

Double-strand-break repair proteins interact with and recruit Sir proteins to ectopic sites in the genome. Recruitment results in gene silencing, which depends on Sir proteins, as well as on histone H2A modification. Silencing also results in the localization of the locus to the nuclear periphery.
Abstract
Heterochromatin formation and nuclear organization are important in gene regulation and genome fidelity. Proteins involved in gene silencing localize to sites of damage and some DNA repair proteins localize to heterochromatin, but the biological importance of these correlations remains unclear. In this study, we examined the role of double-strand-break repair proteins in gene silencing and nuclear organization. We find that the ATM kinase Tel1 and the proteins Mre11 and Esc2 can silence a reporter gene dependent on the Sir, as well as on other repair proteins. Furthermore, these proteins aid in the localization of silenced domains to specific compartments in the nucleus. We identify two distinct mechanisms for repair protein–mediated silencing—via direct and indirect interactions with Sir proteins, as well as by tethering loci to the nuclear periphery. This study reveals previously unknown interactions between repair proteins and silencing proteins and suggests insights into the mechanism underlying genome integrity.
INTRODUCTION
Heterochromatin formation is a common mechanism of stable gene repression in eukaryotes and involves the formation of large chromatin domains that are inaccessible to specific proteins, resulting in repression of transcription and recombination of sequences that are present within these domains. In the budding yeast Saccharomyces cerevisiae, heterochromatic structures are observed at the cryptic mating-type loci HML and HMR on chromosome III, as well as in subtelomeric regions of chromosomes.
At HML and HMR, silencer elements flank the genes that are silenced, whereas the telomeric repeats function as silencers for subtelomeric heterochromatic regions. Mutational and binding studies identified the proteins that bind these sequence elements and are necessary for silencing. These include the origin recognition complex (ORC) and the transcription factors Rap1, Abf1, and Sum1, as well as telomere-bound proteins such as Ku (Rusche et al., 2003; Fabre and Spichal, 2014).
The silencer-bound proteins interact with and recruit the Sir repressor proteins to silence and compact loci. ORC interacts with Sir1, which localizes primarily at the silencers. Sir1 and Rap1 interact with and recruit the other Sir proteins (Sir2, Sir3, and Sir4; Luo et al., 2002; Rusche et al., 2002). Once recruited to the silenced domain, Sir2 deacetylates histone tails, enabling stable interaction of the Sir2, Sir3, and Sir4 complex to the deacetylated histone tails and thereby mediating formation of inaccessible chromatin domains (Ghidelli et al., 2001; Johnson et al., 2009; Martino et al., 2009; Oppikofer et al., 2011, 2013). Whereas the Sir proteins spread bidirectionally from the silencers, the silenced domain is restricted to specific regions of the genome by DNA sequence elements called barrier insulators. Insulators are bound by proteins that use enzymatic activities that act on histone tails to disfavor Sir protein binding or aid in the maintenance of nucleosome-free regions (Donze et al., 1999; Oki and Kamakaka, 2005; Dhillon et al., 2009).
Furthermore, silenced domains, including HMR and the telomeres, cluster together at the nuclear periphery, forming silencing foci. The 16 centromeres cluster together at a single site at the nuclear periphery adjacent to the spindle pole body (Jin et al., 1998), whereas the 32 telomeres cluster at several loci at the nuclear periphery stabilized by protein factors that interact with subtelomeric and telomeric sequences (Hediger et al., 2002; Taddei and Gasser, 2004; Therizols et al., 2010; Fabre and Spichal, 2014). Telomeric clustering at the periphery is dependent on the Sir proteins, nuclear pore proteins, and Esc1 and Ku proteins (Fabre and Spichal, 2014). Of interest, whereas the clustering of HML and HMR is dependent on these factors, it is also affected by mutations in double-strand- break repair proteins (Miele et al., 2009; Kirkland and Kamakaka, 2013).
DNA damage is ubiquitous, and a large number of proteins are involved in DNA repair, dependent on the nature of the damage (Ataian and Krebs, 2006; Symington and Gautier, 2011; Krejci et al., 2012; Iyama and Wilson, 2013). There are two major pathways for the repair of double-stranded breaks: homologous recombination (HR) repair and nonhomologous end joining (NHEJ). A large number of the proteins that function in these two pathways have been identified, and the mechanisms by which they function in repair have been elucidated.
NHEJ involves numerous protein complexes, including the Ku complex, the MRX complex, and the DNA ligase complex (Symington and Gautier, 2011). The Ku complex binds the ends of double-strand breaks (Fisher and Zakian, 2005), which leads to recruitment of the MRX complex (Mre11/Rad50/Xrs2; Stracker and Petrini, 2011). The subsequent end-bridging activity allows the Lig4/Lif1 complex to ligate the ends (Symington and Gautier, 2011).
The HR repair pathway also uses numerous proteins in a series of steps leading to DNA repair (Lisby and Rothstein, 2009). In this pathway, upon detection of a double-strand break, the MRX complex binds the break (Kinoshita et al., 2009; Stracker and Petrini 2011). In addition, chromatin-modifying proteins play important roles in HR repair. One of the first steps involves the phosphorylation of histone H2A on serine 129 (γ-H2A) by Tel1 (ATM) or Mec1 (ATR; Flott et al., 2007; Polo and Jackson 2011). Phosphorylation of H2A aids in the recruitment of various proteins, including the resection machinery (Srs2 and Exo1). Resection is followed by homology search and recognition, strand invasion, and repair mediated by the late repair proteins (Rad51, Rad52, Rad54, and Rdh54, among others; Sugawara et al., 2003; Heyer et al., 2006; Keogh et al., 2006; Wu 2008; Kinoshita et al., 2009; Mortensen et al., 2009). In addition, the chromatin remodelers INO80 and SWR, the histone acetyltransferase NuA4, and the structural maintenance of chromatin proteins are also recruited during this process (Unal et al., 2004; Cortes-Ledesma et al., 2007; Lin et al., 2008; De Piccoli et al., 2009; Bose and Gerton, 2010; Wood et al., 2010).
Of interest, Sir proteins are mobilized from telomeres in response to DNA damage (Martin et al., 1999; McAinsh et al., 1999; Mills et al., 1999), and Sir3 and Sir2 localize to double-strand breaks (Martin et al., 1999; Mills et al., 1999; Tamburini and Tyler 2005). In HR repair, histone acetylation at the site of damage is followed by histone deacetylation (Jazayeri et al., 2004; Morrison et al., 2004; Shroff et al., 2004; Unal et al., 2004; van Attikum et al., 2004; Tamburini and Tyler 2005), and it has been suggested that deacetylation may in part be mediated by Sir2. However, it is unclear how the Sir proteins are recruited to double-strand breaks (DSBs) or which specific repair proteins play a role in this process.
In addition to Sir proteins functioning during HR-mediated DNA repair, several labs have shown that some repair proteins localize to silenced chromatin in the nucleus (d'Adda di Fagagna et al., 2004). The Ku proteins interact with telomeric heterochromatin as well as with the HM loci and affect silencing at these loci (Laroche et al., 1998; Fisher and Zakian, 2005; Patterson and Fox, 2008; Vandre et al., 2008; Bystricky et al., 2009). Histone H2A is constitutively phosphorylated at serine 129 (γ-H2A) in silenced chromatin in S. cerevisiae, Schizosaccharomyces pombe (Szilard et al., 2010; Kitada et al., 2011; Kirkland and Kamakaka, 2013), and Drosophila (Andreyeva et al., 2008), and the presence of this histone mark at silenced domains is dependent on the presence of Sir proteins (Kirkland and Kamakaka, 2013). The MRX complex and Tel1 localize to telomeres in competition with Rap1 and Rif1/2 (Takata et al., 2004; Bianchi and Shore, 2007; Hirano and Sugimoto, 2007; Sabourin et al., 2007; Hirano et al., 2009; Ma and Greider, 2009), and mutations in HR repair proteins leads to loss of clustering of silenced domains; these data collectively suggest direct or indirect links between silenced domains and repair proteins (Miele et al., 2009; Kirkland and Kamakaka, 2013).
To clarify the links between HR repair proteins and silencing, we asked whether DSB repair proteins have the ability to interact with the Sir proteins and affect silencing. Using reporter assays, we show that DSB repair proteins can silence a reporter gene when tethered to a silencer. Using various mutants, we identify the genetic pathway involved in DSB repair protein–mediated silencing. We also show that these repair proteins can interact with the Sir proteins and use two distinct pathways for silencing—one involving direct interactions with the Sir proteins, and the second via tethering of the locus to the nuclear periphery.
