

Abstract
It is unknown how dendritic cells (DCs) become specialized as mucosal DCs and maintain intestinal homeostasis. We report that bone marrow cells express the retinoic acid (RA)-synthesizing enzyme ALDH1a2, and provide RA to DC precursors in the bone marrow microenvironment. RA induced bone marrow-derived DCs (BMDCs) to express CCR9 and ALDH1a2, and conferred upon them mucosal DC functions. This was dependent on a narrow time window and stringent dose effect. RA promoted BMDC production of bioactive TGF-β by inhibiting SOCS3 expression and hence enhancing STAT3 activation. These RA effects were evident in vivo, in that vitamin A-deficient mucosal DCs had reduced mucosal function, namely failure to induce Foxp3+ regulatory T cells. Lastly, MyD88 signaling enhanced RA-educated DC ALDH1a2 expression, and was required for TGF-β production. These data indicate that RA plays a critical role in the generation of mucosal DCs from bone marrow as well as in their functional activity.
Introduction
Intestinal immune cells are extensively distributed throughout the intestine (Newberry and Lorenz, 2005). Within the extensive network of intestinal immunity, dendritic cells (DCs) play a critical role in the decision of whether to stimulate immune regulation or activate immune responses to commensal microbiota (Coombes and Powrie, 2008). Lamina propria and mesenteric lymph node (MLN) DCs have been shown to be significantly better than splenic DCs at inducing Foxp3 expression in naïve T cells in the presence of exogenous TGF-β (Mucida et al., 2007; Sun et al., 2007). In addition, CD103+ DCs from both the lamina propria and MLNs are capable of converting naïve T cells into Foxp3+ regulatory T (Treg) cells via a TGF-β and retinoic acid (RA)-dependent mechanism, whereas MLN CD103- DCs promote the differentiation of interferon-γ (IFN-γ)-producing T cells (Annacker et al., 2005; Coombes et al., 2007; Sun et al., 2007). Peyer's patch DCs have been implicated in B cell class switching to immunoglobulin A (IgA) through RA and other DC-derived signals (Mora et al., 2006). Another hallmark of intestinal CD103+ DCs is to promote the expression of the gut-homing receptors α4β7-integrin and CC-chemokine receptor 9 (CCR9) on responding T and B lymphocytes via RA signaling (Johansson-Lindbom et al., 2005; Johansson-Lindbom et al., 2003; Mora et al., 2003). Thus, mucosal DCs differentially regulate intestinal T cell and B cell responses in the steady state. Mucosal DCs also express pattern recognition receptors such as Toll-like receptors (TLRs) to recognize a broad spectrum of phylogenetically conserved microbial motifs and hence shape the innate and adaptive immune responses towards microbiota. However, the mechanisms by which DCs acquire a mucosal DC phenotype and the factors that instruct mucosal DCs to display such distinctive functional properties are unclear.
It has been shown that mice on a vitamin A-deficient diet had reductions in α4β7+ memory T cells in lymphoid organs and dramatic deficiencies of lamina propria T cells in the small intestine (Iwata et al., 2004), implicating a critical role of vitamin A and its metabolites in the homing of lymphocytes to the gut. RA, a metabolite of vitamin A, is produced by retinal dehydrogenases, such as aldehyde dehydrogenase family 1, subfamily A1 and A2 (ALDH1a1 and ALDH1a2). Consistent with their functional properties, MLN CD103+ DCs express higher levels of Aldh1a2 than their CD103- counterparts (Coombes et al., 2007), and Peyer's patch and MLN DCs can convert vitamin A into RA in vitro (Iwata et al., 2004). Accordingly, the induction of gut tropism to T cells can be partially blocked by the retinal dehydrogenase inhibitor, citral (Iwata et al., 2004), and can be effectively inhibited by RA receptor (RAR) inhibitors (Svensson et al., 2008). These findings indicate an important role of RA in mucosal DC functions. However, whether and how RA imprints gut-homing specificity on DCs, and instructs DCs to acquire the functional properties of mucosal DCs, are largely unknown.
In this study, we investigated the effects of RA on mucosal DC generation. We report here that bone marrow cells express ALDH1a2 and provide RA to DC precursors in the bone marrow microenvironment. RA induced bone marrow-derived DCs (BMDCs) to express CCR9 and ALDH1a2, and conferred upon them a mucosal DC phenotype and function. The effect of RA on educating BMDCs was dependent on a narrow time window and stringent dose response. RA inhibited DC expression of Suppressor Of Cytokine Signaling (SOCS) 3 through direct interference with SOCS3 promoter activity, and thus enhanced Signal Transducer and Activator of Transcription (STAT) 3 activation, which promoted bioactive TGF-β production. In cooperation with RA signaling, commensal microbiota stimulation enhanced RA-educated DC (RA-DC) ALDH1a2 expression and was required for RA-DC TGF-β production. Together, our data indicate that RA plays a critical role in the generation of mucosal DCs from the bone marrow as well as in their functional activity.
Results
RA educates DCs to express functional CCR9 and exhibit the phenotype of immature DCs
Expression of gut-homing receptors is likely to be the first step for DCs to reach the intestine and be further educated to acquire the full complement of mucosal DC functions. Because mucosal DCs originate in the bone marrow (Bogunovic et al., 2009; Varol et al., 2009), we first examined bone marrow cell ALDH1a2 expression and activity. ALDH1a2 was detected in normal C57BL/6 mouse bone marrow by Western blot, and bone marrow cells exhibited ALDH activity that was blocked by the ALDH inhibitor DEAB (diethylaminobenzaldehyde; Figure 1A), indicating that the bone marrow microenvironment can provide RA to DC precursors locally.
Figure 1. RA instructs BMDCs to express functional CCR9.
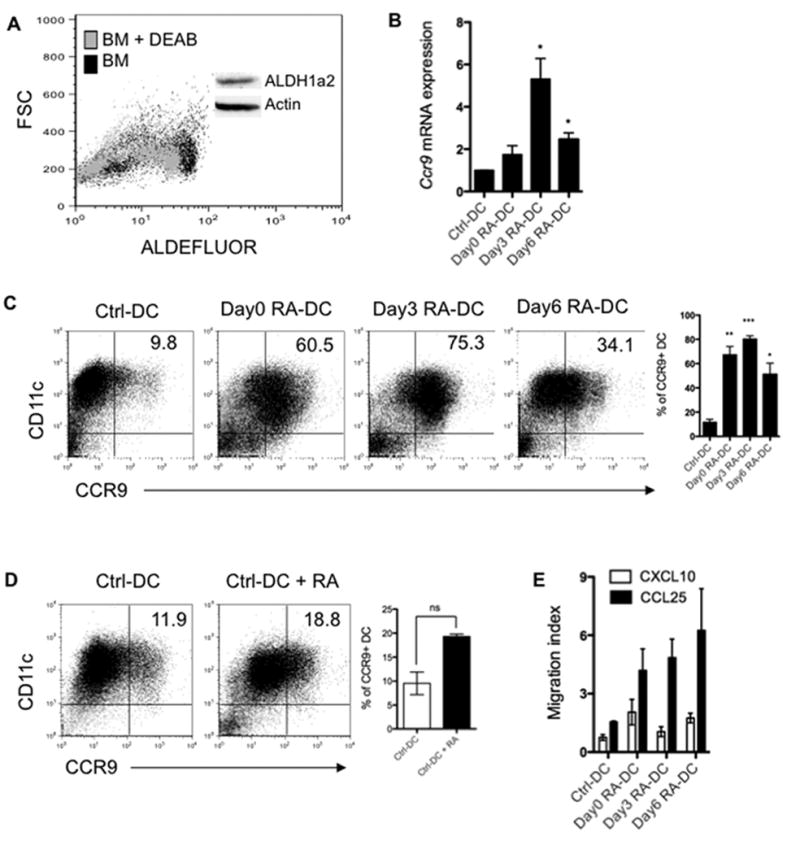
(A) ALDH1a2 expression of freshly isolated normal C57BL/6 bone marrow cells was examined by Western blot (Insert) and ALDH activity was examined by incubating cells with the fluorescent ALDH substrate ALDEFLUOR with or without addition of ALDH inhibitor (DEAB), and assessed by flow cytometry. (B) BMDCs were treated with 1 μM RA from day 0 (day0 RA-DC), day 3 (day3 RA-DC) or day 6 (day6 RA-DC) of an 8-day culture. CCR9 expression was analyzed by quantitative PCR and normalized to Gapdh mRNA expression and (C) by flow cytometry. (D) Day8 control BMDCs were cultured with or without 1 μM RA. CCR9 expression was determined by flow cytometry 48 h later. (E) Chemotactic activity of control BMDCs and RA-DCs toward 10 ng/ml of CCL25 or CXCL10 was analyzed and given as the migration index. Plot numbers represent the percentage of CD11c+ cells in the respective quadrants (C, D). Data are aggregate (B-E; mean ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001 as compared to control BMDCs) or representative (A, C, D) of two (A) or three or more (B-E) experiments.
