

Abstract
We used high-resolution SNP genotyping to identify regions of genomic gain and loss in the genomes of 212 medulloblastomas, malignant pediatric brain tumors. We found focal amplifications of 15 known oncogenes and focal deletions of 20 known tumor suppressor genes (TSG), most not previously implicated in medulloblastoma. Notably, we identified previously unknown amplifications and homozygous deletions, including recurrent, mutually exclusive, highly focal genetic events in genes targeting histone lysine methylation, particularly that of histone 3, lysine 9 (H3K9). Post-translational modification of histone proteins is critical for regulation of gene expression, can participate in determination of stem cell fates and has been implicated in carcinogenesis. Consistent with our genetic data, restoration of expression of genes controlling H3K9 methylation greatly diminishes proliferation of medulloblastoma in vitro. Copy number aberrations of genes with critical roles in writing, reading, removing and blocking the state of histone lysine methylation, particularly at H3K9, suggest that defective control of the histone code contributes to the pathogenesis of medulloblastoma.
Brain tumors, including medulloblastoma, are the most common solid pediatric malignancies and the leading cause of childhood cancer-related deaths. Survivors are often left with serious cognitive and neurological disabilities resulting from the effects of both disease and treatment on the developing central nervous system. Hereditary tumor syndromes have helped identify mutations of genes in the Sonic Hedgehog, Wnt and TP53 signaling pathways in subsets of medulloblastoma1.
A visual overview of our medulloblastoma copy number data on 201 primary tumors and 11 medulloblastoma cell lines reveals known recurrent regions of large-scale gain and loss, including loss of chromosomes 6, 8, 9q, 10q, 11, 16q and 17p and gains of chromosomes 1q, 7 and 17q (Fig. 1a,b and Supplementary Figs. 1 and 2 online)2. Loss of 17p combined with gain of 17q (isochromosome 17q) was identified in ~28% (59/212) of medulloblastomas (Supplementary Fig. 3 online)3. Our SNP array experiments also allow the prediction of inferred loss of heterozygosity (LOH) across the medulloblastoma genome (Supplementary Fig. 4 online). LOH in medulloblastoma can occur secondary to deletion or in the context of uniparental disomy. We demonstrate that mechanisms leading to LOH in medulloblastoma are chromosome-specific, showing monosomy, uniparental disomy or a mixture of the two mechanisms (Supplementary Fig. 4). The underlying biology driving clonal selection secondary to these extremely large regions of gain and loss is unknown, owing to the difficulty of distinguishing drivers from passengers.
Figure 1.
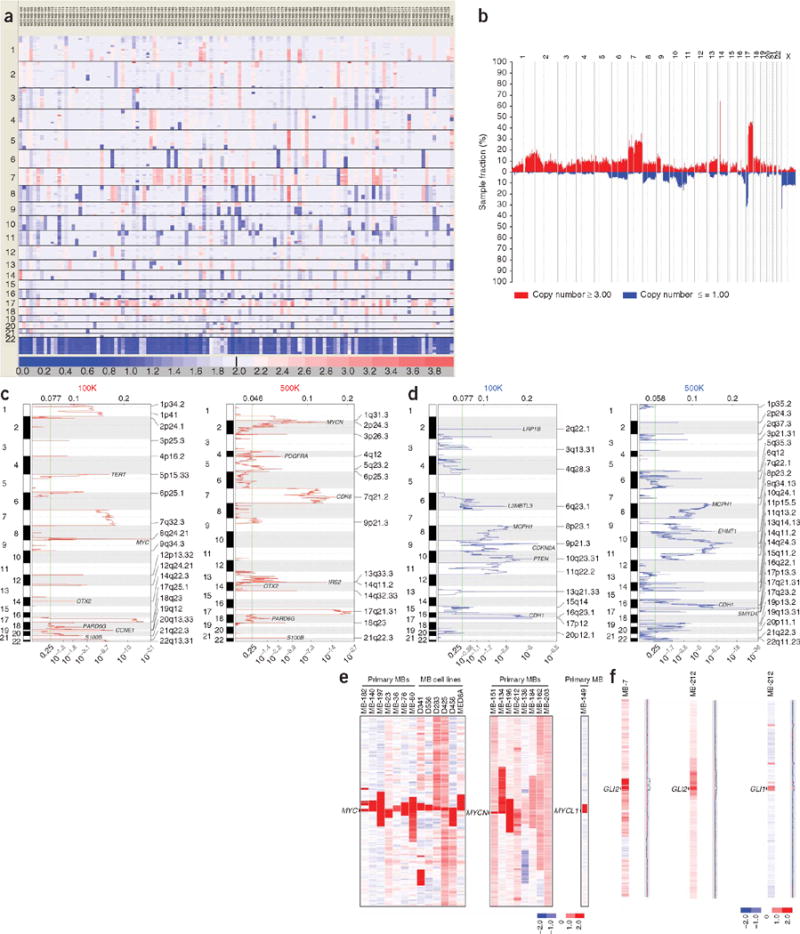
The medulloblastoma genome. (a) Global view of regions of gain and loss across the genome in a series of 123 nonoverlapping medulloblastomas genotyped on the Affymetrix 500K SNP array platform. Output from GenePattern SNP Viewer. Regions of gain are red; regions of loss are blue. (b) Summary plot showing the frequency of regions of gain and loss in the medulloblastoma genome. Output from dChipSNP. Recurrent losses are observed on chromosomes 6, 8, 9q, 10q, 11, 16q, 17p and X. Recurrent gains are observed on chromosome 1q, 7 and 17q. (c) GISTIC output shows significant regions of amplification and gain from nonoverlapping cohorts of medulloblastomas analyzed on the 100K (left) and 500K (right) SNP array platforms. Significant regions are labeled by cytoband, and notable genes are identified. (d) GISTIC output shows significant regions of hemizygous and homozygous deletion from nonoverlapping cohorts of medulloblastomas analyzed on the 100K (left) and 500K (right) SNP array platforms. Significant regions are labeled by cytoband, and notable genes are identified. (e) Amplification of MYC family oncogenes in medulloblastoma (MB) in our cohort of 212 medulloblastomas. Output from dChipSNP. Vertical bars denote MYC family gene loci. (f) Rare amplifications of GLI2 and GLI1 in medulloblastoma, downstream effectors of Sonic Hedgehog signaling. Output from dChipSNP. Vertical bars denote the GLI1 and GLI2 loci.
