

Abstract
Nonsense-mediated RNA decay (NMD) represents an established quality control checkpoint for gene expression that protects cells from consequences of gene mutations and errors during RNA biogenesis that lead to premature termination during translation. Characterization of NMD-sensitive transcriptomes has revealed, however, that NMD targets not only aberrant transcripts but also a broad array of mRNA isoforms expressed from many endogenous genes. NMD is thus emerging as a master regulator that drives both fine and coarse adjustments in steady-state RNA levels in the cell. Importantly, while NMD activity is subject to autoregulation as a means to maintain homeostasis, modulation of the pathway by external cues providesa means to reprogram gene expression and drive important biological processes. Finally, the unanticipated observation that transcripts predicted to lack protein-coding capacity are also sensitive to this translation-dependent surveillance mechanism implicates NMD in regulating RNA function in new and diverse ways.
Keywords: gene regulation, noncoding RNA, nonsense-mediated RNA decay, quality control, translation termination
Introduction
The accurate transmission of genetic information from DNA to protein is crucial for cell survival. Due to its importance, surveillance mechanisms have evolved to monitor various steps throughout the gene expression pipeline and to detect and eliminate intermediates that lack integrity or functionality. One particular quality control checkpoint identifies mRNAs that terminate translation prematurely, such as those harboring nonsense codons within their open reading frames (ORFs). mRNAs identified during this process, termed the nonsense-mediated RNA decay (NMD) pathway, are targeted for rapid degradation thereby preventing accumulation of truncated polypeptides that likely lack activity or could have deleterious consequences for the cell [1, 2].
Recognition and targeting of nonsense-containing mRNA by the NMD pathway requires three proteins, UPF1 (UP-Frameshift 1), UPF2, and UPF3, all of which have close homologs throughout eukarya [3]. In addition to this core machinery, several SMG (Suppressor with Morphogenetic effect on Genitalia) proteins are also required for NMD in metazoa and function primarily to regulate the phosphorylation status of UPF1 during substrate recognition and targeting [3]. How these factors coordinate their activities to distinguish premature translation termination from normal termination and induce degradation of target RNAs continues to be the focus of intense study and is discussed in several excellent recent reviews [4, 5]. Despite incomplete mechanistic detail, it is generally agreed that NMD substrate recognition relies upon differences in mRNA ribonucleoprotein (mRNP) composition between normal mRNAs and those harboring a premature termination codon (PTC). To date, three main mRNP features have been distinctly implicated in promoting NMD (Fig. 1). First, multi-protein exon junction complexes (EJCs) deposited on transcripts during pre-mRNA splicing in higher eukaryotes can enhance association and/or activity of UPF1 on an mRNA when they fail to be displaced by ribosomes that terminate prematurely and upstream of the last exon-exon junction [6]. Second, for organisms or mRNAs in which EJCs are absent or do not contribute to NMD substrate recognition, premature translation termination has been posited to be distinct due to an absence of termination promoting signals from the 3′ UTR, specifically from poly(A)-binding protein PAB1 [7–9]. This scheme, referred to as the faux 3′ UTR model, envisions that a compromised interaction between PAB1 and a prematurely terminating ribosome results in less efficient termination, ribosome pausing, and enhanced interaction between UPF1 and translation release factors (eRFs) that then promote downstream events that drive NMD [10]. Substrate discrimination by NMD, however, can occur independently of PAB1 or its interaction with eRFs in yeast [11–13], indicating that additional features of the mRNP contribute to the mRNA being recognized by the NMD machinery. Indeed, recent evidence indicates that UPF1, an RNA binding protein, itself interacts directly with mRNA and that failure of ribosomes to remove UPF1 from the coding region due to premature termination constitutes an important (and a third) mRNP distinction between normal and PTC-containing mRNA [14–18]. Future studies are required to determine how UPF1 bound to mRNA might initiate events leading to NMD and whether additional mRNP components (including EJCs and PAB1) contribute to UPF1 function in this capacity.
Figure 1.
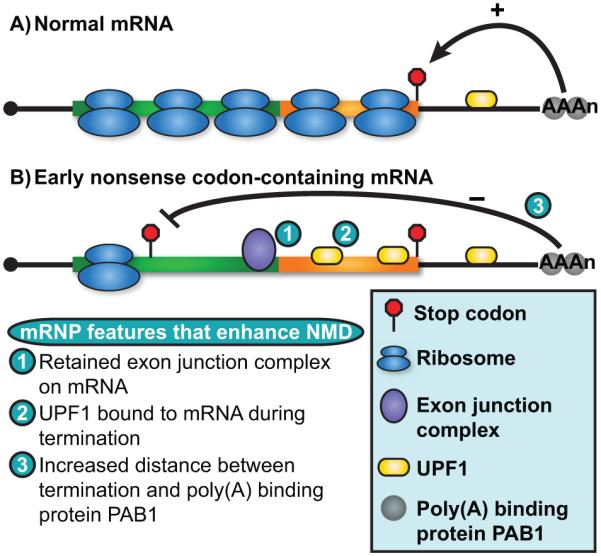
mRNP composition influences NMD substrate recognition. A: Translation of the entire protein-coding region of a normal mRNA by ribosomes serves to remodel the mRNP and clear RNA-binding proteins from this region of the RNA. Termination proximal to the poly(A) tail and poly(A) binding protein PAB1 is postulated to provide positive signals indicating termination is occurring within an appropriate mRNP context. B: mRNAs harboring an early stop codon undergo premature translation termination and, as a consequence, have an increased length of RNA downstream that fails to be remodeled and serves as a platform for a multitude of RNA binding proteins including several known to influence NMD substrate recognition, including the multi-protein exon junction complex deposited at exon junctions (fusion point of green and orange boxes) during pre-mRNA splicing (1) and the NMD factor UPF1 (2). Additionally, ribosomes terminating prematurely and distal to the poly(A) tail and PAB1 would fail to receive termination promoting signals thus indicating that termination is aberrant (3).
While initially branded as a surveillance mechanism to eliminate aberrant mRNA from the cell, recent observations indicate that NMD modulates steady-state levels of a large subset of endogenous transcripts, and that the coupling of NMD to regulated alterations in mRNA transcription, processing, and translation provides a robust approach to rapidly and broadly modulate gene expression. Herein, we describe events during gene expression that lead to production of mRNA isoforms with distinct sensitivities to the NMD pathway, and discuss how NMD might contribute to the function of a new and unexpected class of substrates, long noncoding RNA (lncRNA; highlighted in Box 1). Finally, we discuss how NMD functions to both switch-off and fine-tune mRNA levels to alter gene expression in response to signals and under conditions in which NMD itself is modulated.
Box 1. Long noncoding RNAs emerge as an unexpected class of NMD substrates.
Recent genome-wide expression studies to elucidate NMD substrates have led to the surprising observation that the NMD pathway also modulates levels of a number of long noncoding RNAs (lncRNAs). As a class of RNA predicted to lack protein-coding capacity [106], lncRNAs were a priori anticipated to avoid engagement with the translational machinery and therefore escape translation-dependent surveillance by NMD. In contrast to this expectation, a number of studies profiling polyribosome-associated RNAs and ribosome binding have revealed that many lncRNAs do, in fact, associate with the translation machinery in a number of organisms including budding yeast [31, 35, 107, 108], fission yeast [37], Arabidopsis [104], Drosophila [109], zebrafish [110, 111], mouse [34], and human [111–113]. Although it has been debated whether ribosome binding, particularly as detected by global ribosome profiling, provides conclusive evidence for active translation [114], the most recent study to specifically address this concern support the presence of ribosomes actively engaged in translation on lncRNA transcripts [115]. Moreover, several studies have provided experimental evidence of productive translation through biochemical detection of polypeptides predicted by ribosome profiling to be expressed from lncRNAs [31, 109, 115].
