

Abstract
mibnn2002, found from an allele screen, showed early segmentation defect and severe cell death phenotypes, which are different from previously known mib mutants. Despite distinct morphological phenotypes, the typical mib molecular phenotypes: her4 down-regulation, neurogenic phenotype and cold sensitive dlc expression pattern, still remained. The linkage analysis also indicated that mibnn2002 is a new mib allele. Failure of specification in anterior 7-10 somites is likely due to lack of foxc1a expression in mibnn2002 homozygotes. Somites and somite markers gradually appeared after 7-10 somite stage, suggesting that foxc1a is only essential for the formation of anterior 7-10 somites. Apoptosis began around 16-somite stage with p53 up-regulation. To find the possible links of mib, foxc1a and apoptosis, transcriptome analysis was employed. About 140 genes, including wnt3a, foxc1a and mib, were not detected in the homozygotes. Overexpression of foxc1a mRNA in mibnn2002 homozygotes partially rescued the anterior somite specification. In the process of characterizing mibnn2002 mutation, we integrated the scaffolds containing mib locus into chromosome 2 (or linkage group 2, LG2) based on synteny comparison and transcriptome results. Genomic PCR analysis further supported the conclusion and showed that mibnn2002 has a chromosomal deletion with the size of about 9.6 Mbp.
Mind bomb (Mib), an E3 ubiquitin ligase, regulates Notch signaling pathway by adding ubiquitin onto Notch ligands, such as DeltaC (DlC) and Jagged, on the cell membrane1,2,3,4. So far, Mib has been known as an important regulator in the Notch-related developmental processes, such as inner ear, neuron, somite, lymphocyte, hematopoiesis, retina, intestine, kidney, pancreas, etc3,5,6,7,8,9,10,11,12. The impacts of Mib on these developmental processes are mainly conducted through regulating Notch signaling pathway. Recently, Wnt pathway13, cell death pathway14, hypoxia signaling and deubiquitylases15 have also been demonstrated or proposed to be regulatory targets of Mib. Mib contains two mib/herc2 domains, one ZZ zinc finger domain, two Mib repeats, eight ankyrin repeats and three ring finger (RF) domains1. The most C-terminal RF domain is responsible for Mib E3 ligase activity to ubiquitylate its substrates and further lead ubiquitylated substrates to protein translocation via vesicle trafficking and/or proteasome-dependent degradation. Without ubiquitylation, Notch intracellular domain cannot be released from plasma membrane to induce downstream transcriptional activation, such as her4 that we used to show the Notch activation in this and previous studies1.
Ribs and vertebrae are originally derived from the paraxial mesoderm, where a group of cells locate in presomitic mesoderm (PSM) around the region of tail bud, propagate and be specified progressively into somites – the repeated epithelial segments. Somites give rise to dermatome, myotome and sclerotome, and further become dermis, skeletal muscle, ribs and vertebrae16. The segmentation defect has been found to correlate with the loss of cyclic expression of dlc and/or her1 in the PSM of different Notch-related zebrafish mutants, such as deltaC (bea), deltaD (aei), notch1a (des) and mib1,17,18,19,20,21,22. In these mutants, dlc is desynchronizedly expressed with a salt-and-pepper-like, instead of cyclic, expression pattern, and the posterior somite boundaries are indistinct as those cells are unable to be synchronized by Notch signaling20,23,24,25. On the other hand, there are genes required for somitogenesis in a Notch-independent manner. fss mutants with deficiency in tbx24 gene form no somites26. foxc1a, whose morphants exhibited a similar phenotype to fss mutants, acts in parallel to the fss/tbx24 pathway as an essential factor in zebrafish somitogenesis27.
To further dissect and explore the Mib functions, we carried out an allele screen to identify new mib alleles. Three new mib alleles, mibnn2000, mibnn2001 and mibnn2002, were identified. mibnn2000 and mibnn2001, whose mutations lead to truncation of RF domains, showed segmentation phenotypes similar to previous mib alleles. Interestingly, mibnn2002 had an earlier segmentation defect with severe cell death. her4 and huc expression also suggested the loss of mib activity. We then focused on mibnn2002 mutant for its potential in identifying mib’s novel function. The cell death was mainly distributed around the region of notochord and neural tube with an onset from 16-somite stage (ss) and an up-regulation of p53. While homozygous embryos lost anterior somite segmentation and failed to be specified into somite cell fate as shown by the expression of myoD, dlc and dystrophin, relatively normal-looking posterior somite segments formed with proper somite markers expressed gradually after 14 hours post fertilization (hpf). foxc1a expression was not detected at all stages. The expression of her1 and dlc were down-regulated but still cyclically expressed in the PSM during all developmental stages checked. By linkage analysis, we found a close linkage near the mib locus. Unexpectedly, we uncovered there is a large deletion covering the loci of foxc1a and mib on LG2.
Results
Three new mib alleles, showing similar Notch molecular phenotypes to other mib alleles, were found from allele screen
In order to further understand mib gene functions in the vertebrate development, we carried out an allele screen by non-complementation between F1 progeny of ENU-mutagenized founder fish and heterozygous carriers of mibta52b, a well-known and widely-used mib allele1,17. 3849 potential F1 carriers for new mib alleles were successfully screened by single pair matings to fish that were mibta52b heterozygous carriers. The resulting egg lays were checked around 48 hpf for non-complementation of the mib mutation. Three new alleles: mibnn2000, mibnn2001 and mibnn2002 were identified.
The expression of a Notch downstream gene, her4, and a neuronal marker, huc, were used to indicate the compromise of Notch signaling pathway in mib mutants in previous studies1,7. Besides, the disruption of cyclically expressing dlc was also demonstrated in mib and other Notch-related mutants19,20,21,22. Therefore, the expression patterns of her4, huc and dlc were examined in these new alleles. At 22 °C, the typical down-regulation of her4 and up-regulation of huc were observed in mibnn2000, mibnn2001 and mibnn2002 mutants (Fig. 1A–H). Under the same condition, the dlc expression exhibited a salt-and-pepper (desynchronized) pattern in the anterior PSM and nascent somite region (Fig.1I–L and supplementary Fig. 1). All the analysis suggested that mibnn2000, mibnn2001 and mibnn2002 were new mib alleles that lost the E3 ubiquitin ligase activity7.
Figure 1.

