

The role of myosin IIB in junctional contractility and its mode of regulation are not well understood. It is demonstrated that junctional recruitment of myosin IIB requires the activation of a receptor-type protein tyrosine phosphatase alpha–Src family kinase–Rap1 pathway. This reinforces the concept that E-cadherin–based signaling recruits distinct myosin II paralogues to generate contractile tension.
Abstract
Cell–cell adhesion couples the contractile cortices of epithelial cells together, generating tension to support a range of morphogenetic processes. E-cadherin adhesion plays an active role in generating junctional tension by promoting actin assembly and cortical signaling pathways that regulate myosin II. Multiple myosin II paralogues accumulate at mammalian epithelial cell–cell junctions. Earlier, we found that myosin IIA responds to Rho-ROCK signaling to support junctional tension in MCF-7 cells. Although myosin IIB is also found at the zonula adherens (ZA) in these cells, its role in junctional contractility and its mode of regulation are less well understood. We now demonstrate that myosin IIB contributes to tension at the epithelial ZA. Further, we identify a receptor type-protein tyrosine phosphatase alpha–Src family kinase–Rap1 pathway as responsible for recruiting myosin IIB to the ZA and supporting contractile tension. Overall these findings reinforce the concept that orthogonal E-cadherin–based signaling pathways recruit distinct myosin II paralogues to generate the contractile apparatus at apical epithelial junctions.
INTRODUCTION
Cell–cell adhesion integrates epithelial cells to form mechanically coherent tissues (Gomez et al., 2011; Guillot and Lecuit, 2013). In particular, by physically connecting cells, E-cadherin adhesion provides the mechanical link that couples the contractile cortices of neighboring cells together (Maitre et al., 2012; Levayer and Lecuit, 2013). This generates contractile tension at the junctions, which can influence morphogenetic processes such as neighbor exchange (Rauzi et al., 2010), cell extrusion (Eisenhoffer et al., 2012; Wu et al., 2014), and tissue-level patterning (Martin et al., 2010).
Consistent with this, E-cadherin–based cell–cell junctions are enriched in an actomyosin cortex (Shewan et al., 2005; Miyake et al., 2006; Fernandez-Gonzalez et al., 2009; Smutny et al., 2010). All three mammalian paralogues of nonmuscle myosin II (NMIIA, B, and C) have been reported to localize with F-actin at junctions in tissues and in cultured epithelial cells (Smutny et al., 2010; Ebrahim et al., 2013). In simple polarized epithelia, actomyosin is especially prominent at the zonula adherens (ZA), a specialized adhesive junction that comprises stabilized clusters of E-cadherin distributed as a ring in the juxtaluminal region of cell–cell contacts, just below the tight junction (Ratheesh et al., 2012; Wu et al., 2014). The cortical accumulation of NMII reflects the combined effects at junctions of local actin assembly (Verma et al., 2012) and cell signaling (Smutny et al., 2010; Ratheesh et al., 2012).
The role of cell signaling is exemplified by the recruitment of NMIIA to junctions by the Rho-ROCK signaling pathway in MCF-7 and Caco-2 cell lines (Shewan et al., 2005; Smutny et al., 2010; Ratheesh et al., 2012). Indeed, Rho signaling localized prominently to the ZA, reflecting coordinated regulation of the Rho activator, Ect2, and an inactivator, p190B RhoGAP, by the centralspindlin complex (Ratheesh et al., 2012). Of note, E-cadherin was necessary for Rho signaling at apical junctions (Priya et al., 2013) and localized centralspindlin via an interaction with α-catenin (Ratheesh et al., 2012). This emphasizes that the E-cadherin adhesion system can promote junctional tension both by providing the adhesion that couples contractile cortices together and also by contributing to biogenesis of the junctional actomyosin apparatus itself.
Despite their many similarities, different vertebrate NMII paralogues have distinct biophysical properties, cellular functions, and morphogenetic impacts (Vicente-Manzanares et al., 2009; Dulyaninova and Bresnick, 2013; Heissler and Manstein, 2013). For example, although NMIIA and NMIIB are both found at the ZA of MCF-7 cells, junctional F-actin was affected only by depletion of NMIIB and not by depletion of NMIIA (Smutny et al., 2010). Similarly, tissue-specific ablation of NMIIB in the mouse brain, where it is highly expressed (Kawamoto and Adelstein, 1991), resulted in abnormal neuroepithelial morphogenesis (Tullio et al., 2001; Ma et al., 2007) and defects in neuronal migration (Ma et al., 2004). In contrast, ablation of NMIIA caused severe adhesive defects in the preimplantation mouse embryo, which failed to develop a functional visceral endoderm (Conti et al., 2004; Wang et al., 2011). Recently, careful comparison revealed that, although both NMIIA and NMIIB are expressed in the zebrafish neuroepithelium, each NMII paralogue was responsible for distinct cell shape changes during epithelial folding (Gutzman et al., 2014). Thus the distinct vertebrate NMII proteins are not necessarily functionally redundant.
NMIIB has also been implicated in generating cortical tension during convergent-extension movements (Skoglund et al., 2008). However, less is known about its potential contribution to junctional contractility at cadherin junctions. NMIIB was necessary for ZA integrity (Smutny et al., 2010), but whether this translated to an impact on junctional tension was unclear. Nor do we understand well the signaling pathways that support its recruitment to junctions. We earlier found that the Rho-ROCK pathway, which recruited NMIIA to the ZA in MCF-7 mammary epithelial cells, did not have a similar influence on the junctional accumulation of NMIIB (Smutny et al., 2010; Ratheesh et al., 2012). Instead, junctional NMIIB required both Rap1 and Src family kinase (SFK) signaling (Smutny et al., 2010; McLachlan and Yap, 2011). However, what relationship might exist between these signals was unknown. Accordingly, we sought in the present study to define the contribution of NMIIB to contractile tension at E-cadherin junctions and aimed to elucidate the signaling pathway responsible for its recruitment to the ZA.
RESULTS
NMIIB regulates junctional tension at the ZA of MCF-7 cells
We performed these studies in MCF-7 cells, which form prominent ZA. Earlier confocal analyses showed that both NMIIA and NMIIB concentrate at the ZA in confluent MCF-7 monolayers and contribute to its morphological integrity (McLachlan and Yap, 2011; Smutny et al., 2011). Now, by applying higher-resolution imaging with structured illumination microscopy (SIM), we further found that both paralogues localize in puncta overlying the apical actin rings (Figure 1A), which may correspond to the sarcomeric-like organization of actomyosin seen at epithelial junctions in tissues (Ebrahim et al., 2013).
FIGURE 1:

Myosin IIA and myosin IIB regulate junctional tension at the ZA. (A) Structured illumination images of MCF-7 cells costained for NMIIA or NMIIB and F-actin (phalloidin). Center and right, magnified views showing NMII and F-actin distributions in regions indicated on left panels. (B) Western blot analysis of NMII expression in WT MCF-7 cells, MCF-7 cells infected with a lentivirus encoding for mCherry as an infection marker (Control), or lentiviruses bearing a shRNA sequences against either human NMIIA (NMIIA KD) or against human NMIIB (MNIIB KD). (C and D) Junctional tension measurements using laser nanoscissors. Average recoil profiles (C) and control-normalized average initial velocity recoils (D) for at least 35 cell junctions per condition from three independent experiments. *, p < 0.05; **, p <0.01, one-way ANOVA, Dunnett's multiple-comparison test.
While depletion of NMIIA decreased tension at the ZA (Ratheesh et al., 2012), the potential contribution of NMIIB has not yet been examined. Therefore, to test whether NMIIB can affect junctional tension, we used laser nanoscissors (Konig, 2000) to cut the ZA, identified by expressing E-cadherin–green fluorescent protein (GFP) in E-cadherin RNA interference (RNAi) cells (Smutny et al., 2011; Ratheesh et al., 2012). As previously reported (Ratheesh et al., 2012), laser ablation caused the vertices of junctions to recoil from one another, and we calculated the initial velocity of recoil as an index of the tension present in the contact before ablation (Hutson et al., 2003; Fernandez-Gonzalez et al., 2009; Fernandez-Gonzalez and Zallen, 2013; Ratheesh et al., 2012). We compared monolayers depleted of NMIIB by RNAi with both NMIIA knockdown (KD) cells and control monolayers (Figure 1B). NMIIB KD reduced recoil (Figure 1C) and initial recoil velocities (Figure 1D) after ablation as effectively as did NMIIA KD. This suggested that NMIIB also contributes to the generation of contractile tension at the epithelial ZA. We therefore sought to investigate the molecular mechanisms that control its junctional localization.
