

Abstract
Stroke is one of the leading causes of death and permanent disability in the elderly. However, most of the experimental studies on stroke are based on young animals, and we hypothesised that age can substantially affect the stroke response. The two-vessel occlusion model of global ischemia by occluding the common carotid arteries for 15 min at 40 mmHg of blood pressure was carried out in 3- and 18-month-old male Sprague–Dawley rats. The adhesion molecules E- and P-selectin, cell adhesion molecules (CAMs), both intercellular (ICAM-1) and vascular (VCAM-1), as well as glial fibrillary acidic protein (GFAP), and cleaved caspase-3 were measured at 48 h after ischemia in the cerebral cortex and hippocampus using Western blot, qPCR and immunofluorescence techniques. Diametric expression of GFAP and a different morphological pattern of caspase-3 labelling, although no changes in the cell number, were observed in the neurons of young and old animals. Expression of E-selectin and CAMs was also modified in an age- and ischemia/reperfusion-dependent manner. The hippocampus and cerebral cortex had similar response patterns for most of the markers studied. Our data suggest that old and young animals present different time-courses of neuroinflammation and apoptosis after ischemic damage. On the other hand, these results suggest that neuroinflammation is dependent on age rather than on the different vulnerability described for the hippocampus and cerebral cortex. These differences should be taken into account in searching for therapeutic targets.
Keywords: Ischemia, Age, Inflammation, Apoptosis, GFAP, Selectins, CAMs
Background
Stroke is one of the most important cause of death worldwide according to the World Health Organization (WHO 2011) and the main cause of permanent disability (Donnan et al. 2008). Ischemia causes necrosis in neurons in the first hours after insult in the area where blood flow has been reduced to less than 15 % (ischemic core) (Tamura et al. 1981; Nedergaard et al. 1986; Duverger and MacKenzie 1988). After longer periods, areas with blood flow up to 40 % (penumbra areas) (Ginsberg and Pulsinelli 1994; Hossmann 1994; Back 1998) present delayed cell death, mainly by apoptosis (Mehta et al. 2007). Thus, the goal of neuropharmacological targeting is mainly addressed to preserve or rescue the neurons in the penumbra area of apoptotic delayed cell death (Rami et al. 2008; Fricker et al. 2013). Apoptosis is also tightly related with the strong neuroinflammation elicited by stroke and the impairment of the blood–brain barrier (BBB) (Amor et al. 2010). In the inflammatory response, adhesion molecules play crucial roles since they mediate recruitment and infiltration of neutrophils across the vascular endothelium (Sughrue et al. 2004; Petri et al. 2008). This process requires the sequential action of different selectins and cell adhesion molecules (CAMs) (Stanimirovic and Satoh 2000; Yilmaz and Granger 2008), and we hypothesised that the balance of these molecules could mirror the time-course of this recruitment. In addition, the impairment of the BBB is neutrophil-dependent (Anthony et al. 1997; Perry et al. 1997; Blamire et al. 2000) and, therefore, should be mirrored by the expression of these molecules. This expression could help to identify the BBB permeability at different ages. The impairment of the BBB is also related to astrocyte activation (Ivens et al. 2007; Cacheaux et al. 2009), where quiescent astrocytes become reactive astrocytes. This process modifies these cells substantially, inducing crucial changes in the expression of cytoskeletal molecules, such as the glial fibrillary acidic protein (GFAP) (Sofroniew and Vinters 2010; Colangelo et al. 2014). Reactive astrocytes release soluble factors that are related to the recruitment of leukocytes for crossing the BBB and initiate neuroinflammation in the central nervous system (CNS) (Li et al. 2011).
Ischemia focal models reveal an intrinsic gradient of damage from the ischemic core that has to be considered when a structure is studied (Ayuso et al. 2010). Most of the results from focal models are morphological data based on immunofluorescence and magnetic resonance imaging (MRI) (Dziennis et al. 2011; Liu and McCullough 2012), and many of them are based on the leukocyte response (Stevens et al. 2002; Gelderblom et al. 2009). In addition, despite the fact that strokes mainly occur in the elderly, current studies are mainly performed on young animals and data from old animals are very scarce (Collins et al. 2003; Wasserman et al. 2008). Particularly, studies on transcriptional expression of specific molecules that bind neutrophils (e.g. selectins and CAMs) are very limited and are practically non-existent in old animals (Liu and McCullough 2012), despite their importance for gaining insight into the leukocyte infiltration process.
Age is considered the most relevant factor for stroke risk (Rojas et al. 2007; Rosamond et al. 2008). Since many biochemical parameters decrease with aging (Sinha et al. 2005; Bala et al. 2006; Arumugam et al. 2010; Liu and McCullough 2011; Liu et al. 2012), it would expect a lessened ability of response to the stroke. However, age-dependent differences in the time-course of the stroke-induced response of different molecules could play crucial roles in the discovery of new therapeutic targets or therapies (Anyanwu 2007). In this regard, we present here a study comparing for the first time the ischemic-induced apoptotic damage (delayed cell death), GFAP, and selectin and CAM adhesion molecules involved in leukocyte infiltration and GFAP and show their age dependence.
Material and methods
Animals
Young (3-month-old) and old (18-month-old) male Sprague–Dawley rats, 400 ± 50 and 750 ± 80 g, respectively, were housed at 22 ± 1 °C in a 12-h light/dark controlled environment with free access to food and water. Twenty rats of each age group were divided randomly into ischemic and sham-operated groups. Experiments were performed in accordance to the Guidelines of the Council of the European Union (63/2010/EU), following Spanish regulations (RD 53/2013, BOE 8/2/2013) for the use of laboratory animals. Experimental procedures were also approved by the Scientific Committee of the University of Leon. All efforts were made to minimise animal suffering and to reduce the number of animals used.
