

Septin proteins hetero-oligomerize to form filaments. Mutant septins that subtly misfold a key oligomerization interface retain some function when expressed alone but are excluded from filaments when the wild-type allele is present. Cytosolic chaperones mediate this “quality control” via prolonged interactions with the mutant polypeptides.
Abstract
Septin hetero-oligomers polymerize into cytoskeletal filaments with essential functions in many eukaryotic cell types. Mutations within the oligomerization interface that encompasses the GTP-binding pocket of a septin (its “G interface”) cause thermoinstability of yeast septin hetero-oligomer assembly, and human disease. When coexpressed with its wild-type counterpart, a G interface mutant is excluded from septin filaments, even at moderate temperatures. We show that this quality control mechanism is specific to G interface mutants, operates during de novo septin hetero-oligomer assembly, and requires specific cytosolic chaperones. Chaperone overexpression lowers the temperature permissive for proliferation of cells expressing a G interface mutant as the sole source of a given septin. Mutations that perturb the septin G interface retard release from these chaperones, imposing a kinetic delay on the availability of nascent septin molecules for higher-order assembly. Unexpectedly, the disaggregase Hsp104 contributes to this delay in a manner that does not require its “unfoldase” activity, indicating a latent “holdase” activity toward mutant septins. These findings provide new roles for chaperone-mediated kinetic partitioning of non-native proteins and may help explain the etiology of septin-linked human diseases.
INTRODUCTION
Newly translated polypeptides extruded into the cytosol face a number of challenges in acquiring their native folds, including a densely crowded molecular environment and, for N-terminal sequences, the absence of C-terminal sequences until translation is completed. Exposed hydrophobic residues normally buried in the core of the native fold make non-native polypeptides susceptible to inappropriate intermolecular interactions. Chaperone proteins promote de novo folding in part by transiently associating with hydrophobic patches on nascent proteins (Kim et al., 2013). Cycles of binding to and release from chaperones provide a kinetic partitioning mechanism that reduces the effective concentration of non-native folding intermediates, thereby disfavoring inappropriate intermolecular associations (Kim et al., 2013).
For multisubunit protein complexes, the challenges in acquiring their native conformation are even greater. In many cases, the constituent subunits only achieve their native folds in the context of the complex. For example, to form the α-tubulin–β-tubulin heterodimer (Tian et al., 1997), a series of cytosolic chaperones engage the newly translated polypeptides and direct conformational changes that coordinate binding (and/or hydrolysis) of GTP by tubulins and promote heterodimerization (Lundin et al., 2010). The prefoldin (PFD)/GimC complex acts at an early step to facilitate delivery of nascent α or β tubulin to the cytosolic chaperonin containing TCP1 (CCT)/TRiC (Vainberg et al., 1998). Association of unfolded α tubulin with CCT is necessary and sufficient for folding of the GTP-binding pocket (Tian et al., 1995). After a quasi-native conformation is achieved inside the sheltered environment of the CCT “cage,” a monomeric nucleotide-bound tubulin is released and then engaged by other dedicated folding factors/chaperones (Lopez-Fanarraga et al., 2001). Finally, a round of nucleotide exchange and hydrolysis by β tubulin precedes coupling into α–β heterodimers (Tian et al., 1999) in which the GTP-binding pocket of α tubulin is buried at the α–β interface (Nogales et al., 1998). Tubulin α–β heterodimers are then competent to polymerize head-to-tail into protofilaments, which laterally associate to form microtubules (Downing and Nogales, 2010).
Septin filaments are cytoskeletal polymers also assembled by polymerization of stable guanine nucleotide–binding hetero-oligomers (Sellin et al., 2011a). In yeast, each of four different subunits assembles into a linear, nonpolar hetero-octameric rod, namely Cdc11–Cdc12–Cdc3–Cdc10–Cdc10–Cdc3–Cdc12–Cdc11, with two alternating subunit–subunit interfaces. The Cdc12–Cdc3 and Cdc10–Cdc10 junctions, as well as the Cdc11–Cdc11 association that is responsible for end-to-end polymerization of the rods, are so-called NC interfaces because these subunit–subunit interactions are mediated by residues located within both the N and C termini of the globular domains of each monomer. By contrast, the Cdc11–Cdc12 and Cdc3–Cdc10 junctions are so-called G interfaces because these subunit–subunit interactions are mediated by residues ordered by the binding of guanine nucleotide in a pocket in each septin buried at this surface (Sirajuddin et al., 2007). Indeed, genetic (Versele and Thorner, 2004; Sirajuddin et al., 2009; Weems et al., 2014) and biochemical (Versele and Thorner, 2004; Sirajuddin et al., 2009; Zent and Wittinghofer, 2014) evidence suggests that GTP binding to all subunits and GTP hydrolysis by some subunits promote conformational changes that stabilize all of the subunit–subunit interfaces within the hetero-octamer. Consistent with the importance of nucleotide binding for the formation of functional hetero-octamers, the vast majority of septin mutations that confer a temperature-sensitive (TS) phenotype map to the G interface, suggesting that, at nonpermissive temperature, the mutant proteins are “trapped” in non-native conformations incompatible with assembly of functional higher-order structures (Weems et al., 2014). For those septins capable of hydrolyzing GTP to GDP, hydrolysis is likely coupled to formation of the G dimer interface (Gasper et al., 2009). Thus all available evidence points to a critically important role for GTP in promoting the stable folding of newly translated septins and/or maintaining their conformational competence for assembly into both hetero-octamers and higher-order structures. Such a role for small molecules in promoting or stabilizing protein folding and the assembly of oligomeric proteins was first articulated more than 40 years ago (Alpers and Paulus, 1971). However, other processes and factors in the pathway for achieving the native state of septins were unclear. In this regard, tubulin maturation again provides an instructive paradigm.
If, as appears to be the case, nucleotide binding and hydrolysis are required for final folding and release of β tubulin from its cognate chaperones, then this requirement imposes a straightforward mechanism for quality control (QC): incorrectly folded β tubulin will be defective in its binding and/or hydrolysis of GTP and hence will remain “stuck” in a stable complex with its folding factors. Such sequestration prevents misfolded protomers from participating in tubulin heterodimer assembly (Tian et al., 1999). In agreement with this view, mutations designed to block GTP binding by β tubulin prevent its colocalization with α tubulin and its incorporation into microtubules in vivo, resulting instead in formation of a diffuse and/or punctate localization pattern (Zabala et al., 1996). By contrast, β-tubulin mutations that inhibit heterodimer assembly for other reasons—that is, without affecting the nucleotide-binding pocket —prevent formation of β tubulin–chaperone complexes and lead to rapid proteolytic turnover of the mutant protein (Hoyle et al., 2001; Wang et al., 2006). However, overproduction of wild-type α tubulin allows mutant β tubulin to evade destruction and undergo incorporation into heterodimers, which then dominantly interfere with wild-type heterodimers (Hoyle et al., 2001). Thus maintaining a limiting supply of the cognate interaction partner also appears to be a contributing factor in this chaperone-mediated mechanism for intrinsic nucleotide-dependent QC.
Previous observations in yeast hinted that a similar QC mechanism might govern incorporation of septins into higher-order structures. Specifically, fluorescently tagged septins with substitution mutations in and around the GTP-binding pocket were not localized to the filamentous septin collar at the bud neck if produced from a plasmid in cells also expressing the endogenous wild-type allele of the same septin (Cid et al., 1998; Nagaraj et al., 2008). The extent of exclusion of such a mutant septin from the collar correlates directly with the extent to which the mutant septin causes TS growth when no wild-type allele is present (Nagaraj et al., 2008). However, preferential incorporation of the wild-type septin occurs even at temperatures at which, on its own, the mutant septin would localize to the collar and be functional (Cid et al., 1998; Nagaraj et al., 2008). Long before the structure and assembly of the yeast hetero-octamer was elucidated (Bertin et al., 2008), Cid et al. (1998) suggested that the mutant protein is less able than the wild-type to recognize its “attachment site” at the bud neck. Nagaraj et al. (2008) later postulated that the mutant protein incorporates normally into hetero-octamers but that when fully wild-type septin complexes are available, the mutant-containing complexes are somehow discriminated against for incorporation into the filamentous structures at the bud neck. Considering that the master polarity regulator Cdc42 marks the site for yeast bud emergence by driving assembly of an initial septin ring (Caviston et al., 2003; Kadota et al., 2004; Iwase et al., 2006; Okada et al., 2013), Cdc42-dependent factors might preferentially recruit nucleotide-replete septin hetero-octamers at the expense of those containing nucleotide-free subunits, which presumably have non-optimal conformation and/or stability (Nagaraj et al., 2008).
Here we use a combined approach of genetics, cell biology, and biochemistry to investigate the cellular and molecular requirements for budding yeast septin QC. We focus on five main questions: 1) Do alternative alleles of a single septin compete for a limiting number of interaction partners during septin hetero-oligomerization? 2) Which structural features of a mutant septin render it “aberrant” from the perspective of QC? 3) Does QC act on septin monomers or septin hetero-oligomers? 4) What is the fate of a mutant septin excluded by QC? 5) Which cellular factors mediate septin QC?
RESULTS
Alternative septin alleles compete for occupancy of subunit positions within the septin hetero-octamer
Septin rings at the bud necks of yeast cells are not in a state of dynamic equilibrium. Fluorescence recovery after photobleaching studies demonstrate that, apart from brief periods of “fluidity” during assembly, disassembly, and structural transitions (i.e., ring splitting), septin rings are stable structures in which the constituent subunits are immobile (Caviston et al., 2003; Dobbelaere et al., 2003). Similarly, the hetero-octameric building blocks of septin rings appear to be constructed in a series of rapid and effectively irreversible steps in which conformational changes accompanying initial dimerization (Sirajuddin et al., 2009; Kim et al., 2012; Weems et al., 2014) drive tetramerization and ultimately the assembly of octamers in which there is no exchange of subunits with a cytosolic pool (Sellin et al., 2011a). In fact, all available evidence suggests that there is no free pool of monomeric septins in wild-type yeast cells (Frazier et al., 1998; Vrabioiu et al., 2004; Farkasovsky et al., 2005; Bridges et al., 2014), which indicates that the stoichiometry of septin subunits is normally carefully controlled. These features of de novo higher-order septin assembly create a scenario in which two alleles of a single septin subunit might compete for occupancy of one of the four subunit positions within hetero-octamers. We predicted that this competition underlies the QC phenomenon.
Yeast cells expressing cdc10(D182N) as the sole source of Cdc10 are TS (Hartwell, 1971; McMurray et al., 2011b; Weems et al., 2014). In septins for which atomic-level structures are available, as in other related GTPases (Wittinghofer and Vetter, 2011), the Asp residue found in position 182 in Cdc10 lies within the nucleotide-binding pocket (NBP) and contacts the guanine base of GTP (Sirajuddin et al., 2007). Substitution to Asn, which is also found in a mutant human septin linked to male infertility (Kuo et al., 2012), severely reduces the affinity of the mutant protein for GTP (Kuo et al., 2012), as it does in other small GTPases (Hwang and Miller, 1987). We thus predicted that Cdc10(D182N), like other NBP mutants that cause TS phenotypes (Cid et al., 1998; Nagaraj et al., 2008), would be subject to QC-mediated exclusion from the bud necks of cells expressing wild-type Cdc10. Indeed, when expressed ectopically from a plasmid in otherwise wild-type haploid (CDC10+) (see below) or diploid (CDC10+/CDC10+) cells, Cdc10(D182N) was not incorporated into the septin filaments at the bud neck at any temperature (Figure 1A and unpublished data). We used this mutant septin to assess the effectiveness of QC in cells in which the stoichiometry between Cdc10(D182N), wild-type Cdc10, and the other septin subunits was manipulated by altering the number of copies per cell of the corresponding genes. Incorporation of the green fluorescent protein (GFP)–tagged NBP mutant Cdc10 into septin rings at the bud necks of individual cells was quantified in micrographs by performing line scans (Figure 1A). In confirmation of our prediction, Cdc10(D182N)-GFP was found at the bud neck in diploid cells carrying the cdc10(D182N) allele at one copy of the CDC10 locus and to an even greater extent in CDC10+/cdc10∆ cells (Figure 1B). These results demonstrate that simply making available one extra copy of each of the other septin-encoding genes—which would otherwise produce a limiting supply of hetero-oligomerization “partner” proteins—is sufficient to allow a mutant septin to evade QC and “compete” with the wild type.
FIGURE 1:

Alternative alleles of a given septin subunit compete to occupy a limiting number of positions within hetero-octamers. (A) “Quality control” of higher-order septin assembly in budding yeast. Left, schematic illustration of the localization of a GFP-tagged wild-type (CDC10+) or mutant (cdc10(D182N)-GFP) septin protein after introduction of the plasmid-encoded gene into wild-type (CDC10+/CDC10+) diploid cells. Middle, transmitted light and GFP fluorescence (inverted grayscale) micrographs of BY4743 cells carrying the cdc10(D182N)-GFP plasmid pCdc10-1-GFP, grown to mid log phase at 22°C. Right, schematic illustration of line scans of fluorescence micrographs, with actual data from individual cells. An eight-pixel-wide line was drawn perpendicular to the axis of the septin ring and used to plot a profile of fluorescence signal. The height of the peak (for neck localization) or depth of the trough (for neck exclusion) was calculated as shown. (B–E) Bud neck fluorescence for the indicated plasmid-encoded, fluorescently tagged mutant septin (bracketed genotype) expressed in cells of the indicated chromosomal genotype (genotype without brackets). Error bars, mean with SEM; n, number of cells analyzed. Septin hetero-octamers are illustrated below as in A, with multiple polypeptides competing for occupancy of the same position shown horizontally adjacent to each other. Leftmost, septin subunits identified by numbers: 1, Shs1; 3, Cdc3; 10, Cdc10; 11, Cdc11; 12, Cdc12. Fluorescently tagged subunits are solid green, absent subunits are indicated with dashed lines, and asterisks and daggers indicate G interface/NBP mutations. Shs1 is shown in blue. Strains: BY4743 (CDC10+/CDC10+), BY4741 (CDC12+), CBY07236 (cdc3(G365R)), CBY06417 (cdc10(D182N)), CBY08756 (cdc11(G32E)), CBY05110 (cdc12(G247E)), and JTY3631 (shs1∆). Plasmids: pLA10 ([CDC10-GFP]); pCdc10-1-GFP ([cdc10(D182N)-GFP]); pML51 ([cdc11(K172A D174A)-YFP]); pML114, which encodes Cdc12(G44V K47E T48N)-YFP ([cdc12*-YFP]); and YCpH-cdc12-1-GFP, which encodes Cdc12(G247E)-GFP ([G247E-GFP]). (F) Quantitative immunoblotting to compare levels of Cdc10-GFP expressed from the endogenous CDC10 locus was performed with whole-cell protein extracts of CDC10-GFP strains carrying the wild-type (CDC12+) or cdc12(G247E) allele at the CDC12 locus. After separation of proteins by 4–20% gradient SDS–PAGE and transfer to PVDF, immunoblot analysis was performed using antibodies recognizing GFP and appropriate fluorescently labeled secondary antibodies (top blot). Right, molecular weights of the Li-Cor Chameleon Duo Pre-stained Protein Ladder (928-60000; Li-Cor) indicated with arrows. After scanning and quantifying the GFP signal, the membrane was exposed to antibodies recognizing the loading control Zwf1 (glucose-6-phosphate dehydrogenase) and appropriate secondary antibodies (bottom blot). Left, arrows and labels indicate Cdc10-GFP and Zwf1; gray arrow, Cdc10-GFP signal detected in the Zwf1 scan. Signal for each band was quantified by subtracting the background signal from an equivalent area from a signal-free part of the same lane, then dividing the Cdc10-GFP signal by the Zwf1 signal; each of these values was normalized to this value for the first CDC12+ sample and plotted below. CDC12+ strains were MMY0166 and MMY0167, and cdc12(G247E) strains were MMY0168, MMY0169, and MMY0170.