A major strength of high-resolution SNP platforms is their ability to identify extremely focal regions of amplification and deletion. After eliminating known copy number variants (Supplementary Table 1 online), we identified 139 amplifications and 61 homozygous deletions targeting at least one RefSeq gene (Supplementary Table 2 online). The bioinformatic tool GISTIC identified a number of loci as significantly amplified or deleted, including known medulloblastoma-associated genes such as MYC, MYCN, OTX2, TERT, PDGFRA and CDK6 (Fig. 1c,d and Supplementary Table 3 online)4. We found amplification of other known oncogenes that were not shown previously to be amplified in medulloblastoma (Table 1a). We identified 12 recurrent amplifications, including GLI2, encoding a Hedgehog effector (Fig. 1f), and MYST3, encoding a histone lysine acetyltransferase (Supplementary Table 4 online). Of 20 regions of recurrent focal hemizygous deletion targeting a single gene, 7 (35%) targeted known TSGs (Table 1b). Analysis of 61 homozygous deletions identified 20 known candidate TSGs (Supplementary Table 5 online); 6 of 61 (~10%) homozygous deletions were recurrent (Table 1c). GISTIC also identified significant regions of deletion targeting loci not previously implicated in medulloblastoma, including genes involved in histone lysine methylation such as EHMT1, L3MBTL3 and SMYD4 (Fig. 1d and Supplementary Table 3).
Table 1.
Copy number aberrations in medulloblastoma
| (a) Known oncogenes amplified in medulloblastoma | ||||
|---|---|---|---|---|
| Cytoband | Minimal common region (Mb) | No. samples | Copy number range (HMM) | Known proto-oncogene(s) |
| 1p34.3–p34.2 | 39.432–41.579 | 1 | 9.0 | MYCL1 |
| 1p34.2 | 41.614–43.710 | 1 | 5.3 | MPL |
| 2p24.3 | 16.007–16.156 | 3 | 5.0–14.2 | MYCN |
| 4q12 | 54.826–55.233 | 3 | 5.0–9.7 | PDGFRA |
| 5p13.3–p13.2 | 30.872–36.852 | 1 | 5.0 | SKP2 |
| 7q21.2 | 91.890–92.222 | 1 | 18.3 | CDK6 |
| 8p11.21 | 41.593–42.547 | 2 | 5.1–6.2 | MYST3 |
| 8q24.21 | 128.809–128.809a | 15 | 5.0–17.3 | MYCa |
| 11q24.3–q25 | 127.695–130.314 | 1 | 5.1 | FLI1 |
| 12q13.3–12q14.1 | 55.617–56.498 | 1 | 5.0 | GLI1, CDK4 |
| 13q31.2–q31.3 | 88.641–91.540 | 1 | 10.8 | hsa-mir-17–92 |
| 13q33.3–q34 | 109.098–109.910 | 2 | 5.0–8.0 | IRS2 |
| 14q22.3 | 56.392–56.451a | 2 | 14.9–17.0 | OTX2 |
| 19p13.3–p13.2 | 6.690–7.094 | 1 | 5.0 | VAV1 |
| 19q12 | 34.174–36.744 | 1 | 13.4 | CCNE1 |
| (b) Regions of recurrent, focal hemizygous deletion targeting a single RefSeq | |||||
|---|---|---|---|---|---|
| Cytoband | Minimal common region (Mb) | No. samples | Gene | Homozygously deleted? | Known tumor suppressor? |
| 1p36.22 | 10.242–10.355 | 5 | KIF1B | No | ✔ |
| 1q21.2 | 147.822–147.832 | 5 | BNIPL | No | No |
| 2q22.1 | 141.565–141.930 | 5 | LRP1B | ✔ | ✔ |
| 2q33.2 | 203.686–203.847 | 3 | NBEAL1 | No | No |
| 3p14.3 | 56.730–57.017 | 6 | ARHGEF3 | No | No |
| 3q22.3 | 137.538–137.847 | 3 | STAG1 | No | No |
| 7q11.21 | 64.172–64.513 | 4 | ZNF92 | No | No |
| 8q23.3 | 114.088–114.359 | 4 | CSMD3 | No | No |
| 9p24.3 | 0.517–0.729 | 4 | KANK1 | No | ✔ |
| 9p22.3 | 15.287–15.361 | 3 | TTC39B | No | No |
| 9p21.3 | 21.913–21.972 | 4 | CDKN2A | ✔ | ✔ |
| 11p15.4 | 9.717–9.725 | 8 | SWAP70 | No | No |
| 11q14.1 | 85.109–85.127 | 5 | SYTL2 | ✔ | No |
| 11q24.3 | 128.560–129.016 | 3 | BARX2 | No | No |
| 11q25 | 132.521–132.570 | 3 | OPCML | No | ✔ |
| 16p13.3 | 3.928–4.115 | 3 | ADCY9 | No | No |
| 16q23.1 | 77.223–77.294 | 8 | WWOX | ✔ | ✔ |
| 16q23.2 | 79.891–79.932 | 7 | GAN | No | No |
| 16q23.3 | 81.387–81.559 | 3 | CDH13 | No | ✔ |
| 17p13.1 | 9.125–9.349 | 4 | STX8 | No | No |
| (c) Regions of recurrent homozygous deletion in medulloblastoma | ||||||
|---|---|---|---|---|---|---|
| Cytoband | Minimal common region (Mb) | No. cell lines | No. primary tumors | No. RefSeq | Coding region targeted? | Notable gene(s) |
| 2q22.1 | 141.617–141.909; 142.051–142.090 |
4 | 0 | 1 | ✔ | LRP1B |
| 9p21.3 | 21.913–21.972 | 4 | 0 | 1 | ✔ | CDKN2A |
| 9p21.3 | 23.751–23.765 | 2 | 1 | 1 | ✔ | ELAVL2 |
| 9q34.3 | 137.870–137.927 | 0 | 2 | 1 | ✔ | EHMT1 |
| 11q22.1 | 99.091–99.119 | 0 | 2 | 1 | No | CNTN5 |
| Xp22.33 | 0.677–1.797 | 0 | 2 | 7 | ✔ | CSF2RA, IL3RA |
Minimal common region maps to a single SNP based on array coverage. SNP is adjacent to MYC locus.