Transient global ischemia
Animals were anaesthetised in an anaesthesia induction box supplied with 4 % halothane (Sigma-Aldrich) at 3 L/min in 100 % oxygen. After induction, anaesthesia was maintained with 1.5 to 2.5 % halothane at 800 mL/min in 100 % oxygen using a rat face mask. Trimetaphan (kindly provided by Roche Applied Science) was used as the main hypotensor agent (15 mg/mL, 0.3 mg/min), administered through the femoral artery, to obtain moderate hypotension (40–50 mmHg). This prevents blood flow through the circle of Willis in the two-vessel occlusion models of global ischemia. Arterial tension was also modulated by changing the halothane concentration, which has intrinsic hypotensive effects (Bendel et al. 2005). Body temperature was controlled with a rectal probe and maintained at 36 ± 1 °C during surgery using a feedback-regulated heating pad. Both common carotid arteries were exposed and occluded with atraumatic aneurysm clips for 15 min of transient global ischemia. Then, animal arterial blood pressure was left to recover, the femoral artery catheter was removed and the animal was sutured.
After recovering consciousness, rats were maintained in standard conditions for 48 h (reperfusion time). Procedures in sham-operated rats were performed exactly as for ischemic animals except that carotid arteries were not clamped.
RNA and protein studies
Forty-eight hours after the ischemic insult, animals were decapitated, their brains quickly removed and transferred into a brain rodent matrix (ASI instruments) at 4 °C in order to obtain 2 mm thick sagittal slices at a distance of 1 mm to the medial line. The Cornu Ammonis 1 (CA1) hippocampal region, Cornu Ammonis 3 (CA3) hippocampal region and cerebral cortex (CX) were dissected from those slices under a microscope, frozen in dry ice and stored at −80 °C. Total RNA and protein of each of these regions were extracted using the TriPure™ isolation reagent (Roche Applied Science) following the manufacturer’s instructions and then stored at −80 °C.
Reverse transcription and qPCR
RNA integrity was assessed using the Experion RNA HighSens Analysis Kit (Bio-Rad Laboratories) following the manufacturer’s instructions. Possible contamination with DNA was prevented by incubation with DNase (Sigma-Aldrich) and checked by PCR.
The concentration of the total RNA in each sample was determined by measuring its absorbance (260/280 nm) using a NanoDrop ND-3300 spectrophotometer (NanoDrop Technologies). Six hundred nanograms of total RNA of each sample was used as a template for reverse transcription using the High Capacity complementary DNA (cDNA) Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s instructions. The cDNA obtained was used as a template for the quantitative real-time PCR (qPCR) assays. Primers were designed using Primer Express software (Applied Biosystems), and those with efficiencies lower than 90 % were discarded. Forward and reverse primers used in this study (efficiency values between 90 and 110 %) are shown in Table 1.
Table 1.
Sequences of the primers used for RT-qPCR and GenBank accession numbers
| Gene | Forward primer | Reverse primer | Accession number |
|---|---|---|---|
| P-selectin | acaggcagccctccaatgtgtg | atttgacggctctgcacacggg | [NM_013114.1] |
| E-selectin | tgcttcccgtctttgccacacc | tccgtccttgctcttctgtgcg | [NM_ 017211.2] |
| VCAM-1 | tgctcctgacttgcagcaccac | tgtcatcgtcacagcagcaccc | [NM_ 012889.1] |
| ICAM-1 | tgcagccggaaagcagatggtg | atggacgccacgatcacgaagc | [NM_012967.1] |
| GAPDH | gggcagcccagaacatca | tgaccttgcccacagcct | [NM_017008] |
ICAM intercellular adhesion molecule, VCAM vascular adhesion molecule, GAPDH glyceraldehyde-3-phosphate dehydrogenase
Real-time PCR was performed using a StepOnePlus™ Real-Time PCR System, and SYBR Green PCR Master Mix (Applied Biosystems) was used as the fluorescent DNA-binding dye. Optimal qPCR conditions in our assays were obtained with 2 μL of 1/10 cDNA and 300 nM of primers. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene for normalisation of different transcript values. The normalised messenger RNA (mRNA) levels were expressed as 2−∆Ct (∆Ct = Cttarget − CtGAPDH), and fold changes were compared using the 2−∆∆Ct method (Livak and Schmittgen 2001). All of the qPCR assays in this study were performed according to MIQE Guidelines (Taylor et al. 2010).
Western blot
Proteins were resuspended in 8 M urea and 4 % sodium dodecyl sulphate (SDS) in the presence of a protease inhibitor (complete protease inhibitor cocktail, EDTA-free; Roche Applied Science), and their concentrations were determined using the DC Protein Assay (Bio-Rad) based on the Lowry method. Protein samples (25 μg per lane) were resolved on a 10 % polyacrylamide gel (SDS-PAGE; Bio-Rad) at 110 V for 120 min. Then, proteins were transferred onto a nitrocellulose membrane using a dry transfer system (Invitrogen) at 20 V for 7 min. Nitrocellulose membranes were blocked in 5 % bovine serum albumin and 0.2 % Tween-20 (Sigma-Aldrich) in Tris-buffered saline (TBST) for 60 min at 25 °C. Then, membranes were incubated overnight, at 4 °C, with the primary antibodies (Table 2).
Table 2.
Primary antibodies and concentration used
| Primary antibodies | Manufacturer | Concentration |
|---|---|---|
| P-selectin raised in rabbit | Abcam | 1 μg/mL |
| E-selectin raised in rabbit | Abcam | 0.67 μg/mL |
| ICAM-1 raised in rabbit | Abcam | 0.75 μg/mL |
| GFAP raised in rabbit | Dako | 1 μg/mL |
| β-Actin raised in mouse | Sigma-Aldrich | 0.2 μg/mL |
ICAM intercellular adhesion molecule, GFAP glial fibrillary acidic protein
Primary antibodies were labelled with their appropriate secondary anti-rabbit or anti-mouse antibodies complexed with horseradish peroxidase (Dako) at a dilution of 1:3000. After incubation in Chemiluminescence Luminol Reagent (Life Technologies), the nitrocellulose membranes were exposed onto the proper films (ECL films, Amersham) to obtain images of the protein bands labelled with the enzyme. Densitometry analysis of the bands was performed with ImageJ 1.46r (ImageJ software).
