

Abstract
Background
Despite a significant global burden of disease, there is still no vaccine against shigellosis widely available. One aim of the European Union funded STOPENTERICS consortium is to develop vaccine candidates against Shigella. Given the importance of translational vaccine coverage, here we aimed to characterise the Shigella strains being used by the consortium by whole genome sequencing, and report on the stability of strains cultured in different laboratories or through serial passage.
Methods
We sequenced, de novo assembled and annotated 20 Shigella strains being used by the consortium. These comprised 16 different isolates belonging to 7 serotypes, and 4 derivative strains. Derivative strains from common isolates were manipulated in different laboratories or had undergone multiple passages in the same laboratory. Strains were mapped against reference genomes to detect SNP variation and phylogenetic analysis was performed.
Results
The genomes assembled into similar total lengths (range 4.14–4.83 Mbp) and had similar numbers of predicted coding sequences (average of 4,400). Mapping analysis showed the genetic stability of strains through serial passages and culturing in different laboratories, as well as varying levels of similarity to published reference genomes. Phylogenetic analysis revealed the presence of three main clades among the strains and published references, one containing the Shigella flexneri serotype 6 strains, a second containing the remaining S. flexneri serotypes and a third comprised of Shigella sonnei strains.
Conclusions
This work increases the number of the publically available Shigella genomes available and specifically provides information on strains being used for vaccine development by STOPENTERICS. It also provides information on the variability among strains maintained in different laboratories and through serial passage. This work will guide the selection of strains for further vaccine development.
Keywords: Shigella, STOPENTERICS, Genome, Vaccine
Background
Shigella are Gram-negative bacteria that represent the etiologic agent of the shigellosis, a global human health problem, especially in developing countries and in children younger than 5 years. Shigellosis is estimated to cause annually 125 million cases and 100,000 deaths [1], and is one of main causes of traveller’s diarrhea. The genus Shigella comprises four serogroups (Shigella dysenteriae, Shigella sonnei, Shigella flexneri and Shigella boydii) subdivided in 50 different serotypes based on the carbohydrate composition of the O antigen of their lipopolysaccharide [2] and the presence of serotypes varies among different regions and over time [3]. As no vaccines are currently widely available, one of the aims of the European Union-funded STOPENTERICS consortium (Vaccination against Shigella and ETEC: novel antigens, novel approaches) [4] is to develop novel vaccine candidates against Shigella [e.g. the Generalized Modules for Membrane Antigens (GMMA) approach [5, 6]], as well as to improve the immunogenicity of the existing antigens (e.g. synthetic chemistry for glycoconjugates [7]). To this end, partners of the STOPENTERICS consortium have been integrating basic research, particularly genomics, transcriptomics, proteomics, and other high-throughput technologies, with novel vaccine technologies and synthetic chemistry [7]. To assemble Shigella expertise to identify and rapidly take novel vaccine candidates through to clinical trials for effective vaccine development, the research is carried out among different academic institutions (e.g. University of Oxford, Wellcome Trust Sanger Institute, Institut Pasteur) and vaccines companies (Novartis Vaccines Institute for Global Health and Sanofi-Pasteur).
To ensure the congruence of strains between laboratories, and create a public resource for vaccine development and further Shigella research, we whole genome sequenced the Shigella strains used by the STOPENTERICS consortium which are used as they offer most effective breadth of cross-protection against Shigella sp. in endemic areas [8], and report the assembly and annotation of their draft genomes. We assessed the presence of SNPs between strains and against references, as well as defined their phylogenetic relationships, and compared genetic stability of strains maintained in different consortium laboratories and after serial passage.
Methods
Bacterial strains
The Shigella strains analysed in this study and relevant metadata are summarized in Table 1. Strains were serotyped by slide agglutination using commercially available monovalent antisera (Denka Seiken, Japan) to all type specific somatic antigens and the group factor antigens [9].
Table 1.