As another way to test our model, we replaced in haploid cells the genomic wild-type allele of a given septin gene with an NBP mutant or a non-NBP mutant that also renders cells TS (Weems et al., 2014). A plasmid encoding a fluorescently tagged NBP mutant of Cdc3, Cdc10, Cdc11, or Cdc12 was then introduced into these cells. We predicted that QC would be unable to exclude a mutant septin if no wild-type allele was available to occupy the subunit position in question. Indeed, our findings fit with this prediction. For example, Cdc11(K172A D174A)–yellow fluorescent protein (YFP) was located at the bud neck when expressed in the same cell with a different Cdc11 NBP mutant but not with an NBP mutant in any of three other subunits of the hetero-octamer (Figure 1C). Importantly, the number of Cdc11(K172A D174A)-YFP molecules localized at the bud neck was also detectably increased in cells lacking Shs1 (Figure 1C). We showed previously (Garcia et al., 2011) that Shs1 occupies the same (terminal) position at each end of the hetero-octamer as does Cdc11 (Figure 1B) and likely populates a subset of the septin complexes in vivo. Thus eliminating competition by either wild-type Cdc11 or wild-type Shs1 allowed the Cdc11 NBP mutant to be incorporated.
Similarly, a tagged NBP mutant of Cdc3 (G129V K132E T133N) was found at the bud neck in cells carrying a mutant allele at the cognate genomic CDC3 locus (unpublished data). Importantly, the genomic allele carried a mutation (G365R) outside the NBP per se (Weems et al., 2014), suggesting that residue contacts in the normal G interface, and not merely guanine nucleotide binding, contribute to the “competitiveness” of a given subunit during hetero-octamer formation. Further support for this idea was the failure of a GFP-tagged version of Cdc12(G268R)—which, like Cdc3(G365R), substitutes a bulky, positively charged Arg residue for a Gly within the highly conserved “Sep4”/“WG” motif (Pan et al., 2007; Weems et al., 2014)—to localize to the bud neck when expressed in a CDC12+ strain (see later discussion). The Trp residue adjacent to Gly268 is a critical component of the Cdc12 G heterodimer interface with Cdc11 (Sirajuddin et al., 2007; McMurray et al., 2011a).
Interestingly, a tagged NBP mutant of Cdc12, Cdc12(G44V K47E T48N)-YFP, was not detectably incorporated at the bud neck when expressed from a plasmid in cells carrying a chromosomal cdc12(G247E) allele (Figure 1D). This finding suggests that Cdc12(G247E) is able to outcompete Cdc12(G44V K47E T48N)-YFP and is, in fact, consistent with the reported semidominant character of the cdc12(G247E) allele (Hartwell et al., 1973; Weems et al., 2014). According to this logic, Cdc12(G247E) should be able to evade QC and interfere with septin function in the presence of wild-type Cdc12. To test this prediction directly, we expressed plasmid-encoded Cdc12(G247E)-GFP in CDC12+ cells. As predicted, Cdc12(G247E)-GFP was found at bud necks (Figure 1D).
Unexpectedly, in cdc12(G247E) cells, but not in cells with mutations in any other non-Cdc10 subunit, Cdc10(D182N)-GFP was incorporated at the bud neck (Figure 1E). Tagged NBP Cdc3 or Cdc11 mutants were, unlike Cdc10(D182N)-GFP, excluded from the bud neck (unpublished data), demonstrating specificity of this phenotype. We previously observed (de Val et al., 2013) that, when expressed from the chromosomal locus, another type of substitution mutation that reduces the apparent fitness of a given septin subunit (replacement of endogenous Cys with other residues) can lead to an increase or decrease in the level of one or more of the other subunit types. We thus hypothesized that, in cdc12(G247E) cells, the levels of endogenous Cdc10 may be decreased, permitting Cdc10(D182N)-GFP incorporation at the bud neck. To test this idea, we combined by mating and meiosis a CDC10-GFP allele at the CDC10 locus with a cdc12(G247E) allele at the CDC12 locus. We then used quantitative immunoblotting via the GFP epitope on Cdc10 to compare levels of endogenous Cdc10 in three haploid cdc12(G247E) strains with two control CDC12+ strains isolated in the same cross. Cdc10-GFP levels were reduced on average ∼25% in the cdc12(G247E) cells (Figures 1F), providing support for our hypothesis. We interpret the foregoing results to indicate that the amounts of other subunits available for hetero-octamer assembly become limiting when a single subunit is represented by multiple alleles—each expressed at near-endogenous levels—thus establishing the competitive situation.
G interface lability correlates with subunit discrimination in higher-order assembly
All of the mutant septin alleles known to be at a competitive disadvantage when coexpressed with the corresponding WT subunit render cells TS when expressed as the sole source of the mutant septin (Cid et al., 1998; Nagaraj et al., 2008; Figure 1A), suggesting that the mutant septin is subject to a QC mechanism that recognizes a property of the mutant protein that is related to its dysfunction at high temperature. All of the mutants analyzed to date carry NBP substitutions (Cid et al., 1998; Nagaraj et al., 2008), whereas TS-causing mutations outside the NBP have not been studied in detail. To characterize further the properties of mutant septins recognized by QC, we assessed the ability of non-NBP mutants to localize to the bud neck when expressed de novo in the presence of the wild-type allele.
One potent TS allele, cdc12-6, carries mutations located far from the NBP: a frameshift near the end of the open reading frame results in changes to the C-terminal 17 residues (Figure 2A). Immunoblotting showed that the protein encoded by the cdc12-6 allele is ∼1 kDa shorter than the wild-type protein (Haarer and Pringle, 1987). Sequencing identified the mutation as a deletion of an adenine within a stretch of seven adenines 49 bases upstream of the termination codon (B. Haarer and J. Pringle, personal communication), resulting in a single-residue substitution (K391N) and truncation of the last 16 residues (Figure 2A). Of note, despite some later confusion about the nature of the frameshift (Longtine et al., 2000; McMurray and Thorner, 2008a), subsequent resequencing confirmed the original result (M. Longtine and J. Pringle, personal communication). We determined that the K391N mutation alone does not cause TS growth (unpublished data); instead, the truncation is responsible for the TS phenotype (Figure 2B). We found that, when expressed de novo in a CDC12+ strain, this truncation mutant (hereafter Cdc12(∆392-407)-GFP) was readily detected at the bud neck, even at 37°C (Figure 2, C and D). Similarly, and unlike NBP mutants, Cdc12(∆392-407)-GFP was able to incorporate into the septin structures at the bud neck in cdc12(G247E) cells (unpublished data). These findings are consistent with published observations of the bud neck incorporation of various other C-terminal truncation alleles of Cdc3, Cdc11, or Cdc12 (Casamayor and Snyder, 2003; Versele et al., 2004). In addition, internal deletions in Cdc11 (Casamayor and Snyder, 2003) or Cdc10 (e.g., Cdc10(∆13-29); McMurray et al., 2011a) that alter the NC homodimerization interface are also able to localize properly despite the presence of wild-type alleles of the same septins.
FIGURE 2:

Mutant septins with native G interfaces evade exclusion by quality control. (A) Top, schematic layout of Cdc12 protein domains/motifs, including major elements of the GTP-binding domain (G1/P loop, G3/switch II, and G4) and septin–septin interfaces (G3/switch II, septin-unique element [SUE]) and highlighting the hydrophobic heptad repeats within the extreme C- terminus predicted to form a coiled coil (c.c.). Orange bars, approximate locations of homologous residues buried in the human septin G dimer interface (Sirajuddin et al., 2007, 2009). Bottom, primary sequence of the c.c. region, illustrating the mutations in the Cdc12-6, Cdc12(K391N), and Cdc12(∆392-407)-GFP proteins. (B) Cells of the indicated genotype (plasmid-encoded genes are indicated with brackets) carrying a plasmid (YCpcdc12-6-GFP) encoding Cdc12(∆392-407)-GFP were streaked on solid medium selective for the plasmid and incubated at 22 or 37°C for 3 d. Strains were YEF473A (CDC12+) and M-1726 (cdc12-6). (C) As in Figure 1A, transmitted light and fluorescence micrographs of CDC12+ cells (strain YEF473a) carrying plasmid YCpcdc12-6-GFP grown to mid log phase in selective medium at the indicated temperatures. Arrowheads, fluorescence signal enriched at the bud neck. As indicated, 120-ms exposures were taken to prevent signal saturation. (D) As in Figure 1, B–E, for wild-type strain BY4741 and plasmid YCpcdc12-6-GFP (∆392-407) grown at 22°C. (E) As in Figure 1, B–E, for the CDC12+ CDC10-mCherry strain JTY3992 and plasmid pMVB62 ([CDC12-GFP]) or YEpLcdc12(G268R)-GFP ([cdc12(G268R)-GFP]). Purple, the mCherry tag on Cdc10, fluorescence of which was also quantified as a control for bud neck signal.
On the other hand, when tagged with GFP and introduced on a plasmid into CDC12+ cells, the mutant Cdc12(G268R) was efficiently excluded from the bud neck (Figure 2E). By contrast, in the truncation and NC interface deletion mutants, the G interface would be expected to fold properly and have wild-type stability. Finally, we previously proposed that the mutant NBP in Cdc12(G247E), which evades QC (Figure 1D), is able to bind a non-GTP nucleotide and thereby adopts a G interface conformation that is at least quasi-native (Weems et al., 2014). Taken together, these results point to the integrity of the G dimerization interface, not the NC interface or the NBP per se, as the primary feature monitored by QC.
G interface mutants are at a kinetic disadvantage in septin complex assembly
The foregoing experiments did not directly address why G interface mutants are less competent than the corresponding wild-type protein for incorporation into both hetero-octamers and neck filaments. We considered two possibilities. On one hand, the mutants may fold just as rapidly as the normal subunit but have reduced affinity for, or a slower rate of G interface formation with, the corresponding G interface interaction partner. On the other hand, the mutant septin may fold more slowly (or be more prone to misfolding) and hence be sequestered for a more protracted time by the folding machinery and thus not be available at a sufficient concentration to assemble at a significant rate with the limiting supply of G dimerization partners and other septin subunits.
As one means to distinguish between these possibilities, and assuming that the translation/folding capacity of a cell is in large excess, we asked if simply overexpressing a mutant septin could drive by mass action an increase in its localization to the bud neck even when the corresponding wild-type subunit was present. Our previous results with Cdc12(G268R)-GFP suggested that overexpression does not allow a G interface mutant to bypass QC, because the plasmid encoding Cdc12(G268R)-GFP harbored a high-copy (2μ DNA) origin of replication yet resulted in little or no bud neck incorporation compared with Cdc12-GFP produced in the same way (Figure 2E). However, to increase more drastically the total concentration of a mutant septin, we used derepression of the MET17 promoter to drive high-level production of plasmid-encoded Cdc10(D182N)-GFP. Quantitative immunoblotting demonstrated ∼19- or ∼16-fold increases in steady-state levels for wild-type Cdc10-GFP and Cdc10(D182N)-GFP, respectively, relative to expression of these proteins from the CDC10 promoter (Supplemental Figure S1A). Unlike wild-type Cdc10-GFP produced in the identical manner, this manipulation did not result in detectable incorporation of the mutant septin at the bud neck and instead merely increased the cytosolic fluorescent signal in CDC10+ cells (Figure 3A). These findings are inconsistent with a model in which mutant septins are left out of higher-order structures because they have a reduced affinity compared with wild-type septins.
FIGURE 3:

Exclusion of mutant septins by quality control acts before G dimerization and cannot be overcome by mutant overexpression. (A) Left, as in Figure 1A, but with haploid CDC10+ cells (strain BY4742) and plasmids with which Cdc10-GFP (pMETp-Cdc10-GFP) or Cdc10(D182N)-GFP (pPmet-Cdc10-1-GFP) is expressed from the high-level MET17 promoter in growth medium lacking methionine. The 60- and 250-ms exposures are shown for comparison to micrographs in other figures, which always used 250-ms exposures unless specified otherwise. Right, quantification of bud neck signals and illustration of septin subunit composition. (B) As in A, right, with plasmid pLA10K ([CDC10-GFP]) or YCpK-Cdc10-1-GFP ([cdc10*-GFP*]) and cdc3(D210G) strain MMY0131 (CDC10+) or MMY0130 (cdc10(D182N)).
As an independent means to address this same question, we exploited the fact that two G interface mutants, Cdc3(D210G) and Cdc10(D182N), display interallelic complementation when coexpressed as the sole source of these two subunits in the same cell. That is, this double mutant, but neither single mutant, is able to grow normally at high temperature (37°C; Weems et al., 2014). If preference for incorporation were based only on a subunit having the highest affinity for its partner, then Cdc10(D182N)-GFP would be capable of localizing to the bud neck when expressed de novo in cdc3(D210G) CDC10+ cells. Conversely, exclusion of wild-type Cdc10-GFP might be expected when expressed de novo in cdc3(D210G) cdc10(D182N) cells. Neither of these predictions was borne out. In cdc3(D210G) cells transformed with a plasmid expressing cdc10(D182N)-GFP, only diffuse cytoplasmic fluorescence was observed, and, in cdc3(D210G) cdc10(D182N) cells transformed with a plasmid expressing CDC10-GFP, the majority of the fluorescence signal localized to the bud neck (Figure 3B). We conclude that the “quality” of a NBP mutant septin is assessed before the need for it to engage in a G dimer interface during hetero-octamer formation.