Historically, many well-known TSGs were identified through mapping recurrent focal homozygous deletions. Of six recurrent homozygous deletions, only one targeted the coding region of a single RefSeq gene in primary tumors (Table 1c). These recurrent, focal homozygous deletions of EHMT1 on chromosome 9q34.3 were verified in two primary medulloblastomas (Fig. 2a,b); these somatic deletions were not present in matched constitutional DNA (Fig. 2c). EHMT1 is a SET domain–containing histone lysine methyltransferase that dimethylates H3K9, a predominantly repressive chromatin mark (Table 2)5. Expression of EHMT1 transcript was decreased at least twofold in 10% of medulloblastoma samples, particularly tumors monosomic at the EHMT1 locus (Fig. 2d). Immunohistochemical staining for EHMT1 protein expression and H3K9 dimethylation on a tissue microarray of 64 nonoverlapping human medulloblastomas demonstrated that EHMT1 staining was absent in 16 of 64 (25%) medulloblastomas, and nuclear staining for H3K9me2 was not observed in 26 of 64 tumors (~41%) (Fig. 2e). Of 16 tumors with no EHMT1 staining, 15 (~94%) also stained negative for H3K9me2 (P = 0.0024), consistent with a model in which loss of EHMT1 leads to hypomethylation of H3K9 in medulloblastoma.
Figure 2.
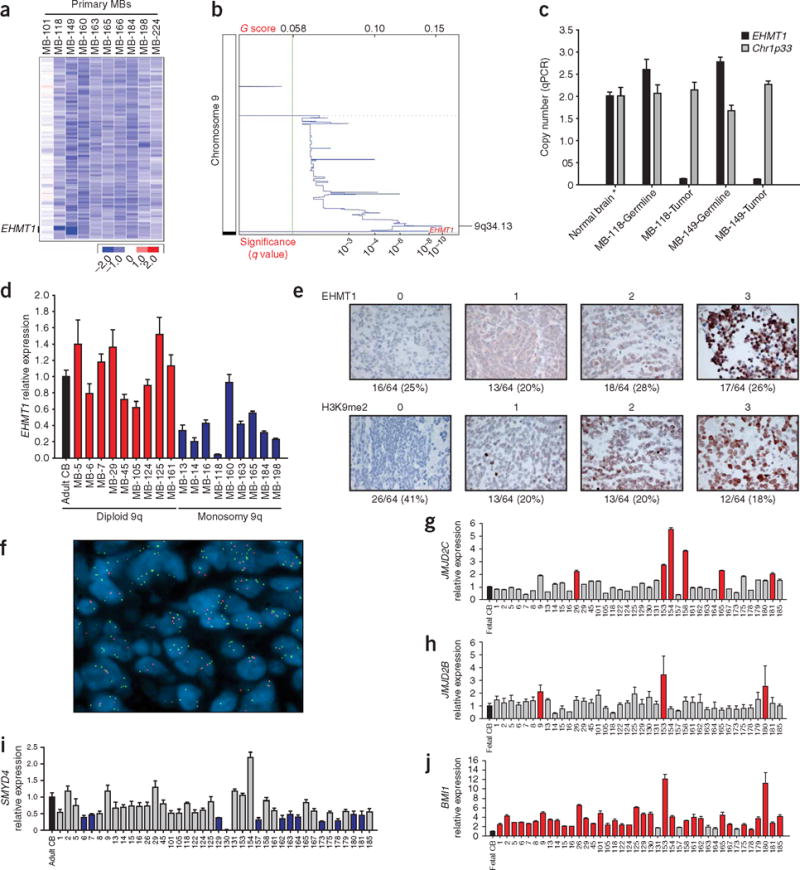
Copy number aberration of genes controlling histone lysine methylation in medulloblastoma. (a) Output from dChipSNP shows focal homozygous deletion limited to EHMT1 in MB-118 and MB-149. MB-101 is diploid for chromosome 9q, whereas MB-160, MB-163, MB-165, MB-166, MB-184, MB-198 and MB-224 are monosomic for 9q. (b) GISTIC output for chromosome 9 (500K SNP array) shows a significant region of focal loss on 9q34 at the EHMT1 locus. (c) Real-time genomic PCR at the EHMT1 locus confirms somatic homozygous deletion in tumor samples, but not in matched constitutional DNA. (d) qRT-PCR for EHMT1 shows significantly decreased expression of EHMT1 in samples with monosomy 9q, as opposed to tumors with diploid chromosome 9q. Two-sample Wilcoxon test, P = 0.0002468. (e) Immunohistochemical staining for EHMT1 expression and H3K9 dimethylation was done on a 64-tumor human medulloblastoma tissue microarray. Staining was graded from 0 to 3 as illustrated. Percentage of tumors in each category is noted below each category. Two-sample test comparing proportions, P = 0.0024. (f) Interphase FISH on paraffin embedded tissues on a medulloblastoma tissue microarray shows amplification of JMJD2C (green) at 9p24.1 as opposed to a control probe (red) at 9q31.2 in a representative medulloblastoma sample. (g) qRT-PCR of JMJD2C shows greater than twofold increased expression in 15% of medulloblastomas as compared to normal fetal cerebellum. Wilcoxon signed rank test, P = 0.1351. (h) qRT-PCR of JMJD2B shows greater than twofold increased expression in 7.5% of medulloblastomas as compared to normal fetal cerebellum. Wilcoxon signed rank test, P = 0.0341. (i) qRT-PCR of SMYD4 shows greater than twofold decreased expression in 30% of medulloblastomas as compared to a normal adult cerebellar control. Wilcoxon signed rank test, P = 2.596e–06. (j) qRT-PCR of BMI1 shows greater than twofold increased expression in >80% of medulloblastomas as compared to normal fetal cerebellum. Wilcoxon signed rank test, P = 9.095e–13. Error bars, ± s.d. See Supplementary Table 6 online for list of primers.