Because it has been shown that RA induces gut-homing receptors on T cells and B cells (Iwata et al., 2004; Mora et al., 2006), the effect of RA on BMDC gut-homing receptor expression was investigated. One μM RA was added from day 0 (day0 RA-DC), day 3 (day3 RA-DC) or day 6 (day6 RA-DC) of an 8-day BMDC culture. Treatment with RA did not induce plasmacytoid DCs as shown by the absence of B220 expression (Figure S1A). When harvested on day 8, BMDC expression of gut-homing receptor CCR9 mRNA was increased up to 1.7-fold, 5.3-fold and 2.5-fold when RA was added from days 0, 3 and 6, respectively (Figure S1A). Consistent with mRNA expression, control BMDCs expressed low levels (9.8%) of CCR9, but treatment with RA greatly enhanced CCR9 expression, in that 60.5% of CD11c+ DCs were CCR9+ when RA was added from day 0, 75.3% when RA was added from day 3, and 34.1% when RA was added from day 6 (Figure 1C), indicating that the induction of gut-homing specificity via RA signaling is not restricted to lymphocytes. However, we did not observe the expression of α4β7-integrin or other chemokine receptors including CCR6, CCR7 or CXCR3 by those BMDCs (Figure S1B). When day8 control BMDCs or splenic DCs were treated with RA for 48 h, RA only moderately augmented CCR9 expression in these fully developed DCs (Figure 1D and Figure S2A). Based on the high-level expression of CCR9 on RA-DCs, we determined the migration capacity of control BMDCs and RA-DCs toward the CCR9 ligand, CCL25/TECK, which is expressed by intestinal cells (Wendland et al., 2007). In vitro transwell migration assays revealed a strong chemotactic response of RA-DCs but not control BMDCs toward CCL25, and no chemotactic response of either control BMDCs or RA-DCs toward irrelevant CXCL10, a ligand for CXCR3 (Figure 1E). These data demonstrate that RA conditions BMDCs to express high levels of functional gut-homing receptor CCR9.
We next characterized the phenotype of RA-DCs. RA greatly decreased the expression of CD11c in a time-dependent manner (Figure S1C). Upon stimulation with anti-CD40 agonist antibody, day3 RA-DCs showed lower levels of CD86 and MHC II than control BMDCs, and the differences were further magnified by stimulation with LPS (Figure S1D), illustrating an immature DC phenotype of RA-DCs.
Narrow time window and stringent dose response to RA for development of functional mucosal-like DCs
Lamina propria DCs (LP-DCs) have been shown to induce Foxp3+ Treg cells (Coombes et al., 2007; Sun et al., 2007). To determine whether RA-DCs functionally mimic LP-DCs, the capability of RA-DCs to induce Foxp3 expression in T cells was investigated. RA-DCs were generated as previously described by adding RA from day 0, day 3 or day 6 of an 8-day culture. CBir1 flagellin peptide-pulsed control BMDCs and RA-DCs were cocultured with FACS-sorted CD4+Foxp3gfp- CBir1 TCR-transgenic (CBir1-Tg) T cells, which are specific for an immunodominant microbial antigen, CBir1 flagellin (Cong et al., 2009a). T cell Foxp3 expression was determined by flow cytometry after 5 days. The maximal induction of Foxp3 on T cells was achieved by culturing with day3 RA-DCs, which induced 10.5% Foxp3+ T cells, whereas control BMDCs failed to induce Foxp3 expression in T cells (Figure 2A). Neither day0 nor day6 RA-DCs induced high levels of Foxp3. When treated with RA for 48 h, neither day8 control BMDCs nor splenic DCs induced Foxp3+ Treg cells from Foxp3- progenitors (Figure 2A and Figure S2B), indicating a narrow time window for the effect of RA on educating DCs to induce T cell Foxp3 expression. These data also excluded the possibility of carryover of residual RA by day3 RA-DCs to achieve the function as none of the other RA-treated DCs induced T cell Foxp3 expression.
Figure 2. Narrow time window and stringent dose response to RA for development of functional mucosal-like DCs.
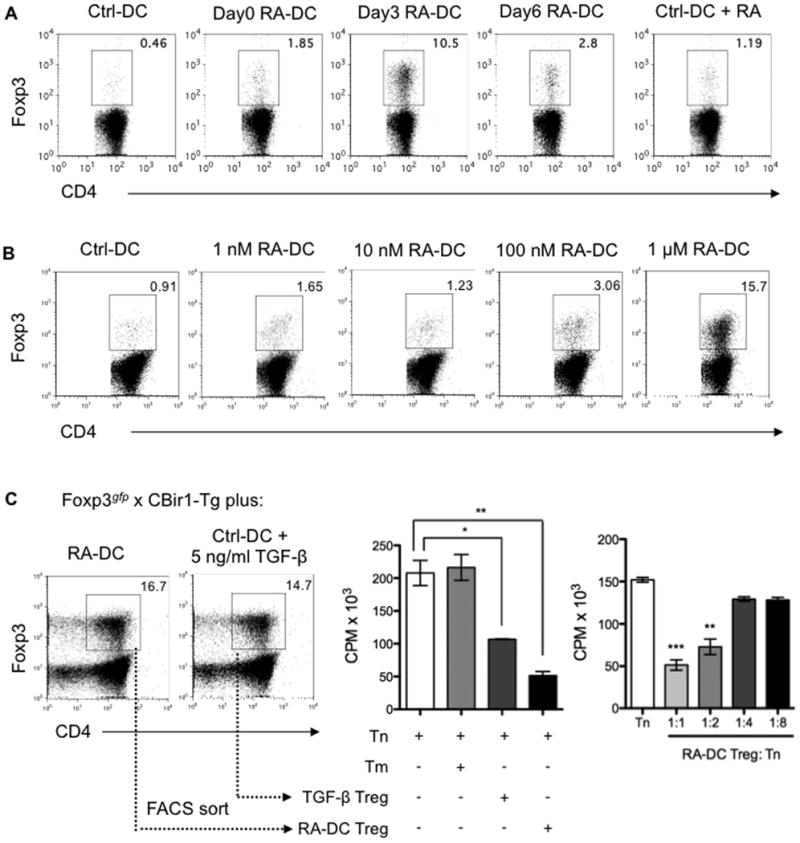
(A) BMDCs generated as described in Figure 1 were pulsed with CBir1 flagellin peptide and then cocultured with FACS-sorted CD4+Foxp3gfp- naïve T cells from Foxp3gfp.CBir1-Tg mice. T cell Foxp3 expression was examined by flow cytometry after 5 days. (B) Doses from 1 nM to 1 μM of RA were added to BMDC cultures from day 3. On day 8, BMDCs pulsed with CBir1 flagellin peptide were cocultured with FACS-sorted CD4+Foxp3gfp- CBir1-Tg naïve T cells. T cell Foxp3 expression was examined by flow cytometry after 5 days. Plots are gated on CD4+ cells, and the percentages of Foxp3+ cells are shown (A, B). (C) Naïve Foxp3gfp.CBir1-Tg CD4+ T cells were cultured with day3 RA-DCs, or with control BMDCs in the presence of 5 ng/ml of TGF-β. CD4+Foxp3gfp+ T cells were sorted by FACS 5 days later. Naïve CBir1-Tg CD4+ T cells were cultured alone (Tn), with memory T cells (Tm), or with FACS-sorted Foxp3gfp+ cells from both conditions at a ratio of 1:1 or as indicated in the presence of CBir1 flagellin peptide-pulsed splenic feeder cells. Tritiated thymidine was added for the last 18 h of a 72-h incubation period for T cell proliferation. Mean CPM of triplicates ± SEM are shown (*, P < 0.05; **, P < 0.01; ***, P < 0.001 as compared to Tn). Data are representative of three experiments.