Based on findings that a subset of lncRNAs are translated, it is perhaps not surprising that the steady-state level of a number of these transcripts responds to inhibition of the NMD pathway. Indeed, a small number of individual lncRNAs in yeast [116, 117] and mammals [118] were previously shown to be sensitive to NMD. What was perhaps unanticipated are findings from genome-wide analyses indicating that as many as 17% of lncRNAs in Arabidopsis, budding yeast, and mouse embryonic stem cells are modulated by NMD [15, 31, 104]. Inspection of NMD-sensitive lncRNAs indicate that many harbor short 5′ roximal ORFs with long regions downstream of the stop codon unprotected by ribosomes [31, 104]. Given that a long 3′ UTR represents a feature sufficient to target mRNA to NMD, translation termination upstream of long ribosome-free regions provides a mechanistic explanation for how these lncRNAs are recognized and targeted by the NMD pathway.
The discovery that a substantial fraction of lncRNAs actively engage the translation machinery has spurred a number of studies aimed at characterizing these RNAs and determining whether their putative protein products have biological roles in the cell. Although experimental validation of protein expression is lacking for a majority of lncRNAs predicted to encoded ORFs, several individual annotated lncRNAs in Drosophila and zebrafish express peptides with documented roles in signaling and development [119–121], providing precedent for functional protein expression from lncRNAs. For these lncRNAs, reclassification of the gene locus as protein-coding may be warranted (see accompanying Figure) [122], and it is likely that future studies will expand this list as biological roles for peptides predicted to be encoded within additional lncRNAs are uncovered.
For lncRNAs whose levels are modulated by NMD, regulation by this pathway will have distinct consequences depending upon the role translation plays in the life of the RNA (see accompanying Figure). For example, for lncRNAs expressed as a consequence of spurious or “noisy” transcription, NMD may work alongside other cellular decay pathways to clear these unproductive transcripts from the cell. In the absence of an active mechanism for retaining these RNAs in the nucleus, the transcripts would be expected to be transported to the cytoplasm and stochastically engage the translation machinery as a consequence of their structural similarity to mRNA [i.e., a 5′ cap structure and 3′ poly(A) tail]. In contrast, the coupling of translation and NMD may serve to modulate the cytoplasmic level and function of a subclass of lncRNAs. For example, mammalian GAS5 lncRNA is constitutively ribosome associated and sensitive to NMD. Inhibition of NMD, however, results in stabilization of GAS5 lncRNA and an increase in its function in growth arrest [118]. In light of observations that the translation status of a lncRNA may be regulated, such as during meiosis in budding and fission yeast [35, 37], a shift in translation would alter the fraction of a lncRNA species targeted to NMD and provide a mechanism to fine-tune its steady-state level. For these lncRNAs, regulation of translation could provide an important means to maintain low basal levels of the RNA in the cytoplasm until its activity is required by the cell, or as a means for clearing the cell of a large percentage of the lncRNA transcripts rapidly and simultaneously [35, 37]. Finally, for some lncRNAs, NMD may help limit accumulation of nonfunctional or deleterious peptides produced during de novo evolution of protein-coding genes [107, 108]. Translation of lncRNAs and their targeting to NMD may permit sampling of new ORFs but with the benefit of minimizing the accumulation of deleterious cryptic peptides.
Finally, NMD may provide an important mechanism to ensure that a lncRNA functions strictly as a regulatory RNA in the nucleus (see accompanying Figure). For lncRNAs whose biological role it is to modulate gene expression though mechanisms such as transcriptional control or chromatin modification, targeting these transcripts for rapid decay by NMD provides an efficient means to reduce steady-state RNA levels in the cytoplasm. NMD would, in effect, limit their accumulation outside the nucleus and establish a concentration gradient in the cell that reflects the molecular function of the transcript [106, 123]. Consistent with the establishment of a gradient, transcriptome analysis of cellular fractions has demonstrated that NMD-sensitive mRNA isoforms are enriched in nuclear versus cytoplasmic compartments relative to normal mRNAs [124]. Rapid degradation of cytoplasmic lncRNAs would, moreover, ensure that a lncRNA population remains predominantly noncoding.
RNA expressed from both mutant and wild-type genes is targeted to NMD
NMD was originally characterized from observations in budding yeast and nematodes that transcripts expressed from genes harboring nonsense or frameshift mutations are rapidly degraded, and that inactivation of a number of UPF or SMG genes selectively stabilizes these mRNAs [2, 19, 20]. These findings were consistent with earlier studies reporting accelerated degradation of PTC-containing b-globin mRNA from thalassemia patients [21], and suggested that a mechanism to recognize mRNAs in which translation of the entire length of the protein coding region is prevented was highly conserved. Classical NMD substrates thus represent transcripts expressed from genes harboring either nonsense mutations in the annotated ORF or nucleotide insertions/deletions that shift the translation reading frame and redirect translation termination to occur upstream of the natural stop codon (Fig. 2A).
Figure 2.

Nonsense codons can be introduced by genetic mutation or by errors or variation in transcription by RNA polymerase II. A: Genomic mutation (red bar) resulting in the introduction of a nonsense codon or frameshift in the open reading frame (ORF; green box) leads to 100% of the mRNA expressed from that locus containing a PTC at a fixed position (left). In contrast, nucleotide misincorporation during transcription by RNA polymerase II generates mRNA harboring PTCs at a frequency of <0.5% at variable positions within the mRNA ORF. B: Variations in transcription initiation site (left) create heterogeneity in the length of the mRNA 5′ UTR, impacting translation initiation and reading frame choice (discussed in detail in the text and see also Fig. 4). Similarly, the use of alternative 3′ end formation sites (right), due to mutations in cleavage and polyadenylation signals or regulated alternative polyadenylation (APA), generates heterogeneity in 3′ UTR length and differential susceptibility of the mRNA to NMD.
In addition to being genetically encoded, PTCs can be introduced into an mRNA co-transcriptionally as a consequence of nucleotide misincorporation by RNA polymerase II (Fig. 2A). While rare, errors in nucleotide addition during transcription occur at an estimated frequency of 10−3–10−4 and would be predicted to introduce a PTC into the ORF of a small percentage (<0.5%) of transcripts expressed from each gene [22]. Importantly, in contrast to genetic mutations in which 100% of expressed transcripts would harbor a PTC, degradation of these transcripts by the NMD pathway would alter steady-state mRNA levels only minimally and without anticipated biological consequence.
While initial studies implicated NMD in degrading transcripts expressed from aberrant genes, additional classes of NMD targets where premature translation termination occurred not due to gene mutation but rather as a consequence of alterations in mRNA processing or translation were promptly uncovered (discussed in detail below). The identification of NMD targets has been greatly facilitated by global gene expression profiling, and studies in a number of model eukaryotes have revealed that up to 10% of genes express transcripts whose steady-state levels change as a consequence of inhibition of the NMD pathway [23, 24]. Critically, these analyses indicate that a majority of substrates targeted to NMD lack mutations in the protein-coding region of their corresponding annotated gene.
Initial transcriptome-wide studies to identify NMD substrates employed microarrays to monitor RNA abundance changes in response to NMD inactivation. Presently, high-throughput RNA sequencing approaches are being employed which provide increased sensitivity in quantifying changes in transcript levels, and the ability to classify RNA at nucleotide resolution and distinguish distinct isoforms expressed from single genes. Despite advances in identifying RNAs whose levels respond to NMD inhibition, steady-state analysis is unable to distinguish mRNAs that are primary substrates of the pathway from secondary targets whose levels change in response to perturbations in RNAs directly targeted to NMD. To overcome this limitation and bias identification towards direct NMD targets, RNAs whose levels are initially and rapidly down-regulated upon re-activation of NMD [25, 26] or which physically associate with UPF1 [15, 16, 18, 25, 27, 28] have been characterized. Additional studies have measured mRNA stability to classify transcripts whose decay rate slows in the absence of NMD—the gold standard for establishing an mRNA as an NMD substrate [28, 29]. Interestingly, these studies have identified a number of RNAs whose stability increases in the absence of NMD despite unchanged steady-state levels [26, 28], highlighting that analyses based solely on mRNA abundance measurements not only over-estimate the number of NMD substrates (by identifying both direct and indirect targets), but also fail to identify bona fide targets of the pathway. To aid in the assembly of a comprehensive list of NMD substrates, transcript profiling has recently been coupled to additional global analyses. For example, transcript-wide UPF1 binding sites have been identified in mammalian cells using cross-linking and immunoprecipitation [15, 16, 18], while analysis of ribosome association and translation by ribosome profiling [30] has, in some cases, provided insight into the molecular rationale for NMD target recognition [15, 31].