Immunofluorescence assays
Animals used for this technique were perfused via the aorta with a saline solution followed by a fixer solution made of 4 % paraformaldehyde (PFA) (Merck) in 50 mM phosphate-buffered saline (PBS; pH 7.4) at 4 °C. The brains were removed, maintained overnight in PFA at 4 °C and then stored until sunk in a cryoprotectant solution made of 30 % sucrose in PBS, pH 7.4. The brains were sectioned into 40 μm thick sagittal slices with a freezer microtome and then were maintained in 0.025 % sodium azide (Sigma-Aldrich) in PBS (pH 7.4) at 4 °C until analysis. Sections were labelled with a rabbit primary antibody against the activated caspase-3 (Cell Signalling) overnight at 4 °C. A biotinylated goat anti-rabbit antibody (Vector; 1:500) was used as the secondary antibody and was labelled with stravidin complexed with DyLight Alexa 592 (Molecular probes; 1:100). Nuclei were labelled with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich; 1:1000) and mounted in 3 % DABCO (Sigma-Aldrich) in glycerol–water (1:1) to preserve the fluorescence. Slices were kept at 4 °C in the dark. Images were obtained using a confocal Nikon TE 2000 EZ.C1 microscope (Nikon).
Quantification of cleaved caspase-3-positive cells was performed on six 40 μm thick equidistant sagittal sections that were between 1 and 4 mm lateral to the middle line per rat. An optical dissector method modified from Zarow et al. (2005) was used. A 60× objective was used to perform the quantification. The counting frame for the dissector was a 35 × 35 μm square, and for the fractionator volume, we used a 30-μm height (discarding 5 μm of both lateral and medial sides that were damaged in the cutting process). In each of the sections, seven equidistant dissectors along the cerebral cortex internal pyramidal layer, seven along CA1 and four along the CA3 pyramidal layers were used per section. Cells were scored as apoptotic when the nucleus was stained by the cleaved caspase-3 antibody and was condensed, or if at least 30 % of the cytosol surrounding the nucleus was labelled. Other staining patterns were considered as artefacts. The results were expressed as the percentage of cleaved caspase-3-positive cells with respect to the total number of cells.
Statistical analysis
Two-way ANOVA tests followed by Bonferroni t test were conducted to detect interactions between age and ischemia. The significance was set at the 95 % confidence level. The statistical analysis was carried out using GraphPad Prism 5 (GraphPad software).
Results
E-selectin mRNA levels on the hippocampus and cerebral cortex
The analysis of the effect of age revealed that the cerebral cortex and CA1 showed significantly lower mRNA levels in the aged sham-operated animals than in the young sham-operated animals. However, we failed to find differences in CA3. In contrast, we only detected an age-dependent effect, with increased E-selectin transcript levels, in the CA3 of injured animals (Fig. 1a). In all of the structures studied, the ratios between old/young sham-operated animals were significantly lower than the ratios for old/young ischemia/reperfusion (I/R)-injured animals.
Fig. 1.

Effect of age on mRNA levels. Fold change (2−ΔΔCt) in (a) E-selectin, (b) VCAM-1 and (c) ICAM-1 mRNA levels in old sham-operated animals (open columns) as compared with young sham-operated animals (represented as a value of 1, dotted line) in the CA1 and CA3 hippocampal structures and the CX. Black columns represent old I/R-injured animals as compared to their respective young I/R animals (represented as a value of 1, dotted line). Age-dependent significant differences are represented by an asterisk, and significant differences between old animals are indicated by a yen sign (p < 0.05, two-way ANOVA, n = 5)
In young animals, we only detected the I/R effect in the CA1 and CA3 hippocampal structures where E-selectin mRNA levels of ischemic animals were significantly lower than those of their respective sham-operated animals. In old animals, E-selectin transcript levels were significantly higher than those of their respective sham-operated animals in all of the structures studies. Also, E-selectin mRNA levels were higher in old injured animals than in young injured animals (Fig. 2a). Two-way ANOVA analysis of E-selectin transcripts revealed significant interactions for both age and ischemia in all of the structures studied.
Fig. 2.
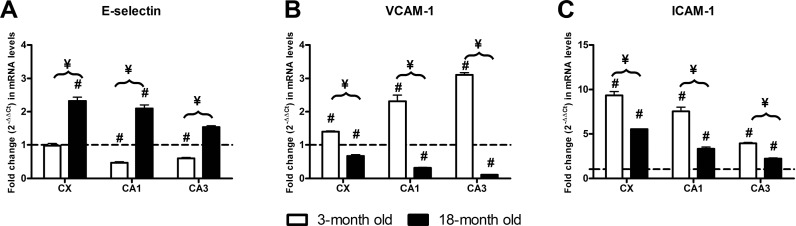
Effect of 48-h I/R on mRNA levels. Fold change (2−ΔΔCt) in (a) E-selectin, (b) VCAM-1 and (c) ICAM-1 mRNA levels between I/R-injured animals when compared with their respective sham-operated animals in the CA1 and CA3 hippocampal structures and the CX. Three-month-old I/R animals (open columns) and 18-month-old animals (black columns) are compared with their respective sham-operated animals (represented as a value of 1, dotted line). I/R-dependent significant differences are represented by a number sign, and significant I/R difference as a consequence of age is represented by a yen sign (p < 0.05, two-way ANOVA, n = 5)
E-selectin protein levels on the hippocampus and cerebral cortex
In the cerebral cortex, E-selectin protein levels were significantly higher in both sham-operated and injured old animals when compared to the young animals. However, in the hippocampus, we failed to detect significant differences in old animals when compared to young animals.
I/R elicited increases in E-selectin in the cerebral cortex and CA3 in young animals, but we could not detect I/R-dependent differences in any of the structures in the old animals (Fig. 3d–f).
Fig. 3.

Effect of age and I/R on selectin protein levels. Representative protein bands of P-selectin (84 kDa) and E-selectin (67 kDa) in the CX (a and d), CA1 (b and e) and CA3 (c and f) in young (open columns) and old animals (black columns). The averages of the densitometric analysis corresponding to five rats (mean ± SEM) normalised to respect to β-actin (40 kDa) are indicated above the bands. Age-dependent significant differences are represented by an asterisk, I/R-dependent significant differences are represented by a number sign and significant interactions between and age and I/R are represented by a yen sign (p < 0.05, two-way ANOVA, n = 5)
P-selectin protein levels on the hippocampus and cerebral cortex
We only detected age-dependent significant P-selectin decreases in the cerebral cortex of sham-operated animals and in the CA3 of old I/R animals when compared with the respective young animals. I/R induced significant changes only in young animals, with significant decreases in P-selectin levels in the cerebral cortex and significant increases in P-selectin in the CA3. Age and I/R showed a significant interaction only in the CA3 of old animals (Fig. 3a–c).
