

Abstract
Although recent studies have reported that Lactobacillus rhamnosus GG (LGG), the most extensively studied probiotic strain, exerts an anti-hyperglycemic effect on several rodent models, the underlying mechanism remains unclear. In this study, twenty male C57BL/KsJ-db/db (db/db) mice were divided into 2 groups, LGG-treated and control group, which received a daily dose of LGG (1 × 108 CFU per mouse) and PBS orally for 4 weeks, respectively. We observed that glucose tolerance was significantly improved in LGG-treated db/db mice. Insulin-stimulated Akt phosphorylation and GLUT4 translocation were higher in skeletal muscle of LGG-treated mice relative to their controls. It was also observed that LGG treatment caused significant reductions in endoplasmic reticulum (ER) stress in skeletal muscle and M1-like macrophage activation in white adipose tissues. Our results indicate that the anti-diabetic effect of LGG in db/db mice is associated with alleviated ER stress and suppressed macrophage activation, resulting in enhanced insulin sensitivity. These findings suggest a therapeutic potential of probiotics for prevention and treatment of type 2 diabetes.
Keywords: probiotics, type 2 diabetic mice, insulin resistance, ER stress, M1-like macrophage activation
Introduction
Metabolic syndrome is a group of risk factors such as obesity, dyslipidemia, hyperglycemia and the proinflammatory state, which is associated with insulin resistance.(1) One of the major goals of treating metabolic syndrome is to reduce the risk of heart disease by controlling hyperglycemia, for which several pharmacologic agents are being used. However, of particular concern is the tendency for most antihyperglycemic treatments to have side effects such as weight gain, hepatotoxicity, and cardiovascular disease.(2) Therefore, alternative therapeutic strategies for the development of agents with reduced side effects are required for more effective prevention and treatment.
Gut microbiota is a complex of microorganism species residing in the mammalian gastrointestinal tract, and recent studies have manifested that gut microbiota is strongly associated with metabolic disorders.(3,4) Studies employing diet-induced obese (DIO) mouse model have suggested that altered gut microbiota composition is related to the pathogenesis of obesity and type 2 diabetes in the host.(5,6) Given the link between the gut microbiota and metabolic disease, it is reasonable to hypothesize that restoration or supplementation of certain bacterial populations may have beneficial effects on metabolic status. Hence, probiotics have emerged as a novel therapeutic agent with the potential to prevent and treat metabolic syndrome.(3–8)
Probiotics are defined as live organisms that, when ingested in adequate amounts, exert health benefits to the host.(9) It has recently been proven that probiotics ameliorate metabolic and infectious diseases by manipulating the intestinal microbiota or affecting the host.(9,10) Lactobacillus rhamnosus GG (LGG), a well-known probiotic strain, has been shown to have beneficial effects on diarrhea,(10–12) atopic dermatitis,(13) and hypercholesterolemia.(14) A few recent studies also showed hypoglycemic effects of LGG in diabetic animal models.(15–17) However, the underlying molecular mechanisms for the reported effect of LGG on insulin sensitivity are still unclear and remain to be elucidated. In the present study, we found that LGG treatment enhanced insulin sensitivity in leptin receptor-deficient (db/db) mice, a type 2 diabetic animal model. It was also observed that both of endoplasmic reticulum (ER) stress in skeletal muscle and M1-like macrophage activation in white adipose tissues were significantly reduced in LGG-treated db/db mice, and as a consequence, the glucose tolerance was improved. These findings indicate profound therapeutic benefits of probiotics for prevention and treatment of type 2 diabetes.
Materials and Methods
Animals
Four-week-old male C57BL/KsJ-db/db (db/db) mice and male C57BL/KsJ (wild-type) mice were purchased from Hyochang Bioscience (Daegu, Korea) and stabilized metabolic conditions with feeding a standard chow diet (2018S, Harlan Laboratories, Indianapolis, IN) for a week. Mice were housed on a 12 h dark/light cycle at a constant temperature of 22 ± 1°C and humidity of 45 ± 10%. After stabilization, mice were divided into 2 groups, LGG-treated and control group, which received a daily dose of LGG (1 × 108 CFU per mouse) and PBS orally for 4 weeks, respectively. The wild-type (WT) mice were orally given PBS.