Transcript 5′ and 3′ end heterogeneity generates substrates for NMD
Alternative gene promoter usage by RNA polymerase II during transcriptional initiation produces RNA isoforms with 5′ untranslated region (UTR) sequences of distinct lengths and nucleotide composition (Fig. 2B). Variation in 5′ UTR identity can, in turn, influence mRNA association with the translation machinery, the site of translation initiation, and the frame read by translating ribosomes. Genome-wide approaches to characterize mRNA 5′ ends, including cap analysis of gene expression (CAGE; [32]) and transcript-leader sequencing [33], have identified transcriptional start sites at nucleotide resolution in a number of organisms and revealed that substantial heterogeneity exists at the 5′ end of mRNAs expressed from many eukaryotic genes. Ribosome profiling and polyribosome association have, moreover, shed light on how 5′ UTR length impacts the engagement of mRNA by ribosomes [15, 34–38]. Specifically, short 5′ UTRs promote inefficient use of 5′ proximal AUG codons resulting in initiation biased towards downstream AUG codons and translation in alternative reading frames [33]. Alternatively, mRNAs with extended 5′ leaders often encode upstream ORFs (uORFs) which, when engaged by the translation machinery, may lead to targeting of the transcript by NMD [25, 33–35, 39, 40]. Thus, 5′ UTR length and composition can dramatically alter the association between the translation machinery and mRNA, and lead to translational recoding and targeting of the mRNA to NMD. Critically, promoter usage and transcriptional start site selection respond to a variety of external cues [40, 41], suggesting that when coupled to NMD, alterations in this early event in mRNA biogenesis can have profound effects on mRNA expression post-transcriptionally.
Cleavage and polyadenylation site choice during 3′ end processing of nascent transcripts also generates heterogeneity in the form of mRNA isoforms with different 3′ UTR lengths that display distinct sensitivities to the NMD pathway (Fig. 2B). Indeed, mutations which alter cis-acting cleavage and polyadenylation signals were first shown in yeast and nematodes to result in mRNAs with extended 3′ UTRs that are rapidly degraded by NMD, despite termination by ribosomes at the natural stop codon [2, 42]. The increased sequence length between the stop codon and 3′ end of the RNA is thought to redefine the natural termination event as premature, perhaps by providing a platform for distinct mRNP associations with the mRNA. Consistent with this, mRNAs with naturally occurring but atypically long 3′ UTRs are targeted by NMD in a number of eukaryotes [15, 26, 27, 39, 43–46].
Although 3′ UTR length represents a common mRNA feature that increases the sensitivity of a transcript to NMD, no straightforward linear correlation between length and mRNA destabilization has emerged, and a number of mRNAs with long 3′ UTRs are immune to NMD [9, 39, 43, 45]. While it is unclear how these transcripts evade recognition by the NMD pathway, recent findings in yeast have revealed that ORF length inversely correlates with sensitivity of an mRNA with a long 3′ UTR to NMD [47]. Additionally, viral RNA structural elements such as the Rous Sarcoma Virus RNA Stability Element provide a means for mRNAs to evade detection by the NMD pathway (reviewed in [48]). In both cases, mechanistic details of how mRNAs with long 3′ UTRs escape recognition by NMD are lacking, and further investigation into this subclass of mRNAs is needed.
Mutation of a canonical 3′ cleavage and polyadenylation site that leads to transcriptional readthrough and production of mRNA isoforms with lengthened 3′ UTRs represents a case of aberrant 3′ UTR variation. Alternative cleavage and polyadenylation site usage (APA), in contrast, represents a highly regulated RNA processing event in eukaryotes that significantly contributes to mRNA 3′ UTR length heterogeneity. Indeed, genome-wide analyses suggest 30–70% of genes undergo APA during transcription termination to produce mRNA isoforms that differ in 3′ UTR length (reviewed in [49]). It has been proposed that APA provides an important strategy for temporal and cell-specific regulation of large subsets of mRNA by modulating inclusion of binding sites in the 3′ UTR for microRNAs and regulatory RNA binding proteins (reviewed in [50, 51]). However, because long 3′ UTRs can trigger NMD, mRNA isoforms with extended 3′ UTRs generated by this processing event would be predicted to have increased sensitivity to NMD, leading to down-regulation when distal cleavage and polyadenylation sites are used. Global shortening of mRNA 3′ UTR length by APA observed in tumorigenic or highly proliferative cells [52] may, in contrast, provide a means for mRNAs to circumvent targeting by NMD and promote cellular proliferation and/or disease. Conceptually, the coupling of APA and NMD provides a clear course to regulate gene expression, however, few specific instances of a single gene producing multiple 3′ UTR isoforms with differential sensitivity to NMD have been documented [53]. Additional studies will therefore be needed to extend our understanding of how APA influences mRNA stability in general, and to appreciate the contribution of APA-associated NMD in gene regulation [54].
Nuclear pre-mRNA splicing generates a rich pool of NMD-sensitive mRNA isoforms
Nuclear pre-mRNA splicing provides the cell with a robust means to enhance protein diversity; however, this step in RNA processing also commonly results in the introduction of PTCs into spliced mRNA (Fig. 3). For example, failure of the spliceosomal machinery to recognize and remove intervening intronic sequences, often as a consequence of weak consensus splice sites, can lead to inclusion of entire introns [55–57]. Moreover, cryptic 5′ or 3′ splice site usage often results in processed mRNA containing partial intronic sequences [58]. Because introns exhibit a bias towards harboring in-frame nonsense codons [59], retention of all or part of an intron in mature mRNA routinely introduces one or more nonsense codons leading to its targeting by NMD. Additionally, intron retention may block export of the RNA from the nucleus, thereby preventing its recognition by NMD in the cytoplasm but nonetheless promoting a reduction in RNA levels due to enhanced turnover by nuclear RNA degradation pathways [39, 56, 60].
Figure 3.
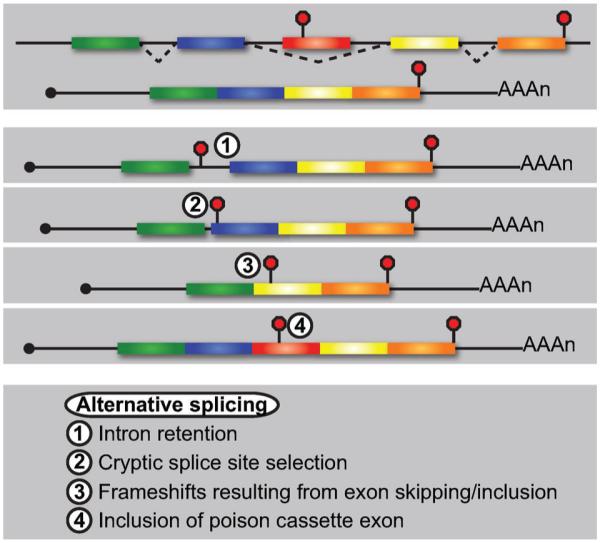
Alternative pre-mRNA splicing generates substrates for NMD by a variety of means. Exon skipping during pre-mRNA splicing generates an mRNA void of early nonsense codons and whose ORF is fully translated (top). Intron retention commonly results in inclusion of an in-frame PTC into the mature mRNA (1). Similarly, alternative 5′ or 3′ splice site usage leads to partial intron retention (2) and/or shifts in the translational reading frame that result in premature translation termination. Skipping (3) or inclusion of an exon of a nucleotide length that is not divisible by 3 results in frameshifts and premature termination. Finally, inclusion of a PTC-containing exon (i.e., a poison cassette exon) introduces a genomically-encoded premature termination codon and targets the splice variant for NMD (4).
