

Abstract
Hibernation is a unique phenotype displayed by a phylogenetically diverse group of organisms including several species of mammals and one species of primate. Here we review evidence for blood and tissue borne signaling molecules in hibernating animals, achievements in isolating and characterizing these molecules, and potential medicinal applications.
Keywords: Heterothermic, Cytoprotective, Tolerance, Hibernation induction trigger, Endogenous factors, Metabolic suppression, Suspended animation, Plasma factors
I. INTRODUCTION
Stroke is the third leading cause of death in the U.S. and the leading cause of disability worldwide, yet the only currently FDA approved therapeutic for stroke, recombinant tissue-type plasminogen activator (rt-PA), is indicated for use in less than 4% of stroke victims. Although the promising, newly developed free radical trapping compound, NXY-059, significantly reduces post-stroke disabilities and has progressed to Phase III clinical trials [1], successful pretrial (i.e. experimental) treatments for stroke have for the most part failed clinically. A dearth of therapeutics exists for stroke as well as other pathologic conditions such as cardiac arrest or neurodegenerative disease.
Hibernation is a unique phenotype thought to have evolved in response to food shortage. Examples of species that hibernate can be found in 9 different orders of mammals, including rodents, bats, carnivores, marsupials and even primates (the fat-tailed dwarf lemur, Cheirogaleus medius) [2, 3]. Several aspects of hibernation physiology such as depressed respiratory and heart rate, hypothermia, increased antioxidant defense, blood hypocoagulability, and suppressed metabolic and immune function expressed during torpor are consistent with neuroprotection and cytoprotection [3, 4]. Prolonged torpor, the phenotype that defines hibernation, is a state in which animals experience a severe (up to 90%) reduction in cerebral perfusion, which is comparable to ischemia, during bouts of torpor [5]. Furthermore, torpid animals rewarm and reenter into euthermia between bouts of torpor for brief periods. During rewarming the animal experiences severe hypoxia without neuronal damage [6].
Tolerance to traumatic brain injury has been shown in hibernating Arctic ground squirrel (AGS, see Table 1 for all hibernating species abbreviations) in vivo [7]. Hibernating thirteen-lined ground squirrel (TLS) tolerate oxygen and glucose deprivation better than non-hibernating TLS in vitro [8], and tissue tolerance persists in longer term culture [9]. Organs harvested from hibernating animals tolerate cold ischemia-warm reperfusion while in the hibernating state [10, 11]. Protection against ischemia-reperfusion injury has been shown in rabbit isolated heart preparations with administration of serum derived from hibernating black bear (BB) [12]. Therefore, factors that may be isolated from tissue or serum of hibernating animals as well as novel molecular mechanisms operative during hibernation may be the key to the profound protective nature of hibernation. Such novel mechanisms in hibernating species could be due to either the presence of previously unidentified factors or the lack of certain factors which could result in unique regulation of a specific pathway. Mammals that hibernate are thus a potential source of multiple therapeutics if endogenous factors presumed to regulate the neuroprotective mechanisms characteristic of the hibernation phenotype can be isolated and identified.
Table 1.
Hibernating Species with Abbreviations Used
| Species Name | Common Name | Abbreviation |
|---|---|---|
| Tamias asiaticus | Asian chipmunk | AC |
| Spermophilus parryii | Arctic ground squirrel | AGS |
| Ursus americanus | Black bear | BB |
| Spermophilus beldingi | Belding’s ground squirrel | BGS |
| Spermophilus citellus | European ground squirrel | EGS |
| Spermophilus lateralis | Golden-mantled ground squirrel | GMGS |
| Spermophilus undulatus | Jackutain ground squirrel | JGS |
| Myotis lucifugus | Little brown bat | LBB |
| Spermophilus richardsonii | Richardson’s ground squirrel | RGS |
| Spermophilus suslicus | Spotted suslik | SS |
| Spermophilus tridecemlineatus | Thirteen-lined ground squirrel | TLS |
| Marmota monax | Eastern woodchuck | WC |
Endogenous substances from organisms with unique phenotypes have been isolated and found to have therapeutic effects in other species. For example, salivary plasminogen activator from the vampire bat (Desmodus rotundus) has been developed into the drug desmoteplase which avoids the deleterious side effects associated with recombinant tissue-type plasminogen activator [13] and is currently in phase II clinical trials for acute stroke [14]. Venomous marine molluscs such as cone snails (genus Conus) produce conotoxins, a class of small peptides with high affinity and specificity for a variety of ion channels with therapeutic applications [15]. For example, ω-conotoxin MVIIA, derived from Conns magnus, targets voltage-gated Ca2+ channels and is FDA approved for treatment of severe chronic inflammatory and neuropathic pain associated with cancer and AIDS [16].
Serum and tissue factors and unique mechanisms in hibernating species have received considerable investigation over the years (Tables 2 and 3), although no molecular structures have been described for compounds isolated from a hibernating species nor has any therapeutic yet been derived from a hibernating species. Thus, the objective of this review is to summarize evidence for bioactive serum or tissue factors isolated from hibernating animals to highlight relevant areas for future pursuit towards discovery of novel therapeutics. We review selected investigations of mammalian species that hibernate in which tissue or serum factors have been isolated, characterized or identified. Our goal, therefore, is to point out potential tissues which could be sources of compounds or factors which could prove to be bioactive since no such molecules have been characterized from hibernating species to date. In addition, we review protective mechanisms in hibernating species that may be regulated by factors that if isolated may have therapeutic value.
Table 2.
Evidence for Tissue/Blood Borne Molecules in Torpid Animals that Induce a Protective Mechanism
| Source Species | Source Tissue | Pharmacologic Effect | Target Species | Target Tissue | Administered | Reference |
|---|---|---|---|---|---|---|
| TLS | brown fat | decreases primary immune response | hamster | spleen fragments | cell culture | [139] |
| AL | brain extract | decreases metabolism and Tb*to4°C | rat | systemic | iv | [23,24] |
| TLS | brain extract | decrease metabolism | rat | systemic | iv | [26] |
| AL | brain extract | decreases metabolism and Tb | mouse | systemic | iv | [25] |
| TLS | brain extract | decreases cell division | Chinese hamster | ovary cells (CHO) | cell culture | [87] |
| JGS, AGS | brain and small intestine epitheluim | decreases brain temperature and awake state; increases slow wave sleep | mouse | brain | ip | [27–29] |
| JGS | plasma | decreases metabolism | mouse | systemic | ip | [31] |
| JGS | urine | decreases Tb | mouse | systemic | ip | [31] |
| TLS | plasma | increases intercellular adhesion molecule-1 (ICAM-1) expression and monocyte adhesion | rat | cerebral microvascular endothelial cells | cell culture | [141] |
| BB | serum | increases protection from ischemia-reperfusion | rabbit | heart | perfuse | [12] |
| TLS, WC | plasma | decreases cell proliferation suggesting mechanism of immunosuppression | mouse | spleenocytes | cell culture | [140] |
Tb = body temperature; AL = African lungfish
Table 3.