Table 2.
Newly identified genetic events converge on H3K9 in medulloblastoma
| Gene | Cytoband | Function | Observation | No. tumors | Frequency (%) | LOH or polysomy (%) |
|---|---|---|---|---|---|---|
| EHMT1 | 9q34.3 | Histone lysine methyltransferase | Homozygous deletion | 2/122 | 1.6 | 14 |
| SMYD4 | 17p13.3 | Histone lysine methyltransferase | Homozygous deletion | 1/212 | 0.5 | 39 |
| Focal hemizygous deletion | 3/212 | 1.4 | ||||
| L3MBTL2 | 22q13.2 | Polycomb group | Focal hemizygous deletion | 4/212 | 1.9 | 9 |
| L3MBTL3 | 6q22–q23 | Polycomb group | Homozygous deletion | 1/212 | 0.5 | 7.5 |
| SCML2 | Xp22.13 | Polycomb group | Homozygous deletion | 1/212 | 0.5 | 16 |
| Focal hemizygous deletion | 2/212 | 0.9 | ||||
| JMJD2C | 9p24.1 | Histone lysine demethylase | Focal gain | 1/212 | 0.5 | 11 |
| Amplification on TMA (FISH) | 6/82 | 7.3 | ||||
| JMJD2B | 19p13.3 | Histone lysine demethylase | Amplification | 1/212 | 0.5 | 13 |
| Focal gain | 3/212 | 1.4 | ||||
| Amplification on TMA (FISH) | 1/82 | 1.2 | ||||
| MYST3 | 8p11.21 | Histone lysine acetyltransferase | Amplification | 2/212 | 0.9 | 7 |
Total focal events: 19.1%
Three additional homozygous deletions targeted genes with known roles in histone lysine methylation (total: 5/61 (~8%) homozygous deletions). We found focal homozygous and hemizygous deletions of the Polycomb genes L3MBTL3, L3MBTL2 and SCML2 in a subset of medulloblastomas (Table 2). The MBT (malignant brain tumor) domains of these Polycomb proteins function to bind and interpret the degree of histone lysine methylation, particularly at H3K9 (ref. 6). An intragenic homozygous deletion targeted SMYD4, encoding a histone lysine methyltransferase, at 17p13.3, the region most frequently deleted in medulloblastoma (Table 2 and Supplementary Fig. 3). Reanalysis of published data on 244 acute lymphoblastic leukemias and 371 lung adenocarcinomas genotyped on similar platforms did not show any homozygous deletions of EHMT1, L3MBTL3, SCML2 or SMYD4, suggesting that these events are specific to medulloblastoma7,8.
Whereas EHMT1 and SMYD4 methylate histone lysine moieties, Jumonji family proteins function as histone lysine demethylases9. Our SNP array analysis detected amplification and focal gain of JMJD2B (Table 2). Subsequent FISH experiments on a tissue microarray of 88 nonoverlapping medulloblastomas revealed recurrent amplifications (copy number 5–10) of JMJD2B, as well as the known oncogene JMJD2C (Fig. 2f and Table 2)9. These proteins demethylate H3K9 and perhaps H3K36 (ref. 10). We found increased expression of JMJD2C or JMJD2B in 15% and 7.5% of medulloblastomas, respectively (Fig. 2g,h). Similarly, we found recurrent amplification of the histone lysine acetyltransferase gene MYST3 (Table 2), which we predict to result in H3K9 hypomethylation, as H3K9 methylation by EHMT1 is blocked by H3K9 acetylation11. SMYD4 showed twofold decreased expression in 30% of medulloblastomas as compared to normal controls (Fig. 2i). As with the focal genetic events described in Table 2, we predict that these expression patterns should result in hypomethylation of H3K9.
Although individually uncommon, collectively, these focal genetic events targeting genes that control histone lysine methylation were found in 19% of medulloblastomas (Table 2). Copy number aberrations targeting chromatin genes listed in Table 2 were mutually exclusive in our dataset (exact test of a binomial proportion, P = 2.2 × 10−16), suggesting that they have a common function. Published literature and our results (see below) provide strong links between H3K9 methylation and EHMT1, L3MBTL3, L3MBTL2, SCML2, JMJD2C, JMJD2B and MYST3 (refs. 5,6,9,11–14). Lack of nuclear staining for H3K9me2 was seen in 41% of medulloblastomas (Fig. 2e). Thus, proper control of the histone code, particularly methylation at H3K9, is important in the pathogenesis of some medulloblastomas. There was no enrichment for sex, age group or histological subtype in the medulloblastomas with copy number aberrations in chromatin genes analyzed by SNP array or the non-overlapping group analyzed on the tissue microarray (Supplementary Table 2).