Differential incorporation of exons during pre-mRNA splicing produces transcripts lacking single or multiple exons or isoforms with exonic sequences not typically present in the mature mRNA; these events can lead to the introduction of a PTC into the mature mRNA in a number of ways (Fig. 3). For example, exon skipping may cause shifts in the reading frame that introduce PTCs and target the isoform to NMD. Alternatively, inclusion of a cassette exon harboring an in-frame nonsense codon—commonly referred to as a poison exon—generates mRNA isoforms efficiently recognized by the NMD pathway when included upstream of the last exon [61].
Although once believed to serve only to eliminate unproductive RNAs generated as a consequence of stochastic errors in splicing, the targeting of mRNA isoforms generated by alternative splicing to NMD is now thought to provide the cell with a potent means for down-regulating gene expression in response to developmental, environmental, or cell-type specific cues (reviewed in [61, 62]). Indeed, the coupling of regulated alternative splicing and NMD (AS-NMD) has been shown to play an important role in mRNA isoform-specific expression across metazoa [39, 45, 60, 63–70]. Alternative pre-mRNA splicing may, in fact, represent the most prominent means of generating NMD substrates in higher eukaryotes, with an estimated one-third of all alternative splicing events producing an mRNA isoform containing an in-frame PTC [67, 71].
Interestingly, a number of splicing factor mRNAs are themselves targets of regulation by AS-NMD as part of an auto-regulatory cycle conserved throughout metazoa that enables NMD to broadly impact expression of alternative splice isoforms indirectly (reviewed in [72]) [44–46, 63, 66, 67, 69, 73]. For example, transcripts for several SR (serine/arginine-rich) proteins mediating alternative splicing harbor poison cassette exons encoded within ultraconserved elements which, when maintained in the mature RNA due to elevated SR protein levels, lead to targeting of the isoform to NMD and down-regulation of protein expression [69, 74]. Moreover, the tissue-specific splicing regulators PTBP1 and PTBP2 participate in alternative splicing of their own transcripts and promote production of mRNA isoforms targeted to NMD [75, 76]. The regulation of splicing factor expression by AS-NMD reveals that the NMD pathway is an important participant in a negative feedback circuit that buffers the overall extent of alternative splicing in the cell.
Translational recoding alters the site of translation termination
Translation of a reading frame distinct from the annotated protein-coding region within an mRNA can be established during either translation initiation or ribosome elongation, and almost invariably directs ribosomes to terminate translation prematurely relative to the position of the natural stop codon (Fig. 4). Initiation of translation at an out-of-frame start codon may occur either upstream or downstream of the annotated start. Indeed, high GC content within 5′ UTRs can promote initiation at upstream, non-AUG codons due to slowed 48 S ribosomal complex scanning caused by RNA secondary structure [77]. Alternatively, scanning 48 S complexes may fail to recognize and initiate translation at the most 5′ proximal AUG as a consequence of a weak or unfavorable nucleotide context surrounding the start codon, a process termed leaky scanning [78]. Ribosome profiling has recently revealed that mRNAs from >1% of human genes are translated in more than one reading frame, a frequency much higher than previously appreciated [34, 36]. Whether mRNA decoding in alternative reading frames occurs due to initiation at out-of-frame AUG start codons or through the use of near cognate, non-AUG codons, which has been suggested to be prevalent in yeast and mammalian cells [34, 35, 38], remains unclear. In light of the consideration that translation of alternative ORFs commonly results in premature translation termination and targeting of the transcript for rapid decay by NMD, experimental detection of dual decoding events by ribosome profiling is likely hampered by low steady-state levels of these mRNAs. Translation initiation in alternative reading frames may, therefore, still be under-estimated and represent a relatively prevalent mechanism for targeting mRNA to the NMD pathway.
Figure 4.
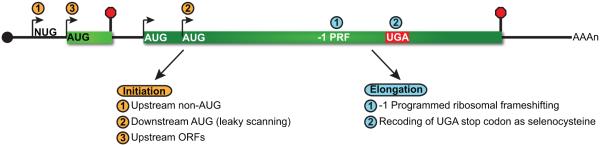
Translational recoding can impact the position of translation termination. The position of translation initiation by 80 S ribosomes can be influenced by a variety of means and impacts the frame read by the translational machinery (left). The use of upstream, non-AUG codons for translation initiation (1) or downstream AUG codons due to leaky scanning (2), can result in translation of alternative reading frames and termination upstream of the natural stop codon. Additionally, translation of upstream open reading frames (uORFs) within the 5′ UTR (3) also leads to premature termination and targeting of the mRNA to NMD. Translational recoding during translation elongation (right) can occur as a consequence of programmed ribosomal frameshifting (1) or incorporation of selenocysteine at upstream UGA in selenoprotein mRNAs (2). The former event often directs translating ribosomes to nonsense codons in the −1 reading frame, while the latter facilitates bypass of the PTC and averts targeting of the mRNA to the NMD pathway. The extent by which translational recoding influences the susceptibility of an mRNA to NMD will depend on the frequency of the recoding event.
Translation may also initiate at alternate ORFs encoded within 5′ UTRs of mRNA (i.e., uORF; Fig. 4). uORF translation provides a mechanism to modulate expression of the downstream annotated ORF through a variety of means (reviewed in [79]), including decreased mRNA stability through NMD. Since uORF decoding leads to translation termination upstream of the natural stop codon, it is perhaps unsurprising that uORF-containing mRNAs are enriched as NMD substrates throughout eukaryotes [15, 29, 44, 46, 64, 66, 68, 70, 80, 81]. Interestingly, it is likely that the number of regulatory uORFs encoded within mRNA 5′ UTRs has also been underestimated, since these reading frames have traditionally been defined based on translation initiation at canonical AUG start codons, which does not account for translation initiation at near-cognate start codons [34, 35, 38].
In addition to initiating translation in an alternate frame, translational recoding can occur during ribosome elongation though programmed ribosomal frameshifting (PRF; Fig. 4). In the most well-documented form of PRF, elongating ribosomes stall and shift backwards a single nucleotide into the −1 frame as a consequence of two cis-acting RNA signals that include a “slippery sequence” and downstream pseudoknot structure [82]. −1 PRF signals are common within eukaryotes, with recent predictions indicating 8–12% of genes contain at least one set of −1 PRF signals [83]. Critically, the vast majority of eukaryotic −1 PRF events direct translation termination to occur upstream of the natural stop codon [84] causing destabilization of the mRNA through NMD [85–87]. While computational prediction of −1 PRF signal motifs identifies transcripts with increased likelihood of promoting translational frameshifting, ribosome profiling is being used to facilitate identification of signals that function in vivo and provide insight into the fraction of ribosomes which shift in response to these signals and under varying conditions [36, 86]. This latter information is critical for estimating the proportion of transcripts from a gene harboring a −1 PRF signal that will be targeted to the NMD pathway.
Finally, in contrast to examples where the frame read by elongating ribosomes is altered, translational recoding also occurs when a stop codon directs incorporation of an amino acid rather than serving as a translation termination signal. For selenocysteine-containing proteins, an early UGA stop codon within their mRNAs—in conjunction with additional cis- and trans-acting factors—serves redundantly to also direct incorporation of the non-standard amino acid selenocysteine (Sec; Fig. 4). Under conditions when environmental selenium is readily available, charged selenocysteine tRNA (Sec-tRNA) is abundant and efficiently competes with translation release factors to decode UGA and suppress early termination. In contrast, in low selenium conditions, limiting charged Sec-tRNA levels result in increased translation termination and enhanced targeting of the mRNA to NMD [88, 89]. The proportion of a selenoprotein mRNA sensitive to NMD, therefore, is modulated by competition between translation termination and selenocysteine incorporation, dictated by selenium levels in the environment and the abundance of charged Sec-tRNA in the cell. Importantly, stability of selenoprotein mRNA is influenced largely by the position of the Sec UGA codon within the mRNA ORF, and mRNAs display the greatest sensitivity to NMD when the UGA is present upstream of the last exon-exon junction [89]. Indeed, differential sensitivity of selenocysteine mRNAs to NMD based on Sec UGA codon position contributes to preferential expression of a subset of selenoproteins in conditions of low selenium. These largely NMD-insensitive mRNAs thus maintain a high relative steady-state level, facilitating competition for limiting Sec-tRNA and favoring expression of selenoproteins whose function may be most needed by the cell [89].