Achievements in Isolating and Characterizing Factors from Torpid Animals with Therapeutic Potential
| Source Species | Source Tissue | Peptide/Protein | Pharmacologic Effect | Target Species/Tissue | Slate of Target Species | Administered | Reference |
|---|---|---|---|---|---|---|---|
| JGS | brain | KKT = neokyotorphin | causes arousal from torpor; increases cardiac activity and respiration | JGS | early in bout | ip | [30] |
| JGS | brain | KT = kyotorphin (fragment of NKT) | inhibitory: decreases heart rate | JGS | awakened mid-bout | ip | [31] |
| JGS | brain | KT and NKT | alters behavior and EEG | rat | n/a | icv | [34] |
| JGS | brain | TSKYR | increases then decreases evoked activity | rat/medial septal slices | n/a | in culture media | [35] |
| DY | blocks evoked activity | ||||||
| JGS | brain | TSKYR | increases duration of inhibition (of evoked neuron activity) | JGS/medial septal slices | torpid | in culture media | [33] |
| TSKYR | shortens inhibition | awake | |||||
| TSKY | increases duration of inhibition | awake | |||||
| DY | decreases duration of inhibition | torpid | |||||
| AC, TLS | blood/plasma | 25kDa, 27kDa, 20kDa, 55kDa proteins | not tested; found as a 140KDa complex | not tested | n/a | n/a | [121] |
| TLS | brain | pp98 | not tested; associated with membrane fraction | not tested | n/a | n/a | [72] |
II. PROTECTIVE MECHANISMS IN HIBERNATING ANIMALS LIKELY REGULATED BY FACTORS WITH THERAPEUTIC POTENTIAL
During hibernation, unique regulation of metabolism, blood clotting, immune response, antioxidant defense, synaptic remodeling, protein synthesis, cell cycle, and ion homeostasis take place. Each of these potentially neuroprotective aspects of hibernation could have implications for development into therapeutics for non-hibernating species. The search for compounds in hibernating animals that mediate protective mechanisms could lead to development of small molecules that mimic these mechanisms. It is not assumed that all aspects of hibernation are protective: some of these mechanisms may be induction mechanisms or mechanisms that are in response to the induction of hibernation. In fact, many therapeutics, either currently approved or in clinical trials, work through mechanisms that resemble what is seen in hibernation. Indeed molecules that mimic neuroprotective aspects of hibernation have been found outside of hibernating animals (that may or may not be related to hibernation [17]; nonetheless, the idea to search for a molecule that suppresses metabolism was conceived from knowledge of hibernating species.
Some studies have described bioactivity of tissue extracts and blood from hibernating animals while other studies have isolated, characterized, or identified specific factors. In addition, currently presumed yet unidentified factors that mediate protective aspects of the hibernation phenotype hold promise for discovery and development of therapeutics as illustrated (Fig. 1). Enhanced antioxidant defense has been reviewed previously [4, 18].
Fig. 1.
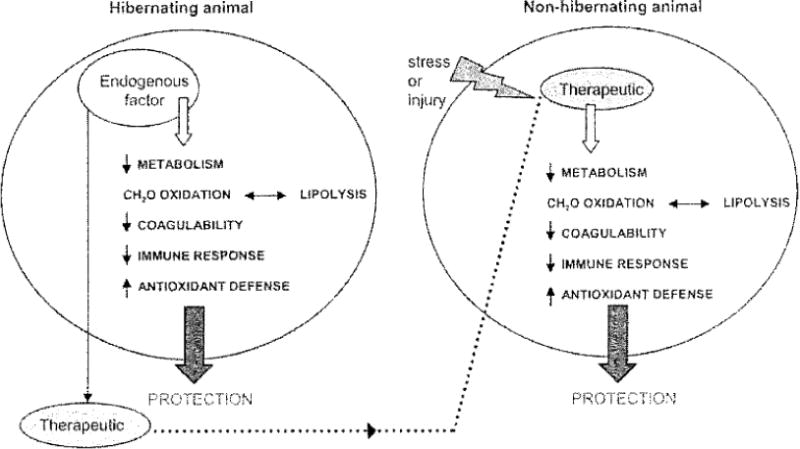
Model of a factor (blood borne or tissue borne) endogenous to torpid animals that regulates a protective mechanism. If isolated the factor could be developed into a therapeutic for non-hibernating species.
A. Decreased Metabolism
i. Therapeutic Potential of a Factor Involved in Decreasing Metabolism
Decreasing metabolism may prevent the deleterious energy deficit normally incurred during acute conditions such as stroke, cardiac arrest, and traumatic brain injury. Preservation of energy balance maintains ion homeostasis and membrane potential, thus avoiding depolarization, excitotoxicity and consequent excess intracellular Ca2+ that yield death or damage. Additionally, induction of a hypometabolic state, such as suspended animation [18, 19], would allow for a broader window for clinical intervention following injury. Suspended animation induced with carbon monoxide in fact provides protection from hypoxia in C. elegans [20].
ii. Evidence for Tissue/Blood Borne Molecules in Hibernation Associated with Decreased Metabolism
Suppression of metabolism is a hallmark of hibernation. The calculated cerebral glucose utilization and whole animal oxygen consumption in deeply hibernating ground squirrels decreases to only 1 to 2% of values of active animals [21]. Moreover, consistent with a pronounced decrease in oxygen consumption and energy demand, oxygen-hemoglobin saturation as well as phospho-creatine and brain glucose increase during torpor [6, 22].
Existence of hypometabolic factors is supported by observations that extracts from brain tissue from animals in torpor decrease metabolism in non-hibernating species Brain extracts from the hibernating African lungfish (AL), Protopterus aethiopicus, and hibernating TLS administered iv decrease metabolism and core body temperature in rats and mice [23–26]. Endogenous substances from brain and tissue of internal organs of AGS and Jakutian ground squirrel (JGS) administered to mice ip decrease brain temperature and wakefulness and increase slow wave sleep [27–29].
The presence of hypometabolic factors in plasma from animals in torpor is also supported by observations that administration of plasma or even urine decreases metabolism and core body temperature in non-hibernating species. Plasma collected from JGS at the beginning of a torpor bout decreases metabolism when injected ip into albino mice [30]. JGS mine injected ip into mice causes hypothermia suggesting that “active” factors in hibernating animals are excreted into urine [31]. Table 2 summarizes evidence for tissue or blood borne molecules in torpid animals that induce metabolic depression or other protective responses.
iii. Achievements in Isolating and Characterizing Molecules Associated with Decreased Metabolism
Brain peptides isolated from hibernating animals decrease metabolism or energy demanding neuronal activity in non-hibernating animals. Table 3 summarizes achievements in isolating and characterizing factors from torpid animals that induce metabolic depression or other protective aspects or hibernation.