Summary results assembly, annotation and mapping
| Name in the study | True name, country of infection, year of isolation | Sample run accession | Sample accession | De novo assembly genomic size | Contigs number | Average Contigs Length | N50 | CDS detected | Reference used for mapping | Number of SNPs detected | % of reference mapped |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ss_53G | Korea, 2000 | ERS387232 | ERR477376 | 4,698,814 | 402 | 11,688.59 | 29,856 | 4,495 | Ss 53G | 2 | 92.86 |
| Ss_53G_p | Korea, 2000 | ERS387243 | ERR477387 | 4,832,559 | 406 | 11,902.85 | 28,177 | 4,655 | Ss 53G | 2 | 92.80 |
| Ss_25931 | Unknown | ERS387235 | ERR477379 | 4,799,852 | 426 | 11,267.26 | 27,765 | 4,578 | Ss 53G | 630 | 89.51 |
| Sf 1a_Sh07.3008 | Sh07-3008, Cameroon, 2007 | ERS445026 | ERR573382 | 4,139,080 | 265 | 15,619.17 | 34,771 | 4,044 | Sf 2a 2457T | 3,459 | 86.58 |
| Sf 1b_Sh04.7434 | Sh04-7432, Tunisia, 2004 | ERS445024 | ERR573380 | 4,402,078 | 314 | 14,019.36 | 34,552 | 4,342 | Sf 2a 2457T | 2,935 | 87.63 |
| Sf 1b_Sh04.9462 | Sh04-9462, Cameroon, 2004 | ERS445025 | ERR573381 | 4,272,358 | 280 | 15,258.42 | 34,756 | 4,206 | Sf 2a 2457T | 3,207 | 87.36 |
| Sf 2a_2457T | Japan, 1954 | ERS387233 | ERR477377 | 4,681,429 | 344 | 13,608.81 | 35,441 | 4,583 | Sf 2a 2457T | 195 | 93.72 |
| Sf 2a_2457T_p | Japan, 1954 | ERS387242 | ERR477386 | 4,697,211 | 343 | 13,694.49 | 35,151 | 4,605 | Sf 2a 2457T | 192 | 93.88 |
| Sf 3a_6865_1 | Unknown | ERS387236 | ERR477380 | 4,665,099 | 335 | 13,925.67 | 35,495 | 4,550 | Sf 2a 2457T | 7,543 | 86.79 |
| Sf 3a_6865_2 | Unknown | ERS445023 | ERR573379 | 4,704,030 | 330 | 14,254.64 | 35,991 | 4,580 | Sf 2a 2457T | 7,708 | 87.08 |
| Sf 5a_M90T | Unknown <1980 | ERS387234 | ERR477378 | 4,486,899 | 327 | 13,721.40 | 32,160 | 4,391 | Sf 5a M90T | 25 | 97.82 |
| Sf 6_Sh10.5302_1 | 201005302, Madagascar, 2010 | ERS387237 | ERR477381 | 4,414,146 | 446 | 9,897.19 | 22,838 | 4,269 | Sb Sb227 | 4,408 | 89.30 |
| Sf 6_Sh10.5302_2 | 201005302, Madagascar, 2010 | ERS445029 | ERR573385 | 4,351,336 | 426 | 10,214.4 | 22,774 | 4,168 | Sb Sb227 | 4,406 | 89.10 |
| Sf 6_Sh10.3933 | 201003933, Nigeria, 2010 | ERS387238 | ERR477382 | 4,508,368 | 433 | 10,411.94 | 23,090 | 4,386 | Sb Sb227 | 4,456 | 89.33 |
| Sf 6_Sh10.8537 | 201008537, Egypt, 2010 | ERS387239 | ERR477383 | 4,524,547 | 425 | 10,645.99 | 23,238 | 4,398 | Sb Sb227 | 4,451 | 89.77 |
| Sf 6_Sh10.6306 | 201006306, India, 2010 | ERS387240 | ERR477384 | 4,481,178 | 439 | 10,207.69 | 23,066 | 4,367 | Sb Sb227 | 4,467 | 89.44 |
| Sf 6_Sh10.6237 | 201006237, Mexico, 2010 | ERS387241 | ERR477385 | 4,528,968 | 434 | 10,435.41 | 24,012 | 4,397 | Sb Sb227 | 4,389 | 89.39 |
| Sf 6_NCDC.2924-71 | NCDC 2924-71, Unknown, 1971 | ERS445027 | ERR573383 | 4,392,208 | 413 | 10,634.89 | 22,784 | 4,246 | Sb Sb227 | 4,288 | 89.66 |
| Sf 6_Sc544 | Unknown, <1977 | ERS445028 | ERR573384 | 4,430,667 | 415 | 10,676.31 | 22,494 | 4,302 | Sb Sb227 | 4,296 | 88.82 |
| Sf 6_Sh11.10088 | 201110088, France (Reunion Island), 2011 | ERS445030 | ERR573386 | 4,547,256 | 423 | 10,750.01 | 23,991 | 4,428 | Sb Sb227 | 4,483 | 89.88 |
DNA extraction and genome sequencing
Bacterial cultures were grown over night in liquid Luria–Bertani (LB) media to an optical density (measured at 600 nm) of approximately three. Genomic DNA was isolated using the Wizard kit (Promega, Madison, WI, USA) according to manufacturer’s instructions. Purified DNA was then sequenced at the Wellcome Trust Sanger Institute (WTSI). Paired end libraries 150 bp in length were generated and sequenced on the Illumina MiSeq instrument (San Diego, CA, USA) according to in house protocols [10, 11], with an approximately 500 bp insert size. Sequence data for each of the strains were deposited in the European Nucleotide Archive (accession numbers in Table 1).