The new alleles show similar Notch molecular phenotypes to those of previous mib mutants.All embryos were raised at 22 °C and flat-mounted WISH performed at 22 hpf. A–D are flat-mounted her4 WISH. (A) WT embryos expressed her4 in brain and neural tube; (B) mibnn2000, (C) mibnn2001 and (D) mibnn2002 mutants all showed a down-regulation of her4 expression. E–H are flat-mounted huc WISH. (E) WT embryos expressed huc in cells of brain and neural tube; (F) mibnn2000, (G) mibnn2001 and (H) mibnn2002 mutants all showed an up-regulation of huc expression. I–L are flat-mounted dlc WISH. (I) WT embryos expressed dlc in PSM, formed somites and cell clusters in the hindbrain; (J) mibnn2000, (K) mibnn2001 and (L) mibnn2002 mutants all showed a salt-and-pepper dlc pattern in the nascent somites and anterior PSM. Note that the dlc expression in formed somites was lost in (L) mibnn2002 mutants.
mib nn2002 mutants showed distinct morphological phenotypes
Both mibnn2000 and mibnn2001 mutants showed typical mib segmentation defects at 22 °C: showing 11-14 and 14-17 visible somites, respectively, compared to about 20 somites (equivalent to 19 hpf at 28.5 °C) in wild-type at 30 hpf (Fig. 2A–C). Besides, mibnn2000 and mibnn2001 exhibited morphological phenotypes similar to those of mibta52b and mibm132 1,17, including brain and pigmentation defects, curly tail and hemorrhage at 3 dpf (Fig. 2D–F). Compared with the later boundary formation defect in mibnn2000, mibnn2001and previously-analyzed mib mutants, mibnn2002 showed somite segmentation defect at an earlier stage (Fig. 3). The V-shaped somite boundaries were not observed in mibnn2002 before 14 hpf (Fig. 3G). The somite-like structures could be observed in mibnn2002 mutants after 17 hpf, though the somites were not V-shaped and boundaries did not form sharply (Fig. 3H and Fig. 5F). Necrotic tissues appeared in mibnn2002 mutants after 24 hpf (Fig. 3I) and the phenotype became worse after 48 hpf with a great amount of cell death, edema and laterally curly tail with no observable pigmentation (Fig. 3J). Because of the distinct morphological phenotypes and similar Notch-related molecular phenotypes to other mib alleles, we then focused on analyzing mibnn2002 mutants as it might reveal a novel function of mib.
Figure 2.

Morphological phenotypes of mibnn2000 and mibnn2001 mutants. A–C are the lateral views of 30 hpf embryos raised at 22 °C. While (A) WT embryos showed about 20 somites at 30 hpf, (B) mibnn2000 homozygotes showed about 11 recognizable somites and (C) mibnn2001 homozygotes showed about 14 somites. D–F are lateral views of 3 dpf embryos at 28.5 °C. Compared to (D) WT embryos, (E) mibnn2000 and (F) mibnn2001 mutants showed a curly tail (arrows), hemorrhage (arrowhead) and a reduction of tail pigmentation (more prominent in E).
Figure 3.

mibnn2002 mutants show phenotypes distinct from those of typical mib mutants. A–E are lateral views of WT embryos. F–J are lateral views of mibnn2002 homozygotes. A and F are at 12 hpf; B and G are at 14 hpf; C and H are at 17 hpf; D and I are at 24 hpf; E and J are at 48 hpf. No visible somites can be observed from the lateral view of mibnn2002 homozygotes at (F) 12 hpf and (G) 14 hpf. Visible somite-like structures can be discerned from the lateral view of mibnn2002 homozygotes at (H) 17 hpf and (I) 24 hpf. Cell death can be spotted in mibnn2002 homozygotes at (I) 24 hpf and (J) 48 hpf. Edema was obvious in mibnn2002 homozygotes at (J) 48 hpf.
Figure 4.

Somite/boundary markers are lost in mibnn2002 mutants. (A) WT embryos and (B) mibnn2002 mutants were stained with tbx24 at 12 hpf. (C) WT embryos and (D) mibnn2002 mutants were stained with papc at 12 hpf. (E) WT embryos and (F) mibnn2002 mutants were stained with myoD at 14 hpf. (G) WT embryos and (H) mibnn2002 mutants were stained with fgf8a at 14 hpf. (I) WT embryos and (J) mibnn2002 mutants were stained with dystrophin (dmd) at 14 hpf. (K) WT embryos and (L) mibnn2002 mutants were stained with tcf15 at 14 hpf, where DsRed filter was used to improve the signal to noise ratio of NBT/BCIP staining under transmitted light. All embryos were mounted in dorsal view with head to the top. tbx24 and papc were expressed in PSM, though the anterior segmental pattern was not observed in the region of nascent somites in mibnn2002 mutants. The expression of myoD, fgf8a, and dystrophin were not detected in the region where somites normally formed. tcf15 was weakly expressed in mibnn2002 mutants and no segmental pattern can be discerned.
Figure 5.

Somite segmentation and specification recover without foxc1a expression. A–F are dorsal views of WT embryos and mibnn2002 mutants. Small round-shaped structures can be observed in (B) mibnn2002 homozygotes at 12 hpf. The newly-generated somite-like structures increased when mibnn2002 homozygotes grew to (D) 14 hpf and (F) 17 hpf. G–N are flat mounts of WT embryos and mibnn2002 mutants stained with dlc. (H) The expression of dlc in the region where somites normally formed was not observed in mibnn2002 homozygotes at 12 hpf. (J, L and N) dlc progressively appeared after 14 hpf in the newly-generated somites. O–R are dorsal views of WT embryos and mibnn2002 mutants stained with tbx24. The segmental tbx24 (marked with red asterisks) appeared progressively in mibnn2002 homozygotes. S–V are flat mounts of WT embryos and mibnn2002 mutants stained with myoD. W and X are lateral views of WT embryos and mibnn2002 mutants stained with myoD. The expression of myoD in formed somites started to emerge after 14 hpf as what was observed in dlc. As embryos developed, (V) the expression of myoD progressively appeared in the region where somites normally formed, except the anterior segments. (X) The cells that expressed myoD still split into dorsal and ventral myotomes even where somite boundaries were not formed. Y and Z are lateral views of WT embryos and mibnn2002 mutants stained with dystrophin (dmd) at 22 hpf. The expression of dystrophin can be observed in formed somites. Note that the somite gap delineated by dystrophin in formed somite region is similar between (Y) WT embryos and (Z) mibnn2002 mutants. a–f are dorsal views of WT embryos and mibnn2002 mutants stained with foxc1a. The expression of foxc1a in mibnn2002 homozygotes are not detected in all the stages examined (b 12 hpf; d 14 hpf and f 17 hpf).
Noticeably, average 22% of embryos from incrosses of mibnn2002 carriers exhibited early segmentation phenotype (supplementary Table 1). The pre-selected embryos with early segmentation phenotype showed down-regulated her4, slightly up-regulated huc and severe cell death phenotypes (data not shown). Furthermore, about a quarter of transheterozygotes (mibnn2002/mibta52b) exhibited typical mib phenotypes (supplementary Table 2), such as late segmentation defect (supplementary Fig. 2), curly tail and less pigmentation (supplementary Fig. 3). The impact of maternal mib on the transheterozygotes also can be observed (supplementary Fig. 3, embryos laid from female mibta52b carriers show a more severe mib phenotype, as observed previously7). These results demonstrated that mibnn2002 mutation follows the patterns of autosomal recessive inheritance with high penetrance.
The cell death in mibnn2002 mutants began after 17 hpf by acridine orange staining and mainly distributed around the regions of neural tube and notochord (supplementary Fig. 4). It was demonstrated to be apoptosis by using TUNEL assay and the apoptotic cell number was significantly higher in mibnn2002 mutants than that in the control embryos (supplementary Fig. 5). The expression of p53 was further found to be highly up-regulated in mibnn2002 mutants (supplementary Fig. 6), which suggests that cell death might be related to genome instability.