RPTPα regulates junctional tension and NMIIB junctional localization
We previously found that the accumulation of NMIIB at the ZA required SFK and protein tyrosine phosphatase (PTP) activity (McLachlan and Yap, 2011). Furthermore, E-cadherin adhesion can activate SFK signaling by a vanadate-sensitive pathway (McLachlan et al., 2007; McLachlan and Yap, 2011), consistent with evidence that PTPs can be upstream of SFKs (Roskoski, 2005). This suggested that a PTP-SFK pathway might participate in the junctional recruitment of NMIIB (Stoker, 2005; Sallee et al., 2006; McLachlan and Yap, 2007).
To begin to pursue this possibility, we first examined PTPs whose catalytic activity has been reported to activate SFKs (Roskoski, 2005; Haj et al., 2012; Monteleone et al., 2012; Murchie et al., 2014). We tested receptor PTP alpha (RPTPα), receptor PTP sigma (RPTPσ), and PTP1B. RPTPα and RPTPσ have been identified at cell–cell junctions in earlier studies (Murchie et al., 2014; Truffi et al., 2014), and we confirmed their coaccumulation with F-actin at the ZA in MCF-7 cells (Figure 2A). In contrast, junctional localization of PTP1B was less evident, the protein staining instead in the perinuclear region and cytoplasm of MCF-7 cells, possibly associated with the endoplasmic reticulum (Haj et al., 2012; Monteleone et al., 2012). These results suggested that either RPTPσ and/or RPTPα could participate in SFK activation in the context of cell–cell adhesion. Of note, we also recently found that RPTPα regulates the integrity of E-cadherin cell–cell contacts and the process of initial contact formation (Truffi et al., 2014).
FIGURE 2:

RPTPα regulates junctional tension and myosin IIB localization. (A) Immunofluorescence for F-actin and PTP1B, RPTPα, or RPTPσ protein phosphatases in confluent MCF-7 cell monolayers. Images were focused at the level of the circumferential actin ring that corresponds to the ZA. (B) Western blot and (C) immunofluorescence analysis for RPTPα in MCF-7 cells treated with RPTPα siRNA. (D and E) Immunofluorescence (inverted color scale) analysis of zonula occludens protein 1 (ZO-1) (D) and junctional linearity measurements (E) in control and RPTPα KD cells. Arrows in D point toward linear junctions (Ctrl siRNA) and wavy junctions in RPTPα-depleted cells (RPTPα siRNA). (F) Normalized average initial recoil values following laser nanoablation of the ZA in control and RPTPα KD cells. (G) Western blot and (H) immunofluorescence analysis of NMIIA and NMIIB in control and RPTPα KD cells. In H, cells were also stained for ZO-1, and GAPDH was used as an internal loading control in Western blotting. (I) Quantitative analysis of junctional NMIIB using line-scan analysis in control, RPTPα KD, or RPTPα KD cells reconstituted with HA-tagged siRNA-resistant WT or C433S,C723S RPTPα. Data in E, F, and I are means ± SEM for at least 50 contacts per condition. ****, p < 0.0001, two-tailed t test (E and F) or one-way ANOVA, Dunnett's multiple-comparison test (I).
Accordingly, we focused on whether RPTPα could affect junctional contractility. First, we tested how depletion of RPTPα by RNAi (Figure 2, B and C) affected junctional morphology (Figure 2, D and E). Whereas control cells displayed junctions that were straight and continuous, those in RPTPα small interfering RNA (siRNA) cells were wavier (Figure 2D), a difference that was quantitatively confirmed using a previously reported linearity index (Figure 2E; Otani et al., 2006; McLachlan and Yap, 2011). This was consistent with our earlier finding that global phosphatase inhibition decreased junctional linearity (McLachlan and Yap, 2011). As this morphological change may reflect changes in apical junctional tension (Otani et al., 2006), we then used laser nanoscissors to measure tension more directly (Figure 2F). We found that RPTPα KD significantly decreased initial recoil velocity, suggesting that apical junctional tension was reduced by depletion of this PTP. Altogether these findings suggested that RPTPα supports contractility at the apical ZA.
We then examined the impact of RPTPα KD on junctional myosin II (Figure 2, G–I, and Supplemental Figure 1A). Interestingly, RPTPα depletion significantly reduced NMIIB staining at the ZA but not that of NMIIA (Figure 2H). Moreover, the reduction in junctional NMIIB was rescued by expression of hemagglutinin (HA)-tagged wild-type (WT) siRNA-resistant mouse RPTPα (Figure 2I), confirming that it was due to depletion of this protein. Of note, however, junctional NMIIB was not restored by expression of the phosphatase-deficient mutant RPTPαC433S,C723S (Buist et al., 2000; Figure 2I), which localized to junctions as effectively as did WT RPTPα (Supplemental Figure 1A). As total protein levels of these myosins were unchanged (Figure 2G), this suggested that RPTPα activity could regulate the junctional recruitment of NMIIB. This differential impact on myosin II paralogue localization was similar to what we had earlier observed when PTP activity was inhibited by vanadate (McLachlan and Yap, 2011). Overall these findings identify a contribution of RPTPα to supporting the contractile apparatus of the ZA, presumably through the recruitment of NMIIB.
RPTPα regulates SFK signaling at junctions
PTPs can activate SFK signaling by dephosphorylating an inhibitory tyrosine residue found in many SFKs (Y530 in human Src; Roskoski, 2005). As its phosphatase activity was necessary for RPTPα to induce the junctional accumulation of NMIIB (Figure 2I), we then asked whether this might reflect an impact on junctional SFK signaling. We first immunostained MCF-7 cells using an antibody that recognizes another tyrosine residue (Y419 in human Src) that is phosphorylated when many SFKs are active (pY419 SFK; Ren et al., 2009; McLachlan and Yap, 2011). We found that the amount of junctional pY419 SFK was significantly lower in RPTPα KD cells than in controls, despite total cellular levels of pY419 SFK being similar in immunoblots (Figure 3, A–C). This suggested that RPTPα might be necessary to support SFK signaling at the ZA.
FIGURE 3:

Junctional SFK signaling is regulated by RPTPα. (A–C) Analysis of total (A) and junctional (B and C) content of pY419 SFK in control and RPTPα KD MCF-7 cells. In C, quantitative analysis was performed using line-scan analysis. (D and E) FRET measurements of SFK activity in live MCF-7 cells. (D) Images shown are YFP fluorescence distribution (left) and FRET/YFP ratio images (right) of the SFK-Bio-tK in control and RPTPα KD cells. (E) Quantitative analysis of FRET/YFP ratios at cell–cell junctions. Data in C and E are means ± SEM for at least 40 contacts and for at least 25 images (∼90–100 cells) per condition, respectively. ****, p < 0.0001, two-tailed t test.
We then used fluorescence resonance energy transfer (FRET) imaging with specific Src-FRET biosensors to better characterize SFK signaling in live cells. We used an SFK substrate biosensor fused to the membrane-targeting domain of K-Ras (Wang et al., 2005; Seong et al., 2009). This biosensor (SFK-Bio-tK) contains a phosphorylatable substrate for SFKs fused to the SH2 domain of Src and flanked by cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP). When this substrate is unphosphorylated, the CFP and YFP fluorophores are in close proximity, which gives a high FRET signal. However, when the biosensor is phosphorylated, the SH2 domain binds to the phosphorylated substrate to induce a conformational change that reduces energy transfer (Wang et al., 2005). Thus SFK activity is reported by a decrease in FRET. Of note, the substrate domain in this biosensor is responsive to Src, and to a lesser extent to Fyn, but not to Yes, Abl, or ERK1 (Wang et al., 2005).
To test that the biosensor was sensitive enough to report SFK activity at junctions, we performed FRET/YFP emission ratio measurements (an index of FRET) under control conditions, upon treatment with a SFK inhibitor (PP2), or in cells overexpressing a WT c-Src-Cherry construct to increase signaling (Supplemental Figure 1B). We found that PP2 increased the FRET index ratio, indicating decreased activity, at junctions compared with controls, showing that this biosensor reports junctional SFK activity at the ZA. Conversely, overexpression of c-Src-Cherry decreased the FRET index ratio, a change consistent with the predicted increase in Src activity. These results confirmed that the sensor was specific and sensitive enough to report junctional Src activity in live cells. We then found that, under the same conditions, RPTPα KD increased the FRET index ratio, indicating that junctional SFK activity was reduced (Figure 3, D and E). Taken with our pY419 SFK immunofluorescence data, this revealed a significant role for RPTPα in promoting junctional SFK activity.
Identifying SFKs that regulate junctional contractility and ZA integrity
We then pursued the role of SFKs in the regulation of junctional NMIIB and tension. Consistent with our earlier experience (McLachlan and Yap, 2011), PP2 reduced junctional NMIIB recruitment (Figure 4, A and B, and Supplemental Figure 2A). This impact of PP2 on NMIIB was comparable to that seen with RPTPα depletion (Figure 4B), but whether SFK signaling also affected junctional tension was unknown. Using laser nanoscissors, we found that PP2 reduced junctional tension at the ZA to a similar extent as did the ROCK inhibitor, Y27632 (Figure 4C). This implied that SFK activity could contribute to junctional contractility as substantively as the ROCK pathway.