Preferential incorporation of a wild-type septin occurs during de novo filament assembly
Our results strongly suggested that incorporation of a wild-type septin in preference to a G interface mutant occurs before assembly of septin hetero-octamers. If this is the case, then QC should be unable to exert this preference when mutant-containing septin hetero-octamers are assembled before introduction of the wild-type allele. We took advantage of two features of septin dynamics to test this prediction. First, septin complexes disassembled during one round of the cell cycle are stable and reused for assembly of a new septin collar in the next round of the cell cycle, and, second, cytoplasmic mixing of the septin hetero-octamer pools occurs during yeast mating (McMurray and Thorner, 2008b). We reasoned, therefore, that septin complexes assembled independently in two haploid partners before mating should be incorporated together into the neck of the zygote when it buds. However, our earlier results suggested that alterations in the stoichiometry of septin-encoding genes in a diploid cell can abrogate QC by providing more available oligomerization partners with which a GFP-tagged mutant septin can interact during de novo septin hetero-oligomerization. Therefore, to monitor unambiguously in diploid cells the localization of Cdc10(D182N) molecules synthesized and assembled before mating, we fluorescently labeled a SNAP-tagged derivative of this mutant under permissive conditions in haploid cells in which it was the sole source of this septin. The SNAP tag, a derivative of human O6-alkylguanine-DNA alkyltransferase, allows covalent labeling in vivo with derivatives of benzylguanine (Keppler et al., 2004). Labeled cells were mated with wild-type haploid cells, and the fate of the “pulse”-labeled Cdc10(D182N)-SNAP was monitored by fluorescence microscopy (Figure 4).
FIGURE 4:

Mutant septins evade exclusion by quality control if assembled into hetero-octamers before introduction of the wild-type allele. Top, fusion of the SNAP-Tag (SNAP) to the C- terminus of Cdc10(D182N) allows covalent fluorescence pulse labeling of a pool of Cdc10(D182N)-SNAP molecules using BG-DAF. The localization of the labeled mutant Cdc10-SNAP molecules after the introduction of wild-type, untagged Cdc10 via mating was monitored by fluorescence microscopy. Bottom, micrographs taken of budded zygotes generated by mating BG-DAF–labeled cells of strain JTY5169 with CDC10+ cells of strain BY4742. Arrowheads, labeled Cdc10(D182N)-GFP in septin rings at zygote bud necks. Asterisks, labeled Cdc10(D182N)-GFP in septin rings in nonzygote cells. Right, quantification of bud neck fluorescence in budded zygotes, as in Figure 1, B–E.
If the “competition” between wild-type and mutant subunits occurs only during formation of polymerization-competent septin hetero-octamers, then preformed mutant-containing septin complexes should be able to coassemble with wild-type complexes into the collar of septin filaments at the neck of a budding zygote. On the other hand, if wild-type complexes outcompete the preformed mutant complexes during polymerization of the bud neck filaments—as predicted by the Cdc42-mediated model proposed by Nagaraj et al. (2008)—then the mutant-containing septin complexes will be excluded from the collar of filaments that assemble at the neck of the zygote when it buds. We found that, although slightly reduced relative to a wild-type Cdc10-SNAP control, the preformed complexes containing Cdc10(D182N)-SNAP were clearly incorporated at the necks of budded zygotes formed after mating with CDC10+ cells (Figure 4). Thus, in cells coexpressing both Cdc10 and Cdc10(D182N), the observed preference for the wild-type subunit is exerted during formation of septin hetero-octamers, which are then polymerized into the filaments that assemble at the bud neck.
Mutant septins subject to quality control are excluded from septin complexes
Although we observed in some cases a modest decrease in the steady-state amount of cellular protein for those mutant septins excluded by QC as compared with the level of the corresponding wild-type septin (Supplemental Figure S1, A and B; Nagaraj et al., 2008), none could explain the lack of bud neck localization by these mutant proteins when expressed in wild-type cells. Instead, when excluded by QC, mutant septin molecules localize diffusely in the cytosol (see earlier results). If QC acts on a G interface mutant, such as Cdc10(D182N), before it has an opportunity to interact with its normal G dimerization partner (namely, Cdc3), then in cells coexpressing wild-type Cdc10, cytosolic Cdc10(D182N) should not be found in any septin complex that contains Cdc3. To test this prediction, we fractionated lysates prepared from diploid CDC10/cdc10(D182N)-GFP cells in which Cdc10(D182N)-GFP was largely excluded from the bud neck by size exclusion chromatography (SEC) in an appropriate separation medium to resolve septin-containing complexes (Figure 5A). The content of Cdc10(D182N)-GFP and Cdc3 in each fraction was determined by immunoblotting with anti-GFP or anti-Cdc3 antibodies (Supplemental Figure S2A). A minor amount of the Cdc10(D182N)-GFP (calculated Mr of 65 kDa) was found in the void volume and nearby fractions (Mr of ∼650 to >700 kDa). However, the bulk of the Cdc10(D182N)-GFP was found in two prominent peaks (Mrapp of ∼90 and ∼325 kDa, respectively; Figure 5B), with the majority in the smaller species. By contrast, the vast majority of Cdc3 in the same lysate eluted with an Mrapp of ∼650 to >700 kDa (Figure 5B). For the same column, and consistent with a highly elongated rod, purified recombinant septin hetero-octamers (calculated Mr of 382.8 kDa) elute at Mrapp of ∼650 kDa (unpublished data). Thus these data suggest that the Cdc10(D182N)-GFP molecules excluded from the bud neck by septin QC are also excluded from complexes containing other septins.
FIGURE 5:
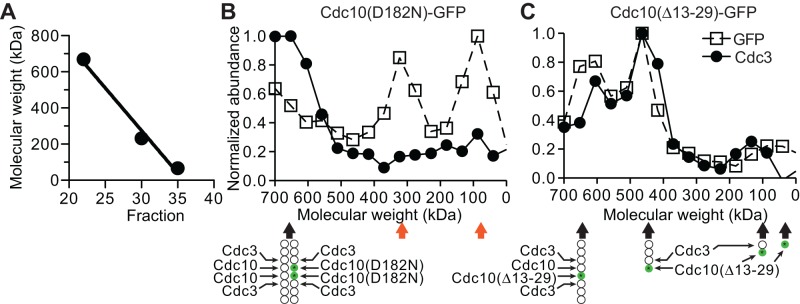
Mutant septins excluded by quality control are not in complex with their G dimer partners. (A) Thyroglobulin (669-kDa tetramer), catalase (232-kDa tetramer), and albumin (67-kDa monomer) from commercial sets of standards (17-0441-01 and 17-0442-01, GE Healthcare) were mixed together and resolved on a Superdex 200 size exclusion column. An apparently linear relationship between elution fraction and native molecular weight (R2 = 0.985) was used to calibrate the column and convert fraction number to estimated molecular weight in B and C. (B) Lysate from a CDC10/cdc10(D182N)-GFP diploid strain made by mating strains BY4742 and JTY3986 was resolved by gel filtration as in A. Fractions were slot-blotted to a nitrocellulose filter, and the GFP or Cdc3 content in each fraction was determined by immunoblotting with mouse anti-GFP or rabbit anti-Cdc3 antibodies. Values shown are normalized to the fraction with the highest signal. Below, illustration of the presumed septin complex composition in the peak Cdc3 fraction. Orange arrows point to fractions populated by Cdc10(D182N)-GFP molecules excluded by QC. (C) As in B, but for cells of strain BY4743 carrying the plasmid pJT3456, which expresses Cdc10(∆13-29)-GFP from the MET17 promoter. Cells were grown in medium lacking uracil (to select for the plasmid) and methionine (to drive high-level expression of the MET17 promoter) for 6 h before lysis. Green circles with asterisks represent Cdc10(∆13-29)-GFP. Note that in A–C, 500 mM NaCl was used to ensure dissociation of septin filaments.
As a control for our ability to detect septin species of various sizes containing both Cdc3 and Cdc10, we fractionated lysates from cells in which Cdc10(∆13-29)-GFP was transiently overexpressed. The deletion removes an element important for formation of the Cdc10–Cdc10 NC homodimer but does not perturb formation of a Cdc3–Cdc10 G interface. As a result, in high-salt buffers as we used here, Cdc10(∆13-29) assembles into Cdc11–Cdc12–Cdc3–Cdc10(∆13-29) heterotetramers and, when wild-type Cdc10 is available, Cdc11–Cdc12–Cdc3–Cdc10(∆13-29)–Cdc10–Cdc3–Cdc12–Cdc11 hetero-octamers (McMurray et al., 2011a). As expected, in such lysates, Cdc10(∆13-29)-GFP was found in four regions: ∼650-kDa peak (the “mixed Cdc10” hetero-octamer); ∼460-kDa peak (hetero-tetramer); ∼150-kDa shoulder (a minor amount of Cdc3–Cdc10(∆13-29)-GFP dimer); and ∼60-kDa peak (minor amount of Cdc10(∆13-29)-GFP monomer; Figure 5C and Supplemental Figure S2A). These results further strengthen our conclusions that the two prominent species observed for NBP mutant Cdc10(D182N)-GFP contain no Cdc3 and thus that QC prevents mutant septins from associating with other septins.
Genetic evidence that misfolded mutant septins are sequestered by cytosolic chaperones
For a mutant tubulin, one aspect of its QC mechanism is prolonged interaction of the aberrant protein with tubulin-specific folding chaperones (Zabala et al., 1996), thereby delaying its entry into the pool of subunits available for hetero-dimerization. Moreover, the “kinetic partitioning” mechanism by which chaperones protect non-native polypeptides from aggregation (Kim et al., 2013) would also be expected to delay proper hetero-oligomerization. By analogy, trapping/partitioning by a chaperone might prevent a septin with an aberrant G interface from associating with its G hetero-dimerization partner. Budding yeast cells respond to the presence of misfolded cytosolic proteins by up-regulating expression of genes encoding a network of interacting chaperones, representing a subset of those controlled by the transcription factor Hsf1 in the heat shock response (Geiler-Samerotte et al., 2011). If a mutant septin with a defect in its G interface has a propensity to misfold, then it seemed possible that the dysfunction observed for such a mutant at high temperature might arise, in part, from its sequestration by cytosolic chaperones, preventing the mutant subunit from contacting its partner subunits. If susceptibility to high-temperature misfolding also reflects partial misfolding at moderate temperatures, then this reasoning fits well with the observed correlation between the severity of a NBP mutation (in terms of the extent to which the mutant septin, if it is the sole source of that subunit present, undergoes heat-exacerbated misfolding and renders a cell heat sensitive) and the degree to which the mutant is excluded from incorporation into septin structures at the bud neck when the wild-type version is present (Casamayor and Snyder, 2003; Nagaraj et al., 2008). As a test of this idea, we predicted that ectopic Hsf1-dependent induction of cytosolic chaperones at moderate temperatures might drive chaperone-mediated sequestration of partially misfolded NBP-mutant septins and inhibit the proliferation of septin NBP mutant cells without affecting proliferation of wild-type cells. Indeed, when we expressed a constitutively active allele of HSF1 (Park et al., 2006) in a TS cdc10(D182N) strain, we observed clear inhibition of colony growth at 27°C, a non–heat-shock temperature otherwise permissive for growth of this septin mutant (Figure 6A, right). The presence of wild-type CDC10 completely suppressed this inhibitory effect (Figure 6A, left).
FIGURE 6:

Overexpression of cytosolic chaperones inhibits the growth of strains carrying G-interface septin mutations. (A) Fivefold serial dilutions of cells of the cdc10(D182N) strain DDY1476 carrying the URA3-marked CDC10 plasmid pMVB57 ([CDC10 URA3]) and a TRP1-marked plasmid—either the empty vector pRS314 ([]) or pRS314-HSF1∆NTA(148-833) ([HSF1∆N]), which encodes a constitutively active allele of HSF1—were spotted on solid medium lacking tryptophan (to select for the TRP1-marked plasmid) and either lacking uracil (to select for pMVB57) or containing uracil and 5-FOA to select against pMVB57. Plates were incubated at 27°C. (B) As in A, but on medium lacking uracil with 2% raffinose (uninducing) or 1% raffinose plus 1% galactose (inducing) as the carbon source and with cells of the genotype indicated at left carrying either an empty URA3-marked plasmid (pRS316; []) or a URA3-marked plasmid with YDJ1 under control of the galactose-inducible GAL1/10 promoter (pPgal-YDJ1; [PGAL–YDJ1]). Strains: BY4741 (wild type), CBY07236 (cdc3(G365R)), CBY06417 (cdc10(D182N)), CBY06427 (cdc11(G29D)), and CBY05110 (cdc12(G247E)). (C) Cells of the indicated genotypes and carrying the indicated plasmid as in B were pregrown overnight at room temperature in liquid medium lacking uracil and containing 2% raffinose and then diluted to an OD600 of 0.1 in 200-μl cultures (12 replicates/genotype) of medium lacking uracil and containing 1% raffinose and 1% galactose. The OD600 of each culture was measured every 5 min for 14 h and used to calculate doubling times. Strains: CBY04956 (cdc3(G365R)), CBY06417 (cdc10(D182N)), CBY08756 (cdc11(G32E)), and CBY05110 (cdc12(G247E). Error bars, SEM. (D) As in B, but with HIS3-marked plasmids—the empty vector pRS313 ([]) or pGALSc104, carrying HSP104 under GAL1/10 promoter control (PGAL–HSP104)—and medium lacking histidine and containing 2% galactose (inducing). (E) As in D, but at 37°C and with growth medium lacked histidine and uracil, the latter to select for the URA3-marked plasmid pLA10 ([CDC10+]) or pMVB145 ([cdc10(S256A)]). Strains were haploid spore clones derived by sporulation from the diploid strains JTY5687, JTY5688, and JTY5690. Each carries a genomic deletion allele of CDC10 and either a wild-type (CLA4+ cdc10∆) or partial deletion allele of CLA4 (cla4 cdc10∆) plus the indicated plasmids.
Ydj1 (an Hsp40) and Hsp104 are two of the 15 chaperones up-regulated in an Hsf1-dependent manner in response to misfolded cytosolic proteins (Geiler-Samerotte et al., 2011). A dominant mutant of Hsp104 was reported to alter bud morphology, cause septin mislocalization, and colocalize with the displaced septins—phenotypes that were alleviated by point mutations in Cdc12—all suggesting that Hsp104 is capable of interacting with septins (Schirmer et al., 2004). Prior work indicates that Hsp104 functionally interacts with Ydj1 (Glover and Lindquist, 1998). Hence we tested if overexpression of either Ydj1 or Hsp104 was able to inhibit growth of septin mutant strains under conditions in which these overproduced chaperones had little or no effect on the growth of wild-type cells. We found that excess Ydj1 (expressed from a GAL promoter) did not affect the proliferation of wild-type cells (Figure 6B) but impeded the growth at 27°C of all five of the septin G dimerization interface mutants tested (Figure 6B), with inhibition of cdc3(G365R) and cdc11(G29D) especially pronounced (Figure 6, B and C).
Similarly, Hsp104 overexpression did not affect wild-type cells (Figure 6D) but inhibited the growth of all tested TS septin mutant strains at 27°C (Figure 6D), with the most obvious effect on cdc10(D182N) cells. We also tested the effect of Hsp104 overexpression at 37°C using a G interface mutant of Cdc10 that does not make cells TS at this temperature. Serine 256 in Cdc10 is adjacent to a critical contact across the G interface (Sirajuddin et al., 2007; McMurray et al., 2011a) and is phosphorylated by the protein kinase Cla4, a modification believed to operate in parallel with GTP binding to promote higher-order septin assembly (Versele and Thorner, 2004). Bulky, positively charged substitutions at the equivalent position in Cdc3 or Cdc12 (e.g., Cdc3(G365R)) render cells inviable at 37°C (Weems et al., 2014), whereas cdc10(S256A) mutants proliferate normally at this temperature (Versele and Thorner, 2004; Figure 6E). At 37°C, Hsp104 overexpression moderately inhibited the growth of cdc10(S256A) cells and had the same effect on cla4∆ and cdc10(S256A) cla4∆ cells (Figure 6E). Thus Hsp104 appears to inhibit the growth of septin-mutant cells to an extent that depends on the extent of mutant septin G interface misfolding.