Brain peptides isolated from hibernating animals result in metabolic alterations following injection into hibernating species in either the hibernating or awake state. A low molecular weight peptide, neokyotorpin (NKT =Thr-Ser-Lys-Tyr-Arg), isolated from JGS brain, is considered a metabolic “activator” since it stimulates cardiac activity and respiration and ultimately arousal from torpor when administered ip to hibernating JGS [30, 31]. KT (a fragment of NKT) inhibits heart rate when given ip to JGS awakened from hibernation and thus is considered a metabolic “inhibitor” [31].
Brain peptides isolated from hibernating animals specifically affect neural activity in hibernating species in a stale dependent manner. Three peptides isolated from JGS bruin influence evoked neuronal activity in medial septal brain slices. TSKYR increases duration of inhibition in slices from hibernating JGS, TSKY increases duration of inhibition in slices from awake JGS, and DY increases activation of responses in slices from hibernating JGS [32, 33].
Additionally neural activity of non-hibernating species is affected by brain peptides isolated from hibernating animals. KT and KKT isolated from JGS brain alter behavior and EEG when administered icv to rat [34], Peptides TSKYR and DY obtained from brain of hibernating JGS affect spontaneous activity of septal neurons; TSKYR increases (but then decreases) activity and DY inhibits activity [35].
iv. Differential Regulation of Known Factors in Hibernating Animals Associated with Decreased Metabolism
Evidence for altered expression of proteins involved in metabolic suppression in hibernating animals supports existence of regulatory factors that if identified could be developed into therapeutics. Expression of the rate limiting enzyme for synthesizing melatonin, arylalkaylaminc-N-acetyltransferase (AA-NAT), increases in the suprachiasmatic nucleus and epithalamic medial habenular nuclei area in hibernating TLS [36]. The suprachiasmatic nucleus is suggested to play an active role in regulation of the hibernation state [37, 38], ICV injection of melatonin into hibernating golden-mantled ground squirrel (GMGS) increases duration of torpor bouts [39]. The ability of melatonin to suppress metabolism, by decreasing core body temperature [40, 41] arid regulating glucose utilization [42] in non-hibernating species, suggests further study of melatonin’s role in hibernation and factors capable of enhancing melatonin synthesis have potential to lead to novel therapeutics. Insulin-like growth factor (IGF) and insulin-like growth factor binding proteins may be involved in the signaling cascade leading to metabolic suppression in torpor. Decreases in serum levels of insulin-like growth factor and insulin-like growth factor binding proteins during torpor in golden-mantled ground squirrel is consistent with downregulation of energy expensive somatic growth [43]. The insulin-like growth factor signaling pathway is involved in regulation of metabolism, development and longevity. Decreased phosphorylation and activity of the insulin-like growth factor regulated serine/threonine kinase, AKT (protein kinase B), occurs during torpor in TLS and suggests that hibernation might use an inducible pathway similar to that seen in dauer larva formation in C. elegans [44].
v. Currently Unidentified Factors that Mediate Regulation of Decreased Metabolism in Hibernating Animals
The search for therapeutics that decrease metabolism is spurred on by the recent finding that hydrogen sulfide (H2S) induces a suspended animation-like state, characterized by decreased metabolism and loss of homeostatic control, in mice [17]. Although H2S may not induce hibernation in a hibernating species, the ability of a molecule such as H2S to alter metabolism in a non-hibernating mammalian species is impetus for further pursuit of an effector molecule that is endogenous to and could be isolated from hibernating animals. The mechanism of the H2S response remains unclear, but could involve inhibition of the electron transport chain as proposed in Blackstone et al., 2005 [17] or a hrypoxie-metabolic response as described in other rodents [45]. A role for H2S as a signaling molecule remains doubtful, however, equal skepticism likely preceded identification of NO as a signaling molecule.
vi. The Search for a “Hibernation Induction Trigger” Leads to Discovery of Cytoprotective Properties of [D-Ala(2),D-Leu(5)]Enkephalin (DADLE) Agonists
Despite many endeavors to characterize and isolate a “Hibernation Induction Trigger” (HIT) from hibernating animals, such a “trigger” has still not been found. Initial experiments suggested that blood from hibernating animals contained “HIT” since transfusions of blood and blood derivatives from hibernating TLS, woodchuck (WC) and AGS seemed to induct hibernation in summer active TLS [46, 47], yet other stimuli such as sham or saline injection similarly induced hibernation in TLS [48]. Additionally such transfusions did not induce hibernation in Richardson’s ground squirrel (RGS) [48, 49], JGS or WC [50]. Studies that claimed to have found “HIT” were conducted in animals that experienced excessively long hibernation bouts, relied on inconsistent methods (i.e. some animals were given ip while other were given iv injections), and yielded irreproducible results as reviewed by Lyman [51].
Although a single “HIT”-like compound has not been found, some evidence suggests plasma albumin fraction and serum albumin fractions have “HIT”-like activity. When plasma albumin fraction was administered to summer active TLS, many of the TLS hibernated within 2 to 6 days [52]. This “HIT”-like compound was further characterized as thermolabile, protease sensitive, and nuclease insensitive [53] and found to have opiate-like properties [54] . A role for opioids in hibernation had been proposed [55], and indeed hypothermia and hypophagia in monkeys caused by icv administration of serum albumin fraction from WC [56] was blocked by the non-selective opiate antagonists naltrexone and naloxone [54]. Tests of mere specific agonists suggested that the δ–opioid receptor was involved in hibernation [57].
Although δ–opioid agonist DADLE infusion in TLS was suggested to induce hibernation comparable to that seen with infusion of plasma albumin fraction [58], results were not convincing since, for example. TLS in these studies did not hibernate until 5 days after administration of DADLE. Subsequently, plasma albumin fraction from hibernating animals (TLS, WC, black bear, or polar bear, Ursus maritimus) was shown to inhibit guinea pig ileum muscle strip contractility and this was partially reversed with naloxone [59]. Other studies of plasma albumin fraction from hibernating animals claiming δ–opioid activity failed to confirm such activity by reversing effects of depressed guinea pig ileum or mouse vas deferens contractility with δ–opioid receptor antagonists or by displacing [3H] DADLE binding [60–63]. Nevertheless, an 88kDa hibernation-related protein (p88 HRP) was isolated from a highly purified plasma albumin fraction from hibernating WC called hibernation related fraction (HRF). This protein was found to be highly homologous with α l–glycoprotein [63] and speculated to be an inhibitor of metalloproteinase [64].