The shaping of gene expression by NMD is both protective and regulatory
The NMD pathway provides cells with an efficient and rapid mechanism to eliminate aberrant mRNA and prevent accumulation of potentially deleterious truncated protein products. In cases where the aberrant mRNA is expressed from a mutant gene, 100% of transcripts will harbor the NMD targeting feature and their degradation acts as a switch to abrogate expression (Fig. 5A). While NMD inarguably plays an important role in mRNA surveillance, aberrant transcripts generated as a consequence of mutation or errors during gene expression are believed to represent only a small percentage of the total cellular pool of transcripts in the cell, and, as is now recognized, only a minority of RNA whose levels are modulated by NMD [26, 46]. Rather, growing evidence indicates that NMD contributes broadly to regulating the expression level of many genes in response to developmental or environmental cues, strongly suggesting that another major role for NMD is to fine-tune gene expression by targeting a subset of transcripts expressed from a single gene. Indeed, the repertoire of RNAs expressed from a gene can be extensively heterogeneous due to variations in expression and/or processing, as described above. Due to this heterogeneity, only a proportion of expressed mRNA will include features that confer sensitivity to NMD (Fig. 5A). Based on the complexity of the gene expression pipeline, it is likely that the majority of eukaryotic genes express some population of mRNAs targeted to NMD, but that the impact of this pathway on transcript levels will be different for each gene, dependent upon the proportion of transcripts that are NMD-sensitive. Modulation of gene expression by NMD is also influenced by the penetrance of the NMD-inducing feature (Fig. 5A). For example, even though 100% of mRNA encoding cis-acting regulatory elements that direct frameshifting are potential targets for NMD, −1 PRF elements often function inefficiently, directing only a proportion of the mRNA population to undergo frameshifting and subsequent premature termination. The efficiency of −1 PRF therefore dictates the level of regulation by NMD [85, 87], and can itself be modulated, as recently shown by microRNA stimulation of −1 PRF in the human HIV-1 co-receptor CCR5 [86]. In this case, fine-tuning of CCR5 expression involves a balance between −1 PRF and NMD that is influenced by cell-specific or temporal changes in microRNA levels.
Figure 5.
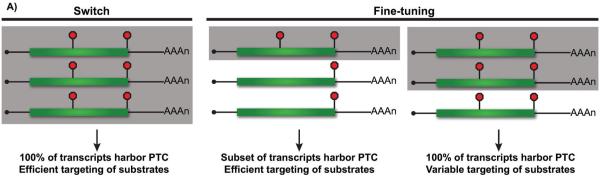
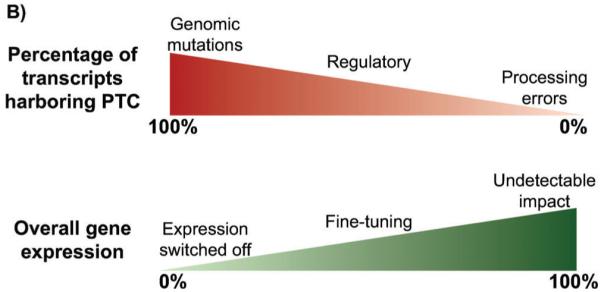
NMD acts as a switch and a dial to modulate gene expression. A: One-hundred percent of transcripts expressed from genes harboring nonsense or frameshift mutations harbor PTCs and are potential substrates for NMD; efficient targeting of these RNAs to this surveillance pathway serves as an effective means to eliminate them from the cell (left; targeted transcripts shaded in gray). In contrast, when only a fraction of expressed transcripts harbor PTCs or are targeted to NMD, the pathway serves to eliminate only a subset of the RNA. Variation in the proportion of an mRNA population targeted to NMD may result from heterogeneity in 5′ or 3′ UTR length or the nature of the spliced isoform (middle), or due to the incomplete penetrance of an NMD feature (right). B: The overall impact of NMD on gene expression (bottom) is dependent upon the fraction of mRNA targeted for degradation (top). When a large percentage of expressed transcripts are targeted, such as for genomic mutations, NMD serves as a switch to ensure minimal expression from the aberrant gene. For stochastic errors in transcription or processing, the overall impact of NMD on gene expression is small and perhaps even experimentally undetectable. In contrast, targeting of mRNA between these extremes provides an important mechanism for fine-tuning gene expression that will have variable phenotypic consequence depending on the gene and overall levels of mRNA (and expressed protein) required by the cell.
An interesting and yet unresolved observation is the unequal influence of NMD on mRNA levels based on the location of the nonsense codon within the ORF. Critically, nonsense codons in a number of eukaryotes exhibit polarity based on position, with premature stop codons proximal to the 5′ end of the mRNA causing a larger reduction in steady-state levels and faster decay of the transcript [2, 8, 19, 90–93]. In contrast, 3′ proximal nonsense codons, including those located in the terminal exon or at the extreme 3′ end of an ORF often have little or no impact on mRNA levels [6]. Although it is unclear through what mechanism the relative position of a nonsense codon affects targeting by NMD, computational modeling coupled with reporter mRNA analysis in yeast has suggested that transcripts with nonsense codons at different positions are each effectively distinguished from normal mRNA, but are degraded by 5′ decapping at position-dependent rates [90]. Recent observations demonstrating UPF1 association with nonsense codon-containing mRNAs in a 3′ UTR length-dependent manner might also provide a basis for nonsense codon-position effects on NMD substrate recognition and/or degradation [14, 15, 17].
Differences in the production of NMD-sensitive mRNA isoforms and their targeting to rapid decay can thus have a broad range of effects on gene expression depending on the proportion of the mRNA population targeted (Fig. 5B). In situations where NMD is acting essentially as a surveillance mechanism to clear products of stochastic errors in mRNA transcription or processing, only a small percentage of all expressed transcripts will be down-regulated and the overall impact on gene expression will be low (and perhaps below experimental detection). In contrast, for genomically-encoded mutations that result in premature translation termination, 100% of the expressed transcripts are substrates for NMD and efficient dampening of expression from the affected gene locus results. Between these two extremes lie many endogenous genes for which an intermediate fraction of expressed transcripts are sensitive to NMD, due to regulated RNA processing events or variable penetrance of the NMD-inducing feature. It is in this range where NMD may primarily serve a regulatory function, either because a substantial percentage of mRNAs expressed from a gene are targeted by NMD or because the level of expression from a gene sits near the threshold of biological impact such that a slight fine-tuning can result in dramatic phenotypic changes. A similar strategy has been proposed for microRNA regulation in higher eukaryotes [94], suggesting that the cell utilizes multiple approaches to modulate gene expression post-transcriptionally as a means to alter biological fate.
The activity of the NMD pathway is subject to external cues and autoregulation
Considering the broad role NMD has in both RNA surveillance and modulating endogenous gene expression, it is not surprising that the NMD machinery itself is exquisitely controlled through both autoregulatory feedback and external cues (reviewed in [72, 95]). Specifically, in a number of metazoans including nematodes, zebrafish, and mammals, transcripts encoding most NMD factors harbor features recognized by the pathway—including long 3′ UTRs, uORFs, or spliced mRNA isoforms with PTCs—and are themselves down-regulated by NMD [46, 96, 97]. Negative feedback regulation of NMD factor expression has been suggested to provide a buffering system to maintain homeostasis of gene expression under conditions of environmental or genetic insults [95, 96], and underscores the importance of maintaining a constant magnitude of NMD for normal cellular function.
