJ, et al. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;204:450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haurigot V, Marco S, Ribera A, et al. Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. The Journal of clinical investigation. 2013 doi: 10.1172/JCI66778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene therapy. 2001;816:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- 41.Burnett BG, Munoz E, Tandon A, et al. Regulation of SMN protein stability. Molecular and cellular biology. 2009;295:1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briese M, Esmaeili B, Fraboulet S, et al. Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum Mol Genet. 2009;181:97–104. doi: 10.1093/hmg/ddn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan YB, Miguel-Aliaga I, Franks C, et al. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet. 2003;1212:1367–1376. doi: 10.1093/hmg/ddg157. [DOI] [PubMed] [Google Scholar]
- 44.McWhorter ML, Monani UR, Burghes AH, et al. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;1625:919–932. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGovern VL, Gavrilina TO, Beattie CE, et al. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nature genetics. 1997;163:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 47.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Human molecular genetics. 1997;68:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 48.Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle & nerve. 2014 doi: 10.1002/mus.24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shababi M, Lorson CL, Rudnik-Schoneborn SS. Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? Journal of anatomy. 2014;2241:15–28. doi: 10.1111/joa.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park GH, Maeno-Hikichi Y, Awano T, et al. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci. 2010;3036:12005–12019. doi: 10.1523/JNEUROSCI.2208-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haidet-Phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;299:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyer K, Ferraiuolo L, Miranda CJ, et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;1112:829–832. doi: 10.1073/pnas.1314085111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGivern JV, Patitucci TN, Nord JA, et al. Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia. 2013;619:1418–1428. doi: 10.1002/glia.22522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mentis GZ, Blivis D, Liu W, et al. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;693:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hunter G, Aghamaleky Sarvestany A, Roche SL, et al. SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Human molecular genetics. 2014;239:2235–2250. doi: 10.1093/hmg/ddt612. [DOI] [PubMed] [Google Scholar]
- 56.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;575:704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finkel RS. Electrophysiological and motor function scale association in a pre-symptomatic infant with spinal muscular atrophy type I. Neuromuscular disorders : NMD. 2013;232:112–115. doi: 10.1016/j.nmd.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Lutz CM, Kariya S, Patruni S, et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest. 2011;1218:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Le TT, McGovern VL, Alwine IE, et al. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet. 2011;2018:3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kariya S, Obis T, Garone C, et al. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest. 2014;1242:785–800. doi: 10.1172/JCI72017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. Loss of motor function in scAAV9-shSMN injected animals
Supplementary Video 2. End stage scAAV9-shSMN injected piglet (PND40).
Supplementary Video 3. Motor function is preserved in treated pre-symptomatic animal.
Supplementary Video 4. Moderate motor function loss in treated symptomatic piglet.