Absence of specific chaperones compromises septin quality control
The preceding results indicated that up-regulation of chaperones under Hsf1 control could be at least partially responsible for the growth inhibition observed in septin mutants at high temperature. Conversely, it has been observed that loss-of-function mutations in factors required for degradation-mediated protein QC, including Ydj1 (Lee et al., 1996), improve the functionality of TS mutants in other classes of proteins (Betting and Seufert, 1996; Bordallo et al., 1998; Gardner et al., 2005). We reasoned that this effect would be manifested as a positive genetic interaction at a semi-permissive or nonpermissive temperature in which the growth rate of the chaperone–septin double-mutant strain is faster than what is predicted from strictly additive growth effects of the individual chaperone and septin mutations. Indeed, others reported from a genome-wide screen that a ydj1∆ mutation displays just such a positive genetic interaction with cdc10(G100E) and that, similarly, loss of a PFD subunit (gim3∆) exhibits a positive genetic interaction with cdc3(G365R) (Costanzo et al., 2010). However, these interactions cannot be considered authentic suppression of the septin mutant phenotype by the chaperone deletion because 1) the chaperone–septin double mutants do not propagate at anywhere near a wild-type growth rate and 2) the chaperone deletion mutants themselves grow at reduced rates, as expected, given that these chaperones are necessary for the de novo folding of many other types of proteins (e.g., tubulin for Gim3 [ Vainberg et al., 1998] and the G1 cyclin Cln3 for Ydj1 [ Vergés et al., 2007]). Nonetheless, together with our overexpression results (Figure 6), these reported genetic relationships point to Hsp104, Ydj1, and PFD as candidates for the chaperones that sequester mutant septins during de novo septin folding.
To test directly if these and other cytosolic chaperones are required for the QC phenomenon by which wild-type septins are preferentially incorporated into higher-order structure, we transformed plasmids encoding a fluorescently tagged mutant septin (Cdc10(D182N)) into strains lacking Ydj1, Gim3, Hsp104, or other chaperones. We observed a clear failure of QC—namely, incorporation of Cdc10(D182N)-GFP at the bud neck—in ydj1∆ cells (Figure 7, A and B). Bud-neck signal in ydj1∆ cells could not be explained by elevated steady-state levels of the mutant septin, as assessed by immunoblotting (Supplemental Figure S1B), and thus reflects an improved ability of the mutant septin molecules to incorporate into septin filaments. By contrast, we did not find Cdc10(D182N)-GFP at the bud neck in hsp104∆ cells (Figure 7B). Given that prior binding of a client protein by an Hsp40 family member, including Ydj1, is typically a prerequisite for productive interaction of that substrate with Hsp104 (Glover and Lindquist, 1998), it is possible that Ydj1-mediated sequestration of the mutant septin explains its exclusion from the bud neck even in hsp104∆ cells. Consistent with that possibility, we found that introducing an hsp104∆ mutation did not improve the growth of any of several temperature-sensitive septin mutants tested (unpublished data). Conversely, deletion of YDJ1 did not prevent excess Hsp104 from inhibiting the growth of cdc10(D182N) cells (unpublished data), which likely reflects an ability of other Hsp40-related proteins (e.g., Apj1; Sahi et al., 2013) to target Hsp104 to mutant septins. However, because Ydj1 is the most abundant Hsp40 in the yeast cytosol (Sahi et al., 2013), its loss should have the strongest effect on mutant septin exclusion. Consistent with this idea, exclusion of Cdc10(D182N)-GFP remained efficient in apj1∆ cells (Figure 7B).
FIGURE 7:

The Hsp40 family member Ydj1 and prefoldin subunit Gim3 are required for septin quality control. (A) As in Figure 1A, but with haploid cells of the indicated genotypes. Strains: BY4741 (wild type); JTY5445 (ydj1∆); MMY0051 (gim3∆). Arrowheads, strong bud neck fluorescence signal. (B) As in Figure 2D, for the strains in A, the hsp104∆ strain JTY4014, the apj1∆ strain MMY0054, the hsp42∆ strain MMY0057, the hsp82∆ strain MMY0058, and the ssb1∆ strain MMY0059. p value, one-tailed unpaired t- test. (C) Cells of the same strains as in A carrying plasmids expressing from the GAL1/10 promoter C-terminally truncated alleles of CDC12 ([PGAL–CDC12∆C]) were resuspended in sterile water and subjected to 10-fold serial dilutions. The residue at position 268 is indicated as wild-type (G; plasmid pMVB160) or substituted to arginine (R; plasmid YCpUG-Cdc12(G268R ∆339-407)). A 5-μl amount of each dilution was spotted on solid synthetic medium containing 10 μg/ml phloxine B to stain dead cells red and dextrose or galactose, as indicated, to repress or induce the GAL1/10 promoter. As indicated, uracil was left out or added to the medium to impose or relieve plasmid selection, respectively. Plates were photographed after 3 d at 30°C.
None of the other chaperone mutants tested phenocopied ydj1∆ (Figure 7B). However, we consistently noticed in the gim3∆ strain a distinct subpopulation of cells with very high Cdc10(D182N)-GFP signal at the bud neck (Figure 7, A and B). Such cells were observed less often in other genotypes (Figure 7B). We wondered if cell-to-cell variations in copy number of the plasmid encoding the mutant septin might explain this phenotypic heterogeneity. Specifically, in cells with high plasmid copy number, high cytoplasmic signal might mask moderate bud neck signals in our line scan analysis (Figure 1A). This scenario predicts that cells with the highest cytoplasmic signals would tend to have calculated bud neck signals close to 0. Instead, such cells were the source of the most-negative bud neck values we calculated (unpublished data). We suspect the lack of penetrance instead reflects a degree of stochasticity and redundancy in the QC pathways, such that a nascent mutant septin must pass multiple distinct “checkpoints” en route toward “escaping” chaperone sequestration, each of which is less than perfectly effective. Indeed, the viability of PFD mutants like gim3∆ has been attributed to folding of the essential proteins tubulin and actin through less efficient, PFD-independent mechanisms (Lundin et al., 2010), providing precedents for “bypass” of this folding pathway.
As an independent test of chaperone requirements in septin QC, we devised a growth-based QC assay. As noted earlier, C-terminally truncated alleles of Cdc12 evade QC, and extensive truncations (e.g., Cdc12(∆339-407)) cause lethality when overexpressed from the GAL1/10 promoter (Versele et al., 2004), presumably because the mutant septin incorporates into hetero-oligomers that block functional filament assembly. We hypothesized that introducing a single G interface mutation into Cdc12(∆339-407) would allow its recognition by septin QC, sequestering the double-mutant septin in the cytosol and allowing proliferation by wild-type cells in which it is overexpressed. As expected, and unlike Cdc12(∆339-407), overexpressed Cdc12(G268R ∆339-407) had no effect on colony growth by wild-type cells (Figure 7C). If QC-mediated exclusion of the mutant septin is indeed responsible for viability in this situation, QC-defective mutants should die. Cells lacking Gim3 exhibited a clear QC defect in this assay (Figure 7C). The gim3∆ growth defect in these conditions did not reflect an inability of the mutant strain to grow on galactose-based media, because when we provided uracil to remove plasmid selection, growth was restored (Figure 7C). A ydj1∆ strain overexpressing Cdc12(G268R ∆339-407) also displayed a growth defect relative to controls (Figure 7C). To sensitize the assay, we included in the medium the vital dye Phloxine B, which stains dead cells red (Nagai, 1963), but ydj1∆ and gim3∆ colonies were not noticeably more red than the wild-type control (Figure 7C). We suspect that, as with Cdc10(∆13-29) (Figure 5B; McMurray et al., 2011a), septin ring assembly tolerates low levels of incorporation of C-terminally truncated Cdc12 molecules, and these mutants become lethal only above a certain threshold level of incorporation, which is frequently achieved by Cdc12(G268R ∆339-407) in gim3∆ mutants, less so in ydj1∆ cells, and not at all in wild-type cells (Figure 7A). These findings implicating Ydj1 and Gim3/PFD in septin QC demonstrate the specificity of the genetic interactions that originally motivated our examination of these factors.
The chaperone Ydj1 interacts specifically with mutant septins during septin QC
All of the preceding evidence predicts that physical interactions between a nascent septin and certain cytosolic chaperones are prolonged when the septin carries a mutation that perturbs folding of the G dimerization interface. When the wild-type allele is present, QC ensures that wild-type molecules are preferentially incorporated into the septin filaments at the bud neck, and mutant septins are left in the cytosol in complexes that are devoid of other septins but presumably contain relevant chaperones. To test these predictions, we examined septin–chaperone interactions in the context of septin QC.
Specifically, we looked for evidence of physical interaction between an excluded mutant septin and a chaperone required for septin QC, Ydj1. Ydj1 was tagged with a tandem affinity purification (TAP) tag consisting of a hexahistidine sequence and a ZZ (immunoglobulin G [IgG]-binding) domain with an intervening cleavage site specific to the human rhinovirus 3C protease (Gelperin et al., 2005) and overexpressed at levels that inhibited the growth of cdc10(D182N) (but not wild-type) cells (Figure 6, B and C). Together with Ydj1-TAP, we coexpressed GFP-tagged wild-type Cdc10 or Cdc10(D182N) in CDC10+ cells. Cdc10(D182N)-GFP molecules were excluded from the bud neck by QC and localized to the cytosol (Figure 8A), where the bulk of endogenous Ydj1 normally resides (Caplan and Douglas, 1991; Huh et al., 2003). As expected, wild-type Cdc10 localized to the bud neck (Figure 8A). Lysates prepared from these cells or control cells in which no Ydj1-TAP was expressed were incubated with IgG-coated beads, from which hexahistidine-tagged Ydj1 and associated proteins were subsequently eluted via protease cleavage. Cdc10(D182N)-GFP, but not wild-type Cdc10-GFP, appeared in the eluate from Ydj1-TAP–expressing cells (Figure 8B), indicating that the chaperone associates specifically with the misfolded septin. Of note, because the IgG-binding activity of the ZZ domain of Ydj1-TAP survives SDS–PAGE and membrane transfer, this ∼64 kDa band reacts with all primary and secondary antibodies. Hence input levels of the GFP-tagged Cdc10 molecules (calculated Mr of 65 kDa) in the lysates could not be reliably compared between the Ydj1-TAP–overexpressing samples. However, boiling the beads in SDS–PAGE sample buffer after protease cleavage demonstrated that in the Cdc10-GFP and Cdc10(D182N)-GFP lysates, the beads captured equivalent amounts of Ydj1-TAP, and that cleavage was somewhat inefficient, as indicated by the presence of full-length Ydj1-TAP in these samples (Figure 8B). Some cleaved Ydj1 also remained associated with the beads, likely due to dimerization with uncleaved Ydj1-TAP, as native Ydj1 is known to homodimerize (Wu et al., 2005). These findings support a model in which association of mutant septins with the cytosolic chaperone Ydj1 contributes to QC-mediated cytosolic sequestration.
FIGURE 8:
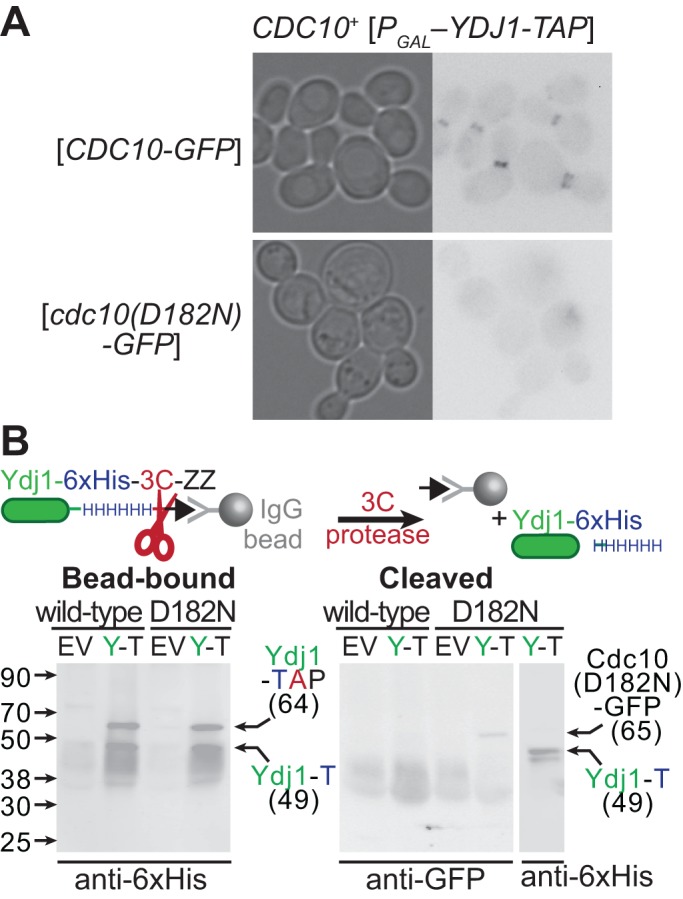
Sequestration of mutant septins by the chaperone Ydj1. (A) Cultures of strain BJ2168 carrying plasmid pLA10K ([CDC10-GFP], wild-type) or YCpK-Cdc10-1-GFP ([cdc10(D182N)-GFP], D182N) and either the empty vector pRS316 (EV) or a plasmid driving TAP-tagged Ydj1 from the GAL1/10-promoter (pPgal-YDJ1, [PGAL–YDJ1-TAP], Y-T) were grown at 30°C in medium lacking uracil and containing 1% raffinose and 1% galactose to induce expression from the GAL1/10 promoter. GFP fluorescence in aliquots of the Ydj1-expressing cells were imaged. (B) The cultures described in A were lysed, and clarified lysates were incubated with IgG-coated beads. After extensive washing, cleaved Ydj1- and Ydj1-bound proteins were eluted from the beads by the addition of GST-tagged 3C protease (3C), which cleaves between the hexahistidine tag (HHHHHH) and IgG-binding domain (ZZ) of Ydj1-TAP, releasing hexahistidine-tagged Ydj1 (Ydj1-T). The beads were boiled in sample buffer (Bead-bound) and, together with the eluate (Cleaved) samples, resolved by SDS–PAGE and transferred to a PVDF membrane. Cdc10(D182N)-GFP was detected using mouse anti-GFP antibodies. Uncleaved Ydj1-TAP and cleaved, hexahistidine-tagged Ydj1 were detected using anti-hexahistidine (anti-6xHis) antibodies (Y1011; UBPBio, Aurora, CO). The leftmost lanes contain a molecular weight ladder (928-60000; Li-Cor Chameleon Duo Pre-stained Protein Ladder). whose contents are labeled on the left with black arrows and sizes. Right, black arrows point to bands migrating with the approximate molecular weight expected for the indicated proteins.