To determine the dose effect of RA on BMDCs, RA at doses from 1 nM to 1 μM was added to BMDC culture from day 3. As shown in Figure 2B, RA educated BMDCs to induce Foxp3+ Treg cells from Foxp3- progenitors in a dose-dependent manner. BMDCs treated with 100 nM RA slightly induced Foxp3+ Treg cells (3.06%), which was increased to 15.7% when BMDCs were treated with 1 μM RA. BMDCs treated with RA at doses of 10 nM or lower did not induce Foxp3+ Treg cells. Collectively, these data indicate a narrow time window (RA added from day 3 of an 8-day culture) and stringent dose effect (1 μM RA) of RA induction of functional mucosal-like DCs.
To determine whether RA-DC-induced Foxp3+ T cells function as conventional Treg cells, their ability to suppress naïve T cell activation was examined. Foxp3+ T cells were generated by RA-DCs or under standard Treg cell-polarizing conditions. FACS-sorted CD4+Foxp3gfp+ CBir1-Tg T cells from both conditions were cultured in triplicate wells with naïve CBir1-Tg CD4+ T cells in the presence of CBir1 flagellin peptide-pulsed splenic feeder cells. Tritiated thymidine was added for the last 18 h of a 72-h incubation period for assessment of T cell proliferation. Foxp3+ T cells generated by RA-DCs were more suppressive than control BMDC-generated Treg cells (Figure 2C).
TGF-β and RA differentially regulate RA-DC induction of CD4+ T cell Foxp3 and CCR9 expression
The underlying mechanisms by which LP-DCs induce Foxp3+ Treg cells are still not completely understood. Consistent with data of others (Coombes et al., 2007; Sun et al., 2007), in the absence of exogenous TGF-β, LP-DCs from both small and large intestine induced higher levels of Foxp3 expression in CD4+ T cells than did splenic DCs (10.7% and 20.8% vs. 4.2%) (Figure S3A). The addition of the TGF-β type I receptor (TGF-β RI) inhibitor SB-505124 or RAR antagonist LE135 inhibited Treg cell conversion by 85% and 55%, respectively (Figure S3B). Similar to LP-DCs, day3 RA-DCs promoted CD4+ T cell Foxp3, CCR9 (Figures 3A and 3B), and α4β7-integrin expression (data not shown). To investigate the role of the TGF-β and RA signaling pathways in the induction of Foxp3+ Treg cells by RA-DCs, we added the TGF-β RI inhibitor or RAR antagonist to block TGF-β or RA signaling in RA-DC-polarized T cells, respectively. TGF-β and RA were found to differentially regulate T cell Foxp3 and CCR9 expression, in that blockade of TGF-β completely inhibited Foxp3 but enhanced CCR9 expression, whereas the RAR antagonist partially downregulated Foxp3 but completely abolished CCR9 expression (Figure 3B).
Figure 3. TGF-β and RA differentially regulate RA-DC induction of CD4+ T cell Foxp3 and CCR9 expression.
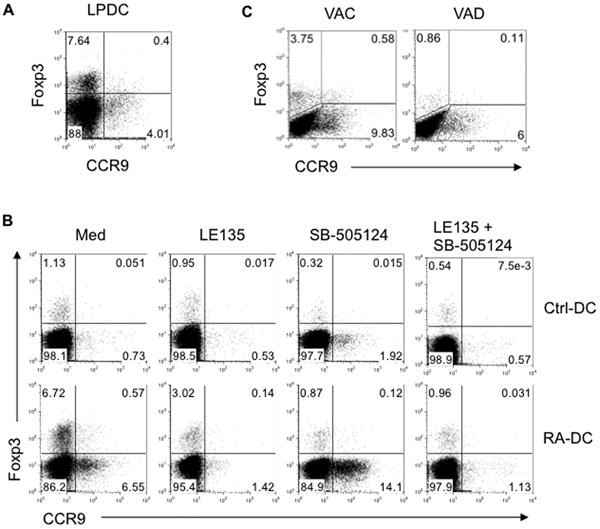
(A) LP-DCs were isolated from C57BL/6 mice and cultured with naïve CBir1-Tg CD4+ T cells in the presence of CBir1 flagellin peptide. Expression of Foxp3 and CCR9 by CD4+ T cells was determined by flow cytometry after 5 days. (B) CBir1 flagellin peptide-pulsed control BMDCs and day3 RA-DCs were cultured with naïve CBir1-Tg CD4+ T cells in the presence or absence of TGF-β RI inhibitor SB-505124, and/or RAR antagonist LE135. Expression of Foxp3 and CCR9 by CD4+ T cells was determined by flow cytometry 5 days later. (C) MLN DCs isolated from control (VAC) or vitamin A-deficient (VAD) mice were pulsed with CBir1 flagellin peptide and cocultured with naïve CBir1-Tg CD4+ T cells. Expression of Foxp3 and CCR9 by CD4+ T cells was determined 5 days later by flow cytometry. Data are representative of two (C) or three (A, B) experiments.
Another notable functional ability of mucosal LP-DCs is to induce IgA-secreting B cells. To determine whether RA-DCs could induce IgA production by B cells, B220+ B cells were isolated from the spleen by magnetic sorting, activated with 10 μg/ml anti-mouse IgM F(ab′)2, and cultured alone, with control BMDCs, or with day3 RA-DCs. Similar to mucosal DCs, RA-DCs induced a significantly higher amount of IgA production by B cells after 4 days, which was blocked by both the TGF-β RI inhibitor and RAR antagonist (Figure S4), implicating an important role of both TGF-β and RA produced by RA-DCs in the induction of IgA-secreting B cells.
To assess the influence of RA on mucosal DC development in vivo, we generated vitamin A-deficient (VAD) mice. MLN-DCs isolated from VAD and control (VAC) mice were pulsed with CBir1 flagellin peptide and cocultured with CBir1-Tg CD4+ T cells. MLN-DCs from VAD mice lost the capability to promote Foxp3+ Treg cell development, and the level of CCR9 expression was lower when T cells were cultured in the presence of MLN-DCs from VAD mice than from VAC mice (Figure 3C). Collectively, our data provide both in vitro and in vivo evidence that RA educates DCs to gain mucosal DC phenotype and function.