Although a “HIT”–like compound from plasma or serum of hibernating animals has not yet been convincingly characterized or isolated, this time of research led to the discovery of cytoprotective properties of serum from hibernating animals and serendipitously led to discovery of cyloprotective properties of DADLE, Pretreatment with serum from hibernating animals (black bear or WC) or DADLE were found to similarly improve postisehemic myocardial metabolism and function in rabbit isolated heart preparations [12]. DADLE has been shown to yield protection in a variety of injurious scenarios such as ischemia-reperfusion in rat liver [65] and promotes neuronal survival [66, 67]. Delta–opioid receptor is involved in preconditioning in heart [68, 69] and in brain [70]. Preliminary evidence suggests DADLE protects against ischemia-reperfusion damage in striatum and cerebral cortexa. The relevance of DADLE to hibernation and neuroprotection has recently been reviewed [71]. Thus, while a true “HIT” has not been discovered, attempts to mimic the putative pharmacological properties of factors isolated from hibernating animals has contributed to development of a novel class of neuroprotectants.
vii. Other Mechanisms Related to Decreased Energy Demand
Synaptic remodeling, decreased protein synthesis, cell cycle regulation, and maintenance of ion homeostasis are mechanisms endogenous to hibernating species and consistent with decreased metabolism. Identification or isolation of the factors that regulate these mechanisms in hibernating animals may also be therapeutically useful in depressing metabolism in a non-hibernating species. For example, the brain and hibernation specific membrane phosphotyrosine protein that has been isolated from TLS, called pp98 according to its molecular weight, may indicate that hibernating animals use tyrosine phosphorylation to transduce a signal to respond to decreased energy [72].
Synaptic regression and remodeling that occurs during hibernation [73–75] is one means by which energy may be conserved during torpor. The mechanisms used by hibernating animals for such profound remodeling have appropriately been considered in applications for neurodegenerative disease [76, 77]. Stimulation of synaptogenesis during arousal, perhaps via the reparative and regulatory roles of growth factors, such as insulin-like growth factor-1 [78], or activation of c-Jun N-terminal kinase (JNK) [79] may yield a better functional outcome. Isolation of molecules that regulate growth factor expression to promote synaptic remodeling in hibernating animals may lead lo efficacious therapeutics for a variety of degenerative conditions.
Suppression of protein synthesis in brain of torpid TLS to only 0.04% of the average rate in summer active TLS [80] is another means by which energy expensive processes are downregulated. Phosphorylation of elongation factor-2 (eEF-2), which is a reversible mechanism related to inhibition of the elongation phase of translation, is increased in liver and brain of hibernating TLS relative to summer active TLS [81]. Translation is depressed and the poly(A) tail length of mRNA is stabilized during torpor in AGS [82]. Discovery of factors that orchestrate suppression of protein synthesis and allow regulated shutdown of cellular function in hibernating animals could protect non-hibernating species during injury and trauma.
Decreasing cellular proliferation is another means of conserving energy. During torpor, cells that normally actively divide become relatively quiescent [83] Attenuation of cellular proliferation in various tissues during torpor occurs in the G1 phase [84], the premitotic phase [85], or in other phases of the cell cycle as reviewed by Lyman [86]. Brain extracts from hibernating TLS decrease cell division in Chinese hamster ovary cells (CHO) [87]. If identified, the factors that regulate entrance into and exit from the cell cycle in hibernating animals may prove to be useful in treating cancer or in avoiding neurodegenerative disease states such as Alzheimer disease which show cell cycle dysregulation [88], Perhaps hibernating animals possess factors that regulate cell cycle such as those described in anoxia induced suspended animation in C. elegans [89].
The energy expensive regulation of ion homeostasis by the ATP-dependent sodium-potassium pump would be unnecessary if ion flux during hibernation were minimized through modulation of ion channels, Downregulation of membrane ion permeability by “channel arrest” hypothesized by Hochachka et al. [90] has been shown in anoxia tolerant species such as the turtle [91–93]. Evidence of channel arrest in torpid TLS suggests modification of Q-type voltage sensitive Ca2+ channels decreases presynaptic Ca2+ accumulation in synaptosomes [94]. Additionally, enhanced Ca2+ homeostasis occurs in cardiac myocytes from the hibernating Daurian suslik, Spermophilus dauricus [95], and preliminary evidence suggests Ca2+ homeostasis is preserved in AGS hippocampus via modulation of NMDA receptorsb. Isolation and characterization of factors from hibernating animals that regulate ion channel arrest may lead to development of novel therapeutics.
B. Metabolic Switch
i. Therapeutic Potential of a Factor Involved in a Metabolic Switch
When energy supply does not match energy demand, as occurs its ischemia or epilepsy, a metabolic crisis ensues in neurons. A therapeutic that switches metabolism from use of carbohydrates to fatty acid oxidation may yield protection. Ketone bodies may act as an alternative energy source during subsequent times of glucose shortage, and increased free fatty acids (FFA) can replace polyunsaturated fatty acids lost during the damage to neuronal membranes that occurs as a result of metabolic crisis.
ii. Successful Experimental or Clinical Evidence of Induction of a Metabolic Switch
Hyperketogenic diets are successful in treatment of epilepsy [96, 97] and have been proposed to benefit patients with Parkinson disease, Alzheimer disease, and other neurodegenerative disorders [98]. Beta-hydroxybutyrate provided as an alternative energy substrate during ischemia yields neuroprotection in rat and mouse [99, 100]. Blood beta-hydroxybutyrate levels increase in TLS exposed to hypoxia, and increased blood beta-hydroxybutyrate levels correlate with prolonged survival times of TLS and rats during hypoxia [101]. Enhanced production of ketones in mice following brief episodes of hypoxia, i.e. preconditioning, yield protection in subsequent hypoxic episodes [102].
Increasing plasma FFA by means of albumin administration, which is currently in clinical trials for stroke, confers protection following middle cerebral artery occlusion [103]. Additionally, human serum albumin protects against oxidant induced neuronal death [104], and a docosahexaenoic acid and albumin complex yields neuroprotection against ischemia while allowing for a lower therapeutic dose of albumin thus decreasing side affects of high dose albumin [105].
iii. Evidence of a Metabolic Switch in Hibernation
Hibernating animals switch from use of carbohydrates to oxidation of stored fatty acids as fuel during torpor. Hibernating Belding’s ground squirrel (BGS) are ketotic compared to non-hibernators [106]. FFA are increased in serum of hibernating European ground squirrel (EGS) [107–109] and decrease during arousal from hibernation [110].
iv. Differential Regulation of Gene Expression in Hibernating Animals Associated with Potential Metabolic Switches
As of yet, no studies have investigated bioactivity of endogenous factors to switch from carbohydrate to fat metabolism. However, studies of differential regulation of gene expression in hibernating animals have uncovered potential metabolic switches. For example, levels of active phosphorylated AKT, peroxisome proliferator-activated receptor (PPAR)γ, and PPAR coaetivator PGC-1 decrease during hibernation in little brown bat (LBB) brain which are likely responsible for metabolic alterations in brain during hibernation such as preferential use of lipids [111].