Mice were fasted for 4 h, and sacrificed. Tissues of liver, epididymal fat, mesenteric fat, and quadriceps muscle were rapidly excised, snap-frozen in liquid nitrogen, and stored at −75°C until processed for experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of Handong Global University (IACUC apporval no. HGU201302).
Glucose tolerance test
After 4 weeks of LGG treatment, mice were fasted for 16 h and followed by intraperitoneal injection of glucose (1 g/kg). Blood samples were obtained by tail-bleeding and blood glucose level was checked by Accu-Check Go (Roche Diagnostics GmbH, Basel, Switzerland) at 0, 15, 30, 60, 90 and 120 min after glucose injection.
Western blot analysis
Immunoblotting was performed as described previously.(17) Protein was extracted using PRO-PREP protein extraction solution (iNtRON Biotechnology, Seongnam, Korea), centrifuged, boiled, separated by 10% SDS-PAGE, and transferred to PVDF membranes. Membranes were blocked, and incubated with primary antibodies against total Akt, phospho (Ser 473) Akt, binding immunoglobulin protein (BiP), and β-actin (Cell signaling technology, Beverly, MA). Membranes were washed, and incubated with anti-rabbit IgG conjugated with horseradish peroxidase (Cell signaling). Signals were detected by enhanced chemiluminescence, and analyzed by AlphaImager 2200 (Protein simple, Santa Clara, CA).
Real-time RT PCR
Total RNA extraction, reverse transcription, and quantitative PCR were performed as described previously.(17) In brief, total RNA was extracted using TRI reagent (Molecular Research Center, Cincinnati, OH) and reverse transcribed with oligo (dT) primer and GoScriptTM reverse transcriptase (Promega, Madison, WI). Gene expression was analyzed using SYBR Premix Ex TaqTM (Tli RNaseH Plus) kit (Takara Bio Inc., Shiga, Japan) on ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA). Quantification of gene transcripts for acetyl-CoA carboxylase (ACC), BiP, CD11b, CD11c, CD36, C/EBP homology protein (CHOP), carnitine palmitoyltransferase 1 (CPT1), diglyceride acyltransferase 1 (DGAT1), F4/80, fatty acid synthase (FAS), glycerol-3-phosphate acyltransferase (GPAT), interleukin-6 (IL-6), monocyte chemotactic protein-1 (MCP-1), protein disulfide-isomerase (PDI), sterol regulatory element-binding protein 1c (SREBP1c), tumor necrosis factor α (TNFα), tribbles-related protein 3 (TRB3), uncoupling protein 2 (UCP2), and UCP3 was performed by using gene-specific primers. Primer sequences are available upon request. Results were presented as mean ± SEM normalized to expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-actin using the ΔΔCt method.
Immunofluorescence
After 4-h fasting and acute insulin stimulation by intraperitoneal insulin administration (5 U/kg) for 5 min, mice were sacrificed, and quadriceps muscle tissue was rapidly excised. Tissues were fixed in 10% formalin, embedded in paraffin, cut at 4-µm thickness, and deparaffinized. Antigens were retrieved by boiling in 10 mM citrate buffer (pH 6.0) for 20 min. To block the nonspecific binding, sections were blocked with buffer (5% fetal bovine serum, 0.3% BSA and 0.25% Tween 20 in PBS) for 2 h at room temperature, and incubated with anti-GLUT4 antibody (Abcam, Cambridge, UK) for 1 h. After washing, sections were incubated with Alexa Fluor 488-conjugated secondary antibody (Life Technologies, Carlsbad, CA) for 30 min. For immunofluorescence, slides were directly mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and examined under a Nikon super-resolution N-STORM fluorescence microscope.
Statistical analysis
Numerical data were presented as mean ± SEM. Differences between groups were assessed by two-tailed Student’s t test. P values <0.05 were considered as statistically significant.