A latent Hsp104 “holdase” activity is sufficient to delay the higher-order assembly of nascent mutant septins
The cellular manifestations of a chaperone-imposed delay in mutant septin availability for oligomerization depend on whether or not a wild-type allele of the same septin is also expressed. When the mutant allele is the only source of the septin, cellular proliferation is inhibited by exposure to elevated temperatures or overexpression of specific chaperones, in each case presumably reflecting an inability of the mutant septin to escape sequestration by the cytosolic proteostasis machinery. We designed an experiment to compare the kinetics of higher-order assembly by wild-type or mutant septin polypeptides during de novo synthesis under conditions in which the mutant proteins are unable to support normal septin function.
Actively growing cultures of cells expressing from the chromosomal CDC10 locus either wild-type Cdc10 or Cdc10(D182N) fused to the SNAP tag were first exposed to unlabeled benzylguanine (BG) to block the ability of the SNAP site to react with any other BG derivative provided thereafter. After 45 min of growth at 30°C—a semipermissive temperature for the proliferation of cdc10(D182N) cells—cells were lysed, and the septin-SNAP molecules synthesized in the 45-min “chase” period after the BG “pulse” were specifically labeled in the lysate with a fluorescent BG derivative conjugated to Oregon green (BG-OG). Native proteins were separated by SEC, and the OG content of each resulting fraction was measured using a fluorescence plate reader.
Control experiments in which no BG or BG-OG was added identified sources of background fluorescence in several fractions, presumably representing molecules with intrinsic fluorescence at 520 nm (Supplemental Figure S2B and Figure 9, A and B, blue arrows). Three new peaks of fluorescence with Mrapp of ∼90, ∼220–355, and ∼670 kDa (calibration of this column is shown in Supplemental Figure S2A) appeared in lysates from CDC10-SNAP cells exposed to the BG-OG pulse-chase regimen at 30°C (Figure 9, A and B). In lysates from cdc10(D182N)-SNAP cells, three differences were apparent: the proportion of total signal present in the ∼670- and ∼190-kDa peaks was reduced, whereas the peak representing 480 kDa was broader, encompassing fractions with Mrapp of ∼500 kDa, and contained more signal (Figure 9, A and B). Immunoblot analysis of the ∼670-, ∼190-, and ∼90-kDa fractions with anti–SNAP-tag antibodies detected wild-type Cdc10-SNAP (calculated Cdc10-SNAP Mr of 57 kDa) only in the ∼670-kDa fraction, consistent with incorporation of this septin into rod-shaped hetero-octamers (Figure 9C). By contrast, Cdc10(D182N)-SNAP was abundant in the highly fluorescent ∼460–485-kDa fractions, particularly in the ∼485-kDa fraction (Figure 9C). We found neither the wild-type nor the mutant SNAP-tagged septin in the ∼90-kDa fraction (Figure 9C), whose strong BG-OG–dependent fluorescence signal we attribute to either proteolyzed fragments of the septin-SNAP fusions that are not recognized by the antibodies or BG-OG binding/attachment to other molecules in the lysate. (At ∼25 kDa, the endogenous yeast O6-alkylguanine-DNA alkyltransferase, Mgt1, is an unlikely suspect; Sassanfar and Samson, 1990). Taken together, these results suggest that, upon exposure to elevated temperatures, newly translated wild-type septins are rapidly incorporated into septin hetero-octamers, whereas G-interface–mutant polypeptides are sequestered in a complex whose size is significantly greater than that of a septin monomer but smaller than that of a septin hetero-octamer.
FIGURE 9:

The chaperone Hsp104 sequesters mutant septins in an ATPase-independent manner. (A) Cells of CDC10-SNAP strain JTY4034 (Cdc10-SNAP) or cdc10(D182N)-SNAP strain JTY5168 (Cdc10(D182N)-SNAP) were exposed to 5 μM BG to inactivate the SNAP tags on existing septin–SNAP fusions. After 45 min of subsequent growth at 30°C in the absence of BG, cells were harvested and lysed. Newly synthesized septin-SNAP proteins were fluorescently labeled in the lysate with BG-OG. Lysates were then resolved by SEC. Note that 500 mM NaCl was used to ensure dissociation of septin filaments. Plotted is the BG-OG fluorescence measured for 200-μl portions of each 2-ml fraction. Values shown are normalized to the fraction with the highest signal. Below, filled arrows point to peak fractions classified by color: blue, fluorescence independent of BG-OG addition (Supplemental Figure S2B); magenta, fluorescence requires BG-OG addition, but fractions contain no detectable SNAP-tagged protein (see C); black, predicted molecular weight is consistent with that of septin hetero-octamers; orange, observed in lysates from cdc10(D182N)-SNAP but not CDC10-SNAP cells. (B) As in A, but considering only fractions corresponding to molecular weights >400 kDa. Values were normalized to the fraction with the highest signal in this molecular weight range. (C) Immunoblot analysis of specific fractions from A and B. Fractions are identified by the relevant genotype of the source strain (CDC10, JTY4034; cdc10(D182N)-SNAP, JTY5168) and by predicted molecular weight, using the same color scheme as in A and B. The membrane was exposed to anti–SNAP-Tag antibodies (P9310S; New England Biolabs) and infrared dye–labeled secondary antibodies. After detection (top), the membrane was exposed to anti-Hsp104 antibodies (ADI-SPA-1040-D; Enzo Life Sciences ) and the same secondary antibodies, then scanned again (bottom). The leftmost lane contains a molecular weight ladder (EZ-Run Prestained Protein Marker; Fisher BioReagents, Pittsburgh, PA), whose contents are labeled on the left with black arrows and sizes (in kilodaltons). Black arrows on the right point to bands migrating with the molecular weight expected for the indicated proteins. Gray arrows point to presumed proteolysis products or cross-reacting bands. (D) As in B, but cells were grown at 27°C during the “chase” period. Plotted values represent measurements made from pools of 100 μl of each of two adjacent fractions. Fractions >650 kDa were not measured. (E) As in B and C, but only strain JTY5169 was used, and fluorescence values were not normalized. In the “no BG-OG” sample, no BG-OG was added to the lysate. In the “Hsp104-overexpressed” sample, the cells carried plasmid pRS316-Pgal-HSP104 and were induced to overexpress Hsp104 5.25 h before BG addition. Columns (gray, Hsp104; green, SNAP) indicate levels (in arbitrary units) of Hsp104 or Cdc10(D182N)-SNAP in the indicated fractions from the Hsp104-overexpressing cells as assessed by immunoblotting (Supplemental Figure S2C). The open orange arrow indicates where BG-OG-labeled Cdc10(D182N)-SNAP molecules were detected in D. Red arrow, peak fraction containing BG-OG-labeled Cdc10(D182N)-SNAP only when Hsp104 was overexpressed. (F) As in Figure 6D, with cdc10(D182N) strain CBY06417, plasmid pRS316, pRS316-Pgal-HSP104, or pRS316-PGAL-HSP104(E285Q E687Q), and media lacking histidine. (G) As in E, showing the same data for overexpression of wild-type Hsp104 in strain JTY5169 overlaid with data from overexpression of Hsp104(E285Q E687Q). The yellow arrow indicates a peak fraction of OG fluorescence specific to the Hsp104(E285Q E687Q)-overexpressing sample. (H) As in C, but with fractions from the Hsp104(E285Q E687Q)-overexpressing sample separated on a 4–20% gradient SDS–PAGE gel before immunoblotting and a different molecular weight ladder (Chameleon Duo Pre-stained Protein Ladder, 928-60000; Li-Cor).
According to our model, sequestration of misfolded septins is mediated by cytosolic chaperones. A size of ∼500 kDa is significantly larger than expected for sequestration of Cdc10(D182N)-SNAP by the PFD complex (∼200 + 57 ≈ 260 kDa) or by a homodimer of Ydj1 with (or without) an associated Hsp70 co-chaperone (90 + 57 ≈ 150 kDa; 220 kDa with an Hsp70). Homohexamers of nucleotide-bound Hsp104, on the other hand, elute during high-salt SEC in approximately this range (Parsell et al., 1994; Figure 9E). Indeed, anti-Hsp104 antibodies detected full-length Hsp104 in the ∼500-kDa Cdc10(D182N)-SNAP–containing fraction but not in the septin hetero-octamer (∼670 kDa) fraction (Figure 9C). We previously showed that Hsp104 overexpression inhibited the growth of cdc10(D182N) cells cultured at 27°C (Figure 6D), which we attributed to sequestration by this chaperone of nascent Cdc10(D182N) proteins even at a temperature otherwise permissive for normal growth of the mutant strain. We repeated the BG-OG pulse-chase experiments at 27°C and found that in this condition, even more nascent molecules of Cdc10(D182N)-SNAP, but not wild-type Cdc10-SNAP, eluted in fractions representing molecular weights of ∼460–580 kDa (Figure 9D).
The major function of Hsp104 in proteostasis is believed to be extensive ATP-dependent unfolding of aggregated proteins to a largely unstructured state, from which de novo folding can begin again. From this perspective, if coelution with Hsp104 of a large fraction of the pool of nascent mutant septin proteins reflected an intermediate step toward complete Hsp104-mediated septin unfolding, then one might expect more-severe cellular consequences, as the unfolded polypeptides would need to reattempt de novo folding, with potentially the same unproductive outcome. However, at low concentrations, Hsp104 is also able to promote in vitro oligomerization of certain of its substrates in a “passive” mechanism that requires hexamerization and ATP binding but does not require ATP hydrolysis (Shorter and Lindquist, 2004). ATP-hydrolysis–dependent substrate unfolding by Hsp104 occurs only on aggregation-prone substrate oligomers or, alternatively, when Hsp104 is present at high concentrations (Shorter and Lindquist, 2004). We wondered if at permissive temperatures (≤30°C), at which Hsp104 expression levels are low and nascent Cdc10(D182N) molecules are only mildly misfolded, Hsp104–Cdc10(D182N) association represents a transient “holding” step along a productive folding pathway toward incorporation into functional septin hetero-octamers. If so, we hypothesized that overexpressing Hsp104 might unleash its catalytic activity and drive mutant septin unfolding.
We repeated the analysis of nascent Cdc10(D182N)-SNAP in cells overexpressing Hsp104 and cultured at 27°C, the same conditions in which we observed inhibition of proliferation upon Hsp104 overexpression (Figure 6D). Fluorescence signal from BG-OG–labeled Cdc10(D182N)-SNAP molecules was no longer detected in the ∼500-kDa fractions (Figure 9E, open orange arrow). Instead, a new peak appeared with Mrapp of ∼290 kDa (Figure 9E, red arrow). Immunoblotting confirmed the presence of Cdc10(D182N)-SNAP molecules in this fraction (Figure 9E, green columns, and Supplemental Figure S2C). Hsp104 behaved as reported in the literature (Parsell et al., 1994), eluting predominantly in fractions with Mrapp of ∼500–600 kDa, with no detectable accumulation in the ∼290-kDa range (Figure 9E, gray columns, and Supplemental Figure S2D). Future studies will be required to identify other constituents of the ∼290-kDa complex(es) containing nascent Cdc10(D182N)-SNAP molecules; size similarity to the ∼325-kDa complex containing QC-excluded Cdc10(D182N)-GFP in our previous experiments (Figure 5B) may not be coincidental. Our findings support a model in which the levels of Hsp104 present in nonstressed cells (i.e., at 27°C) are sufficient only to transiently sequester misfolded septins during de novo synthesis. When Hsp104 levels are elevated, on the other hand, unfolding of nascent mutant septins by the disaggregase precludes their availability for incorporation into higher-order septin assemblies.
According to our model, overexpression of Ydj1 inhibits the proliferation of septin-mutant cells because the mutant septin is kinetically partitioned away from other septins, whereas overexpression of Hsp104 drives unfolding of the mutant septin, in either case preventing efficient higher-order septin assembly. Our model predicts, however, that merely prolonging the interaction between Hsp104 and a nascent septin, without activating Hsp104 unfoldase activity, should be sufficient to impose a kinetic delay on higher-order septin assembly and, consequently, a growth defect in septin-mutant strains. We exploited the fact that ATP-bound Hsp104 has the highest affinity for its substrates, whereas ATP hydrolysis to ADP drives substrate unfolding and release (Shorter and Lindquist, 2004; Bösl et al., 2005; Schaupp et al., 2007). A double-mutant allele of Hsp104 (E285Q E687Q) that contains substitutions in each of its ATP-binding domains lacks ATP hydrolysis activity and binds tightly to substrate proteins (Bösl et al., 2005; Schaupp et al., 2007). If holdase activity is sufficient for Hsp104 to drive mutant septin sequestration, then overexpressing Hsp104(E285Q E687Q) should, like wild-type, unfoldase-competent Hsp104, inhibit the proliferation of cells dependent on a G interface–mutant septin for their septin function. Consistent with previous reports (Moosavi et al., 2010), overexpression of Hsp104(E285Q E687Q) had no effect on the proliferation of wild-type cells (unpublished data). On the other hand, overexpression of wild-type Hsp104 or Hsp104(E285Q E687Q) each inhibited proliferation of cdc10(D182N) cells to an equivalent extent (Figure 9F).
To examine this hypothesis in more detail, we monitored by SEC the elution of nascent Cdc10(D182N)-SNAP molecules upon Hsp104(E285Q E687Q) overexpression. In the same conditions in which no coelution was seen with overexpressed wild-type Hsp104 (Figure 9E), we observed a clear peak of fluorescence in ∼550-kDa fractions that were confirmed by immunoblotting to contain both Hsp104(E285Q E687Q) and Cdc10(D182N)-SNAP (Figure 9G, H). (High fluorescence was also detected in fractions of Mrapp of ∼260 kDa, but these fractions did not contain Cdc10(D182N)-SNAP; Figure 9, G and H). These findings demonstrate that prolonged association of a nascent mutant septin with Hsp104 during de novo synthesis is sufficient to impose a kinetic delay in higher-order septin assembly.
DISCUSSION
Spatiotemporal coordination in the assembly of septin complexes
The evidence suggests that nascent septin subunits may first form via their G interfaces into heterodimers (Kim et al., 2012) that subsequently assemble into heterotetramers via an NC interface and finally into rod-shaped, hetero-octameric protofilaments. The findings presented here suggest that there may be a window in time and/or space during which QC is exerted to favor properly folded subunits over improperly folded ones during protofilament formation. Moreover, our evidence strongly suggests that this bias is imposed by the kinetics of release of a septin protomer from the cytosolic chaperones that supervise its folding.
Apart from a slight increase in CDC10 mRNA during G1, transcript levels of the mitotic septins vary little throughout the yeast cell division cycle (Spellman et al., 1998). It thus seems unlikely that there is a fixed stage or point in the cell cycle at which de novo synthesis, maturation, and assembly of septin protofilaments must occur. If such were the only mechanism to ensure the quality of the septin-based structures assembled, then any slowly maturing (or mutant) molecule synthesized past the assembly point should be available during the next round, and overexpression would further drive incorporation, neither of which we observed.