FIGURE 4:

c-Src and c-Yes contribute to junctional SFK signaling in MCF-7 cells. (A and B) Quantitative immunofluorescence analysis of NMIIB at the ZA in MCF-7 cells treated with control, PP2, RPTPα siRNA, and RPTPα siRNA+PP2 (25 μM, 1 h). Representative images of E-cadherin and NMIIB in control and PP2-treated cells (A) and quantitative line-scan analysis of NMIIB fluorescence at junctions (B). (C) Normalized initial recoil values for laser nanoscissor experiments performed in MCF-7 cells treated with control, PP2 (25 μM, 1 h) and Y-27632 (30 μM, 1 h). (D) Immunofluorescence and (E) Western blot analysis of c-Src and c-Yes kinases in control cells and cells transfected either with a c-Src siRNA or c-Yes siRNA. (F) Quantitative analysis of pY419 SFK at the ZA in control, c-Src KD, and c-Yes KD MCF-7 cells. Data in B, C, and F are means ± SEM for at least 40 contacts per condition. ****, p < 0.0001, two tailed t test (B) and one-way ANOVA, Dunnett's multiple-comparison test (C and F).
Several Src family kinases have been implicated in the regulation of cadherin junctions (Calautti et al., 1998; Owens et al., 2000; Piedra et al., 2003; McLachlan et al., 2007). To test which of these might influence junctional contractility, we probed Western blots for three of the best-characterized family members, c-Src, c-Fyn, and c-Yes. Of these, c-Src and c-Yes were identified in MCF-7 cells, whereas we were unable to detect expression of c-Fyn (unpublished data), as was also recently reported (Elias et al., 2014). Indirect immunofluorescence imaging demonstrated that c-Src localized to the ZA, at the lateral cell–cell junctions located below the ZA, and in a small cytosolic fraction (Figure 4D). c-Yes staining was also evident at the ZA and throughout the lateral junctions (Figure 4D).
To assess the relative contributions of c-Src and c-Yes to junctional SFK activity, we then depleted each of these kinases by siRNA (Figure 4E) and quantitated pY419 SFK immunofluorescence at the ZA (Figure 4F). c-Src siRNA reduced protein levels by > 60%, associated with an ∼40% decrease in junctional pY419 SFK staining, while c-Yes siRNA depleted protein levels by >90%, associated with a decrease in junctional pY419 SFK of ∼50%. Thus both c-Src and c-Yes contributed to the total junctional SFK activity.
We then asked whether these contributions to junctional signaling extended to affect the morphology of the ZA. Whereas E-cadherin prominently accumulated in an apical ZA ring in control cells, E-cadherin immunofluorescence was significantly reduced by either c-Src or c-Yes siRNA (Figure 5, A and B). Total cellular levels of E-cadherin were unaltered (Supplemental Figure 2B), suggesting that these changes at cell–cell contacts reflected an inability of cells to concentrate E-cadherin into an apical ZA. In parallel, F-actin at the ZA was also significantly reduced by c-Src or c-Yes depletion (Figure 5, A and C), changes similar to what we had earlier observed when NMIIB was reduced (Smutny et al., 2010). Indeed, we confirmed that junctional NMIIB was reduced by either c-Src or c-Yes KD (Figure 5D) without a change in the total cellular levels of either NMIIB or actin (Supplemental Figure 2, D and E ). Overall these findings suggest that both c-Src and c-Yes signaling contribute to regulation of the integrity of the ZA and its contiguous actomyosin cytoskeleton.
FIGURE 5:

c-Src and c-Yes regulate the organization of F-actin and E-cadherin at the ZA. (A) Representative confocal images showing the subcellular distribution of E-cadherin and F-actin in control, c-Src KD, and c-Yes KD MCF-7 cells. Quantitative immunofluorescence analysis of junctional E-cadherin (B), F-actin (C), and NMIIB (D) in control, c-Src, and c-Yes KD cells. Data are means ± SEM for at least 100 contacts per condition. ****, p < 0.0001, one-way ANOVA, Dunnett's multiple-comparison test.
SFKs regulate junctional Rap1 signaling
We then sought to investigate the molecular link between SFKs and myosin IIB. One possibility was the GTPase Rap1, whose activity can be regulated by protein kinases (Balzac et al., 2005; Pannekoek et al., 2009). Rap1 localizes to cell–cell junctions (Knox and Brown, 2002; Hogan et al., 2004; Balzac et al., 2005; Li et al., 2010), and we earlier found that it was necessary for the junctional recruitment of NMIIB (Smutny et al., 2010). Accordingly, we asked whether junctional Rap1 signaling was affected by inhibiting SFK activity. First, we assessed Rap1 localization by live-cell imaging of GFP-Rap1 in control and PP2-treated cells (Figure 6A). Control cells displayed stable localization of this reporter at the ZA and to a lesser extent in the perinuclear region. However, this pattern was not altered by treatment with PP2.
FIGURE 6:

Rap1 is a downstream target of junctional SFK signaling. (A) Subcellular distribution of GFP-Rap1WT in Control and PP2-treated cells analyzed by live-cell imaging. (B) FRET/YFP emission ratio images of cells transfected with Raichu-Rap1 FRET biosensor in control cells, PP2-treated cells, and cells transfected with RPTPα siRNA. (C) Quantitative analysis of Rap1 activity (FRET/YFP) emission ratios at the ZA of control, PP2-treated, RPTPα KD, and PP2-treated RPTPα KD cells. Data in C are means ± SEM for at least 25 images (∼90–100 cells) per condition. **, p < 0.01; ****, p < 0.0001, one-way ANOVA. (D) Western blot analysis of p130Cas expression in cells transfected with a control siRNA (Control) or an siRNA against p130Cas (p130 Cas siRNA). GAPDH was used as a loading control. (E and F) Analysis of Rap1 activity at the cell–cell junctions using FRET microscopy (E) and junctional NMIIB accumulation (F) in control (Control siRNA) and p130Cas-depleted cells (p130Cas siRNA). ns, no significant differences, two-tailed t test.
As protein localization does not necessarily reflect the distribution of the GTP-loaded, active form of Rap1 (Nakamura et al., 2005), we then expressed the established Rap1-Raichu FRET reporter in MCF-7 cells to assess potential changes in Rap1 activity (Mochizuki et al., 2001). Control cells showed clear energy transfer at the ZA, confirming that this is a site for Rap1 signaling (Figure 6B). Furthermore, junctional Rap1-Raichu FRET emission ratios were significantly reduced by PP2, suggesting that SFKs are necessary for junctional Rap1 signaling (Figure 6, B and C). In contrast, Rap1KD by siRNA did not affect junctional SFK signaling as assessed using the Src FRET reporter (Supplemental Figure 3). This placed Rap1 downstream of SFK signaling at cadherin junctions.
Because our earlier data identified RPTPα as an activator of junctional SFK signaling, this implied that junctional Rap1 signaling might also be influenced by RPTPα. Indeed, we found that RPTPα KD reduced the Rap1 FRET index to a similar degree as PP2 (Figure 6, B and C). Moreover, PP2 treatment did not further decrease Rap1 activity in RPTPα KD cells (Figure 6C). We therefore conclude that a RPTPα→SFK→Rap1 pathway resides at the ZA.
A number of mechanisms can allow SFK signaling to regulate Rap1 (Kooistra et al., 2007; Pannekoek et al., 2009). In one of these, SFK phosphorylates the adaptor protein, p130Cas, to recruit Rap1 GEFs, such as C3G (Sawada et al., 2006). To test whether p130Cas might be relevant at the ZA, we measured junctional Rap1 activity by FRET when p130Cas was depleted by siRNA (Figure 6D). Neither junctional Rap1 activity nor junctional NMIIB levels (Figure 6, E and F) were affected in p130cas KD cells, despite an ∼70% decrease in p130Cas levels (Figure 6D). This suggested that p130Cas is not a key mediator of SFK signaling to Rap1 at the ZA.
E-cadherin may affect the oligomerization state of RPTPα at the ZA
Finally, we asked how RPTPα activity might be regulated at the ZA. We recently observed that RPTPα was necessary both for steady-state SFK signaling at cell–cell junctions and for E-cadherin ligation to activate SFK (Truffi et al., 2014). Together these results suggested that E-cadherin might also signal through RPTPα to support SFK signaling at the ZA. Consistent with this, we found that SFK activity, as measured by pY419 SFK immunofluorescence, was significantly reduced by E-cadherin siRNA (Figure 7, A and B).