RA upregulates DC ALDH1a2 expression and induces bioactive TGF-β production
Since RA-DCs promoted the development of Foxp3+ Treg cells and IgA-secreting B cells via a TGF-β and RA-dependent mechanism (Figure 3B and Figure S4), we asked whether RA-DCs, similar to mucosal DCs, provide a source of RA and TGF-β to T cells. We first examined the gene expression levels of Aldh1a1 and Aldh1a2 in both control BMDCs and RA-DCs. Aldh1a1 was not detected in either type of DCs (data not shown), while treatment with RA induced substantial expression of Aldh1a2 mRNA (Figure 4A) and protein (Figure 4B) in RA-DCs. Accordingly, RA-DCs exhibited greater ALDH activity than control BMDCs when incubated with the fluorescent ALDH substrate ALDEFLUOR, which was blocked by the inhibitor DEAB (Figure 4C). RA also regulated Aldh1a2 expression in intestinal DCs in vivo: vitamin A-high diet (VAH) fed-mice expressed a significantly higher level of Aldh1a2 than control VAC mice, whereas VAD mice showed a profound defect in Aldh1a2 mRNA expression. Restoration of vitamin A in the VAD mice by switching from the VAD diet to a normal diet restored Aldh1a2 expression levels comparable to the control mice (Figure 4D). These data indicate that ALDH1a2 is robustly induced by RA in DCs both in vitro and in vivo.
Figure 4. RA upregulates DC expression of functional ALDH1a2 and induces bioactive TGF-β production.
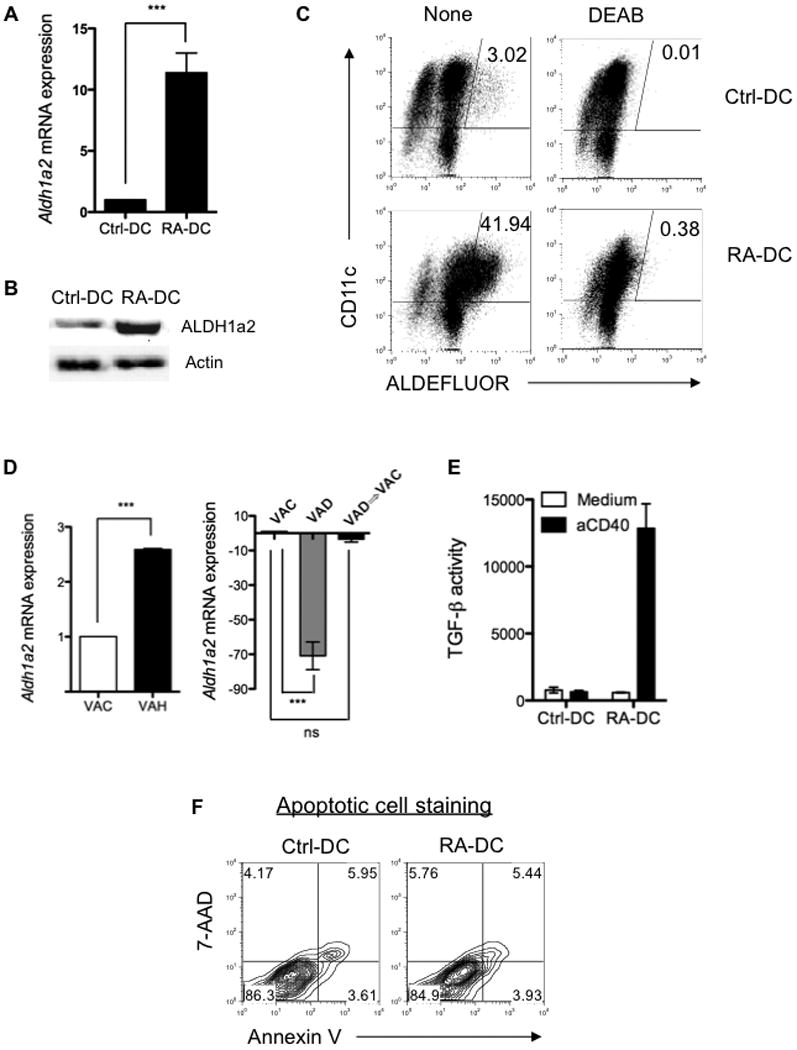
Control BMDCs and day3 RA-DCs were generated as described in Figure 1, and their ALDH1a2 expression was determined by real-time PCR (A) or Western blot (B). (C) ALDH activity of control BMDCs and day3 RA-DCs with or without addition of inhibitor (DEAB) was assessed as described in Figure 1A. (D) Aldh1a2 expression of MLN DCs isolated from VAC mice, vitamin A-high (VAH) diet-fed mice, VAD mice, or mice fed with VAD diet then switched to regular diet (VAD => VAC) was determined by quantitative PCR. (E) Control BMDCs and day3 RA-DC were stimulated with anti-CD40 agonist antibody for 24 h, and bioactive TGF-β production in the supernatants was determined by using the MFB-F11 reporter cell line and shown as reporter chemiluminescence units. (F) Control BMDCs or day3 RA-DCs were stained with 7-AAD and Annexin V, and analyzed by flow cytometry. Data are aggregate (A, D, E; mean ± SEM; ***, P < 0.001) or representative (B, C, F) of two (C, D, F) or at least three (A, B, E) experiments.
To quantify RA-DC TGF-β production, a reporter cell line MFB-F11 (Tesseur et al., 2006) was used. Neither control BMDCs nor RA-DCs secreted any detectable levels of biologically active TGF-β without stimulation (Figure 4E). However, upon stimulation with agonistic anti-CD40 antibody, RA-DCs secreted high levels of bioactive TGF-β. It has been shown that apoptotic cells also secrete TGF-β (Chen et al., 2001; Kushwah et al., 2009). To exclude this possibility, apoptosis of DCs was analyzed by staining with 7-AAD and Annexin V antibody. There was no difference in apoptotic cells between control BMDCs and RA-DCs (Figure 4F). Collectively, these data demonstrate that RA endows DC precursors with mucosal DC functionality by inducing DCs to express ALDH1a2 and to produce biologically active TGF-β.
RA stimulates TGF-β production by suppressing DC SOCS3 promoter activity
We next explored the mechanism by which RA induces DCs to produce bioactive TGF-β. Activated STAT3 has been reported to directly upregulate TGF-β1 promoter activity (Kinjyo et al., 2006; Ogata et al., 2006), and SOCS3 is known to negatively regulate STAT3 activation (Yoshimura, 2006). Therefore, we examined STAT3 activation status in RA-DCs by Western blot analysis. STAT3 activation in RA-DCs was enhanced and prolonged compared to control BMDCs in response to interleukin (IL)-6 and LPS stimulation (Figure 5A). It has also been shown that loss of SOCS3 expression promotes STAT3-mediated TGF-β production in various types of cells, including DCs (Kinjyo et al., 2006; Matsumura et al., 2007; Ogata et al., 2006). To determine the effect of SOCS3 expression on TGF-β production in RA-DCs, we first deleted the Socs3 gene in BMDCs by treating Socs3flox/flox BMDCs with Cre-expressing adenovirus. Socs3flox/flox BMDCs were also treated with control GFP-expressing adenovirus. Deletion of the Socs3 gene was confirmed by genotyping and real-time PCR (Figure 5B). When treated with anti-CD40 agonist antibody or various TLR ligands, SOCS3-/- BMDCs, similar to RA-DCs, produced a substantial amount of bioactive TGF-β (Figure 5C). Accordingly, SOCS3-/- BMDCs promoted de novo generation of Foxp3+ Treg cells, which was completely inhibited by the TGF-β RI inhibitor SB-505124 (Figure 5D). However, the level of CCR9 expression in those T cells was not affected by deletion of the Socs3 gene in DCs, probably because deletion of the Socs3 gene had no effect on Aldh1a2 expression (data not shown). These data confirm a differential role of TGF-β and RA in the induction of Foxp3 and homing receptors as shown in Figure 3.