Despite the tight control of NMD activity, evidence exists that this pathway is modulated under specific cellular conditions (reviewed in [72, 95]). For example, during hypoxia and other stresses, NMD activity is inhibited and, as a consequence, the levels of NMD substrates important for stress response are elevated [72, 98]. Modulation of NMD activity has also been observed during development of the nervous system in vertebrates, likely as a result of direct regulation of the core NMD factor UPF1 by tissue-specific and developmentally-regulated microRNA-128 [99, 100]. In this context, the regulation of NMD impacts expression of a broad range of NMD targets important for appropriate neural differentiation.
While modulation of NMD occurs naturally under certain conditions, inhibition of NMD activity and nonsense-codon recognition has recently been examined as a therapeutic option for a number of diseases. These therapies take two contrasting approaches to modulate NMD, either through inhibition of the activity of the pathway directly [101] or by decreasing the efficiency of translation termination at the PTC [102, 103]. As an example of the former, down-regulation of UPF2 or SMG1 in transformed cells and in vivo tumor mice models has been used to stabilize the heightened number of nonsense-containing mRNA isoforms generated due to genomic dysregulation associated with cancer but which are normally cleared from the cell. Translation of these stabilized mRNAs produces a multitude of novel truncated polypeptides that can act as potent antigens to induce an immune response and inhibit tumor growth [101]. In contrast, pharmacological agents that decrease translational fidelity and/or enhance readthrough at a PTC cause suppression of early translation termination without impacting the NMD pathway directly [102]. This latter approach has resulted in sufficient up-regulation of full-length protein expression and the alleviation of symptoms for cystic fibrosis and muscular dystrophy patients with genetically encoded nonsense mutations in the CFTR or dystrophin genes, respectively [103]. These examples indicate that modulating recognition of mRNAs by the NMD pathway represents a promising therapeutic approach to combat a broad range of human diseases [1, 103].
Conclusions and perspectives
Although NMD has been largely characterized as a surveillance pathway which rapidly eliminates aberrant mRNA species from eukaryotic cells, emerging evidence points to a critical role for NMD in broadly regulating expression of a large percentage of endogenous RNAs. Although it remains unclear which of these activities represents the primary role of NMD in the cell, they are not mutually exclusive, and both are likely important for optimal cellular fitness. As discussed here, the gene expression pipeline provides numerous ways to alter the ratio of NMD-sensitive RNA produced from a single gene locus, allowing the NMD pathway to work as a rheostat to maintain appropriate gene expression for hundreds if not thousands of genes under various conditions. Future studies aimed at identifying substrates of NMD and characterizing the consequences of their targeting will continue to shed light on the biological importance of regulating gene expression through this pathway.
Despite the growing list of substrates and transcript features that play a role in targeting RNAs to NMD, additional targets for this specialized surveillance pathway may exist that have yet to be uncovered. This is highlighted by recent work revealing lncRNAs as a novel category of cellular RNA species regulated by NMD [15, 31, 104] (see also Box 1) and the role of this pathway in restricting viral replication in mammals [105]. The emergence and refinement of technologies to evaluate RNA expression levels at greater sensitivity and translation at nucleotide resolution will be critical for illuminating the complex dynamics between transcription, translation, and mRNA decay, and will greatly increase our ability to identify and classify the entire repertoire of RNA transcripts regulated by NMD. In order to fully appreciate the role of the NMD pathway in shaping gene expression and biological function, however, it will be necessary to expand our survey of NMD-sensitive transcriptomes to a larger range of cells (and organisms) exposed to a multitude of conditions.
Translation of lncRNAs can have different outcomes depending on the biological function of the transcript.

Translation of lncRNAs can produce biologically important peptides (1), suggesting that the genes expressing these transcripts warrant reannotation as protein-coding. lncRNA resulting from spurious transcription are predominantly cleared from the cell through degradation in the nucleus but can be targeted to surveillance by NMD if they escape to the cytoplasm and engage the translation machinery (2). For lncRNAs with regulatory functions in the cytoplasm, translation and targeting to NMD may act to modulate steady-state levels and activity of the RNA; in the absence of targeting by NMD, RNA levels are elevated, favoring enhanced function of that lncRNA (3). For lncRNAs whose function is to regulate gene expression in the nucleus, translation and rapid degradation by NMD in the cytoplasm would establish and maintain a concentration gradient that ensures the lncRNA population is principally nuclear and predominantly noncoding (4). Finally, NMD can assist in clearing the cell of lncRNAs that are sampled by the translation machinery in the process of de novo evolution of protein-coding genes (5). While lncRNA translation is necessary for the sampling of new peptides, rapid elimination of transcripts expressing nonfunctional products is important to protect the cell against deleterious effects.
Acknowledgements
The authors would like to thank Jo Ann Wise for critical reading of the manuscript and apologize to our colleagues for the inability to cite all of the exciting findings in the field due to space limitations. Research on NMD by the authors is funded by the National Institutes of Health (GM095621) and National Science Foundation (MCB1253788); JES was supported by a NIH-funded training fellowship (T32 GM008056).
Abbreviations
- APA
alternative cleavage and polyadenylation
- AS-NMD
alternative splicing coupled to nonsense-mediated RNA decay
- EJC
exon junction complex
- eRF
eukaryotic translation release factor
- lncRNA
long noncoding RNA
- mRNP
mRNA ribonucleoprotein
- NMD
nonsense-mediated RNA decay
- ORF
open reading frame
- PRF
programmed ribosomal frameshifting
- PTC
premature translation termination codon
- Sec
selenocysteine
- uORF
upstream open reading frame
- UTR
untranslated region
Footnotes
The authors have declared no conflict of interest.