FIGURE 7:

E-cadherin associates with RPTPα at cell–cell junctions. (A and B) Immunofluorescence analysis of junctional pY419 SFK in MCF-7 cells transfected with either a control siRNA (Ctrl siRNA) or a siRNA targeted to human E-cadherin (E-cad siRNA). Representative images, together with E-cadherin staining, are shown in A and the corresponding line-scan quantitation in B. (C) Immunoprecipitation analysis of MCF-7 cells using beads coated with a rabbit immunoglobulin G control (Ctrl) or a rabbit polyclonal antibody against RPTPα. Data are Western blots of the immunoprecipitates and whole-cell lysates probed with anti–E-cadherin and RPTPα antibodies. (D) Subcellular distribution of E-cad-GFP and mRPTPα-mCherry constructs expressed in MCF-7 cells and captured by live-cell imaging. (E) Analysis of GFP fluorescence lifetime when fused to E-cadherin and expressed alone (E-cadGFP) or coexpressed with mRPTPα-mCherry or soluble mCherry. Measurements were performed in regions of interest localized at the ZA. (F) mCherry fluorescence anisotropy measurement performed using live cells expressing mCherry alone, mRPTPα-mCherry alone, or mRPTPα-mCherry and E-cad-GFP. Data in B and E are means ± SEM for at least 25 contacts per condition. **, p < 0.01; ****; p < 0.0001, two-tailed t test (B) and one-way ANOVA, Dunnett's multiple-comparison test (E). Data in F are means ± SEM for at least 50 images (∼150 contacts) per condition. *, p < 0.05; ****, p < 0.0001 one-way ANOVA.
Accordingly, we focused on analyzing the relationship between E-cadherin and RPTPα. We found that RPTPα coimmunoprecipitates with endogenous E-cadherin in MCF-7 cells (Figure 7C), indicating that these proteins can interact biochemically. To corroborate this, we performed fluorescence lifetime imaging (FLIM) analysis of GFP in control cells that expressed E-cadherin–GFP alone or in cells that coexpressed E-cadherin–GFP with either mouse RPTPα-mCherry (Truffi et al., 2014) or with cytosolic mCherry. We detected a GFP fluorescence lifetime of ∼2.2 ns when E-cadherin–GFP was expressed alone. However, coexpression of mRPTPα-mCherry, but not of cytoplasmic mCherry, significantly reduced E-cadherin–GFP lifetime due to FRET (Figure 7, D and E). This implied that E-cadherin and RPTPα are in close molecular proximity in MCF-7 cells, as we recently found in Caco-2 colon epithelial cells (Truffi et al., 2014).
The oligomerization state of RPTPα can potentially regulate its activity (den Hertog et al., 1999; Jiang et al., 1999). RPTPα can form homodimers that are inactive because, in their cytoplasmic tails, the membrane proximal region of one monomer can insert into the catalytic site (D1) of the other monomer and vice versa, thereby preventing catalytic activity. This implies that RPTPα can become activated when homodimerization is abrogated (den Hertog et al., 1999; Jiang et al., 1999). We therefore hypothesized that the observed interaction between RPTPα and E-cadherin might reduce RPTPα homodimerization and potentially activate RPTPα. As a first test of this notion, we used fluorescence anisotropy (homo-FRET; Brooks et al., 2014) to test whether overexpression of E-cadherin could affect mRPTPα-Cherry homodimerization at apical cadherin junctions. Fluorescence anisotropy allows the detection of homodimers with high sensitivity, because energy transfer between identical fluorophores that are close to one another causes depolarization of fluorescence emission, thereby reducing anisotropy.
Accordingly, we measured mCherry fluorescence anisotropy in cells expressing cytosolic mCherry, mRPTPα-mCherry, or both mRPTPα-mCherry and E-cadherin–GFP (Figure 7F). Fluorescence anisotropy values for mRPTPα-mCherry alone were lower than those for cytosolic mCherry alone, indicating that RPTPα molecules undergo homo-FRET. However, mRPTPα-Cherry anisotropy was increased when E-cadherin was coexpressed. This indicated that the overexpression of E-cadherin decreased energy transfer between RPTPα molecules, suggesting a decrease in RPTPα homodimers. Thus E-cadherin may directly or indirectly alter the oligomerization status of RPTPα, a potential mechanism for its activation.
DISCUSSION
Cell–cell junctions are contractile apparatuses in which tension is generated to influence morphogenesis (Heisenberg and Bellaiche, 2013; Liang et al., 2014). E-cadherin serves as an active mechanical agent: it contributes to generating junctional tension, both by mechanically coupling the contractile cortices of neighboring cells together (Maitre et al., 2012) and also by participating in biogenesis of the junctional actomyosin apparatus itself (Smutny et al., 2010; Ratheesh et al., 2012; Verma et al., 2012). Multiple myosin II paralogues are expressed in mammalian cells, and all of these can be found at adherens junctions. However, they may be subject to different signaling pathways and potentially have distinct contributions to junctional biology (Smutny et al., 2010, 2011). Earlier, we showed that NMIIB influences ZA integrity (Smutny et al., 2010). We have now extended this analysis to demonstrate that NMIIB, like NMIIA, contributes to the generation of junctional tension at apical ZA. Further, the recruitment of NMIIB and generation of junctional tension is subject to a RPTPα-SFK-Rap1 signaling pathway, distinct from the Rho-ROCK pathway responsible for recruitment of NMIIA (Smutny et al., 2010; Ratheesh et al., 2012). Accepting that we have assayed myosin recruitment as the target of regulation, these findings reinforce the notion that orthogonal signaling pathways converge to regulate different myosin II paralogues at cadherin junctions.
In earlier studies, we found that SFK and Rap1 signaling contributed to recruiting myosin IIB to the apical ZA in MCF-7 cells (Smutny et al., 2010; McLachlan and Yap, 2011). Our current experiments allow us to place these in a signaling pathway downstream of the membrane-bound RPTPα. SFK activity was necessary for Rap1 to be active at the ZA, but inhibition of Rap1 did not affect SFK signaling at junctions. Thus SFK activity could be placed upstream of Rap1. Further, RPTPα, a recognized activator of SFKs (Harder et al., 1998; Su et al., 1999), was necessary for junctional SFK and Rap1 signaling and ultimately supported junctional myosin IIB and contractile tension.
Multiple Src family kinase members are expressed in mammalian epithelial cells, where they can have redundant or nonredundant functions. Our current data suggest that both c-Src and c-Yes contribute to the junctional SFK signaling that recruits NMIIB to support junctional contractility. Similarly, both these kinases influence collective cell movements during gastrulation (van Eekelen et al., 2010) and the mechanical properties of MDCK cells (Andreeva et al., 2014) and zebrafish epithelia (Yoo et al., 2012). Thus RPTPα may regulate multiple kinase targets to support junctional contractility. Downstream, SFK may regulate Rap1 by phosphorylation of the scaffold protein p130Cas and/or the Rap1GEF G3G (Radha et al., 2004; Balzac et al., 2005; Sawada et al., 2006; Asuri et al., 2008; Birukova et al., 2013). However, we were unable to detect p130Cas at cell–cell junctions (unpublished data); nor did depletion of p130Cas affect either junctional Rap1 activity or NMIIB content (Balzac et al., 2005; Sawada et al., 2006; Asuri et al., 2008). Whether C3G and/or other Rap GEFs (Pannekoek et al., 2009) influence junctional contractility has yet to be tested.
Both actin dynamics (Verma et al., 2012; Leerberg et al., 2014; Wu et al., 2014) and myosin recruitment (Ratheesh et al., 2012) contribute to junctional tension. NMIIB may support junctional contractility through its behavior as a high-duty ratio actin-based motor (De La Cruz and Ostap, 2004). In addition, NMIIB can indirectly affect the contractile apparatus by influencing actin assembly. We earlier found that NMIIB depletion reduced steady-state junctional F-actin (Smutny et al., 2010), an effect that is also seen in RPTPα-depleted cells (Truffi et al., 2014). We also recently found that NMIIB can support junctional actin homeostasis through the tension-sensitive recruitment of vinculin, which anchors Mena/VASP proteins at the ZA to promote actin assembly (Leerberg et al., 2014). Thus NMIIB may promote contractility at junctions by regulating the junctional actin cytoskeleton as well as through its intrinsic force-generating capacity.
Furthermore, NMIIB is not the only target of RPTPα-SFK signaling at cadherin junctions. Another is the scaffolding protein cortactin, a known substrate for SFK (Weed and Parsons, 2001), which we recently identified as a target of RPTPα-SFK junctional signaling (Ren et al., 2009; Truffi et al., 2014). Depletion of RPTPα perturbed the integrity of the ZA by decreasing the apical concentration of E-cadherin. This phenocopied the impact of cortactin depletion and, indeed, the ZA of RPTPα KD cells could be restored by expression of a phosphomimetic cortactin mutant (Truffi et al., 2014). RPTPα-SFK signaling may therefore coordinate multiple pathways that ultimately determine the morphological integrity of the ZA and its contractile behavior.