Results
LGG treatment on db/db mice improves glucose tolerance by facilitating the insulin signaling pathway in skeletal muscle
In previous study, we demonstrated that LGG treatment improves glucose tolerance and reduces adiposity in DIO mice.(17) To assess the effect of LGG on glucose tolerance in db/db mice in this study, glucose tolerance test was performed. After 4 weeks of LGG treatment, db/db mice had significantly improved glucose tolerance compared to PBS-treated db/db controls (Fig. 1B). Unlike the results obtained with DIO mice, however, body weight remained unchanged after LGG treatment (Fig. 1A).
Fig. 1.
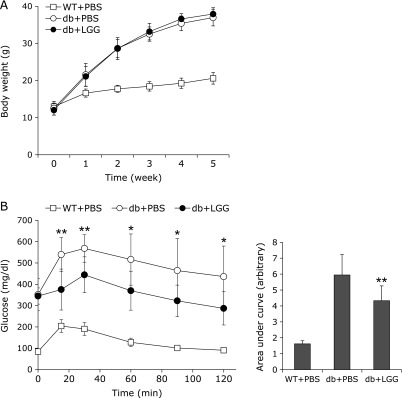
LGG improves glucose tolerance in db/db mice, but body weight remains unchanged. (A) Effect of LGG treatment on body weight and (B) glucose disposal. *p<0.05 and **p<0.01 between PBS- and LGG-treated db/db mice. Data represent means ± SD for 9 or 10 mice per group. WT + PBS: PBS-treated wild-type mice, db + PBS: PBS-treated db/db mice, db + LGG: LGG-treated db/db mice.
To elucidate how glucose disposal was enhanced in LGG-treated db/db mice, we first examined whether LGG treatment augmented insulin signaling in adipose and skeletal muscle tissue. Phosphorylated Akt level was significantly higher in skeletal muscle of LGG-treated mice relative to the PBS-treated controls (Fig. 2A), but not in adipose tissue (data not shown). To verify the insulin-stimulated translocation of GLUT4 to the plasma membrane, immunofluorescence staining was performed on quadriceps muscle. In LGG-treated db/db mice, the translocation of GLUT4 to the plasma membrane upon stimulation with insulin was markedly higher than that seen in PBS-treated controls (Fig. 2B).
Fig. 2.

LGG facilitates insulin signaling pathway in skeletal muscle of db/db mice. (A) Effect of LGG treatment on insulin-stimulated Akt phosphorylation. After 4 h fasting and acute insulin stimulation by intraperitoneal insulin administration (5 U/kg) for 5 min, mice were sacrificed, and quadriceps muscle tissue was rapidly excised. Proteins were extracted from the tissue for SDS-PAGE-immunoblot analysis. Data represent means ± SEM for 4 or 5 mice per group. *p<0.001 between insulin-stimulated PBS- and LGG-treated db/db mice. (B) Effect of LGG treatment on GLUT4 translocation. Paraffin sections of quadriceps muscle were stained with antibody to GLUT4. Immunofluorescence showed the presence of GLUT4 on the plasma membrane. Experiments were repeated three times, with reproducible results. WT + PBS: PBS-treated wild-type mice, db + PBS: PBS-treated db/db mice, db + LGG: LGG-treated db/db mice.
LGG treatment alleviates ER stress by modulating lipid metabolism in skeletal muscle
ER stress in skeletal muscle is one of the main causes involved in the development of insulin resistance.(18) To examine whether the improvement of glucose disposal capacity is due to a reduction of ER stress in insulin-responsive tissues of LGG-treated db/db mice, we analyzed mRNA expression levels of ER stress genes including CHOP, BiP, PDI and TRB3. While there was no difference in mRNA expression levels in adipose tissue and the liver, the expression of ER stress genes was decreased in skeletal muscle of LGG-treated mice (Fig. 3A). The lower level of BiP protein in skeletal muscle of LGG-treated mice relative to PBS-treated controls provided a supporting evidence for the alleviating effect of LGG on ER stress (Fig. 3B).
Fig. 3.