EHMT1 is part of the E2F6 complex that preferentially occupies MYC- and E2F-dependent promoters of cells in G0 rather than in G1, suggesting that this complex contributes to silencing in quiescent cells5. There is strong causative evidence for E2F- and MYC-dependent transcription in the pathogenesis of human and murine medulloblastoma (Fig. 1e)15,16, and in the proliferation and differentiation of neuronal progenitors in the external germinal layer (EGL) of the cerebellum, a putative cell of origin for a large percentage of medulloblastomas17–19. Other members of the E2F6 complex include HP1, and MBT domain–containing Polycomb proteins (L3MBTL)5. The MBT domain is so named as Drosophila l(3)mbt mutants have failure of neuronal differentiation and develop invasive, malignant neuronal neoplasms in the larval brain, reminiscent of medulloblastoma20. The polycomb protein BMI-1, which is overexpressed in most medulloblastomas (Fig. 2j), binds both E2F6, and L3MBTL321. Bmi1−/− mice have a hypoplastic cerebellum22, and knockdown of BMI1 decreases growth of medulloblastoma in vitro and in vivo23.
The medulloblastoma cell line DAOY has a homozygous deletion on chromosome 6 that disrupts three genes: L3MBTL3, SAMD3 and TMEM200A (Table 2 and Fig. 3a). This region of chromosome 6 was identified by GISTIC as a significant region of loss in medulloblastoma (Fig. 3b). If loss of expression of L3MBTL3 provided a clonal advantage, we hypothesized that re-expression of L3MBTL3, but not SAMD3 or TMEM200A, should attenuate the malignant phenotype. Stable re-expression of L3MBTL3 in DAOY results in considerably decreased proliferation as compared to controls when assessed by MTS assay and crystal violet staining (Fig. 3c,d). Overexpression of L3MBTL3 in the medulloblastoma cell line D283 resulted in only a minor phenotypic change by MTS assay (Fig. 3e). There was no apparent difference in the incidence of apoptosis in the L3MBTL3 transfectants (Fig. 3f). Flow cytometry of L3MBTL3-expressing DAOY cells showed a marked reduction in the percentage of cells in G1 phase of the cell cycle as compared to controls (49% versus 66%, respectively) (Fig. 3g), and accumulation of cells in S phase (33% versus 21%, respectively), consistent with the known cell cycle effects of ectopic E2F6 expression24. Knockdown of Drosophila l(3)mbt results in diminished H3K9 dimethylation of E2F-responsive promotors14. Chromatin immunoprecipitation experiments show that L3MBTL3-transfected DAOY cells have increased H3K9 dimethylation in the promotors of three genes known to be targeted by the E2F6 complex (Fig. 3h)5. This demonstrates that re-expression of L3MBTL3 can attenuate the malignant phenotype of a medulloblastoma cell line, alter the state of H3K9 methylation in known targets of the E2F6 complex and, along with our genetic data, support a critical role for histone lysine methylation in the pathogenesis of medulloblastoma.
Figure 3.
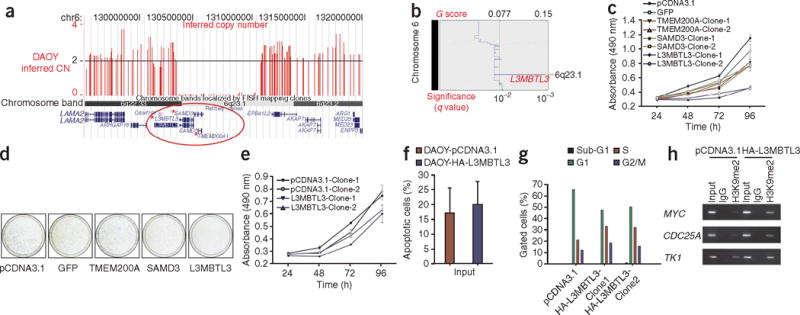
Re-expression of L3MBTL3 in the DAOY medulloblastoma cell line. (a) Output from the UCSC Genome Browser illustrating a homozygous deletion on chromosome 6 that encompasses L3MBTL3, SAMD3 and TMEM200A. Inferred copy number data from SNP array analysis of DAOY was uploaded to the UCSC Genome Browser and is shown in red. (b) GISTIC output for chromosome 6 (100K SNP array) identifies a significant region of extremely focal loss that includes L3MBTL3. (c) Two independent stable transfectants of the DAOY medulloblastoma cell line expressing L3MBTL3 are growth-inhibited as compared to DAOY empty vector, and SAMD3-, TMEM200A- and GFP-expressing controls. (d) Five thousand DAOY cells transfected with either L3MBTL3 or controls were seeded and grown for 7 d. There is greatly reduced growth of the cells re-expressing L3MBTL3 as compared to empty vector control. (e) Overexpression of L3MBTL3 has minimal effect on the growth rate of the D283 medulloblastoma cell line. (f) No sizable difference in the extent of Annexin V labeling is observed in DAOY cells re-expressing L3MBTL3 compared to controls. (g) Flow cytometry analysis of DAOY cells transfected with L3MBTL3 shows a marked reduction in the percentage of cells in G1 as compared to empty vector control. There is also accumulation of cells in S phase of the cell cycle in L3MBTL3 transfectants, as would be predicted in cells with decreased expression from E2F- dependent promoters. (h) Chromatin immunoprecipitation followed by end-point PCR demonstrates that DAOY-L3MBTL3 transfectants show increased H3K9 dimethylation in the promoter regions of the E2F6 target genes MYC, CDC25A and TK1 as compared to controls. Error bars, ± s.d.