Early somite specification defects were identified in mib nn2002 mutants
To discern if the segmentation defect is caused by a failure/delay in boundary formation or early somite specification, we checked the dlc expression first, because the disruption of cyclic expression pattern of Notch components was found highly correlated to the defect of somite boundary formation in Notch-related mutants20,28,29,30,31. The expression of dlc in mibnn2002 mutants under 28.5 °C incubation were examined from 12 hpf (~5 ss) to 19 hpf (20 ss). Unexpectedly, there was no obvious dlc desynchronization detected in the PSM, suggesting that the segmentation defect might be caused by a Notch-independent mechanism (supplementary Fig. 7). This result may be contradictory to the Notch role in somite segmentation at first glance. However, it is consistent with a previously-analyzed mib null allele, mibtfi91, which shows regular boundary formation and normal dlc cycling at 28.5 °C but becomes cold-sensitive and exhibits somite boundary defect and desynchronized dlc expression pattern at 22 °C (compared to Fig. 1L; supplementary Fig. 1; reference 7).
We further used tbx24 and papc, two boundary markers in the region of nascent somites26,32. The expression of tbx24 was steadily expressed in the PSM, only lost in the anterior nascent somites in mibnn2002 mutants at 12 hpf (Fig. 4A–B). Similar phenomenon was also observed by using papc: two anterior nascent somite boundaries were lost in mibnn2002 mutants (Fig. 4C– D). Next, posterior somite marker, myoD18, anterior somite marker, fgf8a33, somite boundary marker, dystrophin34 and tcf15 (paraxis27) were used. The expression of myoD, fgf8a, dystrophin and tcf15 were lost or diminished in the region where somites normally form in mibnn2002 mutants at 14 hpf (Fig. 4E–L). Among these markers, only myoD was detected in the adaxial cells. These results indicate that the early somite segmentation defect in mibnn2002 is caused by a failure in cell fate specification. This also suggests that the early segmentation defect in mibnn2002 is mediated through a Notch-independent pathway.
foxc1a is not essential in later somite segmentation and specification
Although visible somites were hardly observed in mibnn2002 at early stages, the somite-like structures did appear at later stages. To explore the phenotype, we analyzed the somite-like structures of mibnn2002 mutants in more details. Some small round-shaped structures in the region where somites were supposed to form at early stage were found in mibnn2002 mutants (Fig. 5A–B). The number of the small round-shaped structures increased with time in mibnn2002 mutants (Fig. 5A–D). The somites formed in mibnn2002 mutants at a relative late stage have a shape much similar to those observed in wild type (WT) (Fig. 5E–F). In order to understand whether the somites are really formed at later stages, we further examined the expression of dlc, tbx24, myoD and dystrophin at different stages. The expression of dlc, highly correlated to the formation of somite-like structures, was shown in the region of formed somites progressively in mibnn2002 mutants (Fig. 5G–N). The cyclically expressed dlc in the PSM was not altered as described previously (supplementary Fig. 7). Its expression level in the PSM was gradually recovered at later stages, in contrast to the severe down-regulation at early stages (Fig. 5G–N). Similarly, the anterior segmented tbx24 expression pattern was progressively restored (Fig. 5O–R). myoD was also expressed in the formed somites with sharp and striped pattern while the normal-looking somites started appearing after 14 hpf (Fig. 5S–X). The expression of dystrophin also suggested that somites develop into myotome with roughly normal somite boundaries at 22 hpf (Fig. 5Y–Z). All the results suggest that somites are normally specified in mibnn2002 mutants at a relative late somitogenesis stage.
foxc1a has been shown to be an essential factor for somitogenesis and somite segmentation in a morpholino study27. Therefore its expression was examined. Surprisingly, the expression of foxc1a was not detected in mibnn2002mutants at all stages examined (Fig. 5a–f). These data indicated that foxc1a is only essential for anterior somitic cell fate determination, but not required for the later somite specification.
mib nn2000 and mib nn2001 mutants both encode a Mib protein without RF3 domain
To identify the mutations in these three new alleles, the mib mRNA was reversely transcribed, amplified and examined by sequencing. mibnn2000 bore a T to A transversion mutation at the position 2694 from translational start site, resulting in a nonsense mutation C-terminal to RF2 (Fig. 6A and supplementary Fig. 8A). mibnn2001 resulted in a different splicing isoform (mibΔRF3) that has a 143 bp insert after position 2851 from translational start site, leading to a truncated protein without the RF3 domain (Fig. 6A and supplementary Fig. 8B). The characterization and mutation identification of mibnn2000 and mibnn2001 are comparable to the results from mibm132 and mibta52b, which have RF123 domain deletion and a non-functional RF3, respectively1,7.
Figure 6.

mib gene is deleted in mibnn2002 mutants. (A) Diagram of the mutations of different mib alleles. The translation of mib in mibnn2000 and mibnn2001 mutants stops at amino acids 5’ to ring finger domain 3 (RF3). (B) The linkage analysis result of mibnn2002 mutants. The upper part illustrates the locations of mib gene and the SSLP markers. The lower part demonstrates a close linkage of Z4662 and Z13620 to mib, with a recombination frequency of 0/97 and 2/214, respectively. (C) Real-time PCR results of mib expression in mibnn2002 homozygotes and WT embryos. The blue bars represent the expression level of mib in mibnn2002 homozygotes. The purple bars represent the expression level of mib in WT embryos. There is nearly no mib transcript detected in the mibnn2002 homozygotes. (D) WT embryos and (E) mibnn2002 mutants were stained with mib at 14 hpf, respectively. There is no mib mRNA detected in (E) mibnn2002 homozygous. (F) The results of mib genomic PCR were shown with fih (hif1an) as a positive control. AB represents AB WT samples; MU represents mibnn2002 homozygous samples; NE represents negative controls, where distilled water was used instead of genomic extracts as templates. The results showed that no mib genomic fragments are amplified.
mib gene is deleted in mib nn2002 mutants
Difficulty was encountered while we were trying to amplify the coding sequence of mib from mibnn2002 mutants. Linkage analysis was then employed to assure the position of mibnn2002 mutation and it was demonstrated that mibnn2002 mutation is 3.32 cM away from the mib locus in chromosome 2 (LG2) with SSLP marker Z13620 (Fig. 6B). Therefore, mibnn2002 is indeed a new mib allele.
The phenocopy and rescue experiments were then used to further discern two hypotheses of the cause of mibnn2002 mutants. First, mibnn2002 mutation might encode a mutated Mib protein that leads to cell death and early segmentation defect in homozygous embryos. mib morpholino has been previously used in phenocopying mibtfi91 by knocking down mibta52b transcript7. By using the same approach, morpholino can down-regulate the expression of the hypothetical mutated mibnn2002 transcript and phenocopy the mib null allele, mibtfi91. Second, mibnn2002 might bear a mutation in the mib regulatory region that affects its expression level. If so, injecting mib mRNA into mibnn2002 should be able to rescue the mibnn2002 phenotypes to some extent. However, both hypotheses were proved to be incorrect. Although mRNA injection partially restored the expression level of her4 and huc and morpholino injection led to an increase in the number of embryos that exhibit mibta52b-like phenotypes (supplementary Fig. 9), the early segmentation defects and cell death phenotypes in mibnn2002 homozygotes were neither rescued nor alleviated and the ratio of mibnn2002 homozygotes remained unchanged in both rescue and phenocopy experiments (supplementary Fig. 9 and data not shown).