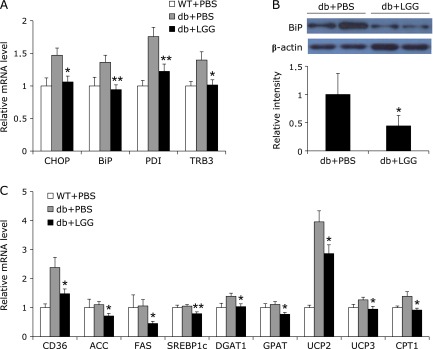
LGG alleviates ER stress by decreasing lipotoxicity in skeletal muscle of db/db mice. (A) Effect of LGG treatment on mRNA expression of ER stress genes in skeletal muscle. Total RNA were extracted from skeletal muscle of mice (n = 9 or 10 per group) and gene expression levels were analyzed. (B) BiP protein levels in skeletal muscle of PBS- and LGG-treated mice (n = 4 or 5 per group). Proteins were extracted from the tissue for SDS-PAGE-immunoblot analysis. (C) Effect of LGG treatment on mRNA expression of genes associated with lipid metabolism in skeletal muscle (9 or 10 mice per group). All genes are normalized with mRNA expression level of β-actin. Data represent means ± SEM. *p<0.05 and **p<0.01 between PBS- and LGG-treated db/db mice. WT + PBS: PBS-treated wild-type mice, db + PBS: PBS-treated db/db mice, db + LGG: LGG-treated db/db mice.
To elucidate a plausible mechanism underlying the reduced ER stress caused by LGG treatment, we analyzed mRNA expression levels of genes associated with uptake, synthesis, and oxidation of fatty acids. The expression of mRNAs of CD36, which is involved in long-chain fatty acid uptake, and lipogenic genes including ACC, FAS, SREBP1c, DGAT, and GPAT, lipid-oxidative genes including UCP2, UCP3, and CPT1 were significantly downregulated in skeletal muscle of LGG-treated db/db mice (Fig. 3C).
LGG treatment reduces macrophage infiltration and activation in white adipose tissues
To determine whether the improvement of glucose tolerance in LGG-treated db/db mice is a consequence of immune modulation, we measured the expression of key proinflammatory cytokines in white adipose tissues, epididymal and mesenteric adipose tissue. However, there were no differences in mRNA expression of TNFα and IL-6 between PBS-treated control and LGG-treated mice (Fig. 4). To further examine the effect of LGG treatment on immune and inflammatory responses, we analyzed gene expression of macrophage markers, since macrophage infiltration and activation is critical to initiate and amplify chronic inflammation. In both epididymal and mesenteric adipose tissues, mRNA expression levels of macrophage markers such as F4/80 and CD11b were lower in LGG-treated mice relative to PBS-treated controls. Moreover, interestingly, mRNA expression of genes involved in monocyte recruitment and M1-like macrophage activation, MCP-1 and CD11c, respectively, was also significantly decreased in adipose tissues of LGG-treated mice.
Fig. 4.

LGG suppresses macrophage infiltration and M1-like activation in white adipose tissues of db/db mice, but the expression of TNFα and IL-6 remains unchanged. Total RNA were extracted from epididymal and mesenteric adipose tissues of mice (n = 4 or 5 per group) and expression level of genes related to macrophage infiltration and activation was analyzed. All genes are normalized with mRNA expression level of GAPDH. Data represent means ± SEM. *p<0.05 and **p<0.01 between PBS- and LGG-treated db/db mice. WT + PBS: PBS-treated wild-type mice, db + PBS: PBS-treated db/db mice, db + LGG: LGG-treated db/db mice, EAT: epididymal adipose tissue, MAT: mesenteric adipose tissue.