RESULTS
Gal4-Mre11–mediated repression at HMR
To investigate the role of DSB repair proteins in gene silencing, we initially sought the consequence of recruiting a specific repair protein to the HMR locus. Two silencers, HMR-E and HMR-I, flank the a1 and a2 genes at HMR. The essential HMR-E silencer contains binding sites for ORC, Rap1, and Abf1 proteins, and the important HMR-I silencer contains binding sites for ORC and Abf1. A strain in which Gal4-binding sites replace the ORC-binding sites at the two silencers is unable to recruit the Sir proteins and is unable to silence the genes present at HMR. Loss of silencing of the a1 gene in an α cell results in an inability of this cell to mate with an a cell and form diploid colonies. Expression and recruitment of Gal4-Sir1 to these modified HMR silencers results in the repression of the a1 gene, which allows the α haploid strain to once again mate with an a strain, forming diploid colonies (Chien et al., 1993). This is due to the fact that the Gal4 DNA–binding domain of Gal4-Sir1 fusion enables this protein to be recruited to the modified silencer even in the absence of the ORC- binding site (Fox et al., 1997). This modified silencer–containing strain can therefore be used to assay silencing after recruitment of DSB repair proteins to the HMR silencers. The Gal4 DNA–binding domain alone is not able to silence (Figure 1B), whereas Gal4-Sir1 is able to robustly silence the a1 gene (Figure 1C), and these serve as negative and positive controls, respectively.
FIGURE 1:

(A) Schematic of the wild-type silenced locus. (B) Schematic of the modified locus used in this study and the mating assay when the Gal4 DNA–binding domain alone is expressed in the cell, resulting in no repression of the a1 gene. (C) Schematic of the modified locus used in this study and the mating assay when Gal4-Sir1 is expressed in the cell, resulting in silencing of the a1 gene.
We first asked whether tethering of the fusion protein Gal4-Mre11 could aid in silencing. Recruitment of Gal4-Mre11 to the silencer was able to silence the reporter gene (Figure 2, WT panels). Comparative serial dilution assays show that Mre11 was not as robust as Gal4-Sir1 in silencing but clearly demonstrate that the repair protein Mre11 has the ability to significantly and reproducibly repress the a1 reporter gene present at HMR.
FIGURE 2:

Gal4-Mre11–mediated silencing. (A) Gal4-Mre11xmediated silencing in wild type and various mutant derivatives of this strain. Tenfold dilutions of MATα strains containing the modified HMR locus were spotted on a YMD-Trp plate (growth control) or a YMD plate containing an a lawn (JRY19a) to assay silencing. (B) Gal4-Mre11– and Gal4-Sir1–mediated silencing in a strain lacking Xrs2.
Mre11-mediated silencing is dependent on Sir proteins
To ascertain whether Mre11-mediated repression was simply localized repression mediated by occlusion/steric hindrance as opposed to gene silencing, we asked whether Gal4-Mre11–mediated gene repression was dependent on the presence of the other Sir proteins. In a sir3Δ background, neither Gal4-Sir1 nor Gal4-Mre11 was able to silence the gene (Figure 2). These results are not specific to Sir3, since these fusion proteins were also not able to silence the reporter gene at HMR in a sir2Δ strain (unpublished data ) or a sir4Δ strain (Figure 2). The loss of silencing in a Sir protein–deficient background demonstrates that Gal4-Mre11–dependent silencing is not due to recombination or resection of the reporter gene either. These results indicate that Mre11-mediated repression of the reporter gene was operating via a bona fide silencing pathway.
Mre11-mediated silencing is partially dependent on Esc2 and histone H2A phosphorylation
To dissect this novel form of gene silencing, we investigated the other factors necessary for Mre11-mediated silencing. In the strain containing the modified HMR locus, we deleted specific genes and asked whether this affected Gal4-Mre11–mediated silencing (Figure 2A). Loss of Mre11, Rad50, and Rad51 had no effect on Gal4-Mre11–mediated silencing. Gal4-Mre11 was also able to silence the a1 gene in the absence of the heterochromatin nuclear tethering proteins Esc1 and Ku70. However, silencing was reduced in the absence of the protein Esc2, which has been implicated in both DNA repair and gene silencing (Dhillon and Kamakaka, 2000; Cuperus and Shore, 2002; Ohya et al., 2008; Mankouri et al., 2009; Miele et al., 2009; Sollier et al., 2009; Choi et al., 2010; Mimura et al., 2010; Yu et al., 2010; Albuquerque et al., 2013).
We next assayed a H2A mutant that could not be phosphorylated by the phosphoinositide 3 (PI3) kinases Mec1 and Tel1 (Downs et al., 2000). Strains that cannot phosphorylate H2A at Ser-129 show a modest decrease in silencing, indicating that γ-H2A was necessary to some degree for Gal4-Mre11–mediated silencing.
The MRX complex is involved in telomere length homeostasis and double-strand break repair. Xrs2 is a member of this complex, interacts with Tel1, and is necessary for the recruitment of Tel1 to double-strand breaks (Ritchie and Petes, 2000; Tsukamoto et al., 2001; Nakada et al., 2003; Shima et al., 2005). Because mutations in H2A affect Mre11-mediated silencing, we asked whether this silencing was dependent on Xrs2. The data show that in the absence of Xrs2, Mre11-mediated silencing was lost, but Sir1-mediated silencing was not significantly affected (Figure 2B).
Gal4-Tel1–mediated silencing
Double-strand breaks in DNA are initially recognized and bound by the MRX complex, as well as by the PI3 kinases. After this, the kinases phosphorylate histone H2A in the vicinity of the break. The PI3 kinase Tel1 functions in the same genetic pathway as Mre11 (Ritchie and Petes, 2000), and it was shown that simply tethering a fragment of ATM was sufficient to phosphorylate H2A, whereas tethering ATR activates the DNA-damage checkpoint in an H2A-dependent manner (Bonilla et al., 2008; Soutoglou and Misteli, 2008). Because Mre11-mediated silencing appeared to be dependent on H2A phosphorylation and Tel1 localizes to telomeres and has kinase-independent functions in telomere maintenance (Ma and Greider 2009), we asked whether Tel1 itself had the ability to recruit Sir proteins and silence the reporter gene at HMR or whether this property was unique to Mre11. We fused full-length wild-type Tel1 to the Gal4 DNA–binding domain and transformed the strain containing the modified HMR locus with this fusion construct. To our surprise, we discovered that Gal4-Tel1 was able to robustly silence the a1 reporter gene (Figure 3). We next asked whether Gal4-Tel1 was able to silence the gene in the absence of Sir proteins. Loss of Sir3 resulted in complete loss of Gal4-Tel1–mediated silencing. This was not a Sir3-specific effect, since we saw similar complete loss of silencing in a sir2Δ strain (Figure 4A), suggesting that Gal4-Tel1–mediated silencing used the entire Sir complex.
FIGURE 3:

Gal4-Tel1–mediated silencing in wild-type and sir3Δ strains. Tethered silencing in the wild-type strain with a C-terminal truncation of Tel1 (Gal4-Tel1 CΔ).
FIGURE 4:

Gal4-Tel1–mediated silencing. (A) Gal4-Tel1–mediated silencing in wild-type and various mutant derivatives of this strain. (B) Gal4-Tel1– and Gal4-Sir1–mediated silencing in a strain lacking Xrs2. (C) Gal4-Tel1–mediated silencing in a strain unable to phosphorylate H2A at serine 129.
The kinase domain of Tel1 resides in the C-terminus of the protein (Greenwell et al., 1995; Khanna et al., 2001). This domain is believed to interact with and phosphorylate the numerous substrates of Tel1 (Smolka et al., 2007; Chen et al., 2010; Gobbini et al., 2013). We truncated the fusion protein such that it lacked this kinase domain. In the absence of this domain, Gal4-Tel1 was unable to silence HMR, suggesting that this domain played a role in silencing (Figure 3). Protein blotting suggests that the truncated protein is expressed to approximately the same extent as wild-type Gal4-Tel1 (Supplemental Figure S1).