We propose that the relevant process that assesses the quality of a septin subunit begins with translation and ends upon completion of a rate-limiting folding event that produces a heterodimerization-competent septin monomer, which co-assembles immediately with a cognate G heterodimerization partner (if one is available), forming pairs that are used in the assembly of heterotetramers and then hetero-octameric protofilaments (Figure 10). Our findings, coupled with prior observations (see later discussion), make the CCT chaperonin the best candidate to mediate final maturation of normal septin folding after nascent subunits undergo cycles of binding to and release from other chaperones upstream in the folding pipeline, in particular PFD, Hsp104, and Ydj1. Mutant septins are presumably delayed in arriving at CCT because they spend more time in a non-native state and hence are slower to be released from PFD, Hsp104, and Ydj1 (Figure 10). For certain other substrates of CCT (e.g., the VHL–elongin BC tumor suppressor complex), release from the chaperonin is believed to be triggered by oligomerization (Feldman et al., 1999; Yam et al., 2008). If the same is true for septins, this could explain the influence of the stoichiometry of partner septins on the capacity of a given septin to incorporate into higher-order ensembles: once all of its partners are incorporated into hetero-octamers, an excess or late-arriving molecule has no route to productive release from CCT and may instead be shunted back to other chaperones.
FIGURE 10:
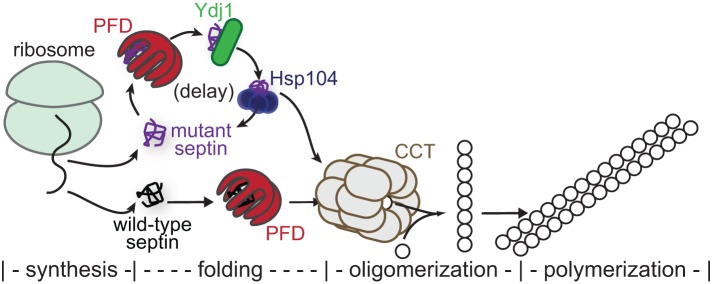
Model for chaperone-mediated quality control of higher-order septin assembly. Nascent septin polypeptides emerging from the ribosome encounter a number of cytosolic chaperones during subsequent de novo folding. Those most relevant to quality control are illustrated here. Wild-type septins efficiently adopt quas-inative conformations, thereby burying hydrophobic residues and escaping chaperone-mediated sequestration. Heterodimerization with other septins—the first oligomerization step toward septin filament assembly—occurs concomitant with exit from the chamber of the cytosolic chaperonin CCT (also called TRiC). Mutant septins that inefficiently fold the G heterodimerization interface are slower to achieve a conformation allowing chaperone release. Interactions with the prefoldin complex (PFD), the Hsp40 chaperone Ydj1, and the disaggregase Hsp104 are particularly prolonged, leading to a delay in availability of the mutant septin for hetero-oligomerization. When expressed at endogenous levels, Hsp104 exerts a passive bind-and-release “holdase” function. On overexpression, Hsp104 “unfoldase” activity may be unleashed on the mutant septin, but “holdase” function is sufficient in these conditions to delay mutant septin availability.
Importantly, the ability of cdc10(D182N) cdc3(D210G) double-mutant cells to proliferate well at high temperatures (Weems et al., 2014) demonstrates that chaperone sequestration of nascent mutant septins must be only transient, with cycles of binding punctuated by periods of accessibility of the mutant septin to other septins. If a partner septin is both available for and conformationally compatible with G dimerization, a mutant septin can eventually find a route to freedom from chaperone-mediated detention.
Quality control acts on mutant septins with specific properties
We propose that the mutant polypeptides recognized by chaperone-mediated QC expose hydrophobic residues that are normally buried, creating high-affinity sites for chaperone binding. The amino acid substitutions that render a mutant septin subject to exclusion by QC rarely introduce hydrophobic residues at surface-exposed positions or replace buried hydrophobic residues with charged/polar residues (a recommended approach for engineering new TS mutants; Varadarajan et al., 1996). Instead, the bulky, charged side chains characteristic of the residues introduced in TS septin mutants most frequently replace neutral residues (e.g., Gly; Nagaraj et al., 2008; Weems et al., 2014) at positions that are predicted to be relatively rigid/immobile within the folded structure (McMurray, 2014) and probably bias the conformational equilibria such that native residues (especially those normally buried) adopt non-native positions.
In this regard, a telling example is an engineered triple mutant of Cdc11 in which three key residues in its P loop (Gly29, Gly32, and Gly34) were all mutated to Ala (Casamayor and Snyder, 2003). This mutant does not cause any TS defect in vivo and is fully competent to localize to the bud neck in cells also expressing wild-type Cdc11 (Casamayor and Snyder, 2003). By contrast, substituting any one of these Gly residues with a bulky, negatively charged residue (Asp or Glu) is sufficient to prevent growth at high temperatures (Nagaraj et al., 2008; Weems et al., 2014), and the G32E mutant is efficiently excluded from the bud neck (the others have not been tested; Nagaraj et al., 2008).
Molecular requirements for Hsp104–septin interactions
The lack of any obvious septin defect in hsp104∆ cells (Schirmer et al., 2004; our unpublished data) indicates that Hsp104 does not have an obligatory role in normal septin biogenesis. Instead, our results suggest that Hsp104 functions primarily to prevent or reverse misfolding by nascent septin polypeptides. Several mechanistic insights into this function can be drawn from the distinct manifestations of Hsp104 involvement in septin folding, depending on cellular Hsp104 concentration and whether or not Hsp104 is able to hydrolyze ATP.
At moderate temperature (27°C) and when expressed at normal levels, Hsp104 transiently associates with nascent mutant Cdc10 molecules, presumably by recognizing a non-native conformation of the mutant polypeptides. In this context, the chaperone–substrate association does not adversely affect septin function in the mutant cells. We speculate that Hsp104 recognizes a specific conformation in mutant septin molecules that is prone to aggregation at high temperature and by this action stabilizes an “on-pathway” folding intermediate favoring a functional folding trajectory. Experiments with purified wild-type human septins provide direct evidence of a nucleotide-free folding intermediate that, at high temperatures, forms amyloid aggregates (Garcia et al., 2007; Pissuti Damalio et al., 2012). Such a “pre-amyloid” conformation appears to be the preferred target of Hsp104 unfoldase activity (Shorter and Lindquist, 2004). Consistent with this idea, overexpressing Hsp104 severely inhibits septin function in cdc10(D182N) cells, likely by unleashing Hsp104 unfoldase activity on the mutant septin protein. Among the TS septin mutants we tested, cdc10(D182N) was the most sensitive to Hsp104 overexpression (Figure 6D), perhaps because among these mutants, cdc10(D182N) is likely the least able to bind nucleotide (Weems et al., 2014) and thus the most susceptible to adopting the “pre-amyloid” conformation.
Of note, our data suggest that Hsp104–Cdc10(D182N) association becomes an impediment to functional septin folding when ATPase-defective Hsp104 is overexpressed, despite a lack of Hsp104 unfoldase activity. We propose that, at high levels, Hsp104(E285Q E687Q) is capable of sequestering nascent Cdc10(D182N) molecules at an earlier point along a putative ordered septin folding pathway (Figure 10). The non-native septin conformation that is stabilized as a result of this interaction is not easily recognized by “downstream” chaperones and therefore embarks on a dysfunctional folding trajectory. Unlike Hsp104(E285Q E687Q), which has no obvious effect on wild-type cells, the adverse cellular effects of overexpressed Hsp104(G217S T499I)—altered in its ATP-binding and “middle” domains—and suppression of those phenotypes by specific mutations in Cdc12 were suggestive of a direct physical association between this double-mutant chaperone and wild-type septins (Schirmer et al., 2004). Hence both substrate recognition and release are likely altered in Hsp104(G217S T499I), leading to its inappropriate sequestration of wild-type septins when overexpressed.
Formation of so-called Q bodies in yeast represents another recently described Hsp104-mediated pathway for sequestration of misfolded proteins (Escusa-Toret et al., 2013). A crucial distinction, however, is that, in those studies, misfolding was induced by thermal stress, and most of the sequestered proteins were incorporated into intracellular inclusion bodies and ultimately degraded (Escusa-Toret et al., 2013). The QC system we characterized allows cells to discriminate between soluble proteins whose conformations must differ in only rather subtle ways. By promoting the functional folding of polypeptides carrying polymorphisms that would otherwise cause misfolding, chaperones—especially Hsp90 family members—are believed to act as buffers against the phenotypic manifestations of underlying genetic variation (Jarosz et al., 2010). By contrast, our study illustrates a distinct buffering mechanism in which chaperone-mediated kinetic partitioning protects cells from dominant phenotypes caused by mutant alleles.
Functional interactions between septins and other chaperones
The only previous reports to examine septin–chaperone associations in any detail focused on wild-type septins and were inconclusive about the significance of those interactions in normal septin biogenesis. First, Dekker et al. (2008) found that all five mitotic septins were among the most abundant proteins associated with CCT, and viable cct mutants lost the septin–CCT interactions and displayed aberrant septin localization. An independent study also demonstrated CCT–septin interactions in vitro with a mammalian septin and in vivo with overexpressed yeast Cdc10 (Yam et al., 2008). However, Dekker et al. (2008) did not consider their findings to be evidence of a role for CCT in de novo septin folding because properly folded recombinant septin complexes can be produced by coexpression of subunits in Escherichia coli (Versele and Thorner, 2004; Farkasovsky et al., 2005; Sirajuddin et al., 2007; Bertin et al., 2008), which lack CCT, and because folded Cdc10 was produced by coupled in vitro transcription-translation with similar efficiency in the absence and presence of added purified CCT (Dekker et al., 2008). However, E. coli expresses an endogenous chaperonin (GroEL-GroES), and the role of GroEL-GroES in production of recombinant septins has not, to our knowledge, been directly tested. Indeed, the recognition specificities of GroEL-GroES overlap those of CCT by >80% (Yam et al., 2008), and GroEL-GroES is capable of promoting the correct folding of a number of other eukaryotic proteins (Weissman et al., 1994; Horst et al., 2007). Although not mentioned by Dekker et al. (2008), GroEL-GroES clearly interacted with Cdc10 in the in vitro translation system and was displaced by the added CCT (Dekker et al., 2008). We thus consider it highly likely that proper de novo septin folding in vivo requires CCT activity and that GroEL-GroES can perform the same role when septins are expressed heterologously in prokaryotes.
Second, a mammalian Hsp40, DNAJB13, colocalizes with the annulus, a ring of septin filaments in the sperm tail, and in vitro interacts directly and stably with human SEPT4 (Guan et al., 2009). DNAJB13 may be important in assembly of the annulus, because it associates with septins in the cytoplasm before annulus formation but dissociates from the ring once its assembly is complete (Guan et al., 2009). Perhaps this Hsp40, like yeast Ydj1, recognizes a quasi-native conformation of one or more of the septins but is released upon septin–septin association during—rather than before, as with Ydj1—hetero-octamer or filament assembly.
Finally, human SEPT5 (formerly designated CDCrel-1) was the first substrate identified for Parkin, a ubiquitin-protein ligase frequently found mutated in patients with familial juvenile (early-onset) Parkinson's disease (PD; Zhang et al., 2000). Other Parkin substrates are misfolded proteins (Olzmann and Chin, 2008), raising the possibility that SEPT5 is especially prone to unfolding and is cleared from neurons in a Parkin-mediated manner. Consistent with this view, SEPT5 overexpression is neurotoxic (Dong et al., 2003; Jung et al., 2008) and, strikingly, HSF1 or Hsp70 co-overexpression protects neurons from SEPT5 toxicity in a rat model of PD, whereas Hsp40 co-overexpression exacerbates the problem (Jung et al., 2008). These studies implicate cytosolic chaperones in septin proteostasis in animal cells, as we have in yeast.
Does septin quality control exist in other cell types?
Distinctions between the fate of septin mutants in yeast and mammals are pertinent to this question. In cultured mammalian cells, endogenous wild-type septins can be efficiently replaced by overexpressed tagged and/or mutant counterparts via a mechanism that appears to involve rapid degradation of superstoichiometric septin subunits, such that there is effectively no free pool of monomeric septins (Sellin et al., 2011b). In yeast, too, all septin subunits normally appear to be incorporated into hetero-octamers, suggestive of carefully controlled stoichiometry. However, unlike in animal cells, experimentally induced imbalance does not result in degradation of excess yeast septins (McMurray et al., 2011a; see also Figure 2A), and the overproduced septin can only replace the endogenous protein if it passes QC. It may be that in mammals, a sequestration-based, chaperone-mediated QC system exists but is easily overwhelmed by the degree of overexpression achieved, whereupon a robust degradation-based mechanism efficiently clears the cytosol of any unincorporated septins (mutant or wild type). In yeast, on the other hand, degradation-based control of septin stoichiometry is inefficient or nonexistent and robust chaperone-mediated sequestration in which “quality” is assessed before higher-order assembly seems to be the predominant mechanism for excluding defective subunits.
The only situation in which a mammalian cell coexpresses at endogenous levels both a G-interface mutant septin and its wild-type counterpart relates to the spermatocytes of heterozygous infertile men (Kuo et al., 2012). Importantly, however, the “heterokaryon-like” character of spermatogenesis might allow mutant septins to incorporate into protofilaments even if a sequestration-based QC system is at play (Weems et al., 2014; Figure 4). In another human disease heterozygous for a septin alteration, hereditary neuralgic amyotrophy, the mutant septin also seems to act dominantly (Kuhlenbäumer et al., 2005; Collie et al., 2010). In this case, the alterations do not lie in the GTP-binding domain at all, but instead are substitutions in (Kuhlenbäumer et al., 2005) or duplications (Collie et al., 2010) of the extended N terminus of SEPT9. Hence a sequestration-based QC mechanism acting on mutants with misfolded G domains would likely be unable to recognize this class of septin mutants. Several types of cancer are also reportedly associated with up-regulation of SEPT9 isoforms with altered N termini (Gonzalez et al., 2007; Connolly et al., 2014). Thus the few human disorders linked to septins seem to prey precisely upon the vulnerabilities of the QC system we describe here.
MATERIALS AND METHODS
Yeast strains, media, plasmids, and genetic manipulations
All yeast strains are listed in Supplemental Table S1 and were manipulated using standard techniques (Amberg et al., 2005). Yeast cells were cultivated in liquid or on solid (2% agar) plates of rich (1% yeast extract, 2% tryptone or peptone [YP]) or synthetic (Drop Out Mix Minus various ingredients; United States Biological, Salem, MA) medium containing total 2% sugar (dextrose, raffinose, or galactose), as appropriate to maintain plasmid selection and induce expression from the GAL1/10 or MET17 promoter. 5-Fluoro-orotic acid (FOA; United States Biological)–containing medium was prepared using proline as the nitrogen source as described elsewhere (McCusker and Davis, 1991). G-418 (Geneticin; Roche Diagnostics, Indianapolis, IN) was added to YP/dextrose (YPD) to final 200 μg/ml of active drug. Sporulation was induced in 1% potassium acetate, 0.05% glucose, 20 mg/l leucine, and 40 mg/l uracil. The bacterial strain DH5α (Agilent Technologies, Santa Clara, CA) was used to propagate plasmids (Supplemental Table S2), of which DNA was obtained as for yeast DNA (see later description) but without glass bead lysis. Yeast transformation was performed using a standard lithium acetate method (Amberg et al., 2005) or Frozen-EZ Yeast Transformation Kit II (Zymo Research, Irvine, CA).