References
- 1.Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. 2010;430:365–77. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- 2.Pulak R, Anderson P. MRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 1993;7:1885–97. doi: 10.1101/gad.7.10.1885. [DOI] [PubMed] [Google Scholar]
- 3.Behm-Ansmant I, Kashima I, Rehwinkel J, Sauliere J, et al. mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett. 2007;581:2845–53. doi: 10.1016/j.febslet.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 4.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–12. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schweingruber C, Rufener SC, Zund D, Yamashita A, et al. Nonsense-mediated mRNA decay - mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta. 2013;1829:612–23. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Lejeune F, Maquat LE. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol. 2005;17:309–15. doi: 10.1016/j.ceb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Amrani N, Ganesan R, Kervestin S, Mangus DA, et al. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432:112–8. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- 8.Eberle AB, Stalder L, Mathys H, Orozco RZ, et al. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol. 2008;6:e92. doi: 10.1371/journal.pbio.0060092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amrani N, Sachs MS, Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol. 2006;7:415–25. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
- 11.Meaux S, van Hoof A, Baker KE. Nonsense-mediated mRNA decay in yeast does not require PAB1 or a poly(A) tail. Mol Cell. 2008;29:134–40. doi: 10.1016/j.molcel.2007.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kervestin S, Li C, Buckingham R, Jacobson A. Testing the faux-UTR model for NMD: analysis of Upf1p and Pab1p competition for binding to eRF3/Sup35p. Biochimie. 2012;94:1560–71. doi: 10.1016/j.biochi.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roque S, Cerciat M, Gaugue I, Mora L, et al. Interaction between the poly(A)-binding protein Pab1 and the eukaryotic release factor eRF3 regulates translation termination but not mRNA decay in Saccharomyces cerevisiae. RNA. 2015;21:124–34. doi: 10.1261/rna.047282.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hogg JR, Goff SP. Upf1 senses 3′ UTR length to potentiate mRNA decay. Cell. 2010;143:379–89. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurt JA, Robertson AD, Burge CB. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013;23:1636–50. doi: 10.1101/gr.157354.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zund D, Gruber AR, Zavolan M, Muhlemann O. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol. 2013;20:936–43. doi: 10.1038/nsmb.2635. [DOI] [PubMed] [Google Scholar]
- 17.Kurosaki T, Maquat LE. Rules that govern UPF1 binding to mRNA 3′ UTRs. Proc Natl Acad Sci USA. 2013;110:3357–62. doi: 10.1073/pnas.1219908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurosaki T, Li W, Hoque M, Popp MW, et al. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 2014;28:1900–16. doi: 10.1101/gad.245506.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leeds P, Peltz SW, Jacobson A, Culbertson MR. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 1991;5:2303–14. doi: 10.1101/gad.5.12a.2303. [DOI] [PubMed] [Google Scholar]
- 20.Leeds P, Wood JM, Lee BS, Culbertson MR. Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:2165–77. doi: 10.1128/mcb.12.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinniburgh AJ, Maquat LE, Schedl T, Rachmilewitz E, et al. mRNA-deficient beta o-thalassemia results from a single nucleotide deletion. Nucleic Acids Res. 1982;10:5421–7. doi: 10.1093/nar/10.18.5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sydow JF, Cramer P. RNA polymerase fidelity and transcriptional proofreading. Curr Opin Struct Biol. 2009;19:732–9. doi: 10.1016/j.sbi.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 23.Rehwinkel J, Raes J, Izaurralde E. Nonsense-mediated mRNA decay: Target genes and functional diversification of effectors. Trends Biochem Sci. 2006;31:639–46. doi: 10.1016/j.tibs.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Peccarelli M, Kebaara BW. Regulation of natural mRNAs by the nonsense-mediated mRNA decay pathway. Eukaryot Cell. 2014;13:1126–35. doi: 10.1128/EC.00090-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johansson MJ, He F, Spatrick P, Li C, et al. Association of yeast Upf1p with direct substrates of the NMD pathway. Proc Natl Acad Sci USA. 2007;104:20872–7. doi: 10.1073/pnas.0709257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapin A, Hu H, Rynearson SG, Hollien J, et al. In vivo determination of direct targets of the nonsense-mediated decay pathway in Drosophila. G3. 2014;4:485–96. doi: 10.1534/g3.113.009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matia-Gonzalez AM, Hasan A, Moe GH, Mata J, et al. Functional characterization of Upf1 targets in Schizosaccharomyces pombe. RNA Biol. 2013;10:1057–65. doi: 10.4161/rna.24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tani H, Imamachi N, Salam KA, Mizutani R, et al. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol. 2012;9:1370–9. doi: 10.4161/rna.22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan Q, Zheng W, Tang S, Liu X, et al. Impact of nonsense-mediated mRNA decay on the global expression profile of budding yeast. PLoS Genet. 2006;2:e203. doi: 10.1371/journal.pgen.0020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, et al. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014;7:1858–66. doi: 10.1016/j.celrep.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi H, Lassmann T, Murata M, Carninci P. 5′ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat Protoc. 2012;7:542–61. doi: 10.1038/nprot.2012.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arribere JA, Gilbert WV. Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res. 2013;23:977–87. doi: 10.1101/gr.150342.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brar GA, Yassour M, Friedman N, Regev A, et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–7. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michel AM, Choudhury KR, Firth AE, Ingolia NT, et al. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012;22:2219–29. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duncan CD, Mata J. The translational landscape of fission-yeast meiosis and sporulation. Nat Struct Mol Biol. 2014;21:641–7. doi: 10.1038/nsmb.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fritsch C, Herrmann A, Nothnagel M, Szafranski K, et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 2012;22:2208–18. doi: 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalyna M, Simpson CG, Syed NH, Lewandowska D, et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012;40:2454–69. doi: 10.1093/nar/gkr932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waern K, Snyder M. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3. 2013;3:343–52. doi: 10.1534/g3.112.003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Law GL, Bickel KS, MacKay VL, Morris DR. The undertranslated transcriptome reveals widespread translational silencing by alternative 5′ transcript leaders. Genome Biol. 2005;6:R111. doi: 10.1186/gb-2005-6-13-r111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muhlrad D, Parker R. Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA. 1999;5:1299–307. doi: 10.1017/s1355838299990829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kebaara BW, Atkin AL. Long 3′-UTRs target wild-type mRNAs for nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Nucleic Acids Res. 2009;37:2771–8. doi: 10.1093/nar/gkp146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramani AK, Nelson AC, Kapranov P, Bell I, et al. High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans. Genome Biol. 2009;10:R101. doi: 10.1186/gb-2009-10-9-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hansen KD, Lareau LF, Blanchette M, Green RE, et al. Genome-wide identification of alternative splice forms down-regulated by nonsense-mediated mRNA decay in Drosophila. PLoS Genet. 2009;5:e1000525. doi: 10.1371/journal.pgen.1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, et al. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA. 2011;17:2108–18. doi: 10.1261/rna.030247.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decourty L, Doyen A, Malabat C, Frachon E, et al. Long open reading frame transcripts escape nonsense-mediated mRNA decay in yeast. Cell Rep. 2014;6:593–8. doi: 10.1016/j.celrep.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 48.Quek BL, Beemon K. Retroviral strategy to stabilize viral RNA. Curr Opin Microbiol. 2014;18:78–82. doi: 10.1016/j.mib.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi Y. Alternative polyadenylation: new insights from global analyses. RNA. 2012;18:2105–17. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011;43:853–66. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 52.Mayr C, Bartel DP. Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–84. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martins R, Proenca D, Silva B, Barbosa C, et al. Alternative polyadenylation and nonsense-mediated decay coordinately regulate the human HFE mRNA levels. PLoS One. 2012;7:e35461. doi: 10.1371/journal.pone.0035461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neve J, Furger A. Alternative polyadenylation: less than meets the eye? Biochem Soc Trans. 2014;42:1190–5. doi: 10.1042/BST20140054. [DOI] [PubMed] [Google Scholar]
- 55.He F, Peltz SW, Donahue JL, Rosbash M, et al. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1-mutant. Proc Natl Acad Sci USA. 1993;90:7034–8. doi: 10.1073/pnas.90.15.7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, et al. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014;24:1774–86. doi: 10.1101/gr.177790.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sayani S, Janis M, Lee CY, Toesca I, et al. Widespread impact of nonsense-mediated mRNA decay on the yeast intronome. Mol Cell. 2008;31:360–70. doi: 10.1016/j.molcel.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawashima T, Douglass S, Gabunilas J, Pellegrini M, et al. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae. PLoS Genet. 2014;10:e1004249. doi: 10.1371/journal.pgen.1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaillon O, Bouhouche K, Gout JF, Aury JM, et al. Translational control of intron splicing in eukaryotes. Nature. 2008;451:359–62. doi: 10.1038/nature06495. [DOI] [PubMed] [Google Scholar]
- 60.Marquez Y, Brown JW, Simpson C, Barta A, et al. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012;22:1184–95. doi: 10.1101/gr.134106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hamid FM, Makeyev EV. Emerging functions of alternative splicing coupled with nonsense-mediated decay. Biochem Soc Trans. 2014;42:1168–73. doi: 10.1042/BST20140066. [DOI] [PubMed] [Google Scholar]