Tel1-mediated silencing is dependent on Esc1/Ku, Esc2, and histone H2A phosphorylation
To better understand the factors involved in Gal4-Tel1–mediated silencing, we deleted proteins involved in DNA repair and chromatin remodeling to identify factors that are necessary for Tel1- mediated silencing. Deletions in genes involved in repair were generated in the strain containing the modified HMR locus. Loss of specific repair proteins, such as Rad54, Rdh54, Rad50, Rad51, Xrs2, and so on, had either no effect or very subtle effects on Tel1-mediated silencing (Figure 4). Similarly, we tested mutants in chromatin-remodeling factors, since these proteins affect DSB repair in heterochromatin (Clapier and Cairns, 2009; Sinha et al., 2009; Deem et al., 2012). Loss of various chromatin-remodeling factors also had no effect on Tel1-mediated silencing. However, loss of Esc1, Ku70, and Esc2 led to a significant reduction of Tel1-mediated silencing, suggesting a dependence on these proteins for Tel1-mediated silencing.
Because one of the earliest substrates of Tel1 in DNA damage repair is histone H2A, we asked whether Tel1-mediated silencing was dependent on H2A phosphorylation (Figure 4C). In a mutant strain in which H2A cannot be phosphorylated, there was a partial loss of silencing. While there are likely additional Tel1 phosphorylated substrates involved in silencing and Tel1 may even have kinase-independent functions in silencing (Ma and Greider, 2009), this result indicates that Tel1-mediated silencing is partially dependent on H2A phosphorylation and is consistent with the demonstration that the catalytic domain of Tel1 might also be necessary.
Tethering Tel1 induces H2A phosphorylation at HMR
Both Gal4-Mre11 and Gal4-Tel1 are able to silence HMR in an H2A-dependent manner, suggesting that these proteins mediate this phenotype via phosphorylation of H2A at HMR. We tested this model by directly measuring H2A phosphorylation at S129 using quantitative chromatin immunoprecipitation (ChIP). Yeast cells with the modified silencers were transformed with Gal4-Tel1, Gal4-Mre11, Gal4-Sir1, or Gal4 DNA-binding domain (DBD) alone and grown under selection before cross-linking and immunoprecipitation with antibodies against γ-H2A. Quantitative ChIP was used to determine the levels of this histone modification at HMR (Figure 5). A probe in the ACT1 gene that lacks γ-H2A was used as a negative control. Analysis of the a1 gene at the modified HMR locus shows clear enrichment of γ-H2A in the strain containing Gal4-Tel1, Gal4-Mre11, and Gal4-Sir1 but not in the strain lacking these fusion proteins. Furthermore, a probe immediately outside the HMR-E silencer is also enriched for γ-H2A in these strains. This suggests that the histone modification at HMR is dependent on Tel1 and Mre11 and furthermore that it spreads in both directions from the recruitment site and is not just found between the modified silencers, consistent with the spread of Sir3 and γ-H2A at the native HMR locus (Kirkland and Kamakaka, 2013).
FIGURE 5:
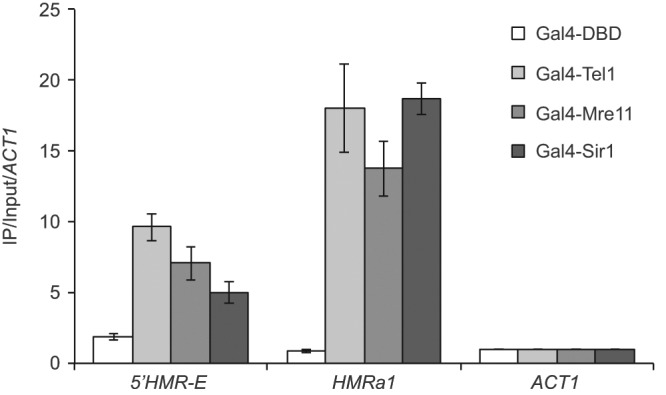
ChIP mapping of γ-H2A at the modified HMR locus. ChIP was performed in a strain with the modified HMR locus expressing either the Gal4 DNA–binding domain alone or Gal4-Sir1, Gal4-Mre11, or Gal4-Tel1. Data are presented as the mean enrichment of IP/input further normalized to an ACT1 amplicon for four IPs from two independent cross-links. Error bars are SD from the mean.
Gal4-Esc2–dependent silencing is independent of Esc1
Both Mre11 and Tel1 required wild-type Esc2 in order to silence robustly the reporter at HMR. Overexpression of Esc2 silences reporter genes in the presence of a mutant Sir1 (Dhillon and Kamakaka, 2000), and Esc2 has been shown to function in DNA repair (Mankouri et al., 2009; Sollier et al., 2009). We therefore wished to know whether Gal4-Esc2 was able to silence genes and whether silencing was dependent on other repair proteins (Figure 6A). Consistent with previous data, Gal4-Esc2 is able to silence the reporter gene at HMR. Whereas Gal4-Esc2–mediated silencing was entirely dependent on Sir2 and Sir3, loss of Mre11, Rad51, Esc1, or Ku70 had no effect on Esc2-mediated silencing.
FIGURE 6:

Gal4-Esc2– and Gal4-Esc1–mediated silencing. (A) Gal4-Esc2–mediated silencing in wild type and various mutant derivatives of this strain. (B) Gal4-Esc2– and Gal4-Sir1–mediated silencing in wild type and a strain lacking Mph1. (C) Gal4-Esc1 mediated silencing in wild type and various mutant derivatives of this strain.
Mph1 and Esc2 interact genetically in repair, and whereas Esc2 plays a role in the resolution of replication-coupled recombination intermediates, Mph1 promotes the formation of these intermediates when replication forks encounter DNA damage (Mankouri et al., 2009; Sollier et al., 2009; Choi et al., 2010). We therefore tested whether Esc2-mediated silencing was altered in the absence of Mph1. Our data show that loss of Mph1 had no effect on Esc2-mediated silencing. These result collectively suggest that Esc2 likely functions downstream of Mre11 and Tel1 in silencing.
Gal4-Esc1–dependent silencing is independent of Esc2
Esc1 interacts with Sir4 and tethers silenced loci to the nuclear periphery (Taddei et al., 2005). Whereas Mre11-mediated silencing was independent of Esc1, Tel1-mediated silencing was partially dependent on Esc1 and Ku70. We therefore asked whether Gal4-Esc1 was sufficient for silencing the reporter gene, consistent with previous results (Andrulis et al., 2002, 2004). Gal4-Esc1 was able to silence HMR to similar levels as Esc2 and Mre11. We investigated Esc1 further and determined that Gal4-Esc1–dependent repression was also independent of Mre11, Rad51, or Esc2 but dependent on the Sir proteins (Figure 6C). These data suggest that there are likely to be two HR repair protein–mediated silencing pathways—one dependent on Esc1/Ku70 and the other on Esc2.
Gal4-Esc2–mediated silencing is stably inherited
The partial silencing at HMR by Mre11, Tel1, and Esc2 could reflect either intermediate levels of expression of the reporter genes in all cells in a colony or a population in which some cells expressed the gene and others did not (Kamakaka, 1997). To distinguish between these two models, we generated a strain with the ADE2 gene under the control of the modified HMR silencers. Yeast colonies lacking ADE2 activity are red, and colonies where the ADE2 gene is active are white. If derepression of HMR::ADE2 were partial in all cells, then the colony color would convert from red to pink. In contrast, if the derepression occurred in only a fraction of the cells and the transcription states were mitotically stable, then there should be a mixture of colonies—some red, some white, and some that exhibit red and white sectors.
In the presence of the Gal4 DNA–binding domain alone, the test strain formed only white colonies, indicating that the ADE2 gene was active. Expression of Gal4-Sir1 repressed the ADE2 gene at HMR, resulting in mostly red colonies with some white colonies (Figure 7). Expression of Gal4-Esc2 and Gal4-Esc1 resulted in colonies that were red with white sectors. These data support a binary mode of silencing by Gal4-Esc2 at HMR and indicated that transcription states of ADE2 at HMR, once established by Gal4-Esc2, were stably inherited for several generations. On the other hand, Gal4-Tel1 generated primarily white/light pink colonies, and Gal4-Mre11 colonies were primarily white, suggesting repression that was unstable and prone to frequent changes.
FIGURE 7:

Silencing of ADE2 at the modified HMR locus by various fusion proteins.