Genomic analysis
Resulting sequencing reads were trimmed using Trimmomatic v0.27 [12] to remove adapters, bases with a PHRED score of <30, and remaining reads with lengths <50 bp.
High quality reads were then mapped to relevant reference strains (Table 1), using SMALT (http://www.sanger.ac.uk/resources/software/smalt/) and Single Nucleotide Polymorphisms (SNPs) were called using Samtools [13]. Nucleotides where mapping quality was below 30 and genotyping quality was below 50 were excluded from further analysis. Mapping coverage of all isolates was approximately 70-fold coverage.
De novo assembly was performed using Velvet Optimiser [14] and contiguous sequences were annotated using Prokka [15]. Clustering and BLAST comparisons were used to determine the presence/absence of genes in annotated assemblies as previously described [16].
To prepare a multiple sequence alignment for phylogenetic analysis, sequencing data from strains in this study and from simulated fastq data created from published reference genomes were mapped to the chromosome of S. flexneri 2457T (GenBank accession: NC_004741.1). The other reference isolates (and their accessions) used in this analysis were: S. sonnei Ss046 (NC_007384.1), S. sonnei 53G (NC_016822.1), S. flexneri 5 M90T (AGNM01000000), S. flexneri 5a 8401 (NC_008258.1), S. flexneri 2a NCTC1 (LM651928), S. flexneri 2a 301 (NC_004337.2), S. flexneri X 2002017 (NC_017328.1) and S. boydii Sb 227 (NC_007613.1). Core genes (n = 2,427) were identified that had 100% mapping coverage in all isolates and phylogenetic analysis was performed using RAxML software v7.0.3 [17] on the 43,349 variable sites (subset from 2,306,256 bp) of these core genes.
In silico molecular serotyping of S. flexneri isolates was performed on de novo assemblies for each isolate (and as in [18]). Briefly, the presence/absence and known differences of the gtr genes (encoding for enzymes responsible of the presence of type specific antigens I, II, IV, V, X, IC), oac genes (encoding for enzymes that mediates O-acetylation modification in serotypes 1b, 3a, 3b, and 4b) and wzx6 (specific for serotype 6) were analyzed, facilitating the differentiation of the six different S. flexneri serotypes.
Results and discussion
Sixteen different Shigella isolates belonging to seven different serotypes were sequenced (listed in Table 1). These included S. sonnei (2 isolates) and different S. flexneri serotypes including 1a, 1b (2 isolates), 2a, 3a, 5a and 6 (eight different isolates) plus four derivative strains from either serial passage (S. sonnei 53G, S. flexneri 2a 2457T) or having been cultivated and the DNA extracted in different laboratories (S. flexneri 3a 6865 and S. flexneri 6 10.5302). Derivative strains from the same isolate, but manipulated in different laboratories of the STOPENTERICS consortium were denoted ‘_1’ and ‘_2’, whereas those that had undergone serial passage (~10 passages) in the same laboratory were denoted ‘_p’. The derivatives allowed us to assess the genetic stability of strains across laboratories and through serial passage.
Results of genomic assembly and annotation were similar for all strains (Table 1). The strains assembled into an average of 381 contigs (range 265–446), with an average contigs length of 12,141 bp (range 9,897–15,619) and an N50 of 28,620 (range 22,494–35,991). The resulting genomic size was similar for all the strains and fell within the range of 4.14–4.83 Mbp. Similarly, automated annotation predicted the presence of an average of 4,400 coding sequences per genome (range 4,044–4,583; Table 1). The serotypes of the Shigella strains were confirmed based on the combinations of gtr and oac genes, encoding the relevant enzymes for the serotype-specific OAg modifications [18] (not shown).
To facilitate strain comparisons and phylogenetic analysis, sequence reads were mapped to existing Shigella reference genomes (Table 1). The percentage of the reference genome covered by mapped reads ranged from 87 to 98% and the number of SNPs varied (Table 1) depending on the isolate. These data showed comparatively few SNPs (<200) when an isolate was compared to a previously published reference of itself (as in the case of S. sonnei 53G, S. flexneri 2a 2457T, S. flexneri 5a M90T). Higher numbers of SNPs were seen where no such reference was available. For example, when an isolate was mapped to a reference genome of a different isolate of the same serotype (e.g. Ss_25931 mapped against Ss_53G) several hundred SNPs were seen, and several thousand SNPs were seen if the isolate was mapped to a reference isolate from a phylogenetic related, but distinct serotype (e.g. S. flexneri six isolates mapped against S. boydii strain Sb227).