Discussion
Recent studies on how gut microbiota affect host metabolism have been performed briskly. Reported data have suggested a possibility to improve host’s metabolic status by modifying gut microbiota through probiotic supplementation.(19–26) Hence, together with their long-term safety and cost-effectiveness, oral delivery of probiotics can be recommended as a therapeutic way. Multiple researches have provided evidences for regulatory effects of probiotics on metabolic and inflammatory responses,(19–26) suggesting that we could consider probiotic therapy as a rational approach to address the problems of metabolic syndrome. In particular, LGG, a probiotic bacterial strain, has been shown to confer health benefits(10–13) and also to prevent metabolic disorders.(14–17) It was reported that LGG administration decreased the level of glycated hemoglobin and ameliorated glucose tolerance through stimulation of insulin secretion in streptozotocin-induced diabetic rats.(15) In KK-Ay mice, a genetic type 2 diabetic model, treated with LGG, fasting and postprandial blood glucose levels were suppressed and glucose tolerance was improved.(16) We also previously showed that LGG improved insulin sensitivity with reduced adiposity in DIO mice through enhancement of adiponectin production.(17) However, much less information is available for the molecular mechanisms underlying the reported effects of LGG. In this study, we observed an improved glucose tolerance in db/db mice treated with LGG, and discovered an interesting mechanism whereby reduced ER stress in skeletal muscle and M1-like macrophage activity in white adipose tissue could be the important links to the insulin sensitizing effect of LGG.
First, the improvement of glucose sensitivity was confirmed with an increased level of phosphorylated Akt and GLUT4 translocation in skeletal muscle of LGG-treated db/db mice (Fig. 2A and B). To explore the underlying mechanism, we focused on the modulation of ER stress, oxidative stress, and macrophage activity caused by LGG treatment.
Upon ER stress, cells activate unfolded protein response to cope with any disruption in normal ER function, resulting in an upregulated transcription of proteins such as BiP, PDI, TRB3, and CHOP. These changes lead to increase protein folding capacity and decrease unfolded protein load, rendering cells adaptable to the stressed condition. Recent studies have shown that ER stress markers were overexpressed in insulin-responsive tissues of obese rodents,(27) which was further confirmed in biopsies of obese patients.(28,29) In this study, we found that expression of ER stress markers including BiP, PDI, TRB3, and CHOP in skeletal muscle of db/db mice was higher than that of their WT counterparts (Fig. 3A). However, the expression of ER stress markers was markedly reduced in skeletal muscle of db/db mice treated with LGG for 4 weeks (Fig. 3A and B), indicating that LGG has an alleviating effect on ER stress.
It is known that ER stress plays an important role in the progression of insulin resistance in tissue-specific manner.(18) Particularly in skeletal muscle, ectopic accumulation of fatty acid, lipotoxicity, is the main cause of insulin resistance. Several researches have demonstrated that ER stress mediates palmitate-induced insulin resistance in skeletal muscle cells.(30–32) Direct exposure of myocytes to palmitate can induce ER stress, and as a result, insulin signaling is interfered through an inactivation of Akt.(33) In this study, we observed a significantly higher expression of genes included in fatty acid uptake and oxidation such as CD36 and CPT1, and slightly higher expression of lipogenic genes such as ACC, FAS, SREBP1c, DGAT1, and GPAT in skeletal muscle of db/db mice relative to WT mice (Fig. 3C), indicating that the insulin resistance in db/db mice is associated with lipotoxicity in skeletal muscle. However, 4-week LGG treatment markedly decreased the expression of genes mentioned above, suggesting that LGG treatment lowers lipotoxicity through modulating the lipid metabolic activity of skeletal muscle. In addition, elevated oxidative stress provoked by lipotoxicity in skeletal muscle under diabetic condition is accompanied by ER stress.(34,35) This is verified by the increase in mRNA expression of UCP2 and UCP3 to attenuate the mitochondrial production of reactive oxygen species.(36) Especially, skeletal muscle UCP2 and UCP3 gene expression has been shown to be up-regulated by free fatty acid flux.(37) It also has been reported that expression of UCP2 and UCP3 mRNA in skeletal muscle of type 2 diabetic patients was significantly reduced by very-low-calorie diet intervention.(38) Consistently, we found that the mRNA expression of UCP2 and UCP3 in skeletal muscle of db/db mice was markedly increased, which was significantly recovered by LGG treatment (Fig. 3C). Taken together, our data suggest that LGG treatment has an insulin-sensitizing effect by alleviating ER and oxidative stress as a consequence of decreased lipotoxicity, followed by enhanced insulin signaling in skeletal muscle.