Proliferating neural progenitor cells in the outer EGL exit the cell cycle and migrate initially to the inner layer of the EGL, to subsequently form the postmitotic, differentiated neurons of the internal granule cell layer (IGL). EHMT1 is expressed in the developing murine cerebellum at the height of EGL cell proliferation (P7) (Fig. 4a,b). H3K9me2 staining in the EGL is seen predominantly in the inner, postmitotic layer of the EGL and not in the progenitor cells of the outer EGL (Fig. 4c). Furthermore, staining for H3K9me2 colocalizes with the cell cycle arrest protein p27Kip1 (Fig. 4d), demonstrating that H3K9 dimethylation occurs primarily in postmitotic cells. Although the P7 EGL has exuberant H3K9me2, there is no detectable staining for H3K9me1, and only rare mitotic cells in the EGL stain for H3K9me3 (Fig. 4c,e,f). Viral transduction of JMJD2C, but not EGFP in cerebellar EGL cells, resulted in high toxicity, as well as diminished H3K9 dimethylation of cerebellar P7 EGL cells in vitro (Fig. 4g). Viral transduction of NIH 3T3 cells and the medulloblastoma cell line UW228 was not accompanied by the same degree of toxicity, suggesting that toxicity is specific to cell type (Fig. 4h,i). Consistent with this, treatment of cerebellar EGL cells with the histone deacetylase inhibitor trichostatin A results in H3K9 hyperacetylation, H3K9 hypomethlation and high occurences of cell death25. Although the role of H3K9 methylation in cerebellar EGL differentiation has not been directly experimentally addressed, P19 cells cannot undergo retinoic acid–induced terminal neuronal differentiation in absence of HP1, the effector of H3K9 methylation25. These data, the known role of H3K9 methylation in embryonic stem cell differentiation, and the known defects in stem/progenitor cell compartments in L3mbtl3, Jmjd2c, Myst3 and Bmi1 mutant mice are consistent with a model in which proliferative cells in the outer EGL undergo methylation of H3K9 at the time of cell cycle exit, resulting in repression of genes that promote a progenitor cell phenotype26. We hypothesize that failure of physiological H3K9 methylation secondary to loss of E2F6 complex members or erasure/blockade of H3K9 methylation results in failure of transcriptional silencing and promotes cellular transformation in the cerebellar EGL.
Figure 4.
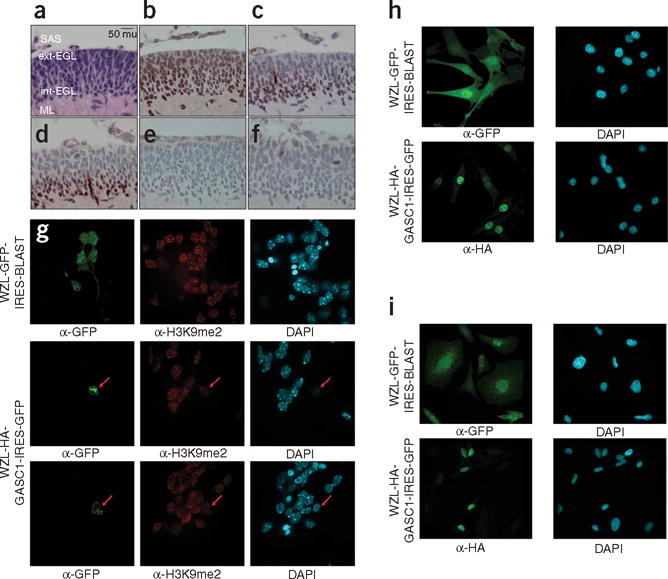
H3K9 in the developing external granule cell layer. (a) Hematoxylin and eosin (H&E) staining of the P7 murine cerebellum. The external layer of the EGL (ext-EGL), the internal layer of the EGL (int-EGL), the molecular layer (ML), and the subarachnoid space (SAS). Original magnification ×400; scale bar, 50 microns (mu). (b) EHMT1 staining of an adjacent section of the external granule cell layer of the cerebellum. EGL cells are a putative cell of origin in medulloblastoma. (c) Dimethylation of histone 3, lysine 9 (H3K9me2) is seen to be more extensive in the inner, postmitotic layer of the cerebellum, with very little staining in the outer, highly proliferative layer of the EGL. (d) Expression of the cell cycle arrest protein p27Kip1 colocalizes with H3K9me2 in the inner EGL. (e) Monomethylation of H3K9 is not seen by immunohistochemistry in the P7 cerebellum. (f) Rare immunohistochemical staining for H3K9me3 is found in a small subset of mitotic cells of the P7 EGL. (g) Retroviral infection of P7 EGL cells with WZL-GFP shows high efficiency of transduction (infection rate >50%), but only rare cells infected with WZL-HA-JMJD2C could be found (infection rate <1%). EGL cells expressing HA-JMJD2C have decreased levels of H3K9 dimethylation. (h) Viral infection of NIH3T3 cells shows high levels of transduction for both WZL-GFP and WZL-HA-JMJD2C. (i) Viral infection of the medulloblastoma cell line UW228 shows high levels of transduction for both WZL-GFP and WZL-JMJD2C. α-GFP, antibody to GFP DAPI, 4,6-diamidino-2-phenylindole.
Our data highlight the genetic heterogeneity of medulloblastoma and support an emerging theme in the literature where mutations of an individual gene are uncommon, but multiple rare genetic events converge on a single common pathway27. Although prior successful targeted therapies for cancer focused on a single mutated gene, therapies based on pathway inhibition may be necessary and effective for some cancers28. Our results link genetic events in brain cancer with epigenetic control of gene expression and strengthen the link between improper control of the histone code and cancer. The recent identification of small molecules targeting histone lysine methylation29, in addition to our genetic and functional data, suggest that manipulating H3K9 methylation should be explored as a targeted therapy for medulloblastoma.
METHODS
Medulloblastoma tumor specimens
We obtained all tumor specimens in accordance with the Research Ethics Board at the Hospital for Sick Children (Toronto, Canada). A total of 201 primary medulloblastomas were obtained as surgically resected, fresh-frozen samples. We obtained tumor specimens from the Co-operative Human Tissue Network (Columbus, OH), the Brain Tumor Tissue Bank (London, Canada) and from our collaborators. Additional clinical details are available in the Supplementary Note online.