Mre11 and Tel1 interact with Sir proteins
Our data suggest that HR repair proteins have the ability to silence when tethered to the HMR silencer. We next wished to know whether this silencing was due to direct interactions between the HR proteins and the Sir proteins. To test whether HR repair proteins interacted with the Sir proteins, we performed a two-hybrid analysis (Figure 8A). Gal4-Mre11, Gal4-Tel1, and Gal4-Esc2 were assayed for interaction with Sir2, Sir3, Sir4, and Esc2 that were fused to the activation domain of Gal4. The two-hybrid analysis showed that whereas the Gal4 DNA–binding domain alone was not able to interact with any of the proteins fused to the activation domain, Gal4-Sir1 interacted specifically with Sir4 and Gal4-Esc2 interacted with Sir2; these results are consistent with previous data (Chien et al., 1991; Cuperus and Shore, 2002). Surprisingly, Gal4-Mre11 showed a weak interaction with Sir2. Of great interest, Gal4-Tel1 interacted robustly with Sir3 and Sir4 and weakly with Esc2, suggesting that Tel1-mediated silencing may be via direct interactions with the Sir proteins. These interactions appear to be direct, since loss of the other Sir proteins did not dramatically alter the interactions (Figure 8B).
FIGURE 8:

Two-hybrid analysis of interactions between Sir proteins and repair proteins. (A) A wild-type strain was transformed with two different 2μ plasmids to coexpress Gal4-DBD fusions and Gal4-AD fusions. Growth on YMD plates lacking histidine was used to monitor interactions. (B) Two-hybrid analysis in strains lacking Sir proteins, using Gal4-Tel1 as bait.
Peripheral tethering of HMR by DSB-repair proteins
One of the characteristic features of silencing is the clustering of these domains at the nuclear periphery, which aids in the efficiency of silencing and is dependent on Esc1 and Ku proteins (Taddei et al., 2009, 2010). We therefore inquired whether silencing mediated by Gal4-Mre11, Gal4-Tel1, and Gal4-Esc2 resulted in the silenced locus moving to the nuclear periphery and whether this movement was dependent on the Sir proteins. We inserted a LacO array adjacent to the modified HMR locus. This strain contained a LacI–green fluorescent protein (GFP) fusion protein that binds the array, thus marking the HMR locus. Furthermore, we marked the nuclear periphery by expressing HDEL-lac-mCherry fusion protein (Madrid et al., 2006; Ruben et al., 2011). The shortest distance between HMR and the nuclear periphery was measured, as was the diameter of the nucleus through the HMR focus. This enabled us to divide the nucleus into two zones of equal surface area—one internal and one peripheral—and we assigned the HMR locus to one of these zones (Figure 9).
FIGURE 9:

Localization of the modified HMR locus in the yeast nucleus. A schematic of a yeast nucleus with two zones of equal surface area and a fluorescent locus are shown in the line diagram. (A–D) Graphs of the percentage of cells in each of the two zones in various strains. The data for Gal4 DNA–binding domain alone are shown simply for ease of comparison. ***p < 0.001 by χ2 test.
In an otherwise wild-type background, expressing just the Gal4 DNA–binding domain resulted in the modified HMR locus residing in the peripheral zone 53.1% of the time and in the internal zone 46.9% of the time. When Gal4-Sir1 or Gal4-Mre11 was expressed, the HMR locus shifted to the peripheral zone 82.7% and 80.4% of the time, respectively. Both of these shifts were statistically significant (p = 5.4e−7 and 9.6e−8, respectively). In a sir3Δ background, Gal4-Sir1 still localized the modified HMR to the periphery, even though the locus was not silenced (zone 1, 79.7%; p = 3.6e−6), revealing a novel role for Sir1 in tethering HMR to the nuclear periphery independent of Sir3. In the absence of Sir3, Gal4-Mre11 was no longer able to localize the HMR locus to the periphery (zone 1, 64.0%; p = 0.10). Gal4-Tel1 also shifted the locus to the nuclear periphery (78.4%), as did Gal4-Esc2 (74.8%). However, Gal4-Tel1– and Gal4-Esc2–mediated localization to the periphery was not altered in the absence of Sir3, unlike that of Gal4-Mre11.
DISCUSSION
Repair of chromosomal breaks is essential for cell survival, and there are numerous pathways involved in the repair of breaks (Lisby and Rothstein, 2009). There are some reports of linkages between repair pathways and silencing proteins. The Ku protein plays a role in both DSB repair and silencing (Fisher and Zakian, 2005). DSBs in yeast cells also result in partial dissociation of Sir proteins from telomeres, which is dependent on PI3 kinases, although the importance of this dissociation is unclear (Martin et al., 1999; McAinsh et al., 1999; Mills et al., 1999). Although initially the Sir proteins were believed to play a direct role in NHEJ repair (Tsukamoto et al., 1997), later studies suggested an indirect role for these proteins in this pathway of repair via their function in silencing the a1 and α2 genes at HMR and HML (Astrom et al., 1999; Lee et al., 1999). On the other hand, histone acetylation is an early step during HR repair (Jazayeri et al., 2004; Morrison et al., 2004; Shroff et al., 2004; Unal et al., 2004; van Attikum et al., 2004; Lin et al., 2008), and histone deacetylation occurs during the later stages of repair (Tamburini and Tyler, 2005), although the function of the Sir proteins in these processes is not clear. We now show that specific repair proteins have the ability to interact with the Sir proteins. Tel1 interacts strongly with Sir4 and Sir3 and weakly with Esc2, which in turn interacts robustly with Sir2. Furthermore, tethering Tel1 to a defective silencer enables silencing of the gene at that locus, and this silencing is dependent on the Sir proteins, demonstrating that Tel1 has the ability to directly or indirectly recruit these proteins to DNA. Data also show that Sir4 is a substrate of Tel1 enzymatic activity (Chen et al., 2010), suggesting that the interaction between these two proteins might be direct. It therefore appears that the localization of Tel1 to sites of damage could potentially result in the recruitment of Sir proteins to these sites, although additional experiments will be necessary to test this model.
Mre11 is one of the first proteins to be recruited to a double-strand break. We have now shown that Mre11 has the ability to recruit the Sir proteins to a modified silencer. Given the weak interaction between Mre11 and Sir proteins, we favor a model in which Mre11 recruits the Sir proteins indirectly via its interactions with other proteins, most likely Tel1.
Esc2 contains two tandem SUMO-like domains that are necessary for its interactions with Sir proteins (Dhillon and Kamakaka, 2000; Cuperus and Shore, 2002; Yu et al., 2010). Recent results show that Esc2 functions with Mms21 in HR repair by preventing the accumulation of recombination intermediates that are generated by Mph1, Mms2, and the SHU complex (Mankouri et al., 2009; Sollier et al., 2009; Choi et al., 2010; Mimura et al., 2010; Albuquerque et al., 2013). The demonstration that both Tel1- and Mre11-mediated silencing is partially dependent on Esc2 and that Esc2 interacts robustly with Sir2 led us to propose that Esc2 is another molecular link between repair proteins and silencing.
Double-strand breaks that are rapidly repaired using the HR repair pathway do not elicit a robust checkpoint response, and the breaks are usually repaired in the interior of the yeast nucleus. However, breaks that are repaired with slower kinetics or ones that were not repairable are recruited to the nuclear periphery in an Mps3-dependent (Oza et al., 2009) and Nup84-dependent (Therizols et al., 2006; Nagai et al., 2008) manner requiring robust checkpoint signaling. In addition, breaks that occur in silenced heterochromatin also need to be tethered to the nuclear pore to be repaired (Therizols et al., 2006). It has been suggested that tethering to the periphery enables telomere-bound complexes to aid in repair (Bennett et al., 2001). It is therefore possible that release of Sir proteins from telomeres upon DNA damage and their recruitment to break sites via direct and indirect interactions with Tel1, Mre11, and Esc2 help target these loci to the nuclear periphery if the break is not repaired rapidly. Consistent with this model is the observation that targeting Nup84 to the HMR locus results in the targeting of this locus to the periphery and its silencing in a Sir-dependent manner (Ruben et al., 2011), although it is unclear whether this Nup84-mediated silencing is dependent on Esc1 and/or Esc2 or other repair proteins.
Mutants in HR repair proteins lead to a reduction in the clustering of HML and HMR (Kirkland and Kamakaka, 2013). Whereas mutants in Mre11 have no noticeable defects in silencing at native HML or HMR, mutants in Ku70 and Esc2 do have small defects in silencing at these loci, and Ku70 localizes to the native HML and HMR domains in wild-type cells (Dhillon and Kamakaka, 2000; Patterson and Fox, 2008; Vandre et al., 2008). Whether Ku70 binds directly to the silencers or localizes to the silenced domain indirectly via interactions with the Sir proteins is not known. We favor the idea, however, that the primary pathway for the recruitment of Sir2, Sir3, and Sir4 to the silenced HML and HMR domains is through silencer-bound Sir1. Although it is possible that DSB repair proteins bind silencers and help recruit Sir proteins, we favor the alternative possibility that the presence of chromatin-bound Sir proteins at these loci recruits the repair proteins (such as Ku and Tel1) to these domains, and the loss of these repair proteins results in effects such as disruption of HML-HMR long-range clustering in the nucleus with only subtle effects on silencing.