By checking the expression of mib with in situ hybridization and real-time PCR, we found that the expression of mib was hardly detected and might be totally lost in mibnn2002 mutants (Fig. 6C–E) and, thereby, started to wonder whether mib was deleted in mibnn2002 mutants. To explore the possibility, genomic PCR was employed to check the existence of mib gene. However, the genomic sequence of mib was separated on two scaffolds, 3504 and 3540, and was not placed in chromosome 2 of Zebrafish Ensembl (Zv9, http://www.ensembl.org/Danio_rerio/Info/Index). Moreover, the 5’ and 3’ genomic sequences of mib were separated by other genes. To further clarify the assembly/continuity of the scaffolds, we first used existing mib mutants, mibtfi91 and mibnn2000, to verify the continuity of the genomic sequence. The results showed that mibtfi91 and mibnn2000 mutations can be identified based on the genomic sequence of scaffolds 3504 and 3540, respectively (supplementary Fig. 10), suggesting that scaffolds 3504 and 3540 are continuous. We assembled the genomic mib sequence from scaffolds 3504 and 3540 and delimited the intron-exon boundaries with the help of cDNA sequence (supplementary File 11). According to the assembled mib genomic sequence, the primers were designed and used to examine the existence of mib in mibnn2002 with fih, a gene not located on LG2, as a positive control. Consistent with our speculation, mib could not be amplified from the genomic DNA of mibnn2002 mutants (Fig. 6F).
mib nn2002 mutants have an arm deletion in chromosome 2, including the loci of mib and foxc1a
In order to dissect the link between mib, foxc1a and the cell death phenotype in mibnn2002 mutants, we performed an RNA-seq analysis on mibnn2002 mutants and AB WT embryos using the Mi-Seq platform. The detailed data will be reported elsewhere. Here, we only present the genes in the chromosome 2. From the RNA-seq, an unexpected result was found. Around 140 transcripts that are located between 19233 and 9578708 in LG2 were not detected (mean expression value <0.5 as set-point) only in the mutants. The full list of LG2 can be found in the supplementary Table 3. Selected transcripts were undergone genomic PCR for confirmation, which included both ends of the truncated region: cldn1 (88773-91785) and dvl3a (9490950-9532978) (Fig. 7, supplementary Fig. 12). The result (summarized in supplementary Table 4) indicated that probably there is an arm deletion in the chromosome 2 of mibnn2002 mutants and the breaking point is between dvl31 and ap2m1a (9532978-9578708) (OR 1133-1144 from zfin mib map ch2, http://zfin.org/cgi-bin/view_mapplet.cgi). To conclude, mibnn2002 mutants have a regional deletion in the genome, where part of the chromosome 2, including mib and foxc1a, is deleted.
Figure 7.
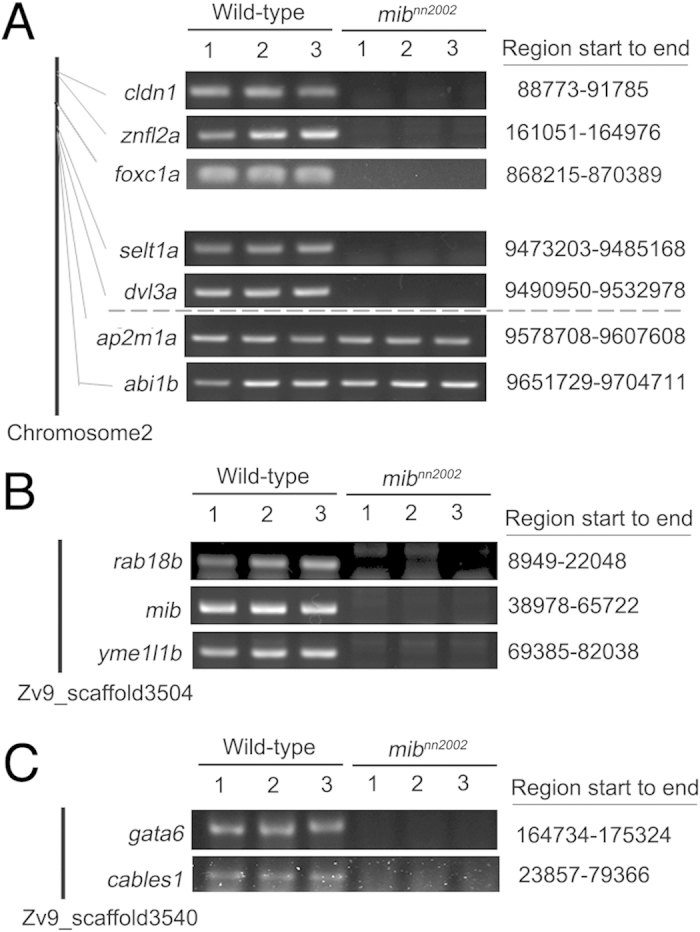
An arm of LG2, including mib gene, is truncated in mibnn2002 mutants. (A) The genomic PCR results of WT embryos and mibnn2002 mutants on LG2. Genes located near the chromosome terminal, cldn1, znfl2a and foxc1a, and near the suspected breaking point, selt1a~abi1b, were examined. The genes located between 88773 ∼ 9532978 bp on LG2 were not amplified in mibnn2002 mutants. (B) The genomic amplification of 5’-mib, rab18b and yme1l1b of scaffold 3504. (C) The genomic amplification of gata6 and cables1 of scaffold 3540. All these genes were not amplified in mibnn2002 mutants (see supplementary Fig. 12 and supplementary Table 4 for a more complete set of data).
The missing genes may play roles in somitogenesis and apoptosis
We further underwent DAVID analysis (see Materials and Methods) on the missing genes in mibnn2002 mutants. 91 genes had the DAVID ID, and 63 of them matched the zebrafish ID. The relatively low matching might be due to the present unknown genes in the list, such as those named as zebrafish gene collection (zgc) and CABZ sequences from clones or whole genome shotgun contigs. Nevertheless, the analysis identified ten enriched annotation clusters in mibnn2002 mutants. For example, somitogenesis and segmentation were identified in the analysis, which matched our molecular and phenotypic data in the mutants. In addition, signalings that are critical in early embryogenesis, such as Hedgehog and Wnt signaling pathways were deregulated (supplementary Table 5). All these results further support the phenotypic analysis of mibnn2002 mutants.
To confirm the causal consequence of foxc1a deficiency in anterior somite segmentation phenotype found in mibnn2002 mutants, foxc1a mRNA was injected into mibnn2002 mutants and found to be able to partially rescue the anterior myoD expression in mibnn2002 mutants at 16 hpf (Fig. 8). Two major rescued features of myoD expression in the anterior somites were segmental (Fig. 8B–E) and laterally-extended (prominently seen in Fig. 8D) myoD pattern, compared to Fig. 5V and Fig. 8A. In the first experiment, 111 embryos were collected for 16 hpf myoD expression pattern analysis. 22 (20% of total embryos) partially rescued or non-rescued like mibnn2002 homozygotes and 23 WT-like siblings were selected for imaging and genotyping. The 22 homozgyote-like embryos and the 23 WT-like embryos were all genotyped as mibnn2002 homozygotes and WT siblings, respectively. Among these 22 homozygotes, 6 foxc1a-overexpressed homozygotes showed a partially rescued myoD expression pattern as shown in Fig. 8B–E (27% of selected homozygotes). To better understand the rescue spectrum, we analyzed all the 38 injected embryos from the second set (supplementary Fig. 13 and data not shown). There were 9 embryos genotyped as homozygotes (supplementary Fig. 13B), 28 as WT and one as unknown (none of the primer sets worked, though the myoD expression pattern suggested that it is a homozygous mutant). Therefore, 24% (9/37) embryos are mibnn2002 homozygous. 4 (44%) homozygotes were partially rescued (supplementary Fig. 13Ac, d, h, i) and 3 (33%) homozygotes were almost fully rescued (supplementary Fig. 13Aa, e, f). These results strengthen the notion that foxc1a is only required for anterior somite segmentation.