Figure 5. RA enhances DC STAT3 activation by suppressing SOCS3 expression through suppression of SOCS3 promoter activity.
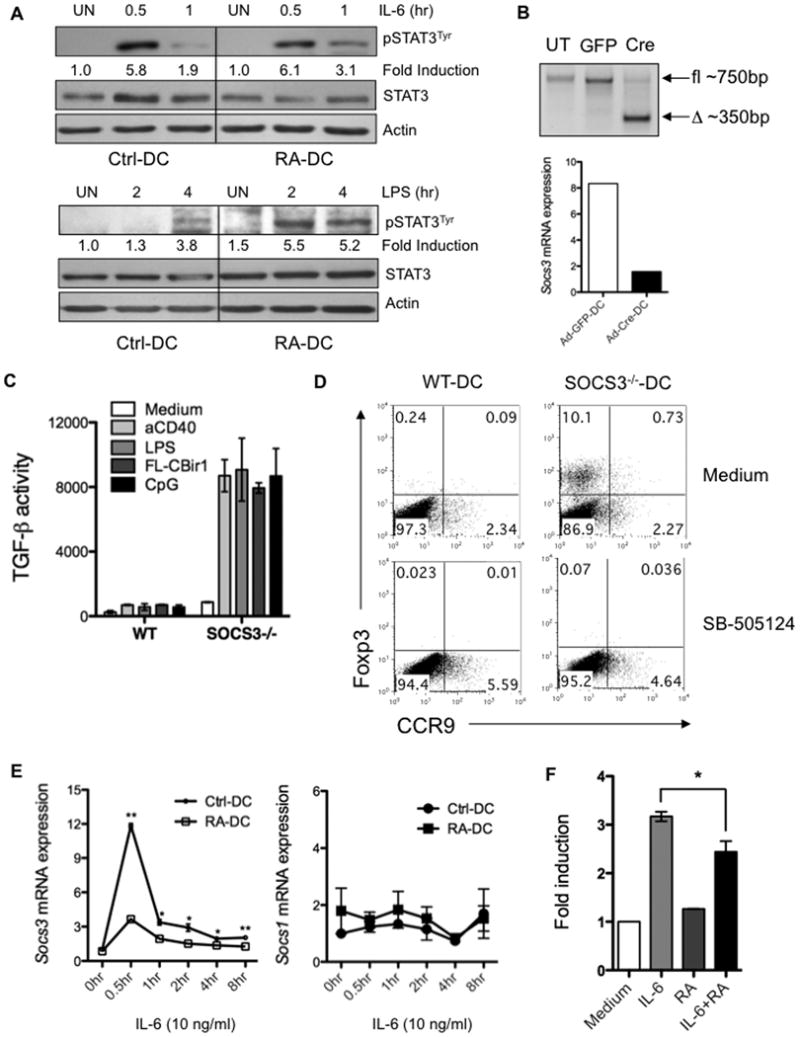
(A) Control BMDCs and day3 RA-DCs were stimulated with IL-6 or LPS for up to 4 h. Cell lysates were immunoblotted with anti-phospho-STAT3Tyr. The membranes were stripped and reblotted with anti-STAT3 and anti-Actin as loading controls. (B-D) BMDCs generated from Socs3flox/flox mice were treated with Cre- (SOCS3-/--DCs) or GFP-expressing adenovirus (WT-DCs) for 48 h. Deletion of the Socs3 gene was confirmed by genotyping and quantitative PCR (B). (C) WT- and SOCS3-/--DCs were stimulated with various TLR ligands or anti-CD40 agonist antibody for 24 h. TGF-β production in the supernatants was determined as described previously. (D) Naïve CBir1-Tg CD4+ T cells were cultured with CBir1 flagellin peptide-pulsed WT- or SOCS3-/--DCs with or without TGF-β RI inhibitor SB-505124. Foxp3 and CCR9 expression by CD4+ T cells was determined. (E) Control BMDCs and day3 RA-DCs were treated with IL-6 for up to 8 h. Socs3 and Socs1 mRNA expression was assessed by quantitative PCR. (F) RAW cells were transfected with the SOCS3 promoter-luciferase reporter construct, and stimulated with IL-6 in the absence or presence of RA. SOCS3 promoter activity was assessed by luciferase activity and given as fold induction by comparing the value of RAW cells treated with medium. Data are aggregate (C, E, F; mean ± SEM; *, P < 0.05; **, P < 0.01) or representative (A, B, D) of two (A) or three or more (B-E) experiments.
To compare Socs3 gene expression in control BMDCs and RA-DCs, we performed real-time PCR at serial time points. IL-6-induced expression of Socs3 peaked at 30 min, and subsided to baseline after 8 h in both DC groups. However, RA-DCs expressed a significantly lower amount of Socs3, but not Socs1, mRNA than control BMDCs at all time points (Figure 5E). Analysis of SOCS3 promoter activity revealed a repressive role of RA on IL-6-induced SOCS3 expression (Figure 5F), indicating that RA suppressed DC SOCS3 expression by affecting SOCS3 promoter activity. Collectively, our data demonstrate that RA promotes DC TGF-β production by suppressing SOCS3 promoter activity, which releases the inhibition of SOCS3 on STAT3 activation.
TLR-MyD88 signaling is required for optimal RA-DC function
Commensal bacteria have a great impact on the mucosal immune system (Cebra, 1999). Once gut-homing receptor-expressing DCs migrate to the intestine, they are exposed to a diverse array of microbiota ligands that can stimulate TLR signaling, mostly through the adapter protein MyD88. To unravel the role of microbial ligands in mucosal DC function, we first compared the function of MyD88-/- mucosal DCs with that of wild type mucosal DCs. As shown in Figure 6A, freshly isolated mucosal DCs from MyD88-/- mice induced fewer Foxp3+ Treg cells than those from wild type mice. Similarly, we observed less induction of T cell Foxp3 expression by RA-DCs from MyD88-/- mice compared to wild type RA-DCs (Figure 6B). To determine whether mucosal DCs need to be activated by TLR-MyD88 signaling to produce maximal amounts of TGF-β, we measured bioactive TGF-β production by wild type RA-DCs upon stimulation by different TLR ligands. LPS, full-length CBir1 flagellin and CpG ODN all stimulated TGF-β production in wild type RA-DCs (Figure 6C). MyD88-/- RA-DCs did not produce detectable levels of bioactive TGF-β in response to stimulation with LPS, flagellin or CpG ODN. In contrast, MyD88-/- RA-DCs did produce bioactive TGF-β in response to anti-CD40 agonist antibody at a comparable level to wild type RA-DCs (Figure 6C). These data indicate that TLR-MyD88 signaling or CD40-CD40L interaction provided by activated T cells is required for optimal TGF-β production by mucosal DCs or RA-DCs and hence, the induction of Foxp3 expression in T cells.
Figure 6. Requirement of TLR-MyD88 signaling for optimal mucosal-like DC development.