ICAM-1 mRNA levels on the hippocampus and cerebral cortex
In the hippocampus and cerebral cortex, intercellular adhesion molecule 1 (ICAM-1) transcripts of old sham-operated animals were significantly higher when compared to young sham-operated animals. In contrast, ICAM-1 transcripts in old injured animals were significantly lower than those of young animals in both the cortex and hippocampus (Fig. 1c).
I/R elicited increases in ICAM transcript levels in all the structures of both young and old animals. However, in young animals, this effect was more noticeable. We found significant interactions between I/R and age in all structures studied (Fig. 2c).
ICAM-1 protein levels on the hippocampus and cerebral cortex
We only found age-dependent significant increases in ICAM-1 protein levels in the CA1 of sham-operated animals. However, old injured animals had higher levels of this protein in the cerebral cortex and CA1 than young injured animals. We only found I/R-dependent increases of ICAM-1 levels in the CA3 in both young and old injured animals (Fig. 4a–c).
Fig. 4.

Effect of age and I/R on ICAM-1 and GFAP protein levels. Representative protein bands of ICAM1 and GFAP in the CX (a and d), CA1 (b and e) and CA3 (c and f) in young (open columns) and old animals (black columns). The averages of the densitometric analysis corresponding to five rats (mean ± SEM) normalised to respect to β-actin (40 kDa) are indicated above the bands. Age-dependent significant differences are represented by an asterisk, I/R-dependent significant differences are represented by a number sign and significant interactions between and age and I/R are represented by a yen sign (p < 0.05, two-way ANOVA, n = 5)
VCAM-1 mRNA levels on the hippocampus and cerebral cortex
Considering the effect of age, vascular adhesion molecule 1 (VCAM-1) transcripts in the cerebral cortex were significantly lower in the old animals when compared to the young animals in both sham-operated and I/R-injured animals. However, VCAM-1 mRNA levels in the hippocampus of old sham-operated animals were significantly higher than those of the young animals. The transcripts in old injured animals were significantly lower than those in young injured animals in both the cerebral cortex and hippocampus (Fig. 1b).
The results of the effect of I/R are shown in Fig. 2b. Significant I/R-dependent increases in VCAM-1 transcript levels were observed in young animals for all of the structures studied when compared with their young sham-operated animals. This is in contrast to the significant decreases observed in old injured animals with respect to old sham-operated animals in all the structures studied.
GFAP protein levels on the hippocampus and cerebral cortex
GFAP mRNA levels at 48 h after reperfusion in young and old animals have been previously described (Montori et al. 2010c), and therefore, we only present the results corresponding to the GFAP protein levels. Regarding the effect of age, young sham-operated animals had significantly lower GFAP levels in the cerebral cortex when compared with the hippocampal structures. In contrast, GFAP levels of old sham-operated animals were lower in the hippocampus than in the cerebral cortex. With respect to the effect of I/R, we observed that GFAP levels decreased 48 h after I/R in young animals in all structures when compared with the sham-operated animals. This is in contrast with the increase in GFAP levels observed in the old injured animals when compared with their respective sham-operated animals (Fig. 4d–f).
Cleaved caspase-3 immunolabelling on the hippocampus and cerebral cortex
In the cerebral cortex, the quantification of the apoptosis was performed in the cerebral cortical layer V pyramidal cells and in the CA1 and CA3 pyramidal cells by determining the percentage of cleaved caspase-3-positive with respect to the total number of cell pyramidal DAPI-stained nuclei. Two different immunocytochemical patterns of cleaved caspase-3 labelling were observed (Fig. 5). One of them was characterised by a cytoplasmic labelling, while the other showed labelling of mainly the nuclear area that correlated with abnormal nuclear morphology (Fig. 6d). The first pattern was significantly higher in the pyramidal cells of old animals than in young animals, while the nuclear staining was significantly higher in young animals than in old animals (Fig. 6d).
Fig. 5.

Different labelling patterns of apoptosis. Labelling of cleaved caspase-3 shows two different patterns: nuclear labelling (arrows), which is predominant in young animals, and cytoplasmic labelling (asterisks), which is predominant in old animals. Nuclear labelling with DAPI and merged images. Bar = 20 μm
Fig. 6.

Age- and I/R-dependent apoptosis. Comparison of the percentage of apoptosis between young and old animals in the CX (a), CA1 (b) and CA3 (c) in sham-operated (open columns) and I/R-injured (black columns) animals. The pattern of staining in both young and old I/R-injured animals in the different structures is also shown (d). Age-dependent significant differences are represented by an asterisk, and I/R-dependent significant differences are represented by a number sign. No significant interactions between age and I/R were found (p < 0.05, two-way ANOVA, n = 5)
Age-dependent effects on apoptosis (measured as the percentage of cleaved caspase-3 cells) were only observed in the CA3 of sham-operated animals and in the CA1 of injured animals. However, a noticeable I/R-dependent increase was observed in all of the structures for both young and old animals (Fig. 6a–c).
Discussion
The main results of this study can be summarised as follows: (a) The effects on the leukocyte transmigration markers, E-selectin, VCAM-1 and ICAM-1 transcription are modified by both ischemia and age, and there is a significant interaction between these two factors; (b) some of the results did not reach significance in the protein analysis of this process, but significant effects of both age and ischemia and its interaction are consistent with the mRNA results; and c) in a similar way, the results of GFAP as a marker of gliosis and the cleaved caspase-3 as a marker of cell death show the effect of age and ischemia and their significant interaction.