Chronic inflammation in white adipose tissue also contributes obesity-induced insulin resistance and type 2 diabetes.(39) Increased macrophage infiltration is associated with obesity, and macrophages which are present in higher numbers in adipose tissue of obese subjects than in that of lean subjects appear to be major sources of inflammatory mediators that are linked to insulin resistance. Adipose tissue macrophages express the antigens F4/80 and CD11b on the surface, and it has been found that F4/80+CD11b+ macrophages accumulate in adipose tissues of diet-induced obese mice.(40) The chemokine MCP-1 is also secreted primarily by macrophages and plays a key role in promoting recruitment of monocyte into adipose tissue, setting up the inflammation and insulin resistance state.(39) In addition to the enhanced macrophage recruitment, obesity drives a skewing of macrophages toward classically activated M1-like phenotype. Two populations of F4/80+ CD11b+ macrophages, M1- and M2-like phenotypes, have been found to reside in adipose tissue. One of which, M1-like population, additionally expresses CD11c and comprises the majority of the increased adipose tissue macrophages in obese subjects.(41) These CD11c+ macrophages play a crucial role in the augmented expression of proinflammatory cytokines in obese adipose tissue, conferring the link between inflammation and insulin resistance.(42) Consistently with this mechanism, we observed in our study that mRNA levels of MCP-1, F4/80, CD11b and CD11c were markedly increased in epididymal and mesenteric adipose tissue of PBS-treated db/db control compared to WT mice (Fig. 4). Interestingly, LGG-treated db/db mice had significantly lower expression of MCP-1, F4/80, CD11b, and CD11c in adipose tissues relative to PBS-treated controls. These results indicate that LGG treatment alleviated inflammation in adipose tissues, providing an additional explanation for insulin-sensitizing effect of LGG. However, LGG treatment had no effect on expression of proinflammatory cytokines such as TNFα and IL-6 in db/db mice, which remains to be elucidated.
Wang et al.(43) showed that administration of probiotics to high-fat diet-fed mice modulated gut microbiota and increased levels of a short-chain fatty acid, acetate, which activated AMPK and eventually improved glucose tolerance.(44) In addition, Kimura et al.(45) reported that the gut microbiota suppressed insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. In their study, adipose-specific Gpr43 transgenic mice fed a high-fat (HF) diet exhibited a decrease in F4/80 expression in adipose tissue, compared with HF diet-fed WT mice. They also observed that, although insulin sensitivity was improved in adipose-specific Gpr43 transgenic mice compared with WT mice, insulin-induced Akt phosphorylation in adipose tissue, but not in skeletal muscle, was markedly suppressed. Administration of acetate to WT mice also significantly suppressed insulin-induced Akt phosphorylation in adipose tissue, but not in skeletal muscle, whereas the effect was not exhibited in adipose tissue of Gpr43 whole-body knockout mice, indicating that GPR43 activation in adipose tissue causes a suppression of insulin signaling in adipose tissue, but not in skeletal muscle. Their results are consistent with our data and provide a plausible explanation of why the insulin sensitivity was improved in skeletal muscle but not in adipose tissue of LGG-treated db/db mice in our study. LGG-mediated modulation of gut microbiota might enhance acetate production to activate GPR43 and suppress fat accumulation in adipose tissue, which reduces fatty acid transport to skeletal muscle and eventually leads to a reduction in lipotoxicity and ER stress in skeletal muscle to improve insulin sensitivity.
Taken together, our results illustrate the insulin-sensitizing effect of LGG in db/db mice that is mediated by alleviation of ER stress in skeletal muscle and M1-like macrophage activation in white adipose tissues. These findings reveal that LGG treatment could be a therapeutic approach for prevention and treatment of type 2 diabetes.
Acknowledgments
This work was supported by the National Research Foundation Grant funded by the Korean Government (MEST, 2012R1A1B 6003789).