Figure 8.

Anterior myoD expression in somites is partially rescued by foxc1a overexpression at 16 hpf.Representative WISH results of (A) non-injected control mibnn2002 mutants, (B–E) 20 pg foxc1a mRNA injected mibnn2002 mutants and (F–J) 20 pg foxc1a mRNA injected WT (siblings) are shown. The rescued somites are marked by arrowheads. myoD expression in the anterior somites of E was rescued symmetrically, while those of B–D were rescued asymmetrically in terms of myoD expression pattern, which may be caused by the unequal segregation of injected foxc1a mRNA. Lower panel shows the genotyping results of the embryos shown in B–J. The genomic fragments of mib 5’ (amplified by tfi91 primer set), mib 3’ (amplified by ta52b primer set) and foxc1a were not detected in B–E, while they were amplified in F–J. fih and vhl were amplified in all samples except for the negative control N. None of the genomic fragments was detected in the negative control.
selt1a and scp2a (related to oxidative damage) and smc6 (related to chromosome structure maintenance) were identified from the deleted genes in mibnn2002 mutants. This finding suggests that DNA damage and chromosome instability may be the cause of p53 up-regulation (supplementary Fig. 6) and further lead to cell death in mibnn2002 mutants. To explore this possibility, γ-H2AX staining was employed, because phosphorylated H2AX (γ-H2AX) can be spotted shortly after the induction of DNA double-stranded breaks (DSBs) and is essential for recruiting several DNA repair proteins to the DNA damage sites35. Indeed, many γ-H2AX foci can be detected in mibnn2002 mutants in contrast to control embryos (supplementary Fig. 14). Moreover, we injected p53 morpholino into mibnn2002 mutants and found that dying cells are decreased, suggesting that cell death in mibnn2002 mutants is p53-dependent (supplementary Fig. 15).
Discussion
Previously, mib was found to be an essential factor in Notch signaling pathway. The loss of mib function will lead to the failure of adding ubiquitin onto Notch ligands and then the down-regulation or loss of Notch downstream gene expression. To further explore the functions of mib, we carried out an allele screen and found three new mib alleles, mibnn2000, mibnn2001 and mibnn2002. mibnn2000 and mibnn2001 mutants exhibited typical mib phenotypes, including disruption in somite boundary formation, curly tail and down-regulation of Notch target genes. Interestingly, mibnn2002 mutants showed defects in early somite boundary formation and cell death. Although mibnn2002 mutants showed similar Notch molecular phenotypes as other mib mutants, their morphological phenotypes were demonstrated to be unrelated to the loss of mib in mibnn2002 mutants, because it was not rescued by mib or mib2 overexpression. Instead, the loss of foxc1a is more likely to be the cause of the anterior segmentation defect. In a previous morpholino study, the down-regulation of foxc1a specifically leads to the loss of myoD expression in the region where somites were supposed to form and does not affect the expression of myoD in the adaxial cells27. The failure of somite segmentation was not caused by the disruption of cyclic dlc expression in the PSM as the dlc is normally and cyclically expressed in the PSM of mibnn2002 mutants. Furthermore, mibnn2002 mutation contained an arm deletion and lost around 140 genes, including mib and foxc1a, on LG2. Therefore, it is possible that the failure of somite specification is caused by the loss of foxc1a. In other words, anterior somite segmentation defect found in mibnn2002 mutants is caused by a Notch-independent pathway.
mibnn2002 is probably a spontaneous deletion allele that lacks mib and other genes, because ENU mutagenesis mainly induces random mutations and produces almost all possible nucleotide substitution changes (transversion and transition), including nonsense mutations (e.g. mibnn2000), mutations that affect transcript splicing (e.g. mibnn2001) and missense mutations36. Interestingly, syut4 has been identified as a spontaneous deletion allele deleting shh gene. Similar to mibnn2002 mutants, syut4 mutants also showed a brain necrosis phenotype, which may be due to other genes affected by the deletion that has not been delimited and characterized37.
In the transcriptome study, three genes related to somite development were found: mib, foxc1a and wnt3a. The participation of mib in the anterior somite formation is excluded, because the null allele (mibtfi91) did not exhibits early boundary formation defect, at least at 28.5 °C7. wnt3a is unlikely responsible for the somite defect, either, for two reasons. First, mouse Wnt3a regulates cyclic Notch signaling through one bHLH transcription factor, Mesogenin1 (Msgn1)38; however, cyclic dlc expression is still observed in mibnn2002 mutants (supplementary Fig. 7). Second, wnt signaling does not appear involved in modulating segmentation clock but wavefront velocity in zebrafish39.
It was believed that foxc1a is crucial for the process of somite segmentation, because morpholino was able to inhibit somite formation before 7 ss but Foxc1a level was restored after 7 ss due to a negative feedback regulation of Foxc1a on transcription of its own gene27. Similar study was also carried out in compound Foxc1; Foxc2 homozygous mice (zebrafish did not have foxc2). It was found that mouse anterior somites (9.5 dpc with about 20 somites; with greater than 60 somites in total after 15 dpc) are not formed at 9.5 dpc; however, the formation of the posterior somite cannot be analyzed because the mice died far too early40. In our study, the expression of foxc1a was lost as it is deleted in the genome of mibnn2002 mutants. Nevertheless, the somite segmentation and boundary formation were relatively well processed without foxc1a at a relative late stage. Importantly, the anterior myoD expression pattern could be partially rescued in mibnn2002 mutants by overexpressing foxc1a. These results, therefore, demonstrate that foxc1a is only required for the formation of anterior somites but not for that of posterior somites.
As mentioned early, the genomic sequence of mib was placed on two scaffolds. Although the two scaffolds were proved to be the sequence encoded mib genomic DNA, they failed to be assembled into one as there are two genes, rab18b and Loc100331480 (PTCHD3 (3 of 3) located between the 5’ and the 3’ parts of mib genomic sequences (supplementary Fig. 10). Through comparing synteny of genomic DNA from different species, including Cavefish, Tilapia, Tetraodon, Stickleback, Platyfish, Fugu, Medaka, Cod, Xenopus, Mouse and Human, none of the mib genomic DNA is separated by rab18b in Ensembl database (data not shown). Besides, there is no overlap between the shotgun sequences, cu694527 and cu681854, which constitute scaffold 3504. Therefore, we propose that the shotgun sequence cu694527 should be removed and then the scaffold 3504 and scaffold 3540 can be assembled into one scaffold (supplementary Fig. 16).