The role of γ-H2A in gene silencing is intriguing and puzzling. The location of the residue in histone H2A, close to the C-terminus, places this site close to the entry and exit sites of DNA in the nucleosome. Although it was originally proposed that phosphorylation of H2A might facilitate chromatin decondensation (Downs et al., 2000), subsequent studies showed that this was not the case (Fink et al., 2007). Proteins containing FHA and BRCT domains, such as INO80, SWR1, and the PI3 kinases, recognize phosphorylated H2A (Kinner et al., 2008). Among the yeast proteins that possess BRCT domains is Rap1, and although Rap1 is a sequence-specific factor, it also binds silenced chromatin and spreads along the domain in a sequence-independent manner, although the mechanism by which this occurs is unknown (Lieb et al., 2001; Valenzuela et al., 2008; Ozaydin and Rine, 2010; Zhang et al., 2011). One possible scenario could involve interactions between γ-H2A present in silenced chromatin and Rap1, which could then further aid in silencing. Phosphorylation of H2A is also necessary for the binding of cohesins to silenced chromatin and the clustering of HML with HMR (Kirkland and Kamakaka, 2013). Thus γ-H2A could alternatively affect silencing via cohesin-mediated clustering of heterochromatic domains. Obviously these are not mutually exclusive scenarios, and other proteins could also interact with γ-H2A and affect silencing. It is intriguing, however, that γ-H2A localizes to silenced chromatin in the distantly related yeast S. cerevisiae and S. pombe as well as in Drosophila (Andreyeva et al., 2008; Rozenzhak et al., 2010; Szilard et al., 2010; Kitada et al., 2011; Kirkland and Kamakaka, 2013), and there are suggestions of a similar distribution of this modification in vertebrates (Fernandez-Capetillo et al., 2003; Ichijima et al., 2005; Shechter et al., 2009).
In conclusion, our data collectively suggest that different DSB repair proteins can interact with the Sir proteins, and these repair proteins, when recruited to sites in the genome, can induce the recruitment of Sir proteins and concomitant silencing at these sites via distinct mechanisms. Mre11 most likely recruits Sir proteins indirectly, and this recruitment of Sir proteins at specific sites in the genome then targets the bound locus to the nuclear periphery. This explains the observation that Mre11-mediated silencing is not altered in the absence of Esc1 and Ku, but is consistent with the demonstration that movement of the Gal4-Mre11–silenced locus to the periphery is dependent on the Sir proteins. On the other hand, Tel1 silences via two distinct mechanisms. It directly interacts with the Sir proteins, as shown by the two-hybrid analysis and the fact that Sir4 is a substrate of this kinase. It also affects silencing via the phosphorylation of H2A. This is evidenced by the observation that cells lacking this histone modification are not able to robustly silence. Finally, Tel1 has the ability to target the locus to the nuclear periphery independent of the Sir proteins. This targeting to a compartment rich in Sir proteins would further favor the formation of silenced chromatin.
It should also be borne in mind that one function of heterochromatin is to reduce recombination of repetitive DNA. The function of Esc2 is the suppression of chromosomal recombination, and it is entirely possible that this function is mediated in part via its ability to bind/recruit Sir2 and the subsequent targeting of Sir protein–bound loci to the nuclear periphery, resulting in the suppression of recombination, as well as allowing the healing of persistent breaks via alternative repair machineries or de novo telomere formation.
MATERIALS AND METHODS
Yeast strains and plasmids
All yeast strains used in this study are in the W303 background except the strains used for the two-hybrid analysis (see Tables 1 and 2). Most gene deletions were performed by transformation and subsequent replacement of the open reading frame with a KanMx cassette. KanMx cassettes were amplified using PCR with primers flanking the deletion using genomic DNA from the deletion collection as template. Strains containing gene deletions marked by auxotrophic markers were built by crossing the test strain (TM47/JKY383) to previously published strains.
TABLE 1:
Plasmids used in this study.
| Plasmid | Description | Source |
|---|---|---|
| pRO1000 | Gal4 (1-147) 2μ URA3 | |
| pRO990 | Gal4-Tel1 CΔ 2μ URA3 | |
| pRO1022 | Gal4-AD-SIR4 2μ LEU2 | |
| GLC370 | Gal4-AD-SIR2 2μ LEU2 | D. Shore (University of Geneva) |
| PM875 | Gal4-AD-SIR3 2μ LEU2 | D. Shore |
| pRO998 | Gal4-AD-ESC2 2μ LEU2 | |
| pAct2.2 | Gal4-AD 2μ LEU2 | |
| pRO83 | Gal4-Sir1 2μ URA3 | |
| pRO963 | Gal4-Tel1 2μ URA3 | |
| pJR1112 | 2μ URA3 | |
| pRO1001 | Gal4-Mre11 2μ TRP1 | |
| pRO1003 | Gal4-Esc1 2μ TRP1 | |
| pRO1005 | Gal4-Sir1 2μ TRP1 | |
| pRO1044 | Gal4-Esc2 2μ TRP1 |
TABLE 2:
Strains used in this study.
| Strain | Description |
|---|---|
| ROY 5008 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ swr1Δ::NatMx |
| ROY 5010 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+rad16Δ::KanMx |
| ROY 5012 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ rad5Δ::KanMx |
| ROY 5014 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ rad54Δ::KanMx |
| ROY 5016 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+rsc2Δ::KanMx |
| ROY 5018 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ chd1Δ::KanMx |
| ROY 5022 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ rdh54Δ::KanMx |
| ROY 5026 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ snf5Δ::KanMx |
| ROY 5028 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ isw1Δ::KanMx |
| ROY 5030 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ ies5Δ::KanMx |
| ROY 5020 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ fun30Δ::KanMx |
| ROY 5024 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ isw2Δ::KanMx |
| ROY 5034 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ sir2Δ::KanMx |
| ROY 5036 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ scs2Δ::KanMx |
| ROY 5348 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ xrs2Δ::LEU2 |
| ROY 5349 | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ xrs2Δ::LEU2 |
| ROY 5351* | MATα HMR::5xGEB-a1-B5xG his3 ura3 leu2 trp1 ADE+ LYS+ mph1Δ::KanMx |
| ROY 5378 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 esc2Δ::KanMx |
| ROY 5379 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 esc2Δ::KanMx |
| ROY 5380 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 mre11Δ::KanMx |
| ROY 5381 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 mre11Δ::KanMx |
| ROY 5382 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 rad51Δ::KanMx |
| ROY 5383 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 hta1S129* hta2S129* |
| ROY 5384 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 hta1S129* hta2S129* |
| ROY 5385 | MATα HMR::5xGEB-a1-B5xG LYS ade2-1 his3 trp1 leu2 ura3 sir4Δ::KanMx |
| ROY 5386 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 rad50Δ::KanMx |
| ROY 5387 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 ku70Δ::HYG |
| ROY 5388 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 esc1::KanMx ku70Δ::HYG |
| ROY 5389 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 ku70Δ::HYG |
| ROY 5390 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 esc1Δ::KanMx |
| ROY 5391 | MATα HMR::5xGEB-a1-B5xG LYS2 ade2-1 his3 trp1 leu2 ura3 esc1Δ::KanMx ku70Δ::HYG |
| ROY 5392 | MATα HMR::5xGEB-a1-B5xG ADE+ LYS+ his3 trp1 leu2 ura3 |
| ROY 5393 | MATα HMR::5xGEB-a1-B5xG ADE+ lys2 his3 trp1 leu2 ura3 sir1Δ::HIS3 |
| ROY 5394 | MATα HMR::5xGEB-a1-B5xG ADE+ LYS+ his3 trp1 leu2 ura3 sir3Δ::LEU2 |
| ROY 5146* | MATa trp1-901 leu2-3112 ura3-52 gal4Δ gal80Δ GAL2p-ADE2 GAL1p-HIS3 metΔ::GAL1p-LacZ |
| ROY 5324* | MATa trp1-901 leu2-3112 ura3-52 gal4Δ gal80Δ GAL2p-ADE2 GAL1p-HIS3 metΔ::GAL1p-LacZ sir2Δ::KanMx |
| ROY 5327* | MATa trp1-901 leu2-3112 ura3-52 gal4Δ gal80Δ GAL2p-ADE2 GAL1p-HIS3 metΔ::GAL1p-LacZ sir3Δ::TRP1 |
| ROY 5332* | MATa trp1-901 leu2-3112 ura3-52 gal4Δ gal80Δ GAL2p-ADE2 GAL1p-HIS3 metΔ::GAL1p-LacZ sir4Δ::KanMx |
| ROY 5395 | MATα HMR::5xGEB-a1-B5xG LacO(64x)GIT1::LEU2 LacI-GFP::ADE2 LYS2 YIP-Lac-HDELdsRED::NatMX |
| ROY 5396 | MATα HMR::5xGEB-a1-B5xG Laco(64x)GIT1::LEU2 LacI-GFP::ADE2 LYS2 YIP-Lac-HDELdsRED::NatMX sir3Δ::HIS3 |
| ROY 5397 | MATα HMR::5xGEB-ADE2p-ADE2-B5xG ade2-1 LYS2 his3 ura3 trp1 leu2 |
| JRY19a | MATa can1 his4-519 leu2-3112 trp1 ura3-52 |
| JRY19@ | MATα can1 his4-519 leu2-3112 trp1 ura3-52 |
*Not isogenic with W303.