Selection of times and markers
The process of brain inflammation that follows I/R starts within the first hours after injury and continues for weeks (Schilling et al. 2003; Tanaka et al. 2003; Yilmaz et al. 2006). However, the maximal recruitment of circulating inflammatory cells in the brain occurs 2 days after ischemia (Stevens et al. 2002; Gelderblom et al. 2009; Jin et al. 2010). Thus, comparison between young and old animals at this time is crucial to determining age-dependent differences. The onset of apoptotic morphology in the core of the lesion has been described to occur between 6 and 12 h, but the development of cell death in the penumbra is difficult to know (Lipton 1999). Some studies on focal ischemia indicate that 48 h after ischemia, 30 % of cells in the ischemic core appear damaged, but show no signs of death, and 15 % of cells are apparently healthy (Li et al. 1998). On the other hand, 48 h after ischemia is considered the limit of the viability for cells in the penumbra area (Meisel et al. 2005; Durukan and Tatlisumak 2007; Kadhim et al. 2008; Lakhan et al. 2009; Kriz and Lalancette-Hebert 2009; Candelario-Jalil 2009). Thus, we chose this time for the study because it seems to be critical in the analysis for both inflammation and cell death.
One of the hallmarks of inflammation in the brain is the activation of glia, and GFAP is probably the best marker of gliosis (Busch and Silver 2007; Rolls et al. 2009). In this regard, gliosis drives a change in morphology that requires the expression of GFAP, which is a necessary marker to evaluate neuroinflammation. Adhesion molecules are markers that should be monitorised since the recruitment of neutrophils requires an adhesion process to allow the cell transmigration across the vascular endothelium (Wang et al. 2007). In this regard, selectins and CAMs are the most relevant molecules involved in the adhesion process (Stanimirovic and Satoh 2000; Wang et al. 2007; Petri et al. 2008). In an attempt to estimate the role of neuroinflammation in apoptosis, we used the effector cleaved caspase-3, which is considered the main marker in apoptosis (He et al. 2006; Wang et al. 2013; Fan et al. 2014).
Cell death
Although we did not find age-dependent changes in the number of apoptotic cells at 48 h after I/R, differences in the apoptotic activity modulated by age have been described at 8 days after global ischemia in the hippocampal CA1 region (He et al. 2006). Our results show differences in the apoptotic labelling, indicating age-dependent differences in the time-course of apoptosis. In this regard, the pattern of a predominant nuclear staining has been described as a later stage in the apoptosis process than the cytoplasmic staining (Eckle et al. 2004). Our data indicate that at 48 h after I/R, old animals exhibit earlier apoptotic stages than young animals, which could represent a higher ratio of cell death in young rats, or age-dependent differences in the time-course of the apoptosis. The similar amounts of the total number of neuronal nuclei for the different conditions and the general similar percentages of apoptotic cells led us to assume that differences in the pattern of caspase labelling mirror differences in the apoptotic time-course rather than the total amount of death due to apoptosis. Studies carried out using a middle cerebral artery occlusion (MCAO) model indicate that cell death in aged animals is higher after 3 days of the injury (Popa-Wagner et al. 2007). However, the time-course of cell death in this model and the one studied here is difficult to compare. The areas studied in the MCAO model are close to the infarct core (Popa-Wagner et al. 2007), while our model simulates only the penumbra area. The distance of a cell to the infarct core is crucial in deciding whether the cell survives or dies, and obviously, the onset of apoptosis can be radically different.
A secondary conclusion of the results of the analysis of the ischemic and sham-operated cell ratios in both young and old animals is that 48 h after I/R is too early to properly measure age-related differences in the caspase-dependent delayed cell death.
The low affinity binding
Our data show that both P- and E-selectins are expressed in the brains of young (3 month) and old (24 month) rats and that there are age-dependent differences between them in the 48-h I/R response. Thus, while E-selectin transcript levels observed in old sham-operated animals are lower than their corresponding young sham-operated animals, 48-h I/R rats reveal an age-dependent increase in transcription. This indicates that E-selectin transcription is maintained at least until the age studied in this report, which is in contrast with our initial hypothesis. In this regard, we thought that age-dependent decreases in the enzymatic activity (Sinha et al. 2005; Bala et al. 2006) could be due to decreases in their expression.
E-selectin has been reported to induce the expression of CD11/CD18 integrins, which are required for the neutrophils to bind with high affinity to VCAMs (Petri et al. 2008). Thus, our results (both in mRNA and protein) support that the low affinity binding (rolling) is maintained in at least the old animals studied here. Protein results also indicate that CA3 and CX, but not CA1, presented and increased I/R-dependent responses in young animals, which correlate with the lower vulnerability of the CA3 and cortex to ischemic damage. However, this difference was not maintained in the older animals, as has been recently reported (Lalonde and Mielke 2014).
P-selectin presented a constitutive expression in peripheral tissues, but not in the cerebral endothelium (Gotsch et al. 1994; Barkalow et al. 1996). The mRNA transcript levels were too low to study accurately, and therefore, we only analysed the P-selectin protein levels. Our data suggest that in old animals, this protein presents a lessened ability of response, in contrast to what was observed for E-selectin. It is also possible that at 48 h after I/R, P-selectin is not at its highest concentration since it is released earlier than E-selectin (Zhang et al. 1998). Therefore, we must note that, although the transcription of the E-selectin gene is maintained, we cannot be sure what happened with P-selectin since it could follow a different time-course. We also found I/R-dependent differences in P-selectin in young animals, both in the cerebral cortex and CA3, but not in CA1, as was observed for E-selectin, although in P-selectin, the CA3 and CX present a diametric response. These types of differences suggest different types of regulation of the cerebrovascular unit in different areas of the brain. Differences in the endothelium along different areas of the brain have already been described (McIntosh and Warnock 2013). Our data support that these differences could be lessened by age.
High affinity binding
Our results indicate an age-dependent diametric response in the CAMs and E-selectin transcripts in sham-operated animals, thus indicating that the transcriptional response of the molecules related with low and high affinity binding of neutrophils is modified by aging. The protein response is not as clear, but it must be taken into account that the method of protein detection is less accurate than qPCR detection. In addition, some overlap in the function of ICAM-1 and selectins (Salas et al. 2006) could explain why the differences are less evident when protein expression was compared. The results for ICAM-1 expression support the idea that transmigration or the high affinity binding is maintained or even increased in all of the structures of older animals after the ischemic damage.