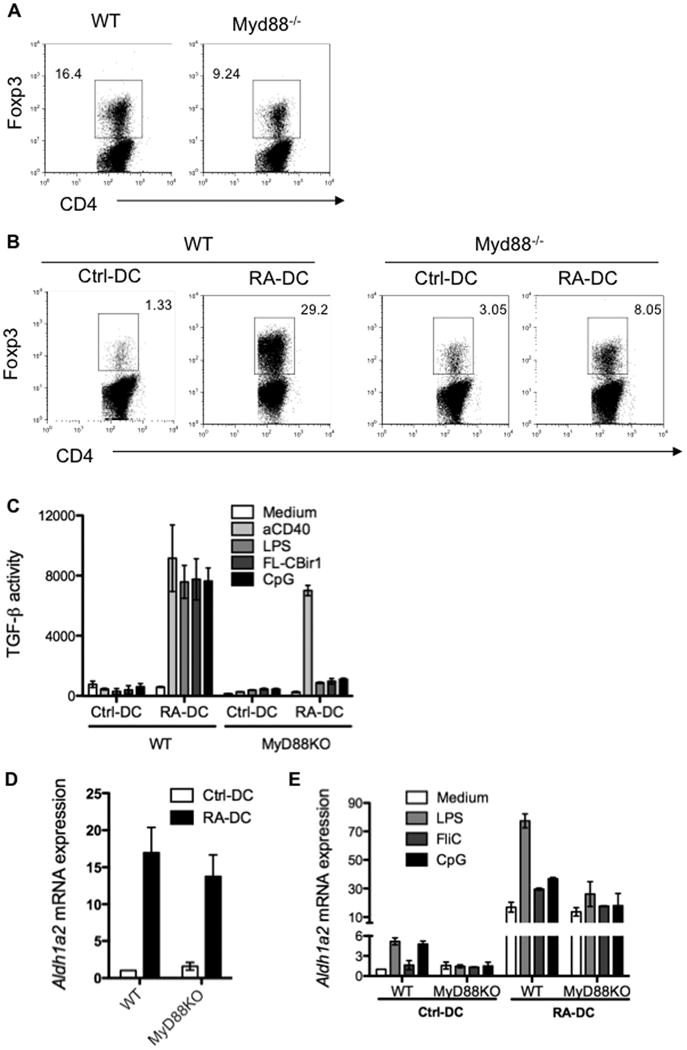
(A) MLN DCs isolated from wild type or MyD88-/- mice were cultured with CBir1-Tg CD4+ T cells. Foxp3 expression by CD4+ T cells was determined 5 days later. (B) Control BMDCs and day3 RA-DCs were generated from wild type or MyD88-/- mice, and cultured with CBir1-Tg CD4+ T cells. Foxp3 expression by CD4+ T cells was determined 5 days later. (C) Wild type and MyD88-/- control BMDCs and day3 RA-DCs were stimulated with various TLR ligands and anti-CD40 agonist antibody. TGF-β production was assessed 24 h later. (D) Control BMDCs and day3 RA-DCs were generated from wild type or MyD88-/- mice, and Aldh1a2 mRNA expression was determined by quantitative PCR before or (E) after being stimulated with various TLR ligands. Bar charts represent aggregate data with mean ± SEM of three or more experiments. Flow plots are representative of at least three independent experiments.
MyD88-/- RA-DCs expressed similar levels of Aldh1a2 mRNA as did wild type RA-DCs (Figure 6D), indicating that RA-mediated induction of Aldh1a2 is independent of TLR-MyD88 signaling. However, when control BMDCs and RA-DCs from wild type and MyD88-/- mice were treated with different TLR ligands, wild type control BMDC Aldh1a2 expression increased up to 5-fold, but wild type RA-DC Aldh1a2 expression increased up to 77-fold. In contrast, control and RA-DCs from MyD88-/- mice had no increase in Aldh1a2 expression above baseline (Figure 6E). Thus, there is an additive effect of TLR-MyD88 signaling in conjunction with RA signaling on DC ALDH1a2 induction. Together, these data indicate that full development of mucosal DC functioning requires microbiota stimulation.
Discussion
Intestinal mucosal DCs have distinct phenotypical and functional properties that allow them to differentially regulate host immune responses towards microbiota and pathogens, which is pivotal to the maintenance of intestinal immune homeostasis (Coombes and Powrie, 2008; Kelsall and Leon, 2005). Several functional aspects of mucosal DCs have been reported, including imprinting lymphocytes with gut-homing tropism, promoting IgA-producing B cells, and inducing the differentiation of Treg cells that are involved in tolerance to soluble oral antigens and commensal bacteria (Coombes and Powrie, 2008). Recent reports demonstrated that common macrophage and DC precursors can give rise to both CD103+ and CD103- DCs, whereas DC precursors or monocytes can differentiate only into CD103+ or CD103- DCs, respectively (Bogunovic et al., 2009; Varol et al., 2009). However, whether and how DC precursors develop into mucosal DCs in the bone marrow is unknown.
Consistent with previous observations (Armstrong et al., 2004; Pearce and Bonnet, 2007), our data show that functionally active ALDH1a2 was present in the bone marrow, suggesting that RA can be produced in bone marrow and affect DC differentiation in that microenvironment. It has been shown that RA imprints gut tropism to T and B lymphocytes by inducing α4β7-integrin and CCR9 expression in these cells (Iwata et al., 2004; Mora et al., 2006). We found that the addition of RA to BMDC cultures greatly promoted BMDC CCR9 expression, and such RA-DCs migrated towards the CCR9 ligand CCL25, which is constitutively expressed in the small intestine (Campbell and Butcher, 2002; Svensson et al., 2002), indicating that RA can also imprint DCs with gut-homing specificity. When cultured with naïve CD4+ T cells, RA-DCs induced α4β7 and CCR9 expression in T cells, and promoted naïve T cell conversion to Foxp3+ Treg cells. RA-DCs also promoted B cell IgA production. Mucosal DC induction of T cell Foxp3 expression was impaired in VAD mice, indicating that vitamin A and its metabolite RA modulate mucosal DC development and function in vivo. Collectively, our data support the notion that mucosal DCs originate in the bone marrow, and RA, produced locally by bone marrow cells, instructs DCs to initiate differentiation to mucosal DCs both phenotypically and functionally.
Notably, the effect of RA on differentiating mucosal-like DCs occurred within a narrow time window and stringent dose response. When a high dose (1 μM) of RA was added from day 3 of an 8-day BMDC culture, it induced DCs to acquire mucosal DC functions with maximum capability. RA did not confer upon fully differentiated BMDCs or resident splenic DCs the ability to induce T cell Foxp3 expression. This time window and dose effect of RA on BMDC development into mucosal-like DCs in vitro implies that in the bone marrow, RA induction of mucosal-like DCs is tightly regulated spatially and temporally. We speculate that DC precursors will develop into mucosal DCs in selected niches within the bone marrow. The identity of niche ALDH-expressing bone marrow cells is unknown at present, but is the subject of further inquiry.
RA and TGF-β are cofactors in mucosal DC function but their relative roles are unclear. Our data demonstrate that blockade of TGF-β signaling completely inhibited RA-DC induction of T cell Foxp3 expression, but enhanced T cell CCR9 expression. Additionally, the RAR antagonist partially downregulated Foxp3 but almost completely abolished CCR9 expression. These data indicate that TGF-β is responsible for the induction of T cell Foxp3 expression, whereas RA induces CCR9 expression and enhances Foxp3 expression.