On the other hand, the position of mib gene was suggested to be located on LG2 through different mappings, such as T51, LN54 and MGH genetic maps, based on the data in ZFIN genetic maps (http://zfin.org/action/mapping/all-panels?record=JUMPTOREFCROSS). However, the genomic sequence of mib was not placed in any chromosome in zebrafish. To unravel the ambiguity, we analyzed and compared the synteny of genes near the mib locus and found that the synteny in zebrafish is much more similar to that in cavefish (supplementary Fig. 17), as in the case of hox gene clusters demonstrated previously41. Therefore, we used the information of these syntenies and the existed Ensembl sequences to rebuild a hypothetical genomic structure of LG2 near the locus of mib (supplementary Fig. 18). In the hypothetical genomic structure of LG2, nearby mib locus are the genes, such as mkxb, rab18b, ptchd3, yme1l1b, gata6 and cables1. The transcripts and genomic sequences of those nearby genes were also not detected in mibnn2002 mutants (Fig. 7; supplementary Fig. 12; supplementary Table 4).
Materials and Methods
Zebrafish
Zebrafish was fed and maintained with the help of Taiwan Zebrafish Core Facility at NHRI (http://www.zebrafish-nthu-nhri.org/chinese/index.php). The maintenance followed the standard operation protocol of the core facility. Embryos were raised at 28.5 °C unless otherwise specified. The allele screen was performed according to a published protocol37. All experimental procedures on zebrafish were approved by the Institutional Animal Care and Use Committee, National Health Research Institutes, Taiwan (NHRI-IACUC-100028) and carried out in accordance with the approved guidelines.
Laid embryos that were used in the whole mount in situ hybridization experiment (WISH) and live imaging were collected within a 15 min interval after mating started. The collected embryos were selected from pooled quality eggs right after the collection and sorted into 50 ∼ 55 embryos per 10 cm dish with E3 egg water to avoid developmental delay. The embryos was staged as described42 and phenotypically checked by comparing with WT or WT-like control embryos (the siblings) that were collected in the same time windows. The homozygotes were identified and collected into different vials according to their developmental stages. Embryos were then fixed in 4% paraformaldehyde and kept in fridge for 1 ∼ 3 days. Afterwards, embryos were dechorionated, dehydrated by methanol and stored in freezer.
Phenocopy, rescue and morpholino experiments
For mib phenocopy and rescue experiments, 10 ng/μl of mib exon1/intron morpholino1 and 1 ng/μl mib1 mRNA or mib2 mRNA were injected into embryos around 1 ∼ 2 cell stage, respectively. Embryos were placed in 28.5 °C incubator before the desired stage.
For foxc1a rescue or overexpression experiments, 20 pg zebrafish foxc1a mRNA was injected into embryos of one-cell stage. Embryos were raised at 28.5 °C before the desired stage.
For p53 knockdown experiments, 15.2 ng p53 morpholino43 was injected into the embryos of one-cell stage. Embryos were raised at 28.5 °C before the desired stage.
Whole mount in situ hybridization (WISH)
RNA probes were produced with digoxigenin or fluorescein labeling mix (Roche). huc, her4, myoD, dlc, tbx24, her1 and mib plasmids were used as templates26,44,45,46,47. The templates of dystrophin, foxc1a, fgf8a, papc, tcf15 and p53 were generated by RT-PCR with primers (listed in supplementary Table 6). The protocol of WISH followed the instruction of previous literatures48,49,50. Embryos were mounted in 95% glycerol (Sigma, cat:15523)/PBST (UniRegion Bio-Tech, cat:UR-PBS001 and Tween20) pH 5.5 or flat-mounted in 95% glycerol/PBST pH 5.5 or 2:1 benzyl benzoate (Sigma, cat:B6630)/benzyl alcohol (Alfa Aesar, cat:041218). Images were captured by Zeiss Axiovision Imager A1 or Zeiss Discovery V8.
Tunel and acridine orange staining
Embryos were placed in 2 μg/ml acridine orange (Sigma, cat:A6014)/Egg water for 30 min, then washed 4 times with a 30 min interval. Embryos anaesthetized by tricaine (Sigma, cat:E10521) were mounted in 4% methylcellulose (Sigma, cat:M0387) with E3 egg water on top and captured by Zeiss Axiovision Imager A1 or Zeiss Discovery V8.
Embryos for Tunel assay (Millipore, cat:S7160) were dehydrated in 70% ethanol overnight after fixation. 10 min ethanol/acetic acid treatment at −20 degree was used to increase membrane permeability. After the pretreatment of equilibration buffer at room temperature for 10 min, embryos were placed in TdT labeling solution for 2 hours and then washed by phosphate buffered saline. Embryos were then mounted in 2% low-temperature agarose and captured by Nikon A1 confocal microscope.
The pixels of green fluorescence were counted and assayed by Image J. The statistic calculation of the pixels was carried out by Excel (Microsoft).
γ-H2AX staining
Laid embryos were collected as mentioned above. The stage of embryos was staged as described42 and doubly checked by WT or WT-like control embryos (the siblings) that were collected in the same time windows. Embryos were dechorionated and sorted into different dishes of mibnn2002 homozygotes and WT siblings at 14 hpf. WT and mibnn2002 homozygotes that did not exposed to UV were moved to room temperate for 15 min. At the same time, the WT-UV control was placed in laminar flow hood with UV on for 15 min. Afterwards, all dishes were moved to 28.5 °C incubator for 1 hour before collected. Collected embryos were dehydrated by −20 °C methanol/acetone (1:1) and stored at −20 °C. Embryos were then sorted into 10 embryos/vial and washed by 1% Triton/PBS several times on shaker. After overnight blocking, embryos were washed by 2% BSA/1% Triton/PBS at 4 °C on shaker, and 1:1000 rabbit anti-human γ-H2AX (Cell signaling, #9718) in 2% BSA/1% Triton/PBS was used for overnight reaction. After intensive wash, 1:1000 goat anti-rabbit Alexa-488 antibody (Molecular probe) in 2% BSA/1% Triton/PBS was used for overnight reaction. After intensive wash, embryos were fixed by 4% paraformaldehyde. Embryos were further deyolked and flat-mounted in 75% glycerol to avoid strong background fluorescence from yolk. Images were taken by Zeiss Axiovision Imager A1.
Genomic PCR and real-time RT-PCR
Genomic DNA was extracted by 50 mM NaOH and 1 M Tris-HCl pH 8.0. One PCR mix (GeneDireX, cat:MB203-0100) and 5x PCR Master Mix II (GeneMark, cat:RP02D) were used with primers (supplementary Table 6). For WISH samples, individual samples were washed repeatedly and sequentially by 1x PBST and double-distilled H2O to avoid glycerol contamination, cross contamination between samples and salt interference before extraction was carried out. PCR protocol followed the instruction of 5x PCR Master Mix II (GeneMark, cat:RP02D).
For the RT-PCR, embryos were collected and the total RNA was extracted using TRIzol (Invitrogen). Reverse transcription followed the instruction of SuperScript III FirstStrand (Invitrogen, cat:18080-051). After reverse transcription, PCRs were conducted using LightCycler 480 real-time PCR detection system using SYBR Green I Master (Roche). The data were normalized using the expression levels of β-actin mRNA. The occurrence of primer dimers and secondary products was inspected using melting curve analysis. Our data indicated that the amplification was specific. There was only one PCR product amplified for individual set of primers. Primer sequences were listed in supplementary Table 6.