Serial dilution mating assays
Strains were transformed with 2μ plasmids containing Gal4 DBD (1–147) or GBD fused in- frame to a protein of interest and selected on yeast minimal dextrose (YMD) –uracil (YMD-U) or –tryptophan (YMD-T), depending on the selection marker of the plasmids. After 2 d, multiple colonies were picked and grown overnight at 30°C (unless otherwise noted) in YMD-U or YMD-T liquid medium. Mating lawn strains were grown overnight in YPD liquid medium at 30°C. They were taken at OD600. For spot assays, 1 OD of mating lawn was plated on YMD plates. Tester strains were diluted to 2 OD/ml and further serially diluted 1:10 (2, 0.2, 0.02, and 0.002 OD/ml). Tester strains were spotted onto YMD mating lawns and YMD-T or YMD-U plates for growth and plasmid retention controls.
Data presentation
Experiments were always performed with a WT strain (TM47) and GBD-only plasmid (negative control) and GBD-Sir1 plasmid (positive control) simultaneously. All experiments were performed a minimum of three independent times by two independent researchers. In some figures the order of strains was changed using image-editing software or representative images are a combination of multiple plates or experiments. In these cases, the figure is given with white line(s) to clearly show this fact.
ADE2 silencing assay
Strains were built in which the ADE2 gene (under the a1 promoter) replaces the a1 coding region. These strains were transformed as described and plated on selection plates containing–uracil or –tryptophan. Colonies were allowed to grow at 30°C for 48 h and then transferred to 4°C for color development.
Microscopy
Microscopy was performed on live cells for all experiments. Cells were grown exponentially in YMD plus amino acids (AA; Leu, Ura, Trp, Lys, Ade, His) to an OD600 of ≤0.6. Cells were rinsed in YMD + AA before imaging and placed on YMD + AA, 1.5% agarose patches on slides, covered, and imaged. Images were acquired on an Olympus xi70 inverted wide-field microscope with DeltaVision precise stage (Applied Precision) using a CoolSNAP HQ2 camera (Photometrics). Optical image stacks of 20 images were acquired with a step size of 200 nm for 400–500 ms in the appropriate wavelength channel. A 100×/1.4 oil objective was used. Acquisition software softWoRx3.7.1 was used for image acquisition and analysis. All images were taken at 25°C. Cropping of images was performed in Photoshop (Adobe).
For zone analysis, 200-nm optical slices were taken on live cells, and only the 10 middle planes of the nucleus were assayed. Images were acquired in the GFP and mCherry channels. The position of the GFP focus in relation to the HDEL-dsRed marked nuclear envelope was determined by first identifying the plane bearing the brightest GFP-LacI focus and then determining the position of the GFP foci in one of two concentric nuclear zones of equal surface area. Three independent trials were performed for each strain, and strains were scored in a blind manner by measuring the distance between the GFP spot (array) and the nuclear membrane (s2p) and the diameter of the nucleus (p2p) in nanometers. A ratio (s2p/p2p) × 2 was calculated and used to designate two zones—the peripheral zone (zone 1) or the interior zone (zone 2)—of approximately equal surface area (B) or volume (D) as previously described. The p values were determined by χ2 test (Ruben et al., 2011).
Acknowledgments
This work was supported by grants from the National Institutes of Health to R.T.K. (GM078068) and J.K. (T32-GM008646). C.S. was supported by a National Institutes of Health T34 Minority Access to Research Careers Fellowship and a Stephen Palais Award; L.B. was supported by a University of California, Santa Cruz, Summer Undergraduate Research Award.
Abbreviations used:
- HR
homologous recombination
- NHEJ
nonhomologous end joining
- ORC
origin recognition complex.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-09-1413) on January 28, 2015.
REFERENCES
- Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, Zhou H. Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication-mediated genome rearrangements. PLoS Genet. 2013;9:e1003670. doi: 10.1371/journal.pgen.1003670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyeva EN, Kolesnikova TD, Belyaeva ES, Glaser RL, Zhimulev IF. Local DNA underreplication correlates with accumulation of phosphorylated H2Av in the Drosophila melanogaster polytene chromosomes. Chromosome Res. 2008;16:851–862. doi: 10.1007/s10577-008-1244-4. [DOI] [PubMed] [Google Scholar]
- Andrulis ED, Zappulla DC, Alexieva-Botcheva K, Evangelista C, Sternglanz R. One-hybrid screens at the Saccharomyces cerevisiae HMR locus identify novel transcriptional silencing factors. Genetics. 2004;166:631–635. doi: 10.1534/genetics.166.1.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrulis ED, Zappulla DC, Ansari A, Perrod S, Laiosa CV, Gartenberg MR, Sternglanz R. Esc1, a nuclear periphery protein required for Sir4-based plasmid anchoring and partitioning. Mol Cell Biol. 2002;22:8292–8301. doi: 10.1128/MCB.22.23.8292-8301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrom SU, Okamura SM, Rine J. Yeast cell-type regulation of DNA repair. Nature. 1999;397:310. doi: 10.1038/16833. [DOI] [PubMed] [Google Scholar]
- Ataian Y, Krebs JE. Five repair pathways in one context: chromatin modification during DNA repair. Biochem Cell Biol. 2006;84:490–504. doi: 10.1139/o06-075. [DOI] [PubMed] [Google Scholar]
- Bennett CB, Snipe JR, Westmoreland JW, Resnick MA. SIR functions are required for the toleration of an unrepaired double-strand break in a dispensable yeast chromosome. Mol Cell Biol. 2001;21:5359–5373. doi: 10.1128/MCB.21.16.5359-5373.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, Shore D. Increased association of telomerase with short telomeres in yeast. Genes Dev. 2007;21:1726–1730. doi: 10.1101/gad.438907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla CY, Melo JA, Toczyski DP. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell. 2008;30:267–276. doi: 10.1016/j.molcel.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose T, Gerton JL. Cohesinopathies, gene expression, and chromatin organization. J Cell Biol. 2010;189:201–210. doi: 10.1083/jcb.200912129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystricky K, Van Attikum H, Montiel MD, Dion V, Gehlen L, Gasser SM. Regulation of nuclear positioning and dynamics of the silent mating type loci by the yeast Ku70/Ku80 complex. Mol Cell Biol. 2009;29:835–848. doi: 10.1128/MCB.01009-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Albuquerque CP, Liang J, Suhandynata RT, Zhou H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J Biol Chem. 2010;285:12803–12812. doi: 10.1074/jbc.M110.106989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CT, Bartel PL, Sternglanz R, Fields S. The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc Natl Acad Sci USA. 1991;88:9578–9582. doi: 10.1073/pnas.88.21.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CT, Buck S, Sternglanz R, Shore D. Targeting of SIR1 protein establishes transcriptional silencing at HM loci and telomeres in yeast. Cell. 1993;75:531–541. doi: 10.1016/0092-8674(93)90387-6. [DOI] [PubMed] [Google Scholar]
- Choi K, Szakal B, Chen YH, Branzei D, Zhao X. The Smc5/6 complex and Esc2 influence multiple replication-associated recombination processes in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2306–2314. doi: 10.1091/mbc.E10-01-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Cortes-Ledesma F, de Piccoli G, Haber JE, Aragon L, Aguilera A. SMC proteins, new players in the maintenance of genomic stability. Cell Cycle. 2007;6:914–918. doi: 10.4161/cc.6.8.4107. [DOI] [PubMed] [Google Scholar]
- Cuperus G, Shore D. Restoration of silencing in Saccharomyces cerevisiae by tethering of a novel Sir2-interacting protein, Esc8. Genetics. 2002;162:633–645. doi: 10.1093/genetics/162.2.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Teo SH, Jackson SP. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004;18:1781–1799. doi: 10.1101/gad.1214504. [DOI] [PubMed] [Google Scholar]
- Deem AK, Li X, Tyler JK. Epigenetic regulation of genomic integrity. Chromosoma. 2012;121:131–151. doi: 10.1007/s00412-011-0358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Piccoli G, Torres-Rosell J, Aragon L. The unnamed complex: what do we know about Smc5-Smc6. Chromosome Res. 2009;17:251–263. doi: 10.1007/s10577-008-9016-8. [DOI] [PubMed] [Google Scholar]