Genomic or plasmid DNA from yeast was isolated by resuspending cells in 250 μl of P1 (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 100 μg/ml RNase A), adding 0.5-mm glass beads to displace an additional ∼250 μl of volume, and vortexing for 3 min before addition of P2 (200 mM NaOH, 1% SDS) and N3 (4.2 M guanidine-HCl, 0.9 M potassium acetate, pH 4.8). Lysates were clarified by centrifugation and applied to Zyppy Plasmid DNA Miniprep columns (Zymo Research); then DNA was washed and eluted according to the manufacturer's instructions. Plasmids were transformed to competent bacterial cells using Mix & Go E. coli Transformation Kit (Zymo Research) cells according to the manufacturer's instructions. PCR was performed with Q5 high-fidelity polymerase (New England Biolabs, Ipswich, MA) according to the manufacturer's instructions. Site-directed mutagenesis was performed using the QuikChange II Site-Directed Mutagenesis kit (Agilent) according to the manufacturer's instructions or by Keyclone Technologies (Lexington, KY) using their methods of choice.
Quantification of culture growth rates
Optical density measurements of yeast cultures were performed in flat-bottom Costar 96-well plates in a Cytation 3 (BioTek, Winooski, VT) imaging plate reader. Growth curve measurements were performed at 5-min intervals using continuous orbital shaking between reads and a temperature set point of 25°C. Temperature began at 25°C and reached 27.7°C by 14 h. Fits to exponential growth equations and derivation of doubling times from the first 14 h of growth were performed with Prism software (GraphPad, La Jolla, CA). The R2 values for individual curve fits ranged from 0.9756 to 0.9990.
SNAP-tag in vivo labeling
For analysis of septin localization in zygotes, a 500-μl YPD culture of the triple-efflux-pump-mutant cdc10-1-SNAP strain JTY5169 was grown to mid log phase at 22°C. A 1 μl amount of 1 mM benzylguanine-diacetylfluorescein (BG-DAF; S9107S, New England Biolabs) dissolved in dimethyl sulfoxide (DMSO) was added, and the culture was rotated in the dark for 30 min. Cells were washed three times with 1 ml of YPD and resuspended in 500 μl of fresh YPD before an additional 30-min rotation, followed by three more YPD washes. Pelleted cells were resuspended in YPD and mixed with an equal number of BY4742 cells from a parallel mid log culture. Mating was allowed to proceed for 6.5 h before cells were pelleted and resuspended in water for imaging.
For biochemical analysis of nascent septin protein complexes, 50-ml cultures of YPD or synthetic raffinose medium lacking uracil (for strains harboring plasmids) were grown at 22°C to mid-log phase. Galactose was added to the synthetic raffinose culture to a final concentration of 2%, and this culture was incubated for an additional 5.25 h. O6-benzylguanine (sc-202747A; Santa Cruz Biotechnology, Santa Cruz, CA) was added from a 1 mM stock in DMSO to a final concentration of 5 μM. Cultures were incubated at 22, 27, or 30°C for 45 min and then pelleted and washed three times with 50 ml of fresh growth medium. Cells were resuspended in 50 ml of fresh growth medium (prewarmed, where appropriate) and shaken for 45 min at the appropriate temperature before cells were pelleted, resuspended in 500 μl of low-salt lysis buffer (125 mM NaCl, 10% glycerol, 2 mM Mg2Cl, 1 mM EDTA, 0.1% Tween-20, and 50 mM sodium phosphate, pH 7.5), transferred to a 2-ml screw-cap microcentrifuge tube, and frozen at −20°C.
Microscopy
All micrographs were captured with an EVOSfl (Advanced Microscopy Group, Bothell, WA) all-in-one microscope equipped with an Olympus (Tokyo, Japan) 60× oil immersion objective and GFP, YFP, and Texas red filters. Cells from mid log cultures were transferred to water, spotted on agarose pads, and visualized by microscopy with the appropriate filter set, using 60-, 120-, or 250-ms exposures at 100% LED intensity.
Size exclusion chromatography of yeast lysates
For GFP-tagged septin experiments, 250-ml cultures of a diploid made by mating BY4742 and JTY3986, or diploid strain BY4743 carrying plasmid pJT3456, were grown in YPD or synthetic uracil-free liquid medium to mid log phase. The BY4742 × JTY3986 culture was pelleted and frozen. The BY4743 [pJT3456] cells were pelleted, washed, and resuspended in 250 ml of medium lacking methionine and uracil. After 6 h of additional incubation, these cells were pelleted and frozen. Cell pellets from the two samples were manipulated in parallel thereafter. After resuspension in 500 μl high-salt lysis buffer (500 mM NaCl, 0.1% Tween-20, 1 mM dithiothreitol [DTT], 50 mM sodium phosphate buffer, pH 7.4), cells were lysed by three rounds of vortexing for 1 min, between which the samples were cooled on ice. After clarifying the lysate for 10 min at 13,000 × g, the supernatant was loaded onto a HiLoad 16/600 Superdex 200 column (GE Healthcare, Little Chalfont, United Kingdom) equilibrated with 500 mM NaCl and 50 mM Tris-HCl, pH 8.0, and resolved at 0.5 ml/ml flow rate in the same buffer. The 2-ml eluate fractions were collected and vacuum filtered onto a nitrocellulose membrane using a slot blot apparatus. The membrane was blocked with Odyssey blocking reagent (Li-Cor Biosciences, Lincoln, NE) and exposed to rabbit anti-Cdc3 (McMurray and Thorner, 2008b) and mouse anti-GFP antibodies (11 814 460 001; Roche ) for 1 h. After washing with Tris-buffered saline with 0.01% Tween-20, the membrane was incubated with fluorescently labeled anti-rabbit (680 nm) and anti-mouse (800 nm) secondary antibodies (Li-Cor) before washing and scanning on a Li-Cor Odyssey device.
For SNAP-tagged septin experiments, glass beads were added to the tube containing frozen cells, and lysis was accomplished by four rounds of vortexing for 1 min, between which the samples were cooled on ice. After clarifying the lysate for 10 min at 13,000 × g, 500 μl of the supernatant was mixed with Oregon green 488 dipivaloyl benzylguanine (Covalys Biosciences, Witterswil, Switzerland) to a final concentration of 3.3 μM. Immediately after 30 min of gentle mixing at 22°C protected from light, the sample was loaded onto a HiLoad 16/600 Sephacryl S300 column (GE Healthcare), equilibrated with 500 mM NaCl and 50 mM Tris-HCl, pH 8.0, and resolved at 0.5 ml/ml flow rate in the same buffer. The 2-ml eluate fractions were collected, of which 200 μl was transferred to the wells of a black, glass-bottom 96-well plate (655891; Greiner Bio-One International, Frickenhausen, Germany). For some experiments, 100-μl aliquots of two adjacent fractions were pooled together. Fluorescence upon excitation with 490-nm light was measured at 520 nm using a gain setting of 100 in the Cytation 3. A 1-ml amount of each peak fraction was mixed with 250 μl of 100% trichloroacetic acid (TCA) and frozen. Thawed precipitates were pelleted for 10 min at 13,000 × g and washed with 1 ml of cold acetone before air drying. Precipitated protein was resuspended in SDS–PAGE sample buffer. In some experiments, 1 ml of selected fractions was transferred directly to nitrocellulose via slot blot. Protein content was analyzed as described.
Ydj1-TAP affinity purification
We lysed 150-ml cultures with glass beads as described in 4 ml of low-salt lysis buffer containing protease inhibitors (Complete EDTA-free; 11 873 580 001, Roche). An aliquot was removed as an “input” sample. Then 25 μl of buffer-washed IgG-coated agarose beads (sc-2345; Santa Cruz Biotechnology) was added and mixed for 2 h at 22°C with end-over-end rotation. Beads were gently pelleted and washed thrice with 1 ml of low-salt lysis buffer, followed by three washes with 100 μl of protease cleavage buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM DTT, and 0.01% NP-40). After resuspension of the beads in 25 μl of cleavage buffer, glutathione S-transferase–tagged 3C protease was added, and cleavage was allowed to proceed overnight at 4°C. Beads were separated from the eluted proteins by gentle centrifugation of the sample through a column filter (UFC30GV00; Millipore, Billerica, MA). The eluate and the beads were mixed with SDS–PAGE sample buffer before boiling and loading on a 4–20% gradient SDS–PAGE gel. After transfer to a polyvinylidene fluoride (PVDF) membrane, proteins were detected as described earlier and in the figure legends.
Preparation of total protein extracts for immunoblotting
The 1-ml cultures were pelleted and frozen at −80°C. Cell pellets were thawed in 150 μl of 1.85 M NaOH and 7.4% β-mercaptoethanol. A 150 μl amount of 50% TCA was added before incubation on ice for 10 min. Precipitated proteins were pelleted at 4°C at maximum speed for 10 min, washed twice with cold acetone (1 ml each time), and resuspended in 80 μl of 0.1 M Tris base/5% SDS. After addition of SDS–PAGE sample buffer, the samples were boiled for 3 min before resolution by SDS–PAGE.
Supplementary Material
Acknowledgments
We thank David Drubin (University of California, Berkeley, CA), John Pringle (Stanford University School of Medicine, Stanford, CA), Mark Longtine (Washington University School of Medicine, St. Louis, MO), and colleagues Chandra Tucker, Aaron M. Johnson, and Jeff Moore of the University of Colorado Anschutz Medical Campus for sharing reagents. Research reported here was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Awards R01GM021841 (to J.T.) and R00GM086603 (to M.A.M.) and by the Alzheimer's Association under Award NIRGD-12-241119 (to M.A.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used:
- CCT
chaperonin containing TCP1
- NBP
nucleotide-binding pocket
- PFD
prefoldin
- QC
quality control
- SEC
size exclusion chromatography
- TS
temperature sensitive.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-11-1531) on February 11, 2015.
*Present address: UCSD Combined Family Medicine and Psychiatry Residency Program, UCSD Medical Center, San Diego, CA 92103.