Expression of a key enzyme that regulates fuel selection, pyruvate dehydrogenase kinase isoenzyme 4 (PDK-4), increases in torpid TLS [112]. Insulin levels decrease during torpor [112] and expression of PDK-4 is inversely related to levels of insulin, FA are ligands for PPARα which in turn activates the PDK-4 gene [113]. Gene expression of pancreatic triacylglycerol lipase, which frees non-esterified FA to be used as fuel, is also upregulated in torpid TLS [114].
A circulating effector molecule has been proposed to induce a metabolic switch via hibernation-associated gene expression [114]. For example, changes in serum insulin and FA may regulate this switch by affecting PDK-4 gene expression [3]. If this molecule could be isolated and developed into a therapeutic, perhaps a metabolic switch could be induced in non-hibernating species.
C. Hypocoagulation
Therapeutic use of factors that regulate hypocoagulability may yield better tissue perfusion during times of low blood flow as well as prevent clot formation that could block vascular flow and lead to stroke. Anticoagulants such as aspirin have long been used as preventative stroke therapy by reducing the likelihood of clot formation. If clot formation does occur and stroke ensues, then a thrombolytic agent is needed to restore blood flow, Growing evidence of significant side effects associated with use of the only clinically approved thrombolytic agent, recombinant tissue- type plasminogen activator [115], in addition to its plethora of centra-indications, calls for a search for alternative thrombolytic agents for stroke.
Blood from hibernating animals possesses unique characteristics such as profound hypocoagnlability [116–119]. Thrombocytopenia and plasma defects (such as decreased levels of Factor VIII and IX and increased prothrombin and partial thromboplastin time) contribute to inhibition of blood clotting during hibernation in TLS [118], yet platelet numbers return to normal values during arousal from hibernation [119]. In hamsters, decreased whole blood viscosity allows for persistent tissue perfusion during cold temperature and low blood flow characteristic of hibernation [120].
Four unique proteins were found in blood of TLS and Asian chipmunk (AC), but not in rodent non-hibernators rat or Formosan tree squirrel, Callosciurus caniceps [121]. These four hibernation proteins (HPs) exist as a 140 kDa complex in the non-hibernating state and are not present in blood during torpor. The 55kDa protein, called HP-55, is highly homologous with α 1-antitrypsin, a member of the serpin :superfamily, part of the major proteolytic cascade, and involved with coagulation, complement and inflammation.
Increased expression of alpha-2 macroglobulin (α2M), a protease inhibitor that decreases coagulation, occurs at miRNA and protein levels in torpid Richardson’s ground squirrel [122, 123], Scrum α2M is also elevated during hibernation in black bear [124], In fact, α2M is a broad spectrum protease inhibitor capable of inactivating matrix metalloproteinases (MMPs) as well as proteins in the clotting cascade. MMPs arc required for proper homeostatic and regulatory mechanisms yet are also destructive in many neurodegenerative conditions [125]. Since α2M clears amyloid-beta, α2M deficiency is thought to lead to Alzheimer disease pathology, but polymorphisms of the α2M gene have not been decisively implicated as causative of Alzheimer disease [126, 127] or Parkinson disease [128]. In cancer, MMPs break down matrix between cells allowing for metastasis. In arthritis and Alzheimer disease, MMPs cause tissue damage, and in ischemia-reperfusion MMPs cause break down of blood-brain-barrier. A proper proteolytic balance of MMP inhibitors and MMPs yields protection while preserving normal function in tissue [129]. Development of α2M as a therapeutic is promising in terms of both its anticoagulant effects and protease binding properties for degenerative disease, ischemia, and cancer. Additionally, isolation of novel factors that regulate hypocoagnlability in hibernating animals may lead to development of therapeutics.
D. Immunosuppression
Immunosuppression may slow tissue destruction that results from inflammatory processes repairing a site of injury following ischemia or traumatic brain injury. In fact atherosclerosis, the leading cause of cardiac disease and stroke, is an inflammatory process [130]. As a therapeutic, suppressing the immune system would be of great value in allowing for clinical intervention before extensive tissue loss occurs.
Current strategics in stroke treatment include modulation of the immune response for example using the immunosuppressive agent FY506 [131] or by antagonism of cytokines such as interleukin-1 [130], Long-term administration of statins decreases risk of stroke and improves outcome following stroke. When administered immediately after an ischemic event, statins improve short-term outcome as recently reviewed [132]. Currently, an immunomodulatory role for statins is being elucidated [133, 134].
Immunomodulation during hibernation includes immune suppression during torpor characterized by leukocytopenia [4, 119, 135] and return of immune function with return of leukocytes during arousal [119]. Accordingly, complement activity was found to be lowest during torpor and highest during arousal in golden mantled ground squirrel [136]. Evidence of immunosuppression during hibernation also includes downregulation of interferons and cytokines in serum of hibernating spotted susliks (SS) produced in response to viral infections [137, 138].
Brown fat extract from TLS depresses the primary immune response in cultured hamster spleen fragments [139]. Plasma from hibernating TLS and WC decreases proliferation in cultured mouse spleenocytes [140]. Plasma from hibernating TLS increases induction of intercellular adhesion molecule-1 (ICAM-I), a protein expressed by endothelial cells that regulates interactions with circulating leukocytes, and increases monoctye adhesion in rat cerebral microvascular endothelial cells [141].
Hibernating animals indeed possess unique factors that are involved in modulation of the immune system; these factors, if isolated, may be of great benefit as therapeutics. By inducing a state in which the immune response is suppressed, tissue loss could be attenuated in neurodegenerative disease and trauma. Torpid AGS brain subjected to a stab-like wound failed to mount an immune response or show signs of tissue damage [7].
III. CONSIDERATIONS FOR DEVELOPMENT OF THERAPEUTICS
Strategies for developing therapeutics may need to include expression of effector pathways that involve both the factor and the target (e.g. growth factor in serum and corresponding receptor in tissue). For example, intrinsic tissue properties of torpid AGS and factors in AGS serum are both necessary for improved survival in AGS hippocampal slices as described in Fig. (2). Similarly, neuroprotection by deferoxamine (chelator of non-protein-bound iron) and allopurinol and oxypurinol (xanthine oxidase inhibitors) requires the presence of either a biood-borne substance or endothelial cells [142]. Lastly, development of effective therapeutics may benefit from use of a ‘cocktail’ where more than one effector pathway is regulated (e.g. decreasing metabolism and immune response) as demonstrated by hibernating animals.