- Dhillon N, Kamakaka RT. A histone variant, Htz1p, and a Sir1p-like protein, Esc2p, mediate silencing at HMR. Mol Cell. 2000;6:769–780. doi: 10.1016/s1097-2765(00)00076-9. [DOI] [PubMed] [Google Scholar]
- Dhillon N, Raab J, Guzzo J, Szyjka SJ, Gangadharan S, Aparicio OM, Andrews B, Kamakaka RT. DNA polymerase epsilon, acetylases and remodellers cooperate to form a specialized chromatin structure at a tRNA insulator. EMBO J. 2009;28:2583–2600. doi: 10.1038/emboj.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donze D, Adams CR, Rine J, Kamakaka RT. The boundaries of the silenced HMR domain in Saccharomyces cerevisiae. Genes Dev. 1999;13:698–708. doi: 10.1101/gad.13.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- Fabre E, Spichal M. Subnuclear architecture of telomeres and subtelomeres in yeast. In: In: Louis EJ, Becker MM, editors. Subtelomeres. Berlin: Springer-Verlag; pp. 13–37. [Google Scholar]
- Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Fink M, Imholz D, Thoma F. Contribution of the serine 129 of histone H2A to chromatin structure. Mol Cell Biol. 2007;27:3589–3600. doi: 10.1128/MCB.02077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher TS, Zakian VA. Ku: a multifunctional protein involved in telomere maintenance. DNA Repair (Amst) 2005;4:1215–1226. doi: 10.1016/j.dnarep.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Flott S, Alabert C, Toh GW, Toth R, Sugawara N, Campbell DG, Haber JE, Pasero P, Rouse J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol Cell Biol. 2007;27:6433–6445. doi: 10.1128/MCB.00135-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CA, Ehrenhofer-Murray AE, Loo S, Rine J. The origin recognition complex, SIR1, and the S phase requirement for silencing. Science. 1997;276:1547–1551. doi: 10.1126/science.276.5318.1547. [DOI] [PubMed] [Google Scholar]
- Ghidelli S, Donze D, Dhillon N, Kamakaka RT. Sir2p exists in two nucleosome-binding complexes with distinct deacetylase activities. EMBO J. 2001;20:4522–4535. doi: 10.1093/emboj/20.16.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbini E, Cesena D, Galbiati A, Lockhart A, Longhese MP. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair (Amst) 2013;12:791–799. doi: 10.1016/j.dnarep.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;8:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- Hediger F, Neumann FR, Van Houwe G, Dubrana K, Gasser SM. Live imaging of telomeres. yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr Biol. 2002;12:2076–2089. doi: 10.1016/s0960-9822(02)01338-6. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Li X, Rolfsmeier M, Zhang XP. Rad54: the Swiss Army knife of homologous recombination. Nucleic Acids Res. 2006;34:4115–4125. doi: 10.1093/nar/gkl481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Fukunaga K, Sugimoto K. Rif1 and rif2 inhibit localization of tel1 to DNA ends. Mol Cell. 2009;33:312–322. doi: 10.1016/j.molcel.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Sugimoto K. Cdc13 telomere capping decreases Mec1 association but does not affect Tel1 association with DNA ends. Mol Biol Cell. 2007;18:2026–2036. doi: 10.1091/mbc.E06-12-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijima Y, Sakasai R, Okita N, Asahina K, Mizutani S, Teraoka H. Phosphorylation of histone H2AX at M phase in human cells without DNA damage response. Biochem Biophys Res Commun. 2005;336:807–812. doi: 10.1016/j.bbrc.2005.08.164. [DOI] [PubMed] [Google Scholar]
- Iyama T, Wilson DM., 3rd DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12:620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, McAinsh AD, Jackson SP. Saccharomyces cerevisiae Sin3p facilitates DNA double-strand break repair. Proc Natl Acad Sci USA. 2004;101:1644–1649. doi: 10.1073/pnas.0304797101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Trelles-Sticken E, Scherthan H, Loidl J. Yeast nuclei display prominent centromere clustering that is reduced in nondividing cells and in meiotic prophase. J Cell Biol. 1998;141:21–29. doi: 10.1083/jcb.141.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A, Li G, Sikorski TW, Buratowski S, Woodcock CL, Moazed D. Reconstitution of heterochromatin-dependent transcriptional gene silencing. Mol Cell. 2009;35:769–781. doi: 10.1016/j.molcel.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakaka RT. Silencers and locus control regions: opposite sides of the same coin. Trends Biochem Sci. 1997;22:124–128. doi: 10.1016/s0968-0004(96)10074-8. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497–501. doi: 10.1038/nature04384. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Lavin MF, Jackson SP, Mulhern TD. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001;8:1052–1065. doi: 10.1038/sj.cdd.4400874. [DOI] [PubMed] [Google Scholar]
- Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, van der Linden E, Sanchez H, Wyman C. RAD50, an SMC family member with multiple roles in DNA break repair: how does ATP affect function. Chromosome Res. 2009;17:277–288. doi: 10.1007/s10577-008-9018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JG, Kamakaka RT. Long-range heterochromatin association is mediated by silencing and double-strand DNA break repair proteins. J Cell Biol. 2013;201:809–826. doi: 10.1083/jcb.201211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Schleker T, Sperling AS, Xie W, Gasser SM, Grunstein M. gammaH2A is a component of yeast heterochromatin required for telomere elongation. Cell Cycle. 2011;10:293–300. doi: 10.4161/cc.10.2.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroche T, Martin SG, Gotta M, Gorham HC, Pryde FE, Louis EJ, Gasser SM. Mutation of yeast Ku genes disrupts the subnuclear organization of telomeres. Curr Biol. 1998;8:653–656. doi: 10.1016/s0960-9822(98)70252-0. [DOI] [PubMed] [Google Scholar]
- Lee SE, Paques F, Sylvan J, Haber JE. Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr Biol. 1999;9:767–770. doi: 10.1016/s0960-9822(99)80339-x. [DOI] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Lin YY, Qi Y, Lu JY, Pan X, Yuan DS, Zhao Y, Bader JS, Boeke JD. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev. 2008;22:2062–2074. doi: 10.1101/gad.1679508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R. Choreography of recombination proteins during the DNA damage response. DNA Repair (Amst) 2009;8:1068–1076. doi: 10.1016/j.dnarep.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Vega-Palas MA, Grunstein M. Rap1-Sir4 binding independent of other Sir, yKu, or histone interactions initiates the assembly of telomeric heterochromatin in yeast. Genes Dev. 2002;16:1528–1539. doi: 10.1101/gad.988802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Greider CW. Kinase-independent functions of TEL1 in telomere maintenance. Mol Cell Biol. 2009;29:5193–5202. doi: 10.1128/MCB.01896-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid AS, Mancuso J, Cande WZ, Weis K. The role of the integral membrane nucleoporins Ndc1p and Pom152p in nuclear pore complex assembly and function. J Cell Biol. 2006;173:361–371. doi: 10.1083/jcb.200506199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Ngo HP, Hickson ID. Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in Saccharomyces cerevisiae. Mol Biol Cell. 2009;20:1683–1694. doi: 10.1091/mbc.E08-08-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- Martino F, Kueng S, Robinson P, Tsai-Pflugfelder M, van Leeuwen F, Ziegler M, Cubizolles F, Cockell MM, Rhodes D, Gasser SM. Reconstitution of yeast silent chromatin: multiple contact sites and O-AADPR binding load SIR complexes onto nucleosomes in vitro. Mol Cell. 2009;33:323–334. doi: 10.1016/j.molcel.2009.01.009. [DOI] [PubMed] [Google Scholar]
- McAinsh AD, Scott-Drew S, Murray JA, Jackson SP. DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Curr Biol. 1999;9:963–966. doi: 10.1016/s0960-9822(99)80424-2. [DOI] [PubMed] [Google Scholar]