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Potenza MV, Mechanick JI. The metabolic syndrome: definition, global impact, and pathophysiology. Nutr Clin Pract. 2009;24:560–577. doi: 10.1177/0884533609342436. [DOI] [PubMed] [Google Scholar]
- 2.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab. 2012;23:205–215. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Delzenne N, Neyrinck AM, Bäckhed F, Cani PD. Targeting gut microbiota in obesity: effects of prebiotics and probiotics. Nat Rev Endocrinol. 2011;7:639–646. doi: 10.1038/nrendo.2011.126. [DOI] [PubMed] [Google Scholar]
- 4.Greiner T, Bäckhed F. Effects of gut microbiota on obesity and glucose homeostasis. Trends Endocrinol Metab. 2011;22:117–123. doi: 10.1016/j.tem.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Rawls JF, Mahowald MA, Ley RE, Gorden JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–433. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gorden JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 7.Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des. 2009;15:1546–1558. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- 8.Shen J, Obin MS, Zhao L. The gut microbiota, obesity and insulin resistance. Mol Aspects Med. 2013;34:39–58. doi: 10.1016/j.mam.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Quigley EM. Gut microbiota and the role of probiotics in therapy. Curr Opin Pharmacol. 2011;11:593–603. doi: 10.1016/j.coph.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Canani RB, Cirillo P, Terrin G, et al. Probiotics for treatment of acute diarrhea in children: randomised clinical trial of five different preparations BMJ2007335340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guandalini S, Pensabene L, Zikri MA, et al. Lactobacillus GG administered in oral rehydration solution to children with acute diarrhea: a multicenter European trial. J Pediatr Gastroenterol Nutr. 2000;30:54–60. doi: 10.1097/00005176-200001000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Österlund P, Ruotsalainen T, Korpela R, et al. Lactobacillus supplementation for diarrhea related to chemotherapy of colorectal cancer: a randomised study. Br J Cancer. 2007;97:1028–1034. doi: 10.1038/sj.bjc.6603990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalliomäki M, Salminen S, Arvilommi H, Kero P, Koskinen P, Isolauri E. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076–1079. doi: 10.1016/S0140-6736(00)04259-8. [DOI] [PubMed] [Google Scholar]
- 14.Kumar M, Rakesh S, Nagpal R, et al. Probiotic Lactobacillus rhamnosus GG and Aloe vera gel improve lipid profiles in hypercholesterolemic rats. Nutrition. 2013;29:574–579. doi: 10.1016/j.nut.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Tabuchi M, Ozaki M, Tamura A, et al. Antidiabetic effect of Lactobacillus GG in streptozotocin-induced diabetic rats. Biosci Biotechnol Biochem. 2003;67:1421–1424. doi: 10.1271/bbb.67.1421. [DOI] [PubMed] [Google Scholar]
- 16.Honda K, Moto M, Uchida N, He F, Hashizume N. Anti-diabetic effects of lactic acid bacteria in normal and type 2 diabetic mice. J Clin Biochem Nutr. 2012;51:96–101. doi: 10.3164/jcbn.11-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SW, Park KY, Kim B, Kim E, Hyun CK. Lactobacillus rhamnosus GG improves insulin sensitivity and reduces adiposity in high-fat diet-fed mice through enhancement of adiponectin production. Biochem Biophys Res Commun. 2013;431:258–263. doi: 10.1016/j.bbrc.2012.12.121. [DOI] [PubMed] [Google Scholar]
- 18.Flamment M, Hajduch E, Ferré P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23:381–390. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Matsuzaki T, Yamazaki R, Hashimoto S, Yokokura T. Antidiabetic effects of an oral administration of Lactobacillus casei in a non-insulin-dependent diabetes mellitus (NIDDM) model using KK-Ay mice. Endocr J. 1997;44:357–365. doi: 10.1507/endocrj.44.357. [DOI] [PubMed] [Google Scholar]
- 20.Aronsson L, Huang Y, Parini P, et al. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4) PLoS One. 2010;5:e13087. doi: 10.1371/journal.pone.0013087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esposito E, Iacono A, Bianco G, et al. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J Nutr. 2009;139:905–911. doi: 10.3945/jn.108.101808. [DOI] [PubMed] [Google Scholar]