Fig. 2.
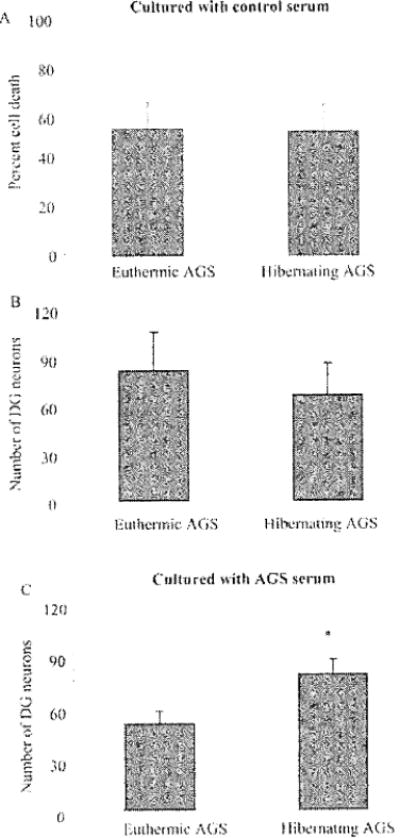
Hippocampal slices from hibernating AGS survive significantly better than slices from euthermic AGS when cultured in the presence of AGS serum. Euthermic AGS include cold-adapted AGS that did not hibernate and AGS in interbout euthermy, A and B) On day 1 of hippocampal slice culture at 37°C, dentate gyms (DG) neurons from hibernating AGS survive the same as euthermic AGS neurons when cultured in media with (control) B-27 Supplement Minus AO (Invitrogen, Carlsbad, CA). Percent cell death is quantified via presidium iodide fluorescence intensity (n=9 slices/3 hibernating AGS and n=9 slices/3 euthermic AGS). Number of neurons in DG arc counted following NeuN immunocytochemistry to identify neurons (n=7 slices/3 hibernating AGS and n=8 slices/3 eutheimie AGS). C) On day 1 of culture at 37°C, DG neurons from hibernating AGS survive better than DG neurons from euthermic AGS when B-27 is replaced with scrum from hibernating or euthermic AGS (n=9 slices/3 hibernating AGS and n=9 slices/3 euthermic AGS), *p< 0.95. Data are expressed as mean ± SEM and were analyzed with two-way ANOVA. Significant (p< 0.05) main effects or interactions were followed by Holm-Sidak pairwise comparison post-hoc analysis using SigmaStat 3.0 (Systat Software, Inc., Point Richmond, CA).
IV. PROTECTIVE MECHANISMS IN HIBERNATING SPECIES EFFICACIOUS IN NON-HIBERNATING SPECIES
Success in regulating protective mechanisms endogenous to non-hibernating species is strongly supported by recent findings in preconditioning and of induction of suspended animation, For example, upregulation of antioxidants in brain and peripheral organs occurs following ischemic preconditioning [143]. Preconditioning results in transcriptional changes involved in suppression of metabolism, immune response, ion channel activity, and blood coagulation, all of which are protective mechanisms endogenous to hibernating species [144]. Hydrogen sulfide inducing a suspended animation-like state, including decreased metabolism, in mice [17] is convincing support that aspects of hibernation can be mimicked in non-hibernating species. In the search for novel and much needed neuroprotective therapeutics, a focused effort to isolate factors that are endogenous to hibernating animals is well warranted.
Acknowledgments
This project was supported by Alaska BRIN (NIH 1P20RR16466) and INBRE (NIH 2P20RR16466) from the RFIP of the National Center tor Research Resources and NIH-NS41069) and by the Alaska SNRP (NIH U54-NS 41069-01 supported by NINDS, NIMH, NCRR and NCMHD).
ABBREVIATIONS
- DADLE
D-Ala(2), D-Leu(5)] enkephalin
- HIT
Hibernation induction trigger
- FFA
Free fatty acids
- PDK-4
Pyruvate dehydrogenase kinase isoenzyme 4
- α2M
Alpha-2 macroglobulin
- MMPs
Matrix metalloproteinases
- DG
Dentate gyrus
Footnotes
Borlongan. C. V.; Oeltgen. P. R.; Su, T. P.; Wang, Y. Delta opioid peptide (DADLE) protects against. ischemia-reperfusion damage in the striatum and cerebral cortex. Soc. Neurosci. Abstr. 1999. 25. 550
Zhao., H. W.; Ross, A.P.; Christian, S.L.; Buchholz, J.N.; Drew, K.L. Suppression of NMDA receptor function in hibernating Arctic Ground Squirrels, 36th American Society for Neurochemistry (ASN) Annual Meeting, June 24–29, 2005.
References
- 1.Green AR, Ashwood T. Curr Drug Targets CNS Neural Disord. 2005;4:109–18. doi: 10.2174/1568007053544156. [DOI] [PubMed] [Google Scholar]
- 2.Dausmann KH, Glos J, Ganzhom JU, Heldmaier G. Nature. 2004;420:825–6. doi: 10.1038/429825a. [DOI] [PubMed] [Google Scholar]
- 3.Carey HV, Andrews MT, Martin SL. Physiol Rev. 2003;83:1153–81. doi: 10.1152/physrev.00008.2003. [DOI] [PubMed] [Google Scholar]
- 4.Drew KL, Rice ME, Kuhn TB, Smith MA. Free Radic Biol Med. 2001;31:563–73. doi: 10.1016/s0891-5849(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 5.Frerichs KU, Kennedy C, Sokoloff L, Hallenbeck JM. J Cereb Blood Flow Metab. 1994;14:193–205. doi: 10.1038/jcbfm.1994.26. [DOI] [PubMed] [Google Scholar]
- 6.Ma YL, Zhu X, Rivera PM, Toien O, Bames BM, Lamaona JC, Smith MA, Drew KL. Am J Physiol Regil Integr Comp Physiol. 2005;289:R1297–306. doi: 10.1152/ajpregu.00260.2005. [DOI] [PubMed] [Google Scholar]
- 7.Zhou F, Zhu X, Castellani RJ, Stimmelmayr R, Perry G, Smith MA, Drew KL. Am J Pathol. 2001;158:2145–51. doi: 10.1016/S0002-9440(10)64686-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frerichs KU, Hallenbeck JM. J Cereb Blood Flow Metab. 1998;18:168–75. doi: 10.1097/00004647-199802000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Ross AP, Christian SL, Zhao HW, Drew KL. J Cereb Blood Flow Metab. 2006 doi: 10.1038/sj.jcbfm.9600271. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 10.Storey KB. J Investig Med. 2004;52:315–22. doi: 10.1136/jim-52-05-31. [DOI] [PubMed] [Google Scholar]