Finally, our findings indicate that E-cadherin supports the junctional SFK signaling pathway, as it does junctional Rho-ROCK signaling (Ratheesh et al., 2012; Priya et al., 2013). Thus E-cadherin depletion reduced SFK activity in our current experiments, as it also reduces Rho signaling (Priya et al., 2013). However, more remains to be learned about how E-cadherin might activate the RPTPα-SFK pathway. RPTPα can be activated by several mechanisms (den Hertog et al., 1995; Tracy et al., 1995; Zheng et al., 2000), but an interesting clue lies in the capacity for homodimerization to inhibit the catalytic activity of RPTPα. It has been shown recently that disruption of this autoinhibitory mechanism may increase Src activity in a significant subset of colon, breast, and liver human tumors (Huang et al., 2011). We found that E-cadherin can physically interact with RPTPα in both immunoprecipitation and FLIM-FRET analyses, suggesting that it may form a heterodimer with RPTPα (Truffi et al., 2014). Further, overexpression of E-cadherin reduced homo-FRET between RPTPα at junctions, as assessed by fluorescence anisotropy. We therefore postulate that interaction with E-cadherin may activate RPTPα by reducing its homodimerization. Whether this is the mechanism responsible for recruiting myosin IIB to junctions is an important question for future research.
MATERIALS AND METHODS
Cell culture
MCF-7 cells were maintained in DMEM (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 1% l-glutamine, 100 U/ml penicillin, and 100 U/ml streptomycin.
Transfection and siRNA knockdown
Cells were transfected with plasmid expression constructs using Lipofectamine 2000 (Invitrogen, Grand Island, NY) according to the manufacturer's instructions and analyzed 24–48 h after transfection. Lipofectamine RNAiMAX (Invitrogen) was used for siRNA transfection. siRNA sequences were control (on-target plus nontargeting pool, UGGUUUACAUGUCGACUAA, UGGUUUACAUGUUGUGUGA, UGGUUUACAUGUUUUCUGA, UGGUUUACAUGUUUUCCUA); siRNA against RPTPα (SASI_Hs01_00169093; Sigma, St. Louis, MO; CUAUCGUGAUGCUAACAGA), c-Src (on-target smart pool siRNA J-003175; Dharmacon, Lafayette, CO; GGGAGAACCUCUAGGCACA, CCAAGGGCCUCAACGUAA, GCAGAGAACCCGAGAGGGA, GCAGUUGUAUGCUGUGGUU); c-Yes (on-target smart pool siRNA J-003184; Dharmacon; CAGAAGACCUUUCAUUUAA, GGAAAGUAUUUGAAGCUUC, GAUCUUCGGGCUGCUAAUA); E-cadherin (on-target smart pool siRNA J-003877; Dharmacon; GGCCUGAAGUGACUCGUAA, GAGAACGCAUUGCCACAUA, GGGACAACGUUUAUUACUA, GACAAUGG-UUCUCCAGUUG; p130Cas (on target smart pool siRNA J-020465; Dharmacon; CCAGAUGGGCAGUACGAGA, GCAAUGCUGCCCACACAUC, GGCCACAGGACAUCUAUGA, GGUCGACAGUGGUGUGUAU). Briefly, 5 μl of Lipofectamine RNAiMAX was added to 250 μl of OptiMEM (Life Technologies) and incubated at room temperature (RT) for 5 min. This was added to 250 μl of the corresponding siRNA (at 0.5–1 μM) and made up to 500 μl with OptiMEM. The Lipofectamine/DNA solution was incubated for 20 min at RT, and then the volume of this solution was increased to 1.25 ml with OptiMEM and used for incubation on cells at 37°C for 5 h before the medium was replaced with DMEM (supplemented with 10% FBS, 1% non-essential amino acids, 1% l-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin). The cells were incubated at 37°C for a further 48 h and were subjected to a second round of siRNA transfection. Cells were analyzed 24 h after this step. In experiments in which siRNA was cotransfected with an additional plasmid (e.g., a biosensor), the plasmid was added to the transfection mix only in the second round of transfection.
Plasmids and shRNA reagents
A lentivirus-based system (Rubinson et al., 2003; Vitriol et al., 2007) was used to functionally silence NMIIA and NMIIB in mammalian cells, as previously described (Smutny et al., 2010). The lentivirus expression vector pLL5.0 and packaging constructs pMDLg/pRRE, RSV-Rev and pMD.G were gifts from Jim Bear (University of North Carolina, Chapel Hill, NC). MCF-7 cells were incubated at 37°C with lentiviral particles in DMEM and polybrene solution (8 μg/ml) and harvested by trypsinization 72 h after infection.
The Src biosensor (SFK-Bio-tK: ECFP-SH2-LInker-SFKsubstrate peptide-YPet) and GFP-Rap1WT were gifts from Yingxiao Wang (University of California–San Diego, La Jolla, CA) and Mark Philips (New York University School of Medicine, New York, NY), respectively. Raichu-Rap1 was a gift from Michiyuki Matsuda (Kyoto University, Japan). pmCherry-N1 was from Clontech (Palo Alto, CA). mRPTPα-mCherry was obtained by cloning mouse RPTPα (Sap et al., 1990) at the N-terminus of mCherry in pmCherry-N1. HA-tagged mouse RPTPα WT and RPTPaC433S,C723S were generously provided by Jeroen den Hertog (Hubretch Institute, The Netherlands) and described previously (Buist et al., 2000). Mouse E-cadherin–GFP cloned into a pll5.0 vector encoding a shRNA against the 3′ UTR sequence of the human CDH1 gene was described previously (Smutny et al., 2011).
Antibodies and immunoprecipitation
Primary antibodies used in this study were as follows: 1) mouse mAb HECD1 against the ectodomain of E-cadherin (a gift from Peggy Wheelock, University of Nebraska, Omaga, NE, with the permission of M. Takeichi); 2) rabbit pAb against myosin IIA (PRB-440P; Covance, Dedham, MA); 3) rabbit pAb against myosin IIB (PRB-445P; Covance); 4) rabbit pAb against GAPDH (2275; R&D Systems, Minneapolis, MN); 5) rabbit pAb against RPTPα (Su et al., 1999); and 6) rabbit pAb from Millipore (07-472; Billerica, MA); 7) phospho-Src family (Y416) antibody (2101; Cell Signaling, Boston, MA); 8) mouse monoclonal (FG6) anti-PTP1B (Calbiochem, Billerica, MA); 9) rabbit pAb against RPTPσ (AF3430; R&D Systems); 10 and 11) rabbit pAb against c-Yes and mAb against c-Src were from Cell Signaling and Santa Cruz (Dallas, TX), respectively; 12) mouse mAb against p130Cas (610271; BD Transduction, San Jose, CA); and 13) mouse anti-HA clone 16B12 (MMS-101P; Covance). F-actin was stained with Alexa Fluor 488-, 546-, 594-, and 647-phalloidin (1:1000 dilution; Invitrogen). Secondary antibodies were species-specific antibodies conjugated with Alexa Fluor 488, 546, 594, or 647 (Invitrogen) for immunofluorescence, or with horseradish peroxidase (Bio-Rad, Hercules, CA) for immunoblotting.
Immunoprecipitation analysis was performed as described previously (Truffi et al., 2014).
Drug treatments
MCF-7 cells were seeded at 30–40% confluency onto coverslips and allowed to recover to 80–90% confluency before drug treatment. Cells were treated for 1 h with PP2 (529573; Calbiochem) or Y-27632 (688000; Merck) at a final concentration of 25 and 30 μM, respectively.
Immunofluorescence imaging
Cells were fixed with 10% trichloroacetic acid on ice for 15 min for RPTPσ staining or fixed in 4% paraformaldehyde in cytoskeletal stabilization buffer (10 mM PIPES pH 6.8, 100 mM KCl, 300 mM sucrose, 2 mM EGTA, 2 mM MgCl2) on ice for 30 min and subsequently treated with 50 mM NH4Cl for 10 min. Permeabilization was then performed with 0.2% saponin/0.1% bovine serum albumin in Tris-buffered saline. Confocal images were taken using a Zeiss LSM-510 META inverted microscope and Zeiss LSM-710 FCS inverted microscope driven by ZEN software (ZEN 2009; Zeiss) and an Elyra microscope system equipped with a C-Apo 63×/1.2-W Korr objective for structured illumination microscopy.
Quantitation of fluorescence at contacts
Quantitative analysis of fluorescence intensity at contacts was performed using the plot profile function of ImageJ. A line of 10 μm in length (averaged over 20 pixels) was positioned orthogonal to, and centered upon, randomly chosen contacts. Numerical values for the fluorescence intensity profile along this line were obtained using the plot profile feature of ImageJ. The baseline for each field of view was corrected by subtracting a constant value from each of the intensity profiles. Average profiles typically yielded peak-shaped curves with sides trending to zero, which were then fitted to a Gaussian function. Peak values for individual contacts were obtained by nonlinear regression. A minimum of 50 contacts from three individual experiments were measured. Statistical analysis was then performed by using a two-tailed t test or one-way analysis of variance (ANOVA) corrected for multiple comparisons, as detailed in the figure captions.