- 62.Sibley CR. Regulation of gene expression through production of unstable mRNA isoforms. Biochem Soc Trans. 2014;42:1196–205. doi: 10.1042/BST20140102. [DOI] [PubMed] [Google Scholar]
- 63.Barberan-Soler S, Lambert NJ, Zahler AM. Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans. RNA. 2009;15:1652–60. doi: 10.1261/rna.1711109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Drechsel G, Kahles A, Kesarwani AK, Stauffer E, et al. Nonsense-mediated decay of alternative precursor mRNA splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell. 2013;25:3726–42. doi: 10.1105/tpc.113.115485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McIlwain DR, Pan Q, Reilly PT, Elia AJ, et al. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA. 2010;107:12186–91. doi: 10.1073/pnas.1007336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, et al. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008;22:1381–96. doi: 10.1101/gad.468808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weischenfeldt J, Waage J, Tian G, Zhao J, et al. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol. 2012;13:R35. doi: 10.1186/gb-2012-13-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, et al. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–8. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- 69.Ni JZ, Grate L, Donohue JP, Preston C, et al. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21:708–18. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wittmann J, Hol EM, Jack HM. HUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol Cell Biol. 2006;26:1272–87. doi: 10.1128/MCB.26.4.1272-1287.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–92. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karam R, Wengrod J, Gardner LB, Wilkinson MF. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta. 2013;1829:624–633. doi: 10.1016/j.bbagrm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 2011;25:373–84. doi: 10.1101/gad.2004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lareau LF, Inada M, Green RE, Wengrod JC, et al. Unproductive splicing of SR genes associated with highly conserved and ultra-conserved DNA elements. Nature. 2007;446:926–9. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 75.Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, et al. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100. doi: 10.1016/s1097-2765(03)00502-1. [DOI] [PubMed] [Google Scholar]
- 76.Boutz PL, Stoilov P, Li Q, Lin CH, et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–52. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Welch EM, Jacobson A. An internal open reading frame triggers nonsense-mediated decay of the yeast SPT10 mRNA. EMBO J. 1999;18:6134–45. doi: 10.1093/emboj/18.21.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barbosa C, Peixeiro I, Romao L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9:e1003529. doi: 10.1371/journal.pgen.1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He F, Li X, Spatrick P, Casillo R, et al. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol Cell. 2003;12:1439–52. doi: 10.1016/s1097-2765(03)00446-5. [DOI] [PubMed] [Google Scholar]
- 81.Rehwinkel J, Letunic I, Raes J, Bork P, et al. Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA. 2005;11:1530–44. doi: 10.1261/rna.2160905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dinman JD. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip Rev RNA. 2012;3:661–73. doi: 10.1002/wrna.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Belew AT, Hepler NL, Jacobs JL, Dinman JD. PRFdb: a database of computationally predicted eukaryotic programmed −1 ribosomal frameshift signals. BMC Genomics. 2008;9:339. doi: 10.1186/1471-2164-9-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jacobs JL, Belew AT, Rakauskaite R, Dinman JD. Identification of functional, endogenous programmed −1 ribosomal frameshift signals in the genome of Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:165–74. doi: 10.1093/nar/gkl1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Advani VM, Belew AT, Dinman JD. Yeast telomere maintenance is globally controlled by programmed ribosomal frameshifting and the nonsense-mediated mRNA decay pathway. Translation. 2013;1:e24418. doi: 10.4161/trla.24418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, et al. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature. 2014;512:265–9. doi: 10.1038/nature13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Belew AT, Advani VM, Dinman JD. Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res. 2011;39:2799–808. doi: 10.1093/nar/gkq1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moriarty PM, Reddy CC, Maquat LE. Selenium deficiency reduces the abundance of mRNA for Se-dependent glutathione peroxidase 1 by a UGA-dependent mechanism likely to be nonsense codon-mediated decay of cytoplasmic mRNA. Mol Cell Biol. 1998;18:2932–9. doi: 10.1128/mcb.18.5.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seyedali A, Berry MJ. Nonsense-mediated decay factors are involved in the regulation of selenoprotein mRNA levels during selenium deficiency. RNA. 2014;20:1248–56. doi: 10.1261/rna.043463.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cao D, Parker R. Computational modeling and experimental analysis of nonsense-mediated decay in yeast. Cell. 2003;113:533–45. doi: 10.1016/s0092-8674(03)00353-2. [DOI] [PubMed] [Google Scholar]
- 91.van Hoof A, Green PJ. Premature nonsense codons decrease the stability of phytohemagglutinin mRNA in a position-dependent manner. Plant J. 1996;10:415–24. doi: 10.1046/j.1365-313x.1996.10030415.x. [DOI] [PubMed] [Google Scholar]
- 92.Wang J, Gudikote JP, Olivas OR, Wilkinson MF. Boundary-independent polar nonsense-mediated decay. EMBO Rep. 2002;3:274–9. doi: 10.1093/embo-reports/kvf036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brogna S. Nonsense mutations in the alcohol dehydrogenase gene of Drosophila melanogaster correlate with an abnormal 3′ end processing of the corresponding pre-mRNA. RNA. 1999;5:562–73. doi: 10.1017/s1355838299981359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 2008;9:831–42. doi: 10.1038/nrg2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang L, Wilkinson MF. Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip Rev RNA. 2012;3:807–28. doi: 10.1002/wrna.1137. [DOI] [PubMed] [Google Scholar]
- 96.Huang L, Lou CH, Chan W, Shum EY, et al. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell. 2011;43:950–61. doi: 10.1016/j.molcel.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Longman D, Hug N, Keith M, Anastasaki C, et al. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013;41:8319–31. doi: 10.1093/nar/gkt585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–41. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bruno IG, Karam R, Huang L, Bhardwaj A, et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell. 2011;42:500–10. doi: 10.1016/j.molcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lou CH, Shao A, Shum EY, Espinoza JL, et al. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 2014;6:748–64. doi: 10.1016/j.celrep.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature. 2010;465:227–30. doi: 10.1038/nature08999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Welch EM, Barton ER, Zhuo J, Tomizawa Y, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 103.Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med. 2013;64:407–25. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kurihara Y, Matsui A, Hanada K, Kawashima M, et al. Genomewide suppression of aberrant mRNA-like noncoding RNAs by NMD in Arabidopsis. Proc Natl Acad Sci USA. 2009;106:2453–8. doi: 10.1073/pnas.0808902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Balistreri G, Horvath P, Schweingruber C, Zund D, et al. The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host Microbe. 2014;16:403–11. doi: 10.1016/j.chom.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 106.Derrien T, Johnson R, Bussotti G, Tanzer A, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carvunis AR, Rolland T, Wapinski I, Calderwood MA, et al. Proto-genes and de novo gene birth. Nature. 2012;487:370–4. doi: 10.1038/nature11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilson BA, Masel J. Putatively noncoding transcripts show extensive association with ribosomes. Genome Biol Evol. 2011;3:1245–52. doi: 10.1093/gbe/evr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aspden JL, Eyre-Walker YC, Phillips RJ, Amin U, et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. eLife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chew GL, Pauli A, Rinn JL, Regev A, et al. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–34. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–93. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ruiz-Orera J, Messeguer X, Subirana JA, Alba MM. Long non-coding RNAs as a source of new peptides. eLife. 2014;3:e03523. doi: 10.7554/eLife.03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Heesch S, van Iterson M, Jacobi J, Boymans S, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15:R6. doi: 10.1186/gb-2014-15-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guttman M, Russell P, Ingolia NT, Weissman JS, et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–51. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, et al. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365–79. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thompson DM, Parker R. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:92–101. doi: 10.1128/MCB.01023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Toesca I, Nery CR, Fernandez CF, Sayani S, et al. Cryptic transcription mediates repression of subtelomeric metal homeostasis genes. PLoS Genet. 2011;7:e1002163. doi: 10.1371/journal.pgen.1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tani H, Torimura M, Akimitsu N. The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS One. 2013;8:e55684. doi: 10.1371/journal.pone.0055684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Magny EG, Pueyo JI, Pearl FM, Cespedes MA, et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341:1116–20. doi: 10.1126/science.1238802. [DOI] [PubMed] [Google Scholar]
- 120.Pauli A, Norris ML, Valen E, Chew GL, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Galindo MI, Pueyo JI, Fouix S, Bishop SA, et al. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:e106. doi: 10.1371/journal.pbio.0050106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cohen SM. Everything old is new again: (linc)RNAs make proteins! EMBO J. 2014;33:937–8. doi: 10.1002/embj.201488303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Niazi F, Valadkhan S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3′ UTRs. RNA. 2012;18:825–43. doi: 10.1261/rna.029520.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen L. A global comparison between nuclear and cytosolic transcriptomes reveals differential compartmentalization of alternative transcript isoforms. Nucleic Acids Res. 2010;38:1086–97. doi: 10.1093/nar/gkp1136. [DOI] [PMC free article] [PubMed] [Google Scholar]