- 22.Kondo S, Xiao JZ, Satoh T, et al. Antiobesity effects of Bifidobacterium breve strain B-3 supplementation in a mouse model with high-fat diet-induced obesity. Biosci Biotechnol Biochem. 2010;74:1656–1661. doi: 10.1271/bbb.100267. [DOI] [PubMed] [Google Scholar]
- 23.Andreasen AS, Larsen N, Pedersen-Skovsgaard T, et al. Effects of Lactobacillus acidophilus NCFM on insulin sensitivity and the systemic inflammatory response in human subjects. Br J Nutr. 2010;104:1831–1838. doi: 10.1017/S0007114510002874. [DOI] [PubMed] [Google Scholar]
- 24.Takemura N, Okubo T, Sonoyama K. Lactobacillus plantarum strain No. 14 reduces adipocyte size in mice fed high-fat diet. Exp Biol Med (Maywood) 2010;235:849–856. doi: 10.1258/ebm.2010.009377. [DOI] [PubMed] [Google Scholar]
- 25.Velayudham A, Dolganiuc A, Ellis M, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology. 2009;49:989–997. doi: 10.1002/hep.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yadav H, Jain S, Sinha PR. Antidiabetic effect of probiotic dahi containing Lactobacillus acidophilus and Lactobacillus casei in high fructose fed rats. Nutrition. 2007;23:62–68. doi: 10.1016/j.nut.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 28.Gregor MF, Yang L, Fabbrini E, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puri P, Mirshahi F, Cheung O, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 30.Deldicque L, Cani PD, Philp A, et al. The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab. 2010;299:E695–E705. doi: 10.1152/ajpendo.00038.2010. [DOI] [PubMed] [Google Scholar]
- 31.Peter A, Weigert C, Staiger H, et al. Individual stearoyl-coA desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes. 2009;58:1757–1765. doi: 10.2337/db09-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rieusset J, Chauvin M, Durand A, et al. Reduction of endoplasmic reticulum stress using chemical chaperones or Grp78 overexpression does not protect muscle cells from palmitate-induced insulin resistance. Biochem Biophys Res Commun. 2012;417:439–445. doi: 10.1016/j.bbrc.2011.11.135. [DOI] [PubMed] [Google Scholar]
- 33.Turban S, Hajduch E. Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett. 2011;585:269–274. doi: 10.1016/j.febslet.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 34.Henriksen EJ, Diamond-Stanic MK, Marchionne EM. Oxidative and the etiology of insulin resistance and type 2 diabetes. Free Radical Bio Med. 2011;51:993–999. doi: 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int J Exp Med. 2010;3:33–40. [PMC free article] [PubMed] [Google Scholar]
- 36.Brand MD, Esteves TC. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005;2:85–93. doi: 10.1016/j.cmet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Samec S, Seydoux J, Dulloo AG. Skeletal muscle UCP3 and UCP2 gene expression in response to inhibition of free fatty acid flux through mitochondrial β-oxidation. Pflugers Arch. 1999;438:452–457. doi: 10.1007/s004249900080. [DOI] [PubMed] [Google Scholar]
- 38.Schrauwen P, Schaart G, Saris WH, et al. The effect of weight reduction on skeletal muscle UCP2 and UCP3 mRNA expression and UCP3 protein content in Type II diabetic subjects. Diabetologia. 2000;43:1408–1416. doi: 10.1007/s001250051547. [DOI] [PubMed] [Google Scholar]
- 39.Olefsky JM, Glass CK. Macrophage, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 40.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–309. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Tang H, Zhang C, et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015;9:1–15. doi: 10.1038/ismej.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carvalho BM, Guadagnini D, Tsukumo DM, et al. Modulation of gut microbiota by antibiotics improves insulin signalling in high-fat fed mice. Diabetologia. 2012;55:2823–2834. doi: 10.1007/s00125-012-2648-4. [DOI] [PubMed] [Google Scholar]
- 45.Kimura I, Ozawa K, Inoue D, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]