Linearity index
The linearity index for each contact was measured as the ratio of the direct linear distance between the vertices and the actual contact length and expressed as percentage values as described previously (McLachlan and Yap, 2011).
FRET measurements
MCF-7 cells were transiently transfected with FRET-based biosensors designed to measure Src (SrcBio-tK) and Rap1 (Raichu-Rap1) activity in live cells. FRET measurements were performed 24 h after transfection. Cells were imaged live on a LSM 710 Zeiss confocal microscope equipped with a chamber incubator at 37°C. Images were acquired with a 63×/1.4 NA oil-immersion objective Plan-Apochromat lens. A first scan was used to simultaneously record donor and FRET channels using a 458-nm laser line, collecting the emission in the donor emission region (BP 470–500 nm) and acceptor emission region (BP 530–560 nm), respectively. A second scan was then used to acquire simultaneously cross-talk and acceptor images using the 514-nm laser line for excitation and collecting the emission in the donor and acceptor emission regions. Scans were acquired sequentially line by line. The FRET index was calculated for every image as the average [FRET/Acceptor] emission ratio for pixels located at cell–cell junctions.
FLIM
FRET-FLIM experiments were carried out using a frequency domain lifetime fluorescence imaging module (Lambert Instruments, Leutingwolde, The Netherlands) attached to an inverted microscope (Olympus IX71) as described previously (Hill et al., 2008). Confluent MCF-7 cells were transiently transfected with mEcad-GFP (donor) alone or cotransfected with mEcad-GFP and mRPTPα-mCherry or with mCherry (using a 1:3 ratio of plasmid DNA). Samples were then excited using a sinusoidally modulated 3-W 470- nm LED at 80 MHz under epi-illumination. For lifetime measurements, a 1 M fluorescein solution (pH 8.0) was used as a reference standard. Cells were imaged with a 60×/1.45 NA oil-immersion objective using a GFP filter set, and lifetime values were obtained on regions corresponding to the ZA.
Fluorescence anisotropy
Fluorescence anisotropy measurements were performed as described recently (Brooks et al., 2014). Briefly, MCF-7 cells were transiently transfected with mCherry or mRPTPα-mCherry or cotransfected with mRPTPα-mCherry and mEcad-GFP (pLL5.0, mE-cadGFP). Cells were imaged live at 37°C by confocal microscopy, and images were acquired on a LSM710 Zeiss confocal microscope equipped with a Plan-Apochromat 40×/1.4 NA oil-immersion objective (Zeiss, Jena, Germany). Images were acquired by using a 561-nm laser excitation line and collecting the corresponding emissions in the range of 580–620 nm using band-pass emission filters. Fluorescence emission was split into the components parallel (Ipar) and perpendicular (Iper) to the plane of excitation by a polarized beam splitter. The fluorescence anisotropy was determined by measuring average values of Ipar and Iper on regions of interest at the cell–cell junctions in Image J and using the following equation:
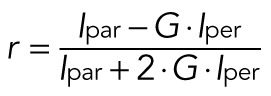 |
where the correction factor G was estimated using r0 = 0.35 of soluble mCherry as reference (Bader et al., 2011; Lindenburg et al., 2013). Data presented are mean r values calculated across the different images (∼50 cells per condition) and their SEs.
Laser nanoscissors
Nanoscissor experiments were performed on a LSM 510 meta Zeiss confocal microscope equipped with a 37°C heating stage as described previously (Caldwell et al., 2015). Images were acquired with a 63×/1.4 NA oil-immersion Plan-Apochromat lens at 2× digital magnification. A total of six frames were acquired with an interval of ∼8 s per frame. The Ti-sapphire laser (Chameleon Ultra; Coherent Scientific, US) was tuned to 790 nm for the ablation of cell–cell contacts labeled with Ecad-GFP. A constant region of interest of 3.8 × 0.6 μm was positioned with the longer axis perpendicular to the cell–cell contact. Contacts were ablated with 30 iterations of 790-nm laser at 24% transmission.
The distance (d) between vertices that define the ablated contact was measured as a fraction of time (t). Distance values after ablation were subtracted from the initial contact length, d(0). The values of d(t) – d(0) were then calculated as a function of time, and initial recoil values for each contact were obtained by nonlinear regression of the data to the following equation:
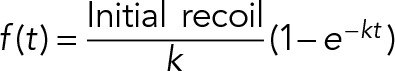 |
Finally, statistical analysis for control-normalized average initial recoil between different groups was performed by ANOVA or t test as described in the corresponding figure caption.
Supplementary Material
Acknowledgments
We thank our lab colleagues for their continuous support and fellowship and our many colleagues who generously provided reagents for this project. We also thank Jeremy Rossy for his help with SIM. This work was supported by project grant funding to A.S.Y. from the National Health and Medical Research Council (NHMRC) Australia (1044041, 1067405) and program grant 1037320 to A.S.Y., R.G.P., and K.G.; the Australian Research Council (DP120104667), the Kids Cancer Project of the Oncology Children's Foundation, a University of Queensland Early Career Research Grant to G.A.G. (2012003354); and the ARC Centre of Excellence in Convergent Bio-Nano Science and Technology to R.G.P. R.W.M. was a recipient of a Queensland Cancer Council PhD scholarship; S.K.W. was supported by a University of Queensland Research Scholarship and R.P. by an ANZ Trustees PhD Scholarship in Medical Research. A.S.Y. and R.G.P. are Research Fellows of the NHMRC (A.S.Y.: 631377 and 1044041; R.G.P.: 1058565). Research in J.S.'s lab was supported by a grant for La Ligue contre le cancer. Optical microscopy was performed at the ACRF/IMB Cancer Biology Imaging Facility, established with the generous support of the Australian Cancer Research Foundation.
Abbreviations used:
- CFP
cyan fluorescent protein
- FBS
fetal bovine serum
- FLIM
fluorescence lifetime imaging
- FRET
fluorescence resonance energy transfer
- GFP
green fluorescent protein
- HA
hemagglutinin
- KD
knockdown
- NMII
nonmuscle myosin II
- PTP
protein tyrosine phosphatase
- RNAi
RNA interference
- RPTPα
receptor protein tyrosine phosphatase alpha
- RPTPσ
receptor protein tyrosine phosphatase sigma
- RT
room temperature
- SFK
Src family kinase
- SIM
structured illumination microscopy
- siRNA
small interfering RNA
- WT
wild type
- YFP
yellow fluorescent protein
- ZA
zonula adherens
- ZO-1
zonula occludens protein 1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-07-1223) on January 28, 2015.
Author contributions: G.A.G., R.W.M., J.S., and A.S.Y. conceived the project; G.A.G. and R.W.M. performed most of the experiments. S.K.W. and R.P. contributed to immunofluorescence experiments and lentivirus production. S.V., B.J.C., and E.M. contributed to Western blot and image analysis. M.B. assisted with fluorescence resonance energy transfer–fluorescence lifetime imaging experiments. K.G. provided assistance and guidance with structured illumination microscopy. R.G.P., K.G., and J.S. contributed to the manuscript editing. G.A.G. and A.S.Y. analyzed the data and wrote the manuscript.
The authors had no financial interests related to this work.