- 11.Green C. Cryo Lett. 2000;21:91–98. [PubMed] [Google Scholar]
- 12.Bolling SF, Tramonlini NL, Kilgore KS, Su TP, Oeligen PR, Harlow HH. Ann Thorac Surg. 1997;64:623–7. doi: 10.1016/s0003-4975(97)00631-0. [DOI] [PubMed] [Google Scholar]
- 13.Liberatore GT, Samson A, Bladin C, Schleuning WD, Medcalf RL. Stroke. 2003;34:537–43. doi: 10.1161/01.str.0000049764.49162.76. [DOI] [PubMed] [Google Scholar]
- 14.Debens T. Curr Opin Mol Ther. 2004;6:567–75. [PubMed] [Google Scholar]
- 15.Terlau H, Olivers BM. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- 16.Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M, Fisher R, Bryce DA, Mangieri EA, Luther RR, Mayo M, McGuire D, Ellis D. JAMA. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- 17.Blackstone E, Morrison M, Roth MB. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 18.Drew KL, Toien O, Rivera PM, Smith MA, Perry G, Rice ME. Comp Biochem Physiol C Toxicol Pharmacol. 2002;133:483–92. doi: 10.1016/s1532-0456(02)00118-7. [DOI] [PubMed] [Google Scholar]
- 19.Bellamy R, Safer P, Tisherman SA, Basford R, Bruttig SP, Capone A, Dubick MA, Ernster L, Hattler BG, Jr, Hochachka P, Klain M, Kochanek PM, Kofke WA, Lancaster JR, McGowan FX, Jr, Oeltgen PR, Severinghaus JW, Taylor MJ, Zar H. Crit Care Med. 1996;24:S24–47. [PubMed] [Google Scholar]
- 20.Nystul TG, Roth MB. Proc Natl Acad Sci USA. 2004;101:9133–6. doi: 10.1073/pnas.0403312101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frerichs KU, Dienel GA, Cruz NF, Sokoloff L, Hallenbeck JM. Am J Physiol. 1995;268:R445–53. doi: 10.1152/ajpregu.1995.268.2.R445. [DOI] [PubMed] [Google Scholar]
- 22.Lust WD, Wheaton AB, Feussner G, Passonneau J. Brain Res. 1989;489:12–20. doi: 10.1016/0006-8993(89)90003-6. [DOI] [PubMed] [Google Scholar]
- 23.Swan H, Jenkins D, Knox K. Nature. 1968;217:671. doi: 10.1038/217671a0. [DOI] [PubMed] [Google Scholar]
- 24.Swan H, Jenkins D, Knox K. Am Nat. 1969;103:247–258. [Google Scholar]
- 25.Swan H, Reinhard FG, Caprio DL, Schatte CL. Cryobiology. 1981;18:598–602. doi: 10.1016/0011-2240(81)90128-0. [DOI] [PubMed] [Google Scholar]
- 26.Swan H, Schatte C. Science. 1977;195:84–5. doi: 10.1126/science.831261. [DOI] [PubMed] [Google Scholar]
- 27.Pastukhov Iu F, Chepkasov IE. Zh Evol Biokhim Fiziol. 1984;20:327–30. [PubMed] [Google Scholar]
- 28.Pastukhov lu F, Chepkasov IE. Fiziol Zh SSSR Im I M Sechenova. 1983;69:1485–90. [PubMed] [Google Scholar]
- 29.Pastukhov lu F, Chepkasov IE. Dokl Akad Nauk SSSR. 1984;275:510–2. [PubMed] [Google Scholar]
- 30.Ignat’ev DA, Sukhova GS, Zagnoiko VI, Sukhov VP, Sviriaev VI. Zh Evol Biokhim Fiziol. 1992;28:459–66. [PubMed] [Google Scholar]
- 31.Ignat’ev DA, Zagnoiko VI, Sukhova GS, Bakaneva VF, Sukhov VP. Zh Obshch Biol. 1995;56:450–69. [PubMed] [Google Scholar]
- 32.Zenchenko KI, Kokoz YM, Ivanov VT, Ziganshin RH, Vinograduva OS. Neuroscience. 2000;96:791–805. doi: 10.1016/s0306-4522(99)00591-6. [DOI] [PubMed] [Google Scholar]
- 33.Zenchenko KI, Kokoz YM, Ivanov VT, Ziganshin RK, Vinogradova OS. Neurosci Behav Physiol. 2001;31:395–403. doi: 10.1023/a:1010436628268. [DOI] [PubMed] [Google Scholar]
- 34.Ignat’ev DA, Vorob’ev VV, Ziganshin R. Zh Vyssh Nerv Deial Im I P Pavlova. 1996;46:1049–58. [PubMed] [Google Scholar]
- 35.Kokoz YM, Zenchenko KI, Alekseev AE, Korystova AF, Lankina DA, Ziganshin RH, Mikhaleva II, Ivanov VT. FEBS Lett. 1997;411:71–6. doi: 10.1016/s0014-5793(97)00607-8. [DOI] [PubMed] [Google Scholar]
- 36.Yu EZ, Hallenbeck JM, Cai D, McCarron RM. Brain Res Mol Brain Res. 2002;102:9–17. doi: 10.1016/s0169-328x(02)00138-9. [DOI] [PubMed] [Google Scholar]
- 37.Kilduff TS, Miller JD, Radeke CM, Sharp FR, Heller HC. J Neurosci. 1900;10:2463–75. doi: 10.1523/JNEUROSCI.10-07-02463.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dark J, Kilduff TS, Heller HC, Licht P, Zuckcr I. Brain Res. 1990;509:111–8. doi: 10.1016/0006-8993(90)90316-4. [DOI] [PubMed] [Google Scholar]
- 39.Stanton TL, Daley JC, Salzman SK. Brain Res. 1987;413:350–5. doi: 10.1016/0006-8993(87)91027-4. [DOI] [PubMed] [Google Scholar]
- 40.Deacon S, Arendt J. Brain Res. 1995;688:77–85. doi: 10.1016/0006-8993(95)96872-i. [DOI] [PubMed] [Google Scholar]
- 41.Satoh K, Mishima K. Clin Neuropharnacol. 2001;24:334–40. doi: 10.1097/00002826-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 42.Derlacz RA, Poplawski P, Napierala M, Jagielski AK, Bryla J. J Pineal, Res. 2005;38:164–9. doi: 10.1111/j.1600-079X.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt KE, Kelley KM. J Exp Zool. 2001;289:66–73. doi: 10.1002/1097-010x(20010101/31)289:1<66::aid-jez7>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 44.Cai D, McCarron RM, Yu EZ, Li Y, Hallenbeck J. Brain Res. 2004;1014:14–21. doi: 10.1016/j.brainres.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 45.Barros RC, Zimmer ME, Branco LG, Milsom WK. J Appl Physiol. 2001;91:603–12. doi: 10.1152/jappl.2001.91.2.603. [DOI] [PubMed] [Google Scholar]