- Miele A, Bystricky K, Dekker J. Yeast silent mating type loci form heterochromatic clusters through silencer protein-dependent long-range interactions. PLoS Genet. 2009;5:e1000478. doi: 10.1371/journal.pgen.1000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- Mimura S, Yamaguchi T, Ishii S, Noro E, Katsura T, Obuse C, Kamura T. Cul8/Rtt101 forms a variety of protein complexes that regulate DNA damage response and transcriptional silencing. J Biol Chem. 2010;285:9858–9867. doi: 10.1074/jbc.M109.082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Mortensen UH, Lisby M, Rothstein R. Rad52. Curr Biol. 2009;19:R676–677. doi: 10.1016/j.cub.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science. 2008;322:597–602. doi: 10.1126/science.1162790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya T, Arai H, Kubota Y, Shinagawa H, Hishida T. A SUMO-like domain protein, Esc2, is required for genome integrity and sister chromatid cohesion in Saccharomyces cerevisiae. Genetics. 2008;180:41–50. doi: 10.1534/genetics.107.086249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki M, Kamakaka RT. Barrier Function at HMR. Mol Cell. 2005;19:707–716. doi: 10.1016/j.molcel.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Oppikofer M, Kueng S, Gasser SM. SIR-nucleosome interactions: structure-function relationships in yeast silent chromatin. Gene. 2013;527:10–25. doi: 10.1016/j.gene.2013.05.088. [DOI] [PubMed] [Google Scholar]
- Oppikofer M, Kueng S, Martino F, Soeroes S, Hancock SM, Chin JW, Fischle W, Gasser SM. A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. EMBO J. 2011;30:2610–2621. doi: 10.1038/emboj.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev. 2009;23:912–927. doi: 10.1101/gad.1782209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaydin B, Rine J. Expanded roles of the origin recognition complex in the architecture and function of silenced chromatin in Saccharomyces cerevisiae. Mol Cell Biol. 2010;30:626–639. doi: 10.1128/MCB.00614-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson EE, Fox CA. The Ku complex in silencing the cryptic mating-type loci of Saccharomyces cerevisiae. Genetics. 2008;180:771–783. doi: 10.1534/genetics.108.091710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie KB, Petes TD. The Mre11p/Rad50p/Xrs2p complex and the Tel1p function in a single pathway for telomere maintenance in yeast. Genetics. 2000;155:475–479. doi: 10.1093/genetics/155.1.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenzhak S, Mejia-Ramirez E, Williams JS, Schaffer L, Hammond JA, Head SR, Russell P. Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 2010;6:e1001032. doi: 10.1371/journal.pgen.1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben GJ, Kirkland JG, Macdonough T, Chen M, Dubey RN, Gartenberg MR, Kamakaka RT. Nucleoporin mediated nuclear positioning and silencing of HMR. PLoS One. 2011;6:e21923. doi: 10.1371/journal.pone.0021923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J. Ordered nucleation and spreading of silenced chromatin in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2207–2222. doi: 10.1091/mbc.E02-03-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- Sabourin M, Tuzon CT, Zakian VA. Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell. 2007;27:550–561. doi: 10.1016/j.molcel.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Chitta RK, Xiao A, Shabanowitz J, Hunt DF, Allis CD. A distinct H2A.X isoform is enriched in Xenopus laevis eggs and early embryos and is phosphorylated in the absence of a checkpoint. Proc Natl Acad Sci USA. 2009;106:749–754. doi: 10.1073/pnas.0812207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima H, Suzuki M, Shinohara M. Isolation and characterization of novel xrs2 mutations in Saccharomyces cerevisiae. Genetics. 2005;170:71–85. doi: 10.1534/genetics.104.037580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M, Watanabe S, Johnson A, Moazed D, Peterson CL. Recombinational repair within heterochromatin requires ATP-dependent chromatin remodeling. Cell. 2009;138:1109–1121. doi: 10.1016/j.cell.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA. 2007;104:10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Driscoll R, Castellucci F, Foiani M, Jackson SP, Branzei D. The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell. 2009;20:1671–1682. doi: 10.1091/mbc.E08-08-0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E, Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008;320:1507–1510. doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, et al. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat Struct Mol Biol. 2010;17:299–305. doi: 10.1038/nsmb.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Gartenberg MR, Neumann FR, Hediger F, Gasser SM. Multiple pathways tether telomeres and silent chromatin at the nuclear periphery: functional implications for sir-mediated repression. Novartis Found Symp. 2005;264:140–156. ; discussion, 156–165, 227–130. [PubMed] [Google Scholar]
- Taddei A, Gasser SM. Multiple pathways for telomere tethering: functional implications of subnuclear position for heterochromatin formation. Biochim Biophys Acta. 2004;1677:120–128. doi: 10.1016/j.bbaexp.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Taddei A, Schober H, Gasser SM. The budding yeast nucleus. Cold Spring Harb Perspect Biol. 2010;2:a000612. doi: 10.1101/cshperspect.a000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Van Houwe G, Nagai S, Erb I, van Nimwegen EJ, Gasser SM. The functional importance of telomere clustering: Global changes in gene expression result from SIR factor dispersion. Genome Res. 2009;19:611–625. doi: 10.1101/gr.083881.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata H, Kanoh Y, Gunge N, Shirahige K, Matsuura A. Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol Cell. 2004;14:515–522. doi: 10.1016/s1097-2765(04)00262-x. [DOI] [PubMed] [Google Scholar]
- Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol Cell Biol. 2005;25:4903–4913. doi: 10.1128/MCB.25.12.4903-4913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therizols P, Duong T, Dujon B, Zimmer C, Fabre E. Chromosome arm length and nuclear constraints determine the dynamic relationship of yeast subtelomeres. Proc Natl Acad Sci USA. 2010;107:2025–2030. doi: 10.1073/pnas.0914187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therizols P, Fairhead C, Cabal GG, Genovesio A, Olivo-Marin JC, Dujon B, Fabre E. Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J Cell Biol. 2006;172:189–199. doi: 10.1083/jcb.200505159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto Y, Kato J, Ikeda H. Silencing factors participate in DNA repair and recombination in Saccharomyces cerevisiae. Nature. 1997;388:900–903. doi: 10.1038/42288. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Taggart AK, Zakian VA. The role of the Mre11-Rad50-Xrs2 complex in telomerase- mediated lengthening of Saccharomyces cerevisiae telomeres. Curr Biol. 2001;11:1328–1335. doi: 10.1016/s0960-9822(01)00372-4. [DOI] [PubMed] [Google Scholar]
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Valenzuela L, Dhillon N, Dubey RN, Gartenberg MR, Kamakaka RT. Long-range communication between the silencers of HMR. Mol Cell Biol. 2008;28:1924–1935. doi: 10.1128/MCB.01647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Vandre CL, Kamakaka RT, Rivier DH. The DNA end-binding protein Ku regulates silencing at the internal HML and HMR loci in Saccharomyces cerevisiae. Genetics. 2008;180:1407–1418. doi: 10.1534/genetics.108.094490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Severson AF, Meyer BJ. Condensin and cohesin complexity: the expanding repertoire of functions. Nat Rev Genet. 2010;11:391–404. doi: 10.1038/nrg2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L. Wrestling off RAD51: a novel role for RecQ helicases. Bioessays. 2008;30:291–295. doi: 10.1002/bies.20735. [DOI] [PubMed] [Google Scholar]
- Yu Q, Kuzmiak H, Olsen L, Kulkarni A, Fink E, Zou Y, Bi X. Saccharomyces cerevisiae Esc2p interacts with Sir2p through a small ubiquitin-like modifier (SUMO)-binding motif and regulates transcriptionally silent chromatin in a locus-dependent manner. J Biol Chem. 2010;285:7525–7536. doi: 10.1074/jbc.M109.016360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang J, Zhang X, Xu C, Tu X. Solution structure of Rap1 BRCT domain from Saccharomyces cerevisiae reveals a novel fold. Biochem Biophys Res Commun. 2011;404:1055–1059. doi: 10.1016/j.bbrc.2010.12.109. [DOI] [PubMed] [Google Scholar]