REFERENCES
- Andreeva A, Lee J, Lohia M, Wu X, Macara IG, Lu X. PTK7-Src signaling at epithelial cell contacts mediates spatial organization of actomyosin and planar cell polarity. Dev Cell. 2014;29:20–33. doi: 10.1016/j.devcel.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuri S, Yan J, Paranavitana NC, Quilliam LA. E-cadherin dis-engagement activates the Rap1 GTPase. J Cell Biochem. 2008;105:1027–1037. doi: 10.1002/jcb.21902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader AN, Hoetzl S, Hofman EG, Voortman J, van Bergen en Henegouwen PM, van Meer G, Gerritsen HC. Homo-FRET imaging as a tool to quantify protein and lipid clustering. Chemphyschem. 2011;12:475–483. doi: 10.1002/cphc.201000801. [DOI] [PubMed] [Google Scholar]
- Balzac F, Avolio M, Degani S, Kaverina I, Torti M, Silengo L, Small JV, Retta SF. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J Cell Sci. 2005;118:4765–4783. doi: 10.1242/jcs.02584. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Tian X, Tian Y, Higginbotham K, Birukov KG. Rap-afadin axis in control of Rho signaling and endothelial barrier recovery. Mol Biol Cell. 2013;24:2678–2688. doi: 10.1091/mbc.E13-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AJ, Dai W, O'Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344:1249783. doi: 10.1126/science.1249783. [DOI] [PubMed] [Google Scholar]
- Buist A, Blanchetot C, den Hertog J. Involvement of the membrane distal catalytic domain in pervanadate-induced tyrosine phosphorylation of receptor protein-tyrosine phosphatase alpha. Biochem Biophys Res Commun. 2000;267:96–102. doi: 10.1006/bbrc.1999.1901. [DOI] [PubMed] [Google Scholar]
- Calautti E, Cabodi S, Stein PL, Hatzfeld M, Kedersha N, Paolo Dotto G. Tyrosine phosphorylation and src family kinases control keratinocyte cell–cell adhesion. J Cell Biol. 1998;141:1449–1465. doi: 10.1083/jcb.141.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell BJ, Lucas C, Kee AJ, Gaus K, Gunning PW, Hardeman EC, Yap AS, Gomez GA. Tropomyosin isoforms support actomyosin biogenesis to generate contractile tension at the epithelial zonula adherens. Cytoskeleton. 2015;71:663–676. doi: 10.1002/cm.21202. [DOI] [PubMed] [Google Scholar]
- Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J Biol Chem. 2004;279:41263–41266. doi: 10.1074/jbc.C400352200. [DOI] [PubMed] [Google Scholar]
- De La Cruz EM, Ostap EM. Relating biochemistry and function in the myosin superfamily. Curr Opin Cell Biol. 2004;16:61–67. doi: 10.1016/j.ceb.2003.11.011. [DOI] [PubMed] [Google Scholar]
- den Hertog J, Blanchetot C, Buist A, Overvoorde J, van der Sar A, Tertoolen LG. Receptor protein-tyrosine phosphatase signalling in development. Int J Dev Biol. 1999;43:723–733. [PubMed] [Google Scholar]
- den Hertog J, Sap J, Pals CE, Schlessinger J, Kruijer W. Stimulation of receptor protein-tyrosine phosphatase alpha activity and phosphorylation by phorbol ester. Cell Growth Differ. 1995;6:303–307. [PubMed] [Google Scholar]
- Dulyaninova NG, Bresnick AR. The heavy chain has its day: regulation of myosin-II assembly. Bioarchitecture. 2013;3:77–85. doi: 10.4161/bioa.26133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahim S, Fujita T, Millis BA, Kozin E, Ma X, Kawamoto S, Baird MA, Davidson M, Yonemura S, Hisa Y, et al. NMII forms a contractile transcellular sarcomeric network to regulate apical cell junctions and tissue geometry. Curr Biol. 2013;23:731–736. doi: 10.1016/j.cub.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484:546–549. doi: 10.1038/nature10999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias D, Vever H, Laenkholm AV, Gjerstorff MF, Yde CW, Lykkesfeldt AE, Ditzel HJ. Gene expression profiling identifies FYN as an important molecule in tamoxifen resistance and a predictor of early recurrence in patients treated with endocrine therapy. Oncogene. 2014 doi: 10.1038/onc.2014.138. doi:10.1038/onc.2014.138. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Simoes Sde M, Roper JC, Eaton S, Zallen JA. myosin II dynamics are regulated by tension in intercalating cells. Dev Cell. 2009;17:736–743. doi: 10.1016/j.devcel.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Zallen JA. Wounded cells drive rapid epidermal repair in the early Drosophila embryo. Mol Biol Cell. 2013;24:3227–3237. doi: 10.1091/mbc.E13-05-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez GA, McLachlan RW, Yap AS. Productive tension: force-sensing and homeostasis of cell–cell junctions. Trends Cell Biol. 2011;21:499–505. doi: 10.1016/j.tcb.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Guillot C, Lecuit T. Mechanics of epithelial tissue homeostasis and morphogenesis. Science. 2013;340:1185–1189. doi: 10.1126/science.1235249. [DOI] [PubMed] [Google Scholar]
- Gutzman JH, Sahu SU, Kwas C. Non-muscle myosin IIA and IIB differentially regulate cell shape changes during zebrafish brain morphogenesis. Dev Biol. 2014;397:103–115. doi: 10.1016/j.ydbio.2014.10.017. [DOI] [PubMed] [Google Scholar]
- Haj FG, Sabet O, Kinkhabwala A, Wimmer-Kleikamp S, Roukos V, Han HM, Grabenbauer M, Bierbaum M, Antony C, Neel BG, Bastiaens PI. Regulation of signaling at regions of cell–cell contact by endoplasmic reticulum-bound protein-tyrosine phosphatase 1B. PLoS One. 2012;7:e36633. doi: 10.1371/journal.pone.0036633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder KW, Moller NP, Peacock JW, Jirik FR. Protein-tyrosine phosphatase alpha regulates Src family kinases and alters cell-substratum adhesion. J Biol Chem. 1998;273:31890–31900. doi: 10.1074/jbc.273.48.31890. [DOI] [PubMed] [Google Scholar]
- Heisenberg CP, Bellaiche Y. Forces in tissue morphogenesis and patterning. Cell. 2013;153:948–962. doi: 10.1016/j.cell.2013.05.008. [DOI] [PubMed] [Google Scholar]
- Heissler SM, Manstein DJ. Nonmuscle myosin-2: mix and match. Cell Mol Life Sci. 2013;70:1–21. doi: 10.1007/s00018-012-1002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–124. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan C, Serpente N, Cogram P, Hosking CR, Bialucha CU, Feller SM, Braga VM, Birchmeier W, Fujita Y. Rap1 regulates the formation of E-cadherin-based cell–cell contacts. Mol Cell Biol. 2004;24:6690–6700. doi: 10.1128/MCB.24.15.6690-6700.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Yao L, Xu R, Wu H, Wang M, White BS, Shalloway D, Zheng X. Activation of Src and transformation by an RPTα splice mutant found in human tumours. EMBO J. 2011;30:3200–3211. doi: 10.1038/emboj.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MS, Tokutake Y, Chang MS, Bloor JW, Venakides S, Kiehart DP, Edwards GS. Forces for morphogenesis investigated with laser microsurgery and quantitative modeling. Science. 2003;300:145–149. doi: 10.1126/science.1079552. [DOI] [PubMed] [Google Scholar]
- Jiang G, den Hertog J, Su J, Noel J, Sap J, Hunter T. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature. 1999;401:606–610. doi: 10.1038/44170. [DOI] [PubMed] [Google Scholar]
- Kawamoto S, Adelstein RS. Chicken nonmuscle myosin heavy chains: differential expression of two mRNAs and evidence for two different polypeptides. J Cell Biol. 1991;112:915–924. doi: 10.1083/jcb.112.5.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox AL, Brown NH. Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science. 2002;295:1285–1288. doi: 10.1126/science.1067549. [DOI] [PubMed] [Google Scholar]
- Konig K. Multiphoton microscopy in life sciences. J Microsc. 2000;200:83–104. doi: 10.1046/j.1365-2818.2000.00738.x. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell–cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- Leerberg JM, Gomez GA, Verma S, Moussa EJ, Wu SK, Priya R, Hoffman BD, Grashoff C, Schwartz MA, Yap AS. Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr Biol. 2014;24:1689–1699. doi: 10.1016/j.cub.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levayer R, Lecuit T. Oscillation and polarity of E-cadherin asymmetries control actomyosin flow patterns during morphogenesis. Dev Cell. 2013;26:162–175. doi: 10.1016/j.devcel.2013.06.020. [DOI] [PubMed] [Google Scholar]
- Li L, Wang S, Jezierski A, Moalim-Nour L, Mohib K, Parks RJ, Retta SF, Wang L. A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem Cells. 2010;28:247–257. doi: 10.1002/stem.289. [DOI] [PubMed] [Google Scholar]
- Liang X, Gomez GA, Yap AS. Current perspectives on cadherin-cytoskeleton interactions and dynamics. Cell Health Cytoskeleton. 2014;7:11–24. [Google Scholar]
- Lindenburg LH, Hessels AM, Ebberink EH, Arts R, Merkx M. Robust red FRET sensors using self-associating fluorescent domains. ACS Chem Biol. 2013;8:2133–2139. doi: 10.1021/cb400427b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Bao J, Adelstein RS. Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II-B-ablated and mutated mice. Mol Biol Cell. 2007;18:2305–2312. doi: 10.1091/mbc.E07-01-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Kawamoto S, Hara Y, Adelstein RS. A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol Biol Cell. 2004;15:2568–2579. doi: 10.1091/mbc.E03-11-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitre JL, Berthoumieux H, Krens SF, Salbreux G, Julicher F, Paluch E, Heisenberg CP. Adhesion functions in cell sorting by mechanically coupling the cortices of adhering cells. Science. 2012;338:253–256. doi: 10.1126/science.1225399. [DOI] [PubMed] [Google Scholar]