STAT3 has been reported to be important in mucosal immune regulation, and STAT3 deficiency in innate cells results in intestinal inflammation (Takeda et al., 1999; Welte et al., 2003). STAT3 can bind to the TGF-β1 promoter and elevate its activity (Kinjyo et al., 2006). Constitutive expression of activated STAT3 in DCs induces TGF-β whereas a dominant negative form of STAT3 suppresses TGF-β production (Kinjyo et al., 2006). SOCS3 regulates immune responses through inhibition of STAT3 activation (Okada et al., 2006; Yoshimura et al., 2007). Thus, STAT3 positively and SOCS3 negatively regulates the production of TGF-β by DCs (Matsumura et al., 2007; Ogata et al., 2006). DCs from SOCS3-/- mice have constitutive activation of STAT3, and produce higher levels of TGF-β than wild type DCs (Matsumura et al., 2007), probably through enhanced STAT3 recruitment to the promoter of TGF-β (Kinjyo et al., 2006). Our data demonstrate decreased levels of SOCS3 expression and an increased and prolonged activation status of STAT3 in RA-DCs. RA suppressed Socs3 gene promoter activity and hence DC SOCS3 expression. Therefore, RA promotes DC TGF-β production by suppressing SOCS3 expression, which releases the inhibition of SOCS3 on STAT3 activation. This is at least one mechanism by which RA induces DCs to produce TGF-β, and thus instructs mucosal DC development. We could not detect CD103 expression by RA-DCs, suggesting that other factors such as epithelial cell-derived signals may contribute to the induction of CD103 in mucosal DCs (Iliev et al., 2009). These data indicate that CD103 is not obligatory for the induction of Foxp3+ Treg cells and T cell gut-homing receptors by mucosal DCs (Jaensson et al., 2008). It is still not known how RA induces DC ALDH1a2 expression and thereby promotes vitamin A conversion to RA itself.
Commensal microbiota critically shape the mucosal immune system, and some organisms may have a greater impact on mucosal immunity than others (Ivanov et al., 2009; Mazmanian et al., 2008; Strober, 2009; Talham et al., 1999). In regard to the effect of gut microflora on DCs, the result of these interactions is rather poorly defined. Our data demonstrate that although RA-mediated induction of Aldh1a2 was independent of TLR-MyD88 signaling, TLR ligation greatly enhanced wild type, but not MyD88-/-, RA-DC Aldh1a2 expression, which revealed a synergistic effect of TLR-MyD88 signaling in conjunction with RA signaling on DC ALDH1a2 induction. Both mucosal DCs and RA-DCs from MyD88-/- mice induced fewer Foxp3+ Treg cells than those from their wild type counterparts. In response to stimulation with different TLR ligands, wild type, but not MyD88-/-, RA-DCs produced a greater amount of bioactive TGF-β. However, MyD88-/- RA-DCs did produce bioactive TGF-β in response to agonistic anti-CD40 antibody at a comparable level to wild type RA-DCs, indicating that TLR-MyD88 signaling and CD40-CD40L interaction are required for optimal TGF-β production by mucosal DCs or RA-DCs, and hence the development of fully functional mucosal DCs.
In summary, our data are consistent with a “multiple-hit-model” for RA-mediated mucosal DC development: mucosal DC precursors encounter RA at the right time in the right microenvironmental niche in the bone marrow, where RA induces mucosal DC precursors to express CCR9. CCR9+ DCs then exit the bone marrow and migrate into the intestine where microbiota via TLR-TLR ligand interactions and signals from epithelial cells further instruct these DCs to develop into fully functional mucosal DCs.
Experimental Procedures
Mice
C57BL/6 and C57BL/6.Foxp3gfp reporter mice were purchased from The Jackson Laboratory. C57BL/6.CBir1 TCR transgenic (CBir1-Tg) mice (Cong et al., 2009a) were bred in the Animal Facility at University of Alabama at Birmingham. Foxp3gfp.CBir1-Tg mice were generated by crossing Foxp3gfp with CBir1-Tg mice. C57BL/6.Socs3flox/flox mice were kindly provided by Dr. Warren Alexander (The Walter and Eliza Hall Institute of Medical Research) (Croker et al., 2003). C57BL/6.Myd88-/- (Myd88-/-) mice were kindly provided by Dr Suzanne Michalek (University of Alabama at Birmingham). All experiments were reviewed and approved by the Institutional Animal Care and Use Committee of University of Alabama at Birmingham.
Antibodies and reagents
Antibodies against mouse phospho-STAT3Tyr705 and STAT3 were purchased from Cell Signaling Technology. Antibodies against mouse ALDH1a2 and Actin were purchased from Santa Cruz Biotechnology. Anti-mouse CD4 (RM4-5), CD11c (HL3), IAb (AF6-120.1), CD80 (16-10A1), CD86 (GL1), CD40 (3/23), α4β7 (DATK32), and the Annexin V-PE Apoptosis Detection Kit were purchased from BD Biosciences. Anti-mouse CCR9 (242503) was purchased from R&D Systems. Anti-mouse Foxp3 (FJK-16s) and intracellular staining kit were purchased from ebioscience.
Live/Dead Fixable stain was purchased from Invitrogen. All-trans RA, SB-505124, and 1-methyl-tryptophan were purchased from Sigma-Aldrich. LE135 was purchased from Tocris Bioscience. Anti-mouse CD40 agonist antibody (FGK45) was affinity purified from hybridoma supernatants provided by Dr. Jan Andersson (Basel Institute for Immunology, Switzerland). Adenovirus expressing GFP and Cre recombinase or GFP alone under control of the CMV promoter was provided by Dr. Rosa Serra (University of Alabama at Birmingham).
Generation and activation of BMDCs
Bone marrow cells were isolated as described previously (Cong et al., 2009b). Briefly, bone marrow cells were suspended at 2.5 × 105/ml in complete RPMI 1640 media containing 10% heat-inactivated FCS (Atlanta Biologicals), 25 mM HEPES buffer, 2 mM sodium pyruvate, 50 mM 2-mercaptoethanol, 100 IU/ml Penicillin, and 100 μg/ml Streptomycin (Cellgro Mediatech). The cells were cultured in the presence of 20 ng/ml GM-CSF (R&D Systems) in 6-well plates at 37°C in 5% CO2 in humid air. RA was added at various time points as described in the text. On day 8, BMDCs were harvested and plated at 1 × 106/ml per well in 24-well plates in the presence of LPS (Sigma-Aldrich), recombinant CBir1 flagellin (Lodes et al., 2004), or CpG ODN (InvivoGen).
Chemotaxis assay
1×106 DCs were resuspended in 100 μl of RPMI medium 1640 and loaded into transwells that were placed in 24-well plates containing 400 μl medium or medium supplemented with 10 μg/ml of CCL25 or CXCL10. After 3 h of incubation at 37°C, the migrated cells were collected and counted. The ratio of the number of DCs that migrated in the presence of chemokine vs. the number of cells that migrated to the PBS control was calculated and given as the migration index.
Quantitative Real-Time PCR
Total RNA was extracted with TriZol reagent and followed by cDNA synthesis with Superscript reverse transcriptase (Invitrogen). Quantitative PCR reactions were performed using TaqMan® Gene Expression Assays for Ccr9, Aldh1a1, Aldh1a2, Socs1, Socs3, and Gapdh (Appliedbiosystems) on Bio-Rad iCycler (Bio-Rad) and all data were normalized to Gapdh mRNA expression.
Isolation of lamina propria DCs and spleen CD4+ T cells
Lamina propria leukocytes were isolated as previously described (Qin et al., 2009). DCs from lamina propria leukocytes were further isolated by magnetic sorting (Miltenyi Biotec). CD4+ T cells were isolated by using anti-mouse CD4-magnetic beads (BD Biosciences).
ALDH activity assay
Cell ALDH activity was determined by using the ALDEFLUOR staining kit (STEMCELL Technologies Inc.) according to the manufacturer's instructions. Briefly, cells were resuspended at 106 cells/ml in ALDEFLUOR assay buffer containing activated ALDEFLUOR substrate with or without the ALDH inhibitor DEAB, and incubated at 37°C for 30 min. ALDEFLUOR-reactive cells were detected in the FITC channel.