100K and 500K GeneChip mapping arrays
Medulloblastoma samples were processed and hybridized to Affymetrix SNP arrays at the Centre for Applied Genomics (TCAG) at the Hospital for Sick Children. We genotyped genomic DNA samples isolated from primary medulloblastomas and cell lines using the Affymetrix 50K Hind 240 and 50K Xba 240, or the 250K Nsp and 250K Sty GeneChip Mapping arrays as directed by the manufacturer. Briefly, 250 ng of DNA was digested with HindIII, XbaI, NspI or StyI (NEB), adaptor-ligated and PCR-amplified using a single primer with AmpliTaq Gold (Applied Biosystems). Amplified PCR products were pooled, concentrated and fragmented with DNase I. Products were subsequently labeled, denatured and hybridized overnight to the respective arrays. Arrays were washed using an Affymetrix fluidics station and scanned using the GeneChip Scanner 3000. We generated CEL files using the Affymetrix GeneChip Operating Software (GCOS) 3.0. See Supplementary Methods online for additional details.
SNP array data processing
Affymetrix CEL files were extracted using the Affymetrix Data Transfer Tool (version 1.1.0). For SNP genotyping, we used the BRLMM Analysis Tool (version 1.0) for individual array platforms using default parameters. Copy number and loss of heterozygosity (LOH) analyses were done using both dChip 2006 and CNAG 2.0. In dChip, we normalized arrays by invariant set normalization and computed signal intensities using PM/MM model-based expression. Raw copy number data was computed using 100 normal control samples as a reference (provided by S.W.S.) and inferred copy numbers were predicted using the hidden Markov model (HMM). We carried out LOH analysis using the HMM considering haplotype method, removing haplotypes consistent with ≥10% of reference samples. In CNAG, nonself analysis was done automatically with the same reference samples as above using a maximum of ten reference samples of the same sex per analysis. Inferred copy number changes and LOH were predicted using the HMM with default parameters.
To identify homozygous deletions, we used the following criteria: (i) ≥3 contiguous SNPs, (ii) size range 1 kb–10 Mb and (iii) mean dChip/CNAG HMM copy number ≤0 or mean dChip raw copy number ≤0.4. To identify amplifications, we used the following criteria: (i) ≥5 contiguous SNPs, (ii) size range 10 kb–10 Mb and (iii) mean dChip/CNAG HMM copy number ≥5. Recurrent, focal single copy losses were reported using the following criteria: (i) ≥3 samples with overlapping interstitial loss (CNAG HMM copy number = 1) and (ii) size range of individual losses 10 kb–5 Mb.
To exclude abnormalities associated with known segmental duplications (LCRs), we compared amplifications and deletions to the LCRs detected in the Human Genome Segmental Duplication Database. Similarly, all amplifications and deletions were compared to known characterized structural variants through comparison with known copy number variants (CNVs) using the Database of Genomic Variants (February 2007). Regions of genomic gain or loss overlapping with known CNVs were eliminated.
To identify regions of statistical significance, raw copy number data was first segmented using GLAD (Gain and Loss Analysis of DNA) and probable CNVs were eliminated on the basis of their overlap with known, recurrent (≥2 samples) CNVs described in the HapMap and POPGEN control populations and/or the Ontario control population. We then analyzed filtered segmented copy number data with GISTIC (Genomic Identification of Significant Targets In Cancer) in GenePattern using default parameters.
Fluorescence in situ hybridization
FISH for JMJD2 family members was carried out on a medulloblastoma tissue microarray as previously published30. BACs used for probes included RP11-1082E7 (JMJD2C, 9p24.1), RP11-235C23 (9q31.2 control), RP11-3214K1 (JMJD2B, 19p13.3), RP11-927F22 (19q13.32 control), RP11-105H7 (19q13.32 control), RP11-5C19 (JMJD2A, 1p34.1), RP11-54H19 (1q22 control) and RP11-336K24 (1q22 control).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipiation of modified histones was done using the Chromatin Immunoprecipitation (ChIP) Assay Kit (Millipore) according to the manufacturer’s instructions. Briefly, 106 medulloblastoma cells were fixed in culture medium with 1% (vol/vol) formaldehyde (VWR International) at 37°C for 10 min, washed twice on ice with cold PBS and collected by centrifugation at 2,000 rpm for 4 min at 4°C. Cells were lysed in SDS lysis buffer for 10 min on ice, sonicated and cleared by centrifugation at 13,000 rpm for 10 min at 4°C. Cell supernatants were diluted in ChIP dilution buffer and pre-cleared for 30 min with Protein A Agarose/Salmon Sperm DNA slurry at 4°C before immunprecipitation with appropriate antibodies overnight at 4°C. Immune complexes were captured by incubation with Protein A Agarose/Salmon Sperm DNA slurry for 1 h at 4°C, before 5 min washes with low salt buffer, high salt buffer, and LiCl buffer, and two 5 min washes with TE buffer. Histone complexes were eluted from primary antibodies by two successive 15-min incubations with elution buffer (1% SDS, 0.1M NaHCO3). Histone-DNA cross-links were reversed by addition of 5M NaCl and heating at 65°C for 4 h, followed by proteinase K digestion for one hour at 45°C. DNA was then recovered by phenol/chloroform extraction and ethanol precipitation with glycogen. Resulting DNA pellets were washed once with 70% ethanol and resuspended in TE buffer. To assess the levels of H3K9me2 at promoters of candidate genes, we carried out end-point PCR reactions using primers targeting the promoter regions of MYC, TK1 and CDC25A.