- Martin AC, Gelbart M, Fernandez-Gonzalez R, Kaschube M, Wieschaus EF. Integration of contractile forces during tissue invagination. J Cell Biol. 2010;188:735–749. doi: 10.1083/jcb.200910099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, Kraemer A, Helwani FM, Kovacs EM, Yap AS. E-cadherin adhesion activates c-Src signaling at cell–cell contacts. Mol Biol Cell. 2007;18:3214–3223. doi: 10.1091/mbc.E06-12-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, Yap AS. Not so simple: the complexity of phosphotyrosine signaling at cadherin adhesive contacts. J Mol Med. 2007;85:545–554. doi: 10.1007/s00109-007-0198-x. [DOI] [PubMed] [Google Scholar]
- McLachlan RW, Yap AS. Protein tyrosine phosphatase activity is necessary for E-cadherin-activated Src signaling. Cytoskeleton. 2011;68:32–43. doi: 10.1002/cm.20492. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Inoue N, Nishimura K, Kinoshita N, Hosoya H, Yonemura S. Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp Cell Res. 2006;312:1637–1650. doi: 10.1016/j.yexcr.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411:1065–1068. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- Monteleone MC, Gonzalez Wusener AE, Burdisso JE, Conde C, Caceres A, Arregui CO. ER-bound protein tyrosine phosphatase PTP1B interacts with Src at the plasma membrane/substrate interface. PLoS One. 2012;7:e38948. doi: 10.1371/journal.pone.0038948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchie R, Guo CH, Persaud A, Muise A, Rotin D. Protein tyrosine phosphatase sigma targets apical junction complex proteins in the intestine and regulates epithelial permeability. Proc Natl Acad Sci USA. 2014;111:693–698. doi: 10.1073/pnas.1315017111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Aoki K, Matsuda M. Monitoring spatio-temporal regulation of Ras and Rho GTPase with GFP-based FRET probes. Methods. 2005;37:146–153. doi: 10.1016/j.ymeth.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Otani T, Ichii T, Aono S, Takeichi M. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J Cell Biol. 2006;175:135–146. doi: 10.1083/jcb.200605012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DW, McLean GW, Wyke AW, Paraskeva C, Parkinson EK, Frame MC, Brunton VG. The catalytic activity of the Src family kinases is required to disrupt cadherin-dependent cell–cell contacts. Mol Biol Cell. 2000;11:51–64. doi: 10.1091/mbc.11.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannekoek WJ, Kooistra MR, Zwartkruis FJ, Bos JL. Cell–cell junction formation: the role of Rap1 and Rap1 guanine nucleotide exchange factors. Biochim Biophys Acta. 2009;1788:790–796. doi: 10.1016/j.bbamem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Piedra J, Miravet S, Castano J, Palmer HG, Heisterkamp N, Garcia de Herreros A, Dunach M. p120 Catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin-α-catenin interaction. Mol Cell Biol. 2003;23:2287–2297. doi: 10.1128/MCB.23.7.2287-2297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R, Yap AS, Gomez GA. E-cadherin supports steady-state Rho signaling at the epithelial zonula adherens. Differentiation. 2013;86:133–140. doi: 10.1016/j.diff.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Radha V, Rajanna A, Swarup G. Phosphorylated guanine nucleotide exchange factor C3G, induced by pervanadate and Src family kinases localizes to the Golgi and subcortical actin cytoskeleton. BMC Cell Biol. 2004;5:31. doi: 10.1186/1471-2121-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A, Gomez GA, Priya R, Verma S, Kovacs EM, Jiang K, Brown NH, Akhmanova A, Stehbens SJ, Yap AS. Centralspindlin and α-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012;14:818–828. doi: 10.1038/ncb2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauzi M, Lenne PF, Lecuit T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 2010;468:1110–1114. doi: 10.1038/nature09566. [DOI] [PubMed] [Google Scholar]
- Ren G, Helwani FM, Verma S, McLachlan RW, Weed SA, Yap AS. Cortactin is a functional target of E-cadherin-activated Src family kinases in MCF7 epithelial monolayers. J Biol Chem. 2009;284:18913–18922. doi: 10.1074/jbc.M109.000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Sallee JL, Wittchen ES, Burridge K. Regulation of cell adhesion by protein-tyrosine phosphatases: II. Cell–cell adhesion. J Biol Chem. 2006;281:16189–16192. doi: 10.1074/jbc.R600003200. [DOI] [PubMed] [Google Scholar]
- Sap J, D'Eustachio P, Givol D, Schlessinger J. Cloning and expression of a widely expressed receptor tyrosine phosphatase. Proc Natl Acad Sci USA. 1990;87:6112–6116. doi: 10.1073/pnas.87.16.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Tamada M, Dubin-Thaler BJ, Cherniavskaya O, Sakai R, Tanaka S, Sheetz MP. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J, Lu S, Ouyang M, Huang H, Zhang J, Frame MC, Wang Y. Visualization of Src activity at different compartments of the plasma membrane by FRET imaging. Chem Biol. 2009;16:48–57. doi: 10.1016/j.chembiol.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewan AM, Maddugoda M, Kraemer A, Stehbens SJ, Verma S, Kovacs EM, Yap AS. myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell–cell contacts. Mol Biol Cell. 2005;16:4531–4542. doi: 10.1091/mbc.E05-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Rolo A, Chen X, Gumbiner BM, Keller R. Convergence and extension at gastrulation require a myosin IIB-dependent cortical actin network. Development. 2008;135:2435–2444. doi: 10.1242/dev.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smutny M, Cox HL, Leerberg JM, Kovacs EM, Conti MA, Ferguson C, Hamilton NA, Parton RG, Adelstein RS, Yap AS. myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 2010;12:696–702. doi: 10.1038/ncb2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smutny M, Wu SK, Gomez GA, Mangold S, Yap AS, Hamilton NA. Multicomponent analysis of junctional movements regulated by myosin II isoforms at the epithelial zonula adherens. PLoS One. 2011;6:e22458. doi: 10.1371/journal.pone.0022458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AW. Protein tyrosine phosphatases and signalling. J Endocrinol. 2005;185:19–33. doi: 10.1677/joe.1.06069. [DOI] [PubMed] [Google Scholar]
- Su J, Muranjan M, Sap J. Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts. Curr Biol. 1999;9:505–511. doi: 10.1016/s0960-9822(99)80234-6. [DOI] [PubMed] [Google Scholar]
- Tracy S, van der Geer P, Hunter T. The receptor-like protein-tyrosine phosphatase, RPTP alpha, is phosphorylated by protein kinase C on two serines close to the inner face of the plasma membrane. J Biol Chem. 1995;270:10587–10594. doi: 10.1074/jbc.270.18.10587. [DOI] [PubMed] [Google Scholar]
- Truffi M, Dubreuil V, Liang X, Vacaresse N, Nigon F, Han SP, Yap AS, Gomez GA, Sap J. RPTPα controls epithelial adherens junctions, linking E-cadherin engagement to c-Src-mediated phosphorylation of cortactin. J Cell Sci. 2014;127:2420–2432. doi: 10.1242/jcs.134379. [DOI] [PubMed] [Google Scholar]
- Tullio AN, Bridgman PC, Tresser NJ, Chan CC, Conti MA, Adelstein RS, Hara Y. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol. 2001;433:62–74. doi: 10.1002/cne.1125. [DOI] [PubMed] [Google Scholar]
- van Eekelen M, Runtuwene V, Overvoorde J, den Hertog J. RPTPα and PTPɛ signaling via Fyn/Yes and RhoA is essential for zebrafish convergence and extension cell movements during gastrulation. Dev Biol. 2010;340:626–639. doi: 10.1016/j.ydbio.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Verma S, Han SP, Michael M, Gomez GA, Yang Z, Teasdale RD, Ratheesh A, Kovacs EM, Ali RG, Yap AS. A WAVE2-Arp2/3 actin nucleator apparatus supports junctional tension at the epithelial zonula adherens. Mol Biol Cell. 2012;23:4601–4610. doi: 10.1091/mbc.E12-08-0574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitriol EA, Uetrecht AC, Shen F, Jacobson K, Bear JE. Enhanced EGFP-chromophore-assisted laser inactivation using deficient cells rescued with functional EGFP-fusion proteins. Proc Natl Acad Sci USA. 2007;104:6702–6707. doi: 10.1073/pnas.0701801104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Ma X, Conti MA, Adelstein RS. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem Soc Transact. 2011;39:1131–1135. doi: 10.1042/BST0391131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- Weed SA, Parsons JT. Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene. 2001;20:6418–6434. doi: 10.1038/sj.onc.1204783. [DOI] [PubMed] [Google Scholar]
- Wu SK, Gomez GA, Michael M, Verma S, Cox HL, Lefevre JG, Parton RG, Hamilton NA, Neufeld Z, Yap AS. Cortical F-actin stabilization generates apical–lateral patterns of junctional contractility that integrate cells into epithelia. Nat Cell Biol. 2014;16:167–178. doi: 10.1038/ncb2900. [DOI] [PubMed] [Google Scholar]
- Yoo SK, Freisinger CM, LeBert DC, Huttenlocher A. Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J Cell Biol. 2012;199:225–234. doi: 10.1083/jcb.201203154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XM, Resnick RJ, Shalloway D. A phosphotyrosine displacement mechanism for activation of Src by PTPalpha. EMBO J. 2000;19:964–978. doi: 10.1093/emboj/19.5.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.