Thus, the response of the selectins and CAMs observed in I/R-injured animals adds further support to the idea that there are age-dependent differences for the molecules involved in low and high affinity binding of the neutrophils. Thus, this study shows that age modifies the response of these adhesion molecules either by altering their transcription or their time-course expression.
GFAP
One of the most noticeable findings in our study was the diametric response of GFAP to the ischemic insult in young and old animals. The results from the young animals confirm the ischemia-dependent decrease in GFAP expression that was previously described 48 h after reperfusion in a 4VO model of global ischemia in young Wistar rats (Zhang et al. 2007). Several studies provide evidence that aging brain reacts stronger to I/R with an early inflammation response (Badan et al. 2003; Popa-Wagner et al. 2007; Buga et al. 2013), and it has been reported that aged Sprague–Dawley rats present an early glial scar in an MCAO model (Popa-Wagner et al. 2006). Also, GFAP reactivity in humans has been described to depend on the age of patient (Dziewulska 1997). All of the above is consistent with our results. Thus, all of the reports show differences in GFAP protein expression between young and old animals.
The hippocampus and cerebral cortex have been reported to have different vulnerabilities to the ischemia (Kirino et al. 1985; Dijkhuizen et al. 1998; Xu et al. 2001). This difference is consistent with differences observed in the expression of different neurotransmitter system genes (Montori et al. 2010a, b, c) or markers of reticulum stress (Llorente et al. 2013). Our results show that changes induced by age modify this structure-dependent vulnerability.
In summary, this study shows different responses in old and young animals at 48 h of I/R, including different patterns of apoptotic labelling, GFAP reactivity and molecules involved in high and low affinity binding of neutrophils. We think that these age-dependent differences represent changes in the time-course response to I/R and should be taken into account in the treatments of or during the development of therapeutic targets for stroke.
Acknowledgments
We wish to thank Marta Fernandez Caso from the University of Leon for technical support and personal help. This study was supported by Junta of Castilla of León (LE184A12-2). Diego Pérez Rodríguez is granted by Junta de Castilla y León (EDU/346/2013)
Conflict of interest
The authors declare that they have no conflict of interests.
Abbreviations
- BBB
Blood–brain barrier
- BSA
Bovine serum albumin
- CA
Cornu Ammonis
- CAM
Cellular adhesion molecules
- DABCO
1,4-Diazabicyclo(2.2.2)octane
- DAPI
4′,6-Diamidino-2-phenylindole
- GFAP
Glial fibrillary acidic protein
- I/R
Ischemia/reperfusion
- ICAM
Intercellular adhesion molecule
- MCAO
Middle cerebral artery occlusion
- MRI
Magnetic resonance imaging
- PFA
Paraformaldehyde
- PBST
Buffer sodium phosphate with Triton X-100
- TBST
Tris-buffered saline 50 mM with Tween-20 0.2 %
- VCAM
Vascular adhesion molecule
Contributor Information
Berta Anuncibay-Soto, Email: b.anuncibaysoto@gmail.com.
Diego Pérez-Rodríguez, Email: dperr@unileon.es.
Irene L Llorente, Email: ilorl@unileon.es.
Marta Regueiro-Purriños, Email: mregf@unileon.es.
José Manuel Gonzalo-Orden, Email: jm.gonzalo.orden@unileon.es.
Arsenio Fernández-López, Email: arsenio.fernandez@unileon.es.
References
- Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony DC, Bolton SJ, Fearn S, Perry VH. Age-related effects of interleukin-1 beta on polymorphonuclear neutrophil-dependent increases in blood–brain barrier permeability in rats. Brain. 1997;120(Pt 3):435–444. doi: 10.1093/brain/120.3.435. [DOI] [PubMed] [Google Scholar]
- Anyanwu EC. Neurochemical changes in the aging process: implications in medication in the elderly. Sci World J. 2007;7:1603–1610. doi: 10.1100/tsw.2007.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam TV, Phillips TM, Cheng A, Morrell CH, Mattson MP, Wan R. Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann Neurol. 2010;67:41–52. doi: 10.1002/ana.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso MI, Garcia-Bonilla L, Martin ME, Salinas M. Assessment of protein expression levels after transient global cerebral ischemia using an antibody microarray analysis. Neurochem Res. 2010;35:1239–1247. doi: 10.1007/s11064-010-0180-9. [DOI] [PubMed] [Google Scholar]
- Back T. Pathophysiology of the ischemic penumbra—revision of a concept. Cell Mol Neurobiol. 1998;18:621–638. doi: 10.1023/A:1020265701407. [DOI] [PubMed] [Google Scholar]
- Badan I, Platt D, Kessler C, Popa-Wagner A. Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology. 2003;49:356–365. doi: 10.1159/000073763. [DOI] [PubMed] [Google Scholar]
- Bala K, Tripathy BC, Sharma D. Neuroprotective and anti-ageing effects of curcumin in aged rat brain regions. Biogerontology. 2006;7:81–89. doi: 10.1007/s10522-006-6495-x. [DOI] [PubMed] [Google Scholar]
- Barkalow FJ, Goodman MJ, Gerritsen ME, Mayadas TN. Brain endothelium lack one of two pathways of P-selectin-mediated neutrophil adhesion. Blood. 1996;88:4585–4593. [PubMed] [Google Scholar]
- Bendel O, Alkass K, Bueters T, von Euler M, von Euler G. Reproducible loss of CA1 neurons following carotid artery occlusion combined with halothane-induced hypotension. Brain Res. 2005;1033:135–142. doi: 10.1016/j.brainres.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Blamire AM, Anthony DC, Rajagopalan B, Sibson NR, Perry VH, Styles P. Interleukin-1beta-induced changes in blood–brain barrier permeability, apparent diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance study. J Neurosci. 2000;20:8153–8159. doi: 10.1523/JNEUROSCI.20-21-08153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buga AM, Di Napoli M, Popa-Wagner A. Preclinical models of stroke in aged animals with or without comorbidities: role of neuroinflammation. Biogerontology. 2013;14:651–662. doi: 10.1007/s10522-013-9465-0. [DOI] [PubMed] [Google Scholar]
- Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E. Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs. 2009;10:644–654. [PubMed] [Google Scholar]
- Colangelo AM, Alberghina L, Papa M (2014) Astrogliosis as a therapeutic target for neurodegenerative diseases. Neurosci Lett [DOI] [PubMed]
- Collins TC, Petersen NJ, Menke TJ, Souchek J, Foster W, Ashton CM. Short-term, intermediate-term, and long-term mortality in patients hospitalized for stroke. J Clin Epidemiol. 2003;56:81–87. doi: 10.1016/S0895-4356(02)00570-X. [DOI] [PubMed] [Google Scholar]
- Dijkhuizen RM, Knollema S, van der Worp HB, Ter Horst GJ, De Wildt DJ, Berkelbach van der Sprenkel JW, Tulleken KA, Nicolay K. Dynamics of cerebral tissue injury and perfusion after temporary hypoxia-ischemia in the rat: evidence for region-specific sensitivity and delayed damage. Stroke. 1998;29:695–704. doi: 10.1161/01.STR.29.3.695. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Duverger D, MacKenzie ET. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, and age. J Cereb Blood Flow Metab. 1988;8:449–461. doi: 10.1038/jcbfm.1988.86. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Mader S, Akiyoshi K, Ren X, Ayala P, Burrows GG, Vandenbark AA, Herson PS, Hurn PD, Offner HA. Therapy with recombinant T-cell receptor ligand reduces infarct size and infiltrating inflammatory cells in brain after middle cerebral artery occlusion in mice. Metab Brain Dis. 2011;26:123–133. doi: 10.1007/s11011-011-9241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewulska D. Age-dependent changes in astroglial reactivity in human ischemic stroke. Immunohistochemical study. Folia Neuropathol. 1997;35:99–106. [PubMed] [Google Scholar]
- Eckle VS, Buchmann A, Bursch W, Schulte-Hermann R, Schwarz M. Immunohistochemical detection of activated caspases in apoptotic hepatocytes in rat liver. Toxicol Pathol. 2004;32:9–15. doi: 10.1080/01926230490260673. [DOI] [PubMed] [Google Scholar]
- Fan W, Dai Y, Xu H, Zhu X, Cai P, Wang L, Sun C, Hu C, Zheng P, Zhao BQ. Caspase-3 modulates regenerative response after stroke. Stem Cells. 2014;32:473–486. doi: 10.1002/stem.1503. [DOI] [PubMed] [Google Scholar]
- Fricker M, Vilalta A, Tolkovsky AM, Brown GC. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J Biol Chem. 2013;288:9145–9152. doi: 10.1074/jbc.M112.427880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Pulsinelli WA. The ischemic penumbra, injury thresholds, and the therapeutic window for acute stroke. Ann Neurol. 1994;36:553–554. doi: 10.1002/ana.410360402. [DOI] [PubMed] [Google Scholar]
- Gotsch U, Jager U, Dominis M, Vestweber D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha in vivo. Cell Adhes Commun. 1994;2:7–14. doi: 10.3109/15419069409014198. [DOI] [PubMed] [Google Scholar]
- He Z, Meschia JF, Brott TG, Dickson DW, McKinney M. Aging is neuroprotective during global ischemia but leads to increased caspase-3 and apoptotic activity in hippocampal neurons. Curr Neurovasc Res. 2006;3:181–186. doi: 10.2174/156720206778018802. [DOI] [PubMed] [Google Scholar]
- Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557–565. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadhim HJ, Duchateau J, Sebire G. Cytokines and brain injury: invited review. J Intensive Care Med. 2008;23:236–249. doi: 10.1177/0885066608318458. [DOI] [PubMed] [Google Scholar]
- Kirino T, Tamura A, Sano K. Selective vulnerability of the hippocampus to ischemia—reversible and irreversible types of ischemic cell damage. Prog Brain Res. 1985;63:39–58. doi: 10.1016/S0079-6123(08)61974-3. [DOI] [PubMed] [Google Scholar]
- Kriz J, Lalancette-Hebert M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009;117:497–509. doi: 10.1007/s00401-009-0496-1. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde CC, Mielke JG. Selective vulnerability of hippocampal sub-fields to oxygen-glucose deprivation is a function of animal age. Brain Res. 2014;1543:271–279. doi: 10.1016/j.brainres.2013.10.056. [DOI] [PubMed] [Google Scholar]
- Li Y, Powers C, Jiang N, Chopp M. Intact, injured, necrotic and apoptotic cells after focal cerebral ischemia in the rat. J Neurol Sci. 1998;156:119–132. doi: 10.1016/S0022-510X(98)00036-7. [DOI] [PubMed] [Google Scholar]
- Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Hoi Yu AC. Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2011;8:67–80. doi: 10.2174/156720511794604543. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu F, McCullough LD. Middle cerebral artery occlusion model in rodents: methods and potential pitfalls. J Biomed Biotechnol. 2011;2011:464701. doi: 10.1155/2011/464701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, McCullough LD. Interactions between age, sex, and hormones in experimental ischemic stroke. Neurochem Int. 2012;61:1255–1265. doi: 10.1016/j.neuint.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Benashski SE, Persky R, Xu Y, Li J, McCullough LD. Age-related changes in AMP-activated protein kinase after stroke. Age (Dordr) 2012;34:157–168. doi: 10.1007/s11357-011-9214-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Llorente IL, Burgin TC, Perez-Rodriguez D, Martinez-Villayandre B, Perez-Garcia CC, Fernandez-Lopez A (2013) Unfolded protein response to global ischemia following 48 h of reperfusion in the rat brain: the effect of age and meloxicam. J Neurochem 127(5):701–710 [DOI] [PubMed]
- McIntosh CT, Warnock JN. Side-specific characterization of aortic valve endothelial cell adhesion molecules under cyclic strain. J Heart Valve Dis. 2013;22:631–639. [PubMed] [Google Scholar]
- Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Montori S, Dos Anjos S, Rios-Granja MA, Perez-Garcia CC, Fernandez-Lopez A, Martinez-Villayandre B. AMPA receptor downregulation induced by ischaemia/reperfusion is attenuated by age and blocked by meloxicam. Neuropathol Appl Neurobiol. 2010;36:436–447. doi: 10.1111/j.1365-2990.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- Montori S, Martinez-Villayandre B, Dos-Anjos S, Llorente IL, Burgin TC, Fernandez-Lopez A. Age-dependent modifications in the mRNA levels of the rat excitatory amino acid transporters (EAATs) at 48 hour reperfusion following global ischemia. Brain Res. 2010;1358:11–19. doi: 10.1016/j.brainres.2010.08.020. [DOI] [PubMed] [Google Scholar]