To assess the genomic stability of isolates held at different laboratories and through serial passage within the same laboratory, we resequenced a number of isolates and compared their mapping results to the relevant reference (Table 1). Two isolates (original and passaged) of S. sonnei 53G had only two SNPs relative to the published reference genome, and these SNPs were the same in both isolates. Similarly, the sequences of original and passaged S. flexneri 2a strain 2457T were very similar, but had 195 and 192 SNPs relative to the published reference genome. Among these SNPs, 188 were common to both isolates and the remaining four and seven sites were not resolved in the other isolate, indicating that the two isolates were likely identical to each other. The level of genetic variation compared to the reference strain was surprising (~200 SNPs) and may have biological significance, showing the utility of obtaining up-to-date genetic information for the exact strain being worked with in a given project. Two strains, Sf 3a_6865 and Sf 6_10.5302, were manipulated for sequencing in separate laboratories in the consortium. These strains differed by only one and two SNPs respectively, indicating that over a 2–3 year time period, isolate genomes remain relatively stable through passage and between laboratories, but may differ significantly from published references.
To assess the phylogenetic relationship of the isolates, we constructed a maximum likelihood phylogenetic tree of a large core genome shared among the strains (Figure 1). Consistent with expectations based on prior evolutionary studies of shigellae [19, 20], the strains were divided into three main clades, with the S. flexneri six strains being phylogenetically removed from the remaining S. flexneri serotypes, and the S. sonnei strains forming a separate clade.
Figure 1.
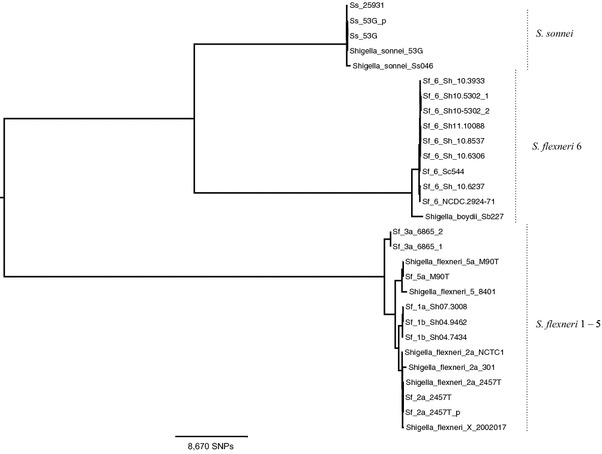
Mid-point rooted maximum likelihood phylogeny of strains based on core genome. Names of strains sequenced in this study are abbreviated and those of reference genomes are given in full.
Conclusions
The work presented here increases the number of publically available Shigella genomes, including for the first time, sequencing data for S. sonnei 25931, two S. flexneri 1b, one S. flexneri 1a, one S. flexneri 3a and 8 S. flexneri six isolates. We provide details on the draft genomes generated from this sequencing data, and report SNP variation in strains maintained in different laboratories and after serial passage. We also described the relatedness of the strains and isolates used by the STOPENTERICS consortium, and have deposited this data as a public resource. Data presented in this work will guide the selection of strains for further development of vaccine and contribute to a growing awareness of diversity in Shigella.
Author’s contributions
OR, KB and NRT analyzed the sequencing data. OR, KB, AP, FXW, FC, PJS, CG and NRT participated on data collection analysis and contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank David Harris (Wellcome Trust Sanger Institute) for sequencing the strains and Mariaelena Caboni (Novartis Vaccines Institute for Global Health) for providing S. sonnei 53G and S. flexneri 2a lines after several passages. The research received funding from the European Union Seventh Framework Programme [FP7/2007–2013] under Grant Agreement 261472 ‘STOPENTERICS’, and Wellcome Trust grant number 098051 also funded authors from the WTSI.
Compliance with ethical guidelines
Competing interests Omar Rossi, Francesco Citiulo and Christiane Gerke are employees of Novartis Vaccines Institute for Global Health. This does not alter the authors’ adherence to all ‘Gut pathogens’ policies on sharing data and materials.
Availability of supporting data
Wellcome Trust Sanger Institute sequence data is available in the European Nucleotide Archive under the accession numbers reported in Table 1.