Supplementary Material
Acknowledgments
We thank the individuals and families who agreed to take part in these studies. We thank S. Egan, J. Ellis and R. Bremner for critical review of the manuscript. We thank R. Wechsler-Reya (Duke), A.M. Kenney (Memorial-Sloan Kettering) and D. Rowitch (University of California San Francisco) for reagents and helpful discussions. We are grateful to K. Helin (University of Copenhagen) for providing JMJD2C expression constructs. We thank P. Paroutis for artwork and S. Archer for editing. We are grateful to C. Marshall and J. Wei for assistance with copy number analysis. This work was supported by the Canadian Cancer Society and the Pediatric Brain Tumor Foundation. M.D.T. is supported by a Sontag Foundation Distinguished Scholar award. Additional research support was obtained from the Hospital for Sick Children Foundation, The Neurosurgery Research and Education Foundation, BRAINCHILD, the 407 Express Toll Route, and the Walker family. M.D.T. was supported by the Laurie Berman fund in Brain Tumor Research, the American Brain Tumor Association and a Clinician-Scientist award from the Canadian Institutes of Health Research. P.A.N. was supported by a Restracomp salary award from the Hospital for Sick Children.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS
P.A.N. coordinated and designed the study, wrote the manuscript, isolated nucleic acids from tumors, performed copy number and LOH analysis and identified regions of interest, validated copy number regions, performed qRT-PCR analyses, generated stable cell lines and retroviruses, and performed in vitro functional assays, immunoblotting, immunofluorescence, and ChIP assays. Y.N. isolated DNA from cell lines, performed copy number and LOH analysis, and validated copy number regions. X.W. built expression constructs and generated stable cell lines. L.F. identified and filtered known CNVs from the datasets. D.W.E. performed FISH and IHC on medulloblastoma TMA. S.C. performed IHC on P7 murine CB and scored medulloblastoma TMAs. S.M. performed GISTIC analysis and validated copy number regions. P.N.K. isolated DNA from tumors, performed copy number and LOH analysis, and validated copy number regions. J.P. built expression constructs, analyzed qRT-PCR data and validated copy number regions. A.D. built expression constructs. Y.S.R. performed mouse experiments. K.Z. validated copy number regions. J.M. performed animal husbandry and mouse experiments. S.W.S. provided technical advice/intellectual contribution. J.S.R. performed statistical analyses. C.G.E. performed IHC on medulloblastoma TMA. W.G., Y.G., B.L., R.G., I.F.P., R.L.H., T.V.M., C.G.C., F.B. and D.B. contributed clinical materials. R.J.G. and J.T.R. provided technical advice, intellectual contribution and helped write the manuscript. M.D.T. designed the study and experiments, interpreted the results, provided supervision, and wrote the manuscript and revisions.
References
- 1.Marino S. Medulloblastoma: developmental mechanisms out of control. Trends Mol Med. 2005;11:17–22. doi: 10.1016/j.molmed.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 2.Reardon DA, et al. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 1997;57:4042–4047. [PubMed] [Google Scholar]
- 3.Bayani J, et al. Molecular cytogenetic analysis of medulloblastomasand supratentorial primitive neuroectodermal tumors by using conventional banding, comparative genomic hybridization, and spectral karyotyping. J Neurosurg. 2000;93:437–448. doi: 10.3171/jns.2000.93.3.0437. [DOI] [PubMed] [Google Scholar]
- 4.Beroukhim R, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F-and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- 6.Klymenko T, et al. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes Dev. 2006;20:1110–1122. doi: 10.1101/gad.377406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 8.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cloos PA, et al. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 10.Shin S, Janknecht R. Diversity within the JMJD2 histone demethylase family. Biochem Biophys Res Commun. 2007;353:973–977. doi: 10.1016/j.bbrc.2006.12.147. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 12.Fodor BD, et al. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howe L, et al. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 2001;15:3144–3154. doi: 10.1101/gad.931401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, Ruhf ML, Perrimon N, Leder P. A genome-wide RNA interference screen identifies putative chromatin regulators essential for E2F repression. Proc Natl Acad Sci USA. 2007;104:9381–9386. doi: 10.1073/pnas.0610279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 16.Olson MV, et al. Transgenic E2F1 expression in the mouse brain induces a human-like bimodal pattern of tumors. Cancer Res. 2007;67:4005–4009. doi: 10.1158/0008-5472.CAN-06-2973. [DOI] [PubMed] [Google Scholar]
- 17.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 18.Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marino S, Hoogervoorst D, Brandner S, Berns A. Rb and p107 are required for normal cerebellar development and granule cell survival but not for Purkinje cell persistence. Development. 2003;130:3359–3368. doi: 10.1242/dev.00553. [DOI] [PubMed] [Google Scholar]
- 20.Gateff E, Loffler T, Wismar J. A temperature-sensitive brain tumor suppressor mutation of Drosophila melanogaster: developmental studies and molecular localization of the gene. Mech Dev. 1993;41:15–31. doi: 10.1016/0925-4773(93)90052-y. [DOI] [PubMed] [Google Scholar]
- 21.Trimarchi JM, Fairchild B, Wen J, Lees JA. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc Natl Acad Sci USA. 2001;98:1519–1524. doi: 10.1073/pnas.041597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung C, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 23.Wiederschain D, et al. Contribution of polycomb homologues Bmi-1 and Mel-18 to medulloblastoma pathogenesis. Mol Cell Biol. 2007;27:4968–4979. doi: 10.1128/MCB.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartwright P, Muller H, Wagener C, Holm K, Helin K. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene. 1998;17:611–623. doi: 10.1038/sj.onc.1201975. [DOI] [PubMed] [Google Scholar]
- 25.Panteleeva I, et al. HP1alpha guides neuronal fate by timing E2F-targeted genes silencing during terminal differentiation. EMBO J. 2007;26:3616–3628. doi: 10.1038/sj.emboj.7601789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas T, et al. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006;20:1175–1186. doi: 10.1101/gad.1382606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romer JT, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Kubicek S, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 30.Thompson MC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.