Genomic DNA extraction of F2 embryos and linkage analysis
F1 heterozygous parents (nn2002/WIK) were crossed to produce F2 embryos for genomic DNA extraction. mibnn2002 embryos were identified at 10 ss, lysed by 50 μl lysis buffer (10 mM Tris-HCl (pH 8.3), 1.0 mM EDTA, 12.5 mM KCl, 0.3% Tween 20, 0.3% NP-40), heated at 98 °C for 10 min, and incubated at 55 °C overnight with 1 mg/ml proteinase K. Proteinase K was then inactivated by incubation at 98 °C for 10 min. Afterwards, typical PCR assays were performed using selected simple-sequence length polymorphisms (SSLPs) markers. Since the mutant has the typical mib mutant phenotype, we selected seven SSLPs markers at chromosome 2, where the mib is located. PCR products were then undergone gel electrophoresis, and recombinant band was counted in each PCR reaction.
RNA preparation and Illumina RNA-seq
Fish heterozygous for the mibnn2002 alleles were crossed to obtain mibnn2002 homozygotes. Total RNA from 15 pooled homozygous mutants and AB wild type at 16 ss was extracted using the mirVanaTM miRNA isolation kit (Applied Biosystems). RNA quality was assessed using the Agilent 2100 Bioanalyzer system and samples with a RNA Integrity Number (RIN) greater than 8 were used for RNA library construction.
Library construction was conducted according to the manufacturer’s protocol (Illumina, TruSeq Stranded mRNA Sample Prep). Briefly, six RNA (cDNA) libraries were constructed (three AB wild-type and three mibnn2002 homozygotes), and each prepared from 300 ng of total RNA. Illumina adaptors (Integrated DNA technology) were ligated to their 5’ and 3’ ends to construct strand-specific cDNA libraries. Sequencing was performed on the Illumina MiSeq sequencer to generate 2.0 Gb of sequencing data per sample. Sequences were extracted from image files using the Illumina pipeline set at default parameters. Low quality, homopolymers and adaptor sequences were removed, and the resulting filtered reads were then aligned with the zebrafish reference genome Zv9 (GCA_000002035.2) and undergone transcriptome analysis by CLC Genomics Workbench 6. Only selected data on chromosome 2 are shown in this study to prove the arm deletion. Data deposit and further bioinformatics analysis will be reported and released in the future (unpublished data).
Bioinformatics analysis
The arm deletion was found after the rearrangement of the chromosome two. Individual reads of wild-type AB fish and mibnn2002 mutant samples (n = 3 for each) were listed (supplementary Table 3). The Database for Annotation, Visualization and Integrated Discovery (DAVID) was used for functional annotation clustering analysis on the missing genes in mibnn2002 mutants with the highest classification stringency as default51,52.
Note
Similar to what we have demonstrated here, we noticed that foxc1a plays an important role in early somitogenesis shown by examining two TALEN-induced foxc1a null alleles52.
Additional Information
How to cite this article: Hsu, C.-H. et al. A new mib allele with a chromosomal deletion covering foxc1a exhibits anterior somite specification defect. Sci. Rep. 5, 10673; doi: 10.1038/srep10673 (2015).
Supplementary Material
Acknowledgments
We would like to thank the staff in the Zebrafish Facility of NHRI for their efforts in maintaining fish stocks. Also, we are grateful to Kwok Yi Lan, Selvamani Balasubramaniam and Qimei Ng for technical help, to Dr. Chiaw-Hwee Lim for the advice in γ-H2AX staining and to Dr. Li-Chuan Tseng for the advice in genotyping mibtfi91. We appreciate the instructive suggestions from Dr. Freek van Eeden and his lab members in the University of Sheffield, UK. We are particularly grateful to Dr. Julian Lewis for the discussion in the somite phenotype of the new mib allele.
Chia-Hao Hsu carried out his thesis research under the auspices of the Graduate Program of Biotechnology in Medicine, National Tsing Hua University and National Health Research Institutes. The work was supported by grants from the National Health Research Institutes, Taiwan (MG-103-PP-13 and MG-103-PP-14) to YJJ, grants from the Ministry of Science and Technology, Taiwan to YYJ (NSC 100-2911-I-400-002 and 100-2311-B-400-001-MY3) and to MSY (NSC 103-2321-B-400-010), grant from the Faculty Research Fund, Hong Kong Baptist University, Hong Kong (FRG1/13-14/016) to WKFT and grant from the Seed Funding Programme for Basic Research, the University of Hong Kong, Hong Kong (201308159001) to KPL. JWL is supported partly by a General Research Fund (GRF461712) to TFC. This paper is dedicated to the memory of Prof. Julian H. Lewis (1946-2014).
Footnotes
Author Contributions Conceived and designed the experiments: Y.J.J. Performed the experiments: C.H.H. J.S.L. W.K.F.T. Analyzed the data: C.H.H. K.P.L. J.W.L. W.K.F.T. Y.J.J. Contributed reagents/materials/analysis tools: M.S.Y. T.F.C. Wrote the paper: C.H.H. W.K.F.T. Y.J.J.