TGF-β bioassay
As described previously (Tesseur et al., 2006), MFB-F11 cells are embryonic fibroblasts from Tgfb-/- mice that are stably transfected with a reporter plasmid consisting of TGF-β responsive Smad-binding elements coupled to a secreted alkaline phosphatase reporter gene (SBE-SEAP). MFB-F11 cells, kindly provided by Dr. Tony Wyss-Coray of Stanford University, were seeded at 20,000 cells per well in 96-well plates the day before assay. After incubation in 50 μl of serum-free DMEM for 2 h, 50 μl supernatant of cell samples was added and incubated for 24 h. Secreted alkaline phosphatase (SEAP) activity shown as chemiluminescence unit was measured using Great EscApe SEAP Chemiluminescence kit 2.0 (Clontech) following manufacturer's instructions and represents biologically active TGF-β activity.
Transient transfection of SOCS3 promoter-reporter and luciferase assay
RAW cells (ATCC) were seeded in 12-well plates (1.5 × 106 cells/well) and transfected with the murine SOCS3 promoter (-1492 to +127 bp) using Lipofectamine 2000 (Invitrogen). Transfected cells were treated with medium, IL-6, and/or RA for 12 h, and the luciferase activity of each sample was normalized to the total protein concentration in each well. Luciferase activity from the untreated sample was arbitrarily set at 1 for calculation of fold induction.
Generation of vitamin A-modified mice
Casein-based diets were purchased from Harlan with vitamin A content modified as follows: vitamin A-deficient (VAD) diet, 0.2 IU/g (TD88407); vitamin A-control (VAC) diet, 4 IU/g (TD96007); and vitamin A-high (VAH) diet, 250 IU/g (TD96008). From the second week of pregnancy, pregnant females were fed with the respective diets and maintained throughout the pregnancy and postnatally through weaning. Progeny were weaned and maintained on the same diet.
Statistical analysis
Levels of significance were determined by Student's t test. P values of < 0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Craig Maynard for assisting in the experiments with vitamin A-modified diet-fed mice and Dr. Donald Buchsbaum for offering the ALDEFLUOR staining kit. We also thank Enid Keyser for assistance in cell sorting by flow cytometry. This work was supported by research grants from NIH grants DK071176, DK079918, AI083484, NS50665, NIH 57563, NMSS RG 3892-A-12, NMSS CA1059-A-13, Digestive Diseases Research Development Center grant DK064400, and NIH RR20136.
Footnotes
No authors have conflicting financial interests.
References
- Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, Agace WW, Parker CM, Powrie F. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med. 2005;202:1051–1061. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong L, Stojkovic M, Dimmick I, Ahmad S, Stojkovic P, Hole N, Lako M. Phenotypic characterization of murine primitive hematopoietic progenitor cells isolated on basis of aldehyde dehydrogenase activity. Stem Cells. 2004;22:1142–1151. doi: 10.1634/stemcells.2004-0170. [DOI] [PubMed] [Google Scholar]
- Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, Liu K, Jakubzick C, Ingersoll MA, Leboeuf M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DJ, Butcher EC. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J Exp Med. 2002;195:135–141. doi: 10.1084/jem.20011502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebra JJ. Influences of microbiota on intestinal immune system development. Am J Clin Nutr. 1999;69:1046S–1051S. doi: 10.1093/ajcn/69.5.1046s. [DOI] [PubMed] [Google Scholar]
- Chen W, Frank ME, Jin W, Wahl SM. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–725. doi: 10.1016/s1074-7613(01)00147-9. [DOI] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A. 2009a;106:19256–19261. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Wang L, Konrad A, Schoeb T, Elson CO. Curcumin induces the tolerogenic dendritic cell that promotes differentiation of intestine-protective regulatory T cells. Eur J Immunol. 2009b;39:3134–3146. doi: 10.1002/eji.200939052. [DOI] [PubMed] [Google Scholar]
- Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Iliev ID, Mileti E, Matteoli G, Chieppa M, Rescigno M. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2009;2:340–350. doi: 10.1038/mi.2009.13. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, Berg PL, Davidsson T, Powrie F, Johansson-Lindbom B, Agace WW. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med. 2008;205:2139–2149. doi: 10.1084/jem.20080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, Agace WW. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med. 2005;202:1063–1073. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson-Lindbom B, Svensson M, Wurbel MA, Malissen B, Marquez G, Agace W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med. 2003;198:963–969. doi: 10.1084/jem.20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsall BL, Leon F. Involvement of intestinal dendritic cells in oral tolerance, immunity to pathogens, and inflammatory bowel disease. Immunol Rev. 2005;206:132–148. doi: 10.1111/j.0105-2896.2005.00292.x. [DOI] [PubMed] [Google Scholar]
- Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, Takaki H, Himeno K, Takaesu G, Kobayashi T, Yoshimura A. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006;203:1021–1031. doi: 10.1084/jem.20052333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwah R, Oliver JR, Zhang J, Siminovitch KA, Hu J. Apoptotic dendritic cells induce tolerance in mice through suppression of dendritic cell maturation and induction of antigen-specific regulatory T cells. J Immunol. 2009;183:7104–7118. doi: 10.4049/jimmunol.0900824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, Hershberg RM. Bacterial flagellin is a dominant antigen in Crohn disease. The Journal of clinical investigation. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Kobayashi T, Ichiyama K, Yoshida R, Hashimoto M, Takimoto T, Tanaka K, Chinen T, Shichita T, Wyss-Coray T, et al. Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol. 2007;179:2170–2179. doi: 10.4049/jimmunol.179.4.2170. [DOI] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- Newberry RD, Lorenz RG. Organizing a mucosal defense. Immunol Rev. 2005;206:6–21. doi: 10.1111/j.0105-2896.2005.00282.x. [DOI] [PubMed] [Google Scholar]
- Ogata H, Chinen T, Yoshida T, Kinjyo I, Takaesu G, Shiraishi H, Iida M, Kobayashi T, Yoshimura A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene. 2006;25:2520–2530. doi: 10.1038/sj.onc.1209281. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Pearce DJ, Bonnet D. The combined use of Hoechst efflux ability and aldehyde dehydrogenase activity to identify murine and human hematopoietic stem cells. Exp Hematol. 2007;35:1437–1446. doi: 10.1016/j.exphem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, Reynolds SL, Weaver CT, Roarty K, Serra R, et al. TGF-beta promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W. The multifaceted influence of the mucosal microflora on mucosal dendritic cell responses. Immunity. 2009;31:377–388. doi: 10.1016/j.immuni.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson M, Johansson-Lindbom B, Zapata F, Jaensson E, Austenaa LM, Blomhoff R, Agace WW. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunol. 2008;1:38–48. doi: 10.1038/mi.2007.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson M, Marsal J, Ericsson A, Carramolino L, Broden T, Marquez G, Agace WW. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. The Journal of clinical investigation. 2002;110:1113–1121. doi: 10.1172/JCI15988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Talham GL, Jiang HQ, Bos NA, Cebra JJ. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun. 1999;67:1992–2000. doi: 10.1128/iai.67.4.1992-2000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Berber E, Zhang H, Wyss-Coray T. Highly sensitive and specific bioassay for measuring bioactive TGF-beta. BMC Cell Biol. 2006;7:15. doi: 10.1186/1471-2121-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol C, Vallon-Eberhard A, Elinav E, Aychek T, Shapira Y, Luche H, Fehling HJ, Hardt WD, Shakhar G, Jung S. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31:502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, Forster R. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A. 2007;104:6347–6352. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A. Signal transduction of inflammatory cytokines and tumor development. Cancer Sci. 2006;97:439–447. doi: 10.1111/j.1349-7006.2006.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.