- Montori S, Dos-Anjos S, Martinez-Villayandre B, Regueiro-Purrinos MM, Gonzalo-Orden JM, Ruano D, Fernandez-Lopez A. Age and meloxicam attenuate the ischemia/reperfusion-induced down-regulation in the NMDA receptor genes. Neurochem Int. 2010;56:878–885. doi: 10.1016/j.neuint.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Gjedde A, Diemer NH. Focal ischemia of the rat brain: autoradiographic determination of cerebral glucose utilization, glucose content, and blood flow. J Cereb Blood Flow Metab. 1986;6:414–424. doi: 10.1038/jcbfm.1986.74. [DOI] [PubMed] [Google Scholar]
- Perry VH, Anthony DC, Bolton SJ, Brown HC. The blood–brain barrier and the inflammatory response. Mol Med Today. 1997;3:335–341. doi: 10.1016/S1357-4310(97)01077-0. [DOI] [PubMed] [Google Scholar]
- Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. 2008;180:6439–6446. doi: 10.4049/jimmunol.180.10.6439. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A, Dinca I, Yalikun S, Walker L, Kroemer H, Kessler C. Accelerated delimitation of the infarct zone by capillary-derived nestin-positive cells in aged rats. Curr Neurovasc Res. 2006;3:3–13. doi: 10.2174/156720206775541732. [DOI] [PubMed] [Google Scholar]
- Popa-Wagner A, Badan I, Walker L, Groppa S, Patrana N, Kessler C. Accelerated infarct development, cytogenesis and apoptosis following transient cerebral ischemia in aged rats. Acta Neuropathol. 2007;113:277–293. doi: 10.1007/s00401-006-0164-7. [DOI] [PubMed] [Google Scholar]
- Rami A, Bechmann I, Stehle JH. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog Neurobiol. 2008;85:273–296. doi: 10.1016/j.pneurobio.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Rojas JI, Zurru MC, Romano M, Patrucco L, Cristiano E. Acute ischemic stroke and transient ischemic attack in the very old-risk factor profile and stroke subtype between patients older than 80 years and patients aged less than 80 years. Eur J Neurol. 2007;14:895–899. doi: 10.1111/j.1468-1331.2007.01841.x. [DOI] [PubMed] [Google Scholar]
- Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat Rev Neurosci. 2009;10:235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y, American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–e146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- Salas A, Shimaoka M, Phan U, Kim M, Springer TA. Transition from rolling to firm adhesion can be mimicked by extension of integrin alphaLbeta2 in an intermediate affinity state. J Biol Chem. 2006;281:10876–10882. doi: 10.1074/jbc.M512472200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183:25–33. doi: 10.1016/S0014-4886(03)00082-7. [DOI] [PubMed] [Google Scholar]
- Sinha N, Baquer NZ, Sharma D. Anti-lipidperoxidative role of exogenous dehydroepiendrosterone (DHEA) administration in normal ageing rat brain. Indian J Exp Biol. 2005;43:420–424. [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanimirovic D, Satoh K. Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 2000;10:113–126. doi: 10.1111/j.1750-3639.2000.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932:110–119. doi: 10.1016/S0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- Sughrue ME, Mehra A, Connolly ES, Jr, D’Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53:497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- Tamura A, Graham DI, McCulloch J, Teasdale GM. Focal cerebral ischaemia in the rat: 2. Regional cerebral blood flow determined by [14C]iodoantipyrine autoradiography following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1:61–69. doi: 10.1038/jcbfm.1981.7. [DOI] [PubMed] [Google Scholar]
- Tanaka R, Komine-Kobayashi M, Mochizuki H, Yamada M, Furuya T, Migita M, Shimada T, Mizuno Y, Urabe T. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117:531–539. doi: 10.1016/S0306-4522(02)00954-5. [DOI] [PubMed] [Google Scholar]
- Taylor S, Wakem M, Dijkman G, Alsarraj M, Nguyen M. A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods. 2010;50:S1–S5. doi: 10.1016/j.ymeth.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Zhang Y, Wu L, Wang Y, Cao Y, He L, Li X, Zhao J. Puerarin protected the brain from cerebral ischemia injury via astrocyte apoptosis inhibition. Neuropharmacology. 2013;79C:282–289. doi: 10.1016/j.neuropharm.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Wasserman JK, Yang H, Schlichter LC. Glial responses, neuron death and lesion resolution after intracerebral hemorrhage in young vs. aged rats. Eur J Neurosci. 2008;28:1316–1328. doi: 10.1111/j.1460-9568.2008.06442.x. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) The top 10 causes of death. Fact sheet number 310. Geneva: WHO; 2011. [Google Scholar]
- Xu XJ, Plesan A, Yu W, Hao JX, Wiesenfeld-Hallin Z. Possible impact of genetic differences on the development of neuropathic pain-like behaviors after unilateral sciatic nerve ischemic injury in rats. Pain. 2001;89:135–145. doi: 10.1016/S0304-3959(00)00356-0. [DOI] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30:783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]
- Zarow C, Vinters HV, Ellis WG, Weiner MW, Mungas D, White L, Chui HC. Correlates of hippocampal neuron number in Alzheimer’s disease and ischemic vascular dementia. Ann Neurol. 2005;57:896–903. doi: 10.1002/ana.20503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998;785:207–214. doi: 10.1016/S0006-8993(97)01343-7. [DOI] [PubMed] [Google Scholar]
- Zhang M, Li WB, Geng JX, Li QJ, Sun XC, Xian XH, Qi J, Li SQ. The upregulation of glial glutamate transporter-1 participates in the induction of brain ischemic tolerance in rats. J Cereb Blood Flow Metab. 2007;27:1352–1368. doi: 10.1038/sj.jcbfm.9600441. [DOI] [PubMed] [Google Scholar]