REFERENCES
- Alpers JB, Paulus H. Allosteric preconditioning: role of allosteric ligands in promoting the maturation of enzymes. Nature. 1971;233:478–480. doi: 10.1038/233478a0. [DOI] [PubMed] [Google Scholar]
- Amberg D, Burke D, Strathern J. Methods in Yeast Genetics, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Bertin A, McMurray MA, Grob P, Park S-S, Garcia G, 3rd, Patanwala I, Ng H-L, Alber T, Thorner J, Nogales E. Saccharomyces cerevisiae septins: supramolecular organization of heterooligomers and the mechanism of filament assembly. Proc Natl Acad Sci USA. 2008;105:8274–8279. doi: 10.1073/pnas.0803330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betting J, Seufert W. A yeast Ubc9 mutant protein with temperature-sensitive in vivo function is subject to conditional proteolysis by a ubiquitin- and proteasome-dependent pathway. J Biol Chem. 1996;271:25790–25796. doi: 10.1074/jbc.271.42.25790. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. Septin assemblies form by diffusion-driven annealing on membranes. Proc Natl Acad Sci USA. 2014;111:2146–2151. doi: 10.1073/pnas.1314138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösl B, Grimminger V, Walter S. Substrate binding to the molecular chaperone Hsp104 and its regulation by nucleotides. J Biol Chem. 2005;280:38170–38176. doi: 10.1074/jbc.M506149200. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Douglas MG. Characterization of YDJ1: a yeast homologue of the bacterial dnaJ protein. J Cell Biol. 1991;114:609–621. doi: 10.1083/jcb.114.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casamayor A, Snyder M. Molecular dissection of a yeast septin: distinct domains are required for septin interaction, localization, and function. Mol Cell Biol. 2003;23:2762–2777. doi: 10.1128/MCB.23.8.2762-2777.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Longtine M, Pringle JR, Bi E. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol Biol Cell. 2003;14:4051–4066. doi: 10.1091/mbc.E03-04-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Adamíková L, Cenamor R, Molina M, Sánchez M, Nombela C. Cell integrity and morphogenesis in a budding yeast septin mutant. Microbiology. 1998;144:3463–3474. doi: 10.1099/00221287-144-12-3463. [DOI] [PubMed] [Google Scholar]
- Collie AMB, Landsverk ML, Ruzzo E, Mefford HC, Buysse K, Adkins JR, Knutzen DM, Barnett K, Brown RH Jr, Parry GJ, et al. Non-recurrent SEPT9 duplications cause hereditary neuralgic amyotrophy. J Med Genet. 2010;47:601–607. doi: 10.1136/jmg.2009.072348. [DOI] [PubMed] [Google Scholar]
- Connolly D, Hoang HG, Adler E, Tazearslan C, Simmons N, Bernard VV, Castaldi M, Oktay MH, Montagna C. Septin 9 amplification and isoform-specific expression in peritumoral and tumor breast tissue. Biol Chem. 2014;395:157–167. doi: 10.1515/hsz-2013-0247. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker C, Stirling PC, McCormack EA, Filmore H, Paul A, Brost RL, Costanzo M, Boone C, Leroux MR, Willison KR. The interaction network of the chaperonin CCT. EMBO J. 2008;27:1827–1839. doi: 10.1038/emboj.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val N, McMurray MA, Lam LH, Hsiung CC-S, Bertin A, Nogales E, Thorner J. Native cysteine residues are dispensable for the structure and function of all five yeast mitotic septins. Proteins. 2013;81:1964–1979. doi: 10.1002/prot.24345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelaere J, Gentry MS, Hallberg RL, Barral Y. Phosphorylation-dependent regulation of septin dynamics during the cell cycle. Dev Cell. 2003;4:345–357. doi: 10.1016/s1534-5807(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Dong Z, Ferger B, Paterna J-C, Vogel D, Furler S, Osinde M, Feldon J, Büeler H. Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proc Natl Acad Sci USA. 2003;100:12438–12443. doi: 10.1073/pnas.2132992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing KH, Nogales E. Cryoelectron microscopy applications in the study of tubulin structure, microtubule architecture, dynamics and assemblies, and interaction of microtubules with motors. Methods Enzymol. 2010;483:121–142. doi: 10.1016/S0076-6879(10)83006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escusa-Toret S, Vonk WIM, Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat Cell Biol. 2013;15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkasovsky M, Herter P, Voss B, Wittinghofer A. Nucleotide binding and filament assembly of recombinant yeast septin complexes. Biol Chem. 2005;386:643–656. doi: 10.1515/BC.2005.075. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J. Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell. 1999;4:1051–1061. doi: 10.1016/s1097-2765(00)80233-6. [DOI] [PubMed] [Google Scholar]
- Frazier JA, Wong ML, Longtine MS, Pringle JR, Mann M, Mitchison TJ, Field C. Polymerization of purified yeast septins: evidence that organized filament arrays may not be required for septin function. J Cell Biol. 1998;143:737–749. doi: 10.1083/jcb.143.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia G, 3rd, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E. Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol. 2011;195:993–1004. doi: 10.1083/jcb.201107123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia W, de Araújo APU, Lara F, Foguel D, Tanaka M, Tanaka T, Garratt RC. An intermediate structure in the thermal unfolding of the GTPase domain of human septin 4 (SEPT4/Bradeion-beta) forms amyloid-like filaments in vitro. Biochemistry. 2007;46:11101–11109. doi: 10.1021/bi700702w. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Nelson ZW, Gottschling DE. Degradation-mediated protein quality control in the nucleus. Cell. 2005;120:803–815. doi: 10.1016/j.cell.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Gasper R, Meyer S, Gotthardt K, Sirajuddin M, Wittinghofer A. It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol. 2009;10:423–429. doi: 10.1038/nrm2689. [DOI] [PubMed] [Google Scholar]
- Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, et al. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez ME, Peterson EA, Privette LM, Loffreda-Wren JL, Kalikin LM, Petty EM. High SEPT9_v1 expression in human breast cancer cells is associated with oncogenic phenotypes. Cancer Res. 2007;67:8554–8564. doi: 10.1158/0008-5472.CAN-07-1474. [DOI] [PubMed] [Google Scholar]
- Guan J, Kinoshita M, Yuan L. Spatiotemporal association of DNAJB13 with the annulus during mouse sperm flagellum development. BMC Dev Biol. 2009;9:23. doi: 10.1186/1471-213X-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer BK, Pringle JR. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol Cell Biol. 1987;7:3678–3687. doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971;69:265–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Mortimer RK, Culotti J, Culotti M. Genetic control of the cell division cycle in yeast: V. Genetic analysis of cdc mutants. Genetics. 1973;74:267–286. doi: 10.1093/genetics/74.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst R, Fenton WA, Englander SW, Wüthrich K, Horwich AL. Folding trajectories of human dihydrofolate reductase inside the GroEL GroES chaperonin cavity and free in solution. Proc Natl Acad Sci USA. 2007;104:20788–20792. doi: 10.1073/pnas.0710042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyle HD, Turner FR, Brunick L, Raff EC. Tubulin sorting during dimerization in vivo. Mol Biol Cell. 2001;12:2185–2194. doi: 10.1091/mbc.12.7.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Hwang YW, Miller DL. A mutation that alters the nucleotide specificity of elongation factor Tu, a GTP regulatory protein. J Biol Chem. 1987;262:13081–13085. [PubMed] [Google Scholar]
- Iwase M, Luo J, Nagaraj S, Longtine M, Kim HB, Haarer BK, Caruso C, Tong Z, Pringle JR, Bi E. Role of a Cdc42p effector pathway in recruitment of the yeast septins to the presumptive bud site. Mol Biol Cell. 2006;17:1110–1125. doi: 10.1091/mbc.E05-08-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Jung AE, Fitzsimons HL, Bland RJ, During MJ, Young D. HSP70 and constitutively active HSF1 mediate protection against CDCrel-1-mediated toxicity. Mol Ther. 2008;16:1048–1055. doi: 10.1038/mt.2008.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota J, Yamamoto T, Yoshiuchi S, Bi E, Tanaka K. Septin ring assembly requires concerted action of polarisome components, a PAK kinase Cla4p, and the actin cytoskeleton in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:5329–5345. doi: 10.1091/mbc.E04-03-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci USA. 2004;101:9955–9959. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Froese CD, Xie H, Trimble WS. Uncovering principles that control septin-septin interactions. J Biol Chem. 2012;287:30406–30413. doi: 10.1074/jbc.M112.387464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Kuhlenbäumer G, Hannibal MC, Nelis E, Schirmacher A, Verpoorten N, Meuleman J, Watts GD, De Vriendt E, Young P, Stögbauer F, et al. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat Genet. 2005;37:1044–1046. doi: 10.1038/ng1649. [DOI] [PubMed] [Google Scholar]
- Kuo Y-C, Lin YH, Chen HI, Wang YY, Chiou YW, Lin HH, Pan HA, Wu CM, Su SM, Hsu CC, et al. SEPT12 mutations cause male infertility with defective sperm annulus. Hum Mutat. 2012;33:710–719. doi: 10.1002/humu.22028. [DOI] [PubMed] [Google Scholar]
- Lee DH, Sherman MY, Goldberg AL. Involvement of the molecular chaperone Ydj1 in the ubiquitin-dependent degradation of short-lived and abnormal proteins in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4773–4781. doi: 10.1128/mcb.16.9.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fanarraga M, Avila J, Guasch A, Coll M, Zabala JC. Review: postchaperonin tubulin folding cofactors and their role in microtubule dynamics. J Struct Biol. 2001;135:219–229. doi: 10.1006/jsbi.2001.4386. [DOI] [PubMed] [Google Scholar]
- Lundin VF, Leroux MR, Stirling PC. Quality control of cytoskeletal proteins and human disease. Trends Biochem Sci. 2010;35:288–297. doi: 10.1016/j.tibs.2009.12.007. [DOI] [PubMed] [Google Scholar]
- McCusker JH, Davis RW. The use of proline as a nitrogen source causes hypersensitivity to, and allows more economical use of 5FOA in Saccharomyces cerevisiae. Yeast. 1991;7:607–608. doi: 10.1002/yea.320070608. [DOI] [PubMed] [Google Scholar]
- McMurray M. Lean forward: genetic analysis of temperature-sensitive mutants unfolds the secrets of oligomeric protein complex assembly. Bioessays. 2014;36:836–846. doi: 10.1002/bies.201400062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Bertin A, Garcia G, 3rd, Lam L, Nogales E, Thorner J. Septin filament formation is essential in budding yeast. Dev Cell. 2011a;20:540–549. doi: 10.1016/j.devcel.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Stefan CJ, Wemmer M, Odorizzi G, Emr SD, Thomer J. Genetic interactions with mutations affecting septin assembly reveal ESeRT functions in budding yeast cytokinesis. Biol Chem. 2011b;392:699–712. doi: 10.1515/BC.2011.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Thorner J. Biochemical properties and supramolecular architecture of septin hetero-oligomers and septin filaments. In: Hall PA, Russell SE, Pringle JR, editors. In: The Septins. Hoboken, NJ: John Wiley & Sons; 2008a. pp. 49–100. [Google Scholar]
- McMurray MA, Thorner J. Septin stability and recycling during dynamic structural transitions in cell division and development. Curr Biol. 2008b;18:1203–1208. doi: 10.1016/j.cub.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosavi B, Wongwigkarn J, Tuite MF. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI+] prion but not for prion propagation. Yeast. 2010;27:167–179. doi: 10.1002/yea.1742. [DOI] [PubMed] [Google Scholar]
- Nagai S. diagnostic color differentiation plates for hereditary respiration deficiency in yeast. J Bacteriol. 1963;86:299–302. doi: 10.1128/jb.86.2.299-302.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj S, Rajendran A, Jackson CE, Longtine MS. Role of nucleotide binding in septin-septin interactions and septin localization in Saccharomyces cerevisiae. Mol Cell Biol. 2008;28:5120–5137. doi: 10.1128/MCB.00786-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH. Structure of the alpha beta tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Okada S, Leda M, Hanna J, Savage NS, Bi E, Goryachev AB. Daughter cell identity emerges from the interplay of Cdc42, septins, and exocytosis. Dev Cell. 2013;26:148–161. doi: 10.1016/j.devcel.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Chin L-S. Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy. 2008;4:85–87. doi: 10.4161/auto.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Malmberg RL, Momany M. Analysis of septins across kingdoms reveals orthology and new motifs. BMC Evol Biol. 2007;7:103. doi: 10.1186/1471-2148-7-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K-W, Hahn J-S, Fan Q, Thiele DJ, Li L. De novo appearance and “strain” formation of yeast prion [PSI+] are regulated by the heat-shock transcription factor. Genetics. 2006;173:35–47. doi: 10.1534/genetics.105.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Lindquist S. Saccharomyces cerevisiae Hsp104 protein. Purification and characterization of ATP-induced structural changes. J Biol Chem. 1994;269:4480–4487. [PubMed] [Google Scholar]
- Pissuti Damalio JC, Garcia W, Alves Macêdo JN, de Almeida Marques I, Andreu JM, Giraldo R, Garratt RC, Ulian Araújo AP. Self assembly of human septin 2 into amyloid filaments. Biochimie. 2012;94:628–636. doi: 10.1016/j.biochi.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Sahi C, Kominek J, Ziegelhoffer T, Yu HY, Baranowski M, Marszalek J, Craig EA. Sequential duplications of an ancient member of the DnaJ-family expanded the functional chaperone network in the eukaryotic cytosol. Mol Biol Evol. 2013;30:985–998. doi: 10.1093/molbev/mst008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassanfar M, Samson L. Identification and preliminary characterization of an O6-methylguanine DNA repair methyltransferase in the yeast Saccharomyces cerevisiae. J Biol Chem. 1990;265:20–25. [PubMed] [Google Scholar]
- Schaupp A, Marcinowski M, Grimminger V, Bösl B, Walter S. Processing of proteins by the molecular chaperone Hsp104. J Mol Biol. 2007;370:674–686. doi: 10.1016/j.jmb.2007.04.070. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Homann OR, Kowal AS, Lindquist S. Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol Biol Cell. 2004;15:2061–2072. doi: 10.1091/mbc.E02-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Holmfeldt P, Stenmark S, Gullberg M. Microtubules support a disk-like septin arrangement at the plasma membrane of mammalian cells. Mol Biol Cell. 2011a;22:4588–4601. doi: 10.1091/mbc.E11-09-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Sandblad L, Stenmark S, Gullberg M. Deciphering the rules governing assembly order of mammalian septin complexes. Mol Biol Cell. 2011b;22:3152–3164. doi: 10.1091/mbc.E11-03-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- Sirajuddin M, Farkasovsky M, Hauer F, Kühlmann D, Macara IG, Weyand M, Stark H, Wittinghofer A. Structural insight into filament formation by mammalian septins. Nature. 2007;449:311–315. doi: 10.1038/nature06052. [DOI] [PubMed] [Google Scholar]
- Sirajuddin M, Farkasovsky M, Zent E, Wittinghofer A. GTP-induced conformational changes in septins and implications for function. Proc Natl Acad Sci USA. 2009;106:16592–16597. doi: 10.1073/pnas.0902858106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Bhamidipati A, Cowan NJ, Lewis SA. Tubulin folding cofactors as GTPase-activating proteins. GTP hydrolysis and the assembly of the alpha/beta-tubulin heterodimer. J Biol Chem. 1999;274:24054–24058. doi: 10.1074/jbc.274.34.24054. [DOI] [PubMed] [Google Scholar]
- Tian G, Lewis SA, Feierbach B, Stearns T, Rommelaere H, Ampe C, Cowan NJ. Tubulin subunits exist in an activated conformational state generated and maintained by protein cofactors. J Cell Biol. 1997;138:821–832. doi: 10.1083/jcb.138.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Vainberg IE, Tap WD, Lewis SA, Cowan NJ. Quasi-native chaperonin-bound intermediates in facilitated protein folding. J Biol Chem. 1995;270:23910–23913. doi: 10.1074/jbc.270.41.23910. [DOI] [PubMed] [Google Scholar]
- Vainberg IE, Lewis SA, Rommelaere H, Ampe C, Vandekerckhove J, Klein HL, Cowan NJ. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell. 1998;93:863–873. doi: 10.1016/s0092-8674(00)81446-4. [DOI] [PubMed] [Google Scholar]
- Varadarajan R, Nagarajaram HA, Ramakrishnan C. A procedure for the prediction of temperature-sensitive mutants of a globular protein based solely on the amino acid sequence. Proc Natl Acad Sci USA. 1996;93:13908–13913. doi: 10.1073/pnas.93.24.13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergés E, Colomina N, Garí E, Gallego C, Aldea M. Cyclin Cln3 is retained at the ER and released by the J chaperone Ydj1 in late G1 to trigger cell cycle entry. Mol Cell. 2007;26:649–662. doi: 10.1016/j.molcel.2007.04.023. [DOI] [PubMed] [Google Scholar]
- Versele M, Gullbrand B, Shulewitz MJ, Cid VJ, Bahmanyar S, Chen RE, Barth P, Alber T, Thorner J. Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell. 2004;15:4568–4583. doi: 10.1091/mbc.E04-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versele M, Thorner J. Septin collar formation in budding yeast requires GTP binding and direct phosphorylation by the PAK, Cla4. J Cell Biol. 2004;164:701–715. doi: 10.1083/jcb.200312070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrabioiu AM, Gerber SA, Gygi SP, Field CM, Mitchison TJ. The majority of the Saccharomyces cerevisiae septin complexes do not exchange guanine nucleotides. J Biol Chem. 2004;279:3111–3118. doi: 10.1074/jbc.M310941200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tian G, Cowan NJ, Cabral F. Mutations affecting beta-tubulin folding and degradation. J Biol Chem. 2006;281:13628–13635. doi: 10.1074/jbc.M513730200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weems AD, Johnson CR, Argueso JL, McMurray MA. Higher-order septin assembly is driven by GTP-promoted conformational changes: evidence from unbiased mutational analysis in Saccharomyces cerevisiae. Genetics. 2014;196:711–727. doi: 10.1534/genetics.114.161182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman JS, Kashi Y, Fenton WA, Horwich AL. GroEL-mediated protein folding proceeds by multiple rounds of binding and release of nonnative forms. Cell. 1994;78:693–702. doi: 10.1016/0092-8674(94)90533-9. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, Vetter IR. Structure-function relationships of the G domain, a canonical switch motif. Annu Rev Biochem. 2011;80:943–971. doi: 10.1146/annurev-biochem-062708-134043. [DOI] [PubMed] [Google Scholar]
- Wu Y, Li J, Jin Z, Fu Z, Sha B. The crystal structure of the C-terminal fragment of yeast Hsp40 Ydj1 reveals novel dimerization motif for Hsp40. J Mol Biol. 2005;346:1005–1011. doi: 10.1016/j.jmb.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Yam AY, Xia Y, Lin H-TJ, Burlingame A, Gerstein M, Frydman J. Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol. 2008;15:1255–1262. doi: 10.1038/nsmb.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabala JC, Fontalba A, Avila J. Tubulin folding is altered by mutations in a putative GTP binding motif. J Cell Sci. 1996;109:1471–1478. doi: 10.1242/jcs.109.6.1471. [DOI] [PubMed] [Google Scholar]
- Zent E, Wittinghofer A. Human septin isoforms and the GDP-GTP cycle. Biol Chem. 2014;395:169–180. doi: 10.1515/hsz-2013-0268. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.