Abbreviations
- SNP
single nucleotide polymorphism
- Ss
Shigella sonnei
- Sf
Shigella flexneri
- Sb
Shigella boydii
Footnotes
Omar Rossi and Kate S Baker contributed equally
Contributor Information
Omar Rossi, Email: omar.rossi@novartis.com.
Kate S Baker, Email: kb14@sanger.ac.uk.
Armelle Phalipon, Email: armelle.phalipon@pasteur.fr.
François-Xavier Weill, Email: fxweill@pasteur.fr.
Francesco Citiulo, Email: francesco.citiulo@novartis.com.
Philippe Sansonetti, Email: philippe.sansonetti@pasteur.fr.
Christiane Gerke, Email: christiane.e.gerke@gsk.com.
Nicholas R Thomson, Email: nrt@sanger.ac.uk.
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Wang Q, et al. Structure and genetics of Shigella O antigens. FEMS Microbiol Rev. 2008;32:627–653. doi: 10.1111/j.1574-6976.2008.00114.x. [DOI] [PubMed] [Google Scholar]
- 3.Levine MM, Kotloff KL, Barry EM, Pasetti MF, Sztein MB. Clinical trials of Shigella vaccines: two steps forward and one step back on a long, hard road. Nat Rev Microbiol. 2007;5:540–553. doi: 10.1038/nrmicro1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.STOPENTERICS, FP7/2007-2013, http://stopenterics.bio-med.ch/cms/default.aspx. Accessed 22 Apr 2015
- 5.Berlanda Scorza F, Colucci AM, Maggiore L, Sanzone S, Rossi O, Ferlenghi I, et al. High yield production process for Shigella outer membrane particles. PLoS One. 2012;7:e35616. doi: 10.1371/journal.pone.0035616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi O, Pesce I, Giannelli C, Aprea S, Caboni M, Citiulo F. Modulation of endotoxicity of Shigella generalized modules for membrane antigens (GMMA) by genetic lipid A modifications: relative activation of TLR4 and TLR2 pathways in different mutants. J Biol Chem. 2014;289:24922–24935. doi: 10.1074/jbc.M114.566570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gauthier C, Chassagne P, Theillet FX, Guerreiro C, Thouron F, Nato F, et al. Non-stoichiometric O-acetylation of Shigellaflexneri 2a O-specific polysaccharide: synthesis and antigenicity. Org Biomol Chem. 2014;12:4218–4232. doi: 10.1039/C3OB42586J. [DOI] [PubMed] [Google Scholar]
- 8.Livio S, Strockbine N, Panchalingam S, Tennant SM, Barry EM, Marohn ME, et al. Shigella isolates from the Global Enteric Multicenter Study (GEMS) Inform Vaccine Development. Clin Infect Dis. 2014;59:933–941. doi: 10.1093/cid/ciu468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlin NI, Lindberg AA. Monoclonal antibodies specific for Shigella flexneri lipopolysaccharides: clones binding to type I and type III: 6, 7, 8 antigens, group 6 antigen, and a core epitope. Infect Immun. 1986;53:103–109. doi: 10.1128/iai.53.1.103-109.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quail MA, Otto TD, Gu Y, Harris SR, Skelly TF, McQuillan JA, et al. Optimal enzymes for amplifying sequencing libraries. Nat Methods. 2012;9:10–11. doi: 10.1038/nmeth.1814. [DOI] [PubMed] [Google Scholar]
- 12.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zerbino DR. Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinform. 2010 doi: 10.1002/0471250953.bi1105s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 16.Baker KS, Mather AE, McGregor H, Coupland P, Langridge GC, Day M, et al. The extant World War 1 dysentery bacillus NCTC1: a genomic analysis. Lancet. 2014;384:1691–1697. doi: 10.1016/S0140-6736(14)61789-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 18.Ashton PM, Baker KS, Gentle A, Wooldridge DJ, Thomson NR, Dallman TJ, et al. Draft genome sequences of the type strains of Shigella flexneri held at Public Health England: comparison of classical phenotypic and novel molecular assays with whole genome sequence. Gut Pathog. 2014;6:7. doi: 10.1186/1757-4749-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pupo GM, Lan R, Reeves PR. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc Natl Acad Sci USA. 2000;97:10567–10572. doi: 10.1073/pnas.180094797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Nie H, Chen L, Zhang X, Yang F, Xu X, et al. Revisiting the molecular evolutionary history of Shigella spp. J Mol Evol. 2007;64:71–79. doi: 10.1007/s00239-006-0052-8. [DOI] [PubMed] [Google Scholar]