References
- Itoh M. et al., Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 4, 67–82 (2003). [DOI] [PubMed] [Google Scholar]
- Zhang C., Li Q., & Jiang Y.-J., Zebrafish Mib and Mib2 are mutual E3 ubiquitin ligases with common and specific Delta substrates. J. Mol. Biol. 366, 1115–1128 (2007). [DOI] [PubMed] [Google Scholar]
- Ma M. & Jiang Y.-J., Jagged2a-Notch signaling mediates cell fate choice in zebrafish pronephric duct. PLoS Genet. 3, e18 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M. et al., Mib-Jag1-Notch signalling regulates patterning and structural roles of the notochord by controlling cell-fate decisions. Development 137, 2527–2537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddon C., Jiang Y.-J., Smithers, L., and Lewis, J., Delta-Notch signalling and the patterning of sensory cell differentiation in the zebrafish ear: evidence from the mind bomb mutant. Development 125, 4637–4644 (1998). [DOI] [PubMed] [Google Scholar]
- Bingham S. et al., Neurogenic phenotype of mind bomb mutants leads to severe patterning defects in the zebrafish hindbrain. Dev. Dyn. 228, 451–463 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Li Q., Lim C.-H., Qiu X., & Jiang Y.-J., The characterization of zebrafish antimorphic mib alleles reveals that Mib and Mind bomb-2 (Mib2) function redundantly. Dev. Biol. 305, 14–27 (2007). [DOI] [PubMed] [Google Scholar]
- Song R. et al., Mind bomb 1 in the lymphopoietic niches is essential for T and marginal zone B cell development. J. Exp. Med. 205, 2525–2536 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M. J. et al., Mind bomb-1 is essential for intraembryonic hematopoiesis in the aortic endothelium and the subaortic patches. Mol. Cell. Biol. 28, 4794–4804 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardos R. L., Lentz S. I., Wolfe M. S., & Raymond P. A., Notch-Delta signaling is required for spatial patterning and Muller glia differentiation in the zebrafish retina. Dev. Biol. 278, 381–395 (2005). [DOI] [PubMed] [Google Scholar]
- Crosnier C. et al., Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development 132, 1093–1104 (2005). [DOI] [PubMed] [Google Scholar]
- Horn S. et al., Mind bomb 1 is required for pancreatic beta-cell formation. Proc Natl Acad Sci USA 109, 7356–7361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt J. D. et al., Mindbomb 1, an E3 ubiquitin ligase, forms a complex with RYK to activate Wnt/beta-catenin signaling. J. Cell Biol. 194, 737–750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. & Gallagher P. J., Mind bomb 1 regulation of cFLIP interactions. Am. J. Physiol., Cell Physiol. 297, C1275–1283 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng L.-C. et al., New classes of Mind bomb-interacting proteins identified from yeast two-hybrid screens. PLoS ONE 9, e93394 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourquié O., Vertebrate somitogenesis. Annu. Rev. Cell Dev. Biol. 17, 311–350 (2001). [DOI] [PubMed] [Google Scholar]
- Jiang Y.-J. et al., Mutations affecting neurogenesis and brain morphology in the zebrafish, Danio rerio. Development 123, 205–216 (1996). [DOI] [PubMed] [Google Scholar]
- van Eeden F. J. M. et al., Mutations affecting somite formation and patterning in the zebrafish Danio rerio. Development 123, 153–164 (1996). [DOI] [PubMed] [Google Scholar]
- Holley S. A., Geisler R., & Nüsslein-Volhard C., Control of her1 expression during zebrafish somitogenesis by a Delta-dependent oscillator and an independent wave-front activity. Genes Dev. 14, 1678–1690 (2000). [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.-J. et al., Notch signalling and the synchronization of the somite segmentation clock. Nature 408, 475–479 (2000). [DOI] [PubMed] [Google Scholar]
- Holley S. A., Jülich D., Rauch G. J., Geisler R., & Nüsslein-Volhard C., her1 and the notch pathway function within the oscillator mechanism that regulates zebrafish somitogenesis. Development 129, 1175–1183 (2002). [DOI] [PubMed] [Google Scholar]
- Jülich D. et al., beamter/deltaC and the role of Notch ligands in the zebrafish somite segmentation, hindbrain neurogenesis and hypochord differentiation. Dev. Biol. 286, 391–404 (2005). [DOI] [PubMed] [Google Scholar]
- Bessho Y., Hirata H., Masamizu Y., & Kageyama R., Periodic repression by the bHLH factor Hes7 is an essential mechanism for the somite segmentation clock. Genes Dev. 17, 1451–1456 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale J. K. et al., Periodic notch inhibition by lunatic fringe underlies the chick segmentation clock. Nature 421, 275–278 (2003). [DOI] [PubMed] [Google Scholar]
- Delaune E. A., Francois P., Shih N. P., & Amacher S. L., Single-cell-resolution imaging of the impact of Notch signaling and mitosis on segmentation clock dynamics. Dev. Cell 23, 995–1005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido M. et al., Tbx24, encoding a T-box protein, is mutated in the zebrafish somite-segmentation mutant fused somites. Nat. Genet. 31, 195–199 (2002). [DOI] [PubMed] [Google Scholar]
- Topczewska J. M. et al., The winged helix transcription factor Foxc1a is essential for somitogenesis in zebrafish. Genes Dev. 15, 2483–2493 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnpenny P. D. et al., Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J. Med. Genet. 40, 333–339 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittock N. V. et al., Mutated MESP2 causes spondylocostal dysostosis in humans. Am. J. Hum. Genet. 74, 1249–1254 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow D. B. et al., Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am .J. Hum. Genet. 78, 28–37 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow D. B., Guillen-Navarro E., Fatkin D., & Dunwoodie S. L., Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum. Mol. Genet. 17, 3761–3766 (2008). [DOI] [PubMed] [Google Scholar]
- Yamamoto A. et al., Zebrafish paraxial protocadherin is a downstream target of spadetail involved in morphogenesis of gastrula mesoderm. Development 125, 3389–3397 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. A., Hammond C. L., & Hughes S. M., Fgf8 drives myogenic progression of a novel lateral fast muscle fibre population in zebrafish. Development 132, 4211–4222 (2005). [DOI] [PubMed] [Google Scholar]
- Bassett D. I. et al., Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 130, 5851–5860 (2003). [DOI] [PubMed] [Google Scholar]
- Paull T. T. et al., A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 (2000). [DOI] [PubMed] [Google Scholar]
- Wienholds E. et al., Efficient target-selected mutagenesis in zebrafish. Genome Res. 13, 2700–2707 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauerte H. E. et al., Sonic hedgehog is not required for the induction of medial floor plate cells in the zebrafish. Development 125, 2983–2993 (1998). [DOI] [PubMed] [Google Scholar]
- Chalamalasetty R. B. et al., The Wnt3a/beta-catenin target gene Mesogenin1 controls the segmentation clock by activating a Notch signalling program. Nat. Commun. 2, 390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajard L. et al., Wnt-regulated dynamics of positional information in zebrafish somitogenesis. Development 141, 1381–1391 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T., Jiang H., Topczewska J. M., & Hogan B. L. M., The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 15, 2470–2482 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amores A. et al., Developmental roles of pufferfish Hox clusters and genome evolution in ray-fin fish. Genome Res. 14, 1–10 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., & Schilling T. F., Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 (1995). [DOI] [PubMed] [Google Scholar]
- Langheinrich U., Hennen E., Stott G. & Vacun G., Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr. Biol. 12, 2023–2028 (2002). [DOI] [PubMed] [Google Scholar]
- Smithers L., Haddon C., Jiang Y.-J., & Lewis J., Sequence and embryonic expression of deltaC in the zebrafish. Mech. Dev. 90, 119–123 (2000). [DOI] [PubMed] [Google Scholar]
- Takke C., Dornseifer P., von Weizsäcker E., & Campos-Ortega J. A., her4, a zebrafish homologue of the Drosophila neurogenic gene E(spl), is a target of NOTCH signalling. Development 126, 1811–1821 (1999). [DOI] [PubMed] [Google Scholar]
- Takke C. & Campos-Ortega J. A., her1, a zebrafish pair-rule like gene, acts downstream of notch signalling to control somite development. Development 126, 3005–3014 (1999). [DOI] [PubMed] [Google Scholar]
- Weinberg E. S. et al., Developmental regulation of zebrafish MyoD in wild-type, no tail and spadetail embryos. Development 122, 271–280 (1996). [DOI] [PubMed] [Google Scholar]
- Qiu X., Xu H., Haddon C., Lewis J., & Jiang Y.-J., Sequence and embryonic expression of three zebrafish fringe genes, lunatic fringe, radical fringe, and manic fringe. Dev. Dyn. 231, 621–630 (2004). [DOI] [PubMed] [Google Scholar]
- Thisse C. & Thisse B., High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 3, 59–69 (2008). [DOI] [PubMed] [Google Scholar]
- Lauter G., Soll I., & Hauptmann G., Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev. Biol. 11, 43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G. Jr. et al., DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003). [PubMed] [Google Scholar]
- Li J. et al., Zebrafish foxc1a plays a crucial role in early somitogenesis by restricting the expression of aldh1a2 directly. J. Biol. Chem. 290, 10216–10228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.