- 46.Dawe AR, Spurrier WA. Science. 1969;163:298–9. doi: 10.1126/science.163.3864.298. [DOI] [PubMed] [Google Scholar]
- 47.Spurrier WA, Folk GE, Jr, Dawe AR. Cryobiology. 1976;13:368–74. doi: 10.1016/0011-2240(76)90120-6. [DOI] [PubMed] [Google Scholar]
- 48.Wang LC, Belke D, Jourdan ML, Lee TF, Westly J, Nurnberger F. Cryobiology. 1988;25:355–62. doi: 10.1016/0011-2240(88)90043-0. [DOI] [PubMed] [Google Scholar]
- 49.Abbotts B, Wang LC, Glass JD. Cryobiology. 1979;16:179–83. doi: 10.1016/0011-2240(79)90029-4. [DOI] [PubMed] [Google Scholar]
- 50.Galster WA. J Thermal Biol. 1978;3:93. [Google Scholar]
- 51.Lyman CP. In: Hibernation and Torpor in Mammals and Birds. Lyman CP, Willis JS, Malan A, Wang LCH, editors. Academic Press; New York: 1982. pp. 294–301. [Google Scholar]
- 52.Oeltgen PR, Bergmann LC, Spurrier WA, Jones SB. Prep Biochem. 1978;8:171–88. doi: 10.1080/00327487808069058. [DOI] [PubMed] [Google Scholar]
- 53.Oeltgen PR, Spurrier WA. In: Survival in the Cold; Hibernation and Other Adaptations. Mussachia XJ, Jansky L, editors. Elsevier; New York: 1981. pp. 139–157. [Google Scholar]
- 54.Oeltgen PR, Walsh JW, Hamann SR, Randall DC, Spurrier WA, Myers RD. Pharmacol Biochem Behav. 1982;17:1271–4. doi: 10.1016/0091-3057(82)90132-0. [DOI] [PubMed] [Google Scholar]
- 55.Beckman AL, Llados-Eekman C, Stanton TL, Adler MW. Science. 1981;212:1527–9. doi: 10.1126/science.7233241. [DOI] [PubMed] [Google Scholar]
- 56.Myers RD, Oeltgen PR, Spurrier WA. Brain Res Bull. 1981;7:691–5. doi: 10.1016/0361-9230(81)90120-9. [DOI] [PubMed] [Google Scholar]
- 57.Oeltgen PR, Welborn JR, Nuchols PA, Spurrier WA, Bruce DS, Su TP. Life Sci. 1987;41:2115–20. doi: 10.1016/0024-3205(87)90529-7. [DOI] [PubMed] [Google Scholar]
- 58.Oeltgen PR, Nilckani SP, Nuchols PA, Spurrier WA, Su TP. Life-Sci. 1988;43:1565–74. doi: 10.1016/0024-3205(88)90406-7. [DOI] [PubMed] [Google Scholar]
- 59.Bruce DS, Ambler DL, Henschel TM, Oeltgen PR, Nilckani SP, Amstrup SC. Pharmacol Biochem Behav. 1992;43:199–203. doi: 10.1016/0091-3057(92)90658-3. [DOI] [PubMed] [Google Scholar]
- 60.Bruce DS, Cox DE, Crane SK, Denholm ML, Dhyanchand RJ, Hampl MJ, Kary JA, Krober AS, Oeltgen PR, Horton ND, Harlow HJ. Pharmacol Biochem Behav. 1997;58:627–30. doi: 10.1016/s0091-3057(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 61.Bruce DS, Bailey EC, Crane SK, Oeltgen PR, Horton ND, Harlow HJ. Pharmacol Biochem Behav. 1997;58:621–5. doi: 10.1016/s0091-3057(97)00063-4. [DOI] [PubMed] [Google Scholar]
- 62.Horton ND, Oeltgen PR, Kaftam DJ, Su TP, Bruce DS, Krober AS, Jones JF. In: Adaptations to the Cold: Tenth International Hibernation Symposium. Geiser F, Hulbert AJ, Nieol SC, editors. University of New England Press: Armidale; 1996. pp. 333–339. [Google Scholar]
- 63.Horton ND, Kaftani DJ, Bruce DS, Bailey EC, Krober AS, Jones JR, Turker M, Khattar N, Su TP, Bolling SF, Oeltgen PR. Comp Biochem Physiol B Biochem Mol Biol. 1998;119:787–805. doi: 10.1016/s0305-0491(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 64.Catancse JJ, Kress LF. Biochemistry. 1992;31:410–8. doi: 10.1021/bi00117a015. [DOI] [PubMed] [Google Scholar]
- 65.Yamanouchi K, Yanaga K, Okudaira S, Eguchi S, Furui J, Kanematsu T. J Surg Res. 2003;114:72–7. doi: 10.1016/s0022-4804(03)00196-3. [DOI] [PubMed] [Google Scholar]
- 66.Su TP. J Biomed Sci. 2000;7:195–9. doi: 10.1007/BF02255466. [DOI] [PubMed] [Google Scholar]
- 67.Hayashi T, Su TP. Eur J Pharmacol. 2003;464:237–9. doi: 10.1016/s0014-2999(03)01419-5. [DOI] [PubMed] [Google Scholar]
- 68.Karck M, Tanaka S, Oeltgen P, Su TS, Bolling SF, Haverich A. Langenbecks Arch Chir Sappl Kongressbd. 1998;115:1–6. [PubMed] [Google Scholar]
- 69.Sigg DC, Coles JA, Jr, Gallagher WJ, Oeltgen PR, Iaizzo PA. Ann Thorac Surg. 2001;72:1576–82. doi: 10.1016/s0003-4975(01)03084-3. [DOI] [PubMed] [Google Scholar]
- 70.Ma MC, Qian H, Ghassemi F, Zhao P, Xia Y. J Biol Chem. 2005;280:16208–18. doi: 10.1074/jbc.M408055200. [DOI] [PubMed] [Google Scholar]
- 71.Borlongan CV, Wang Y, Su TP. Front Biosci. 2004;9:3392–8. doi: 10.2741/1490. [DOI] [PubMed] [Google Scholar]
- 72.Ohtsuki T, Jaffe H, Brenner M, Azzam N, Azzam R, Frerichs KU, Hallenbeck JM. J Cereb Blood Flow Metab. 1998;18:1040–5. doi: 10.1097/00004647-199809000-00014. [DOI] [PubMed] [Google Scholar]
- 73.Popov VI, Bocharova LS. Neuroscience. 1992;48:53–62. doi: 10.1016/0306-4522(92)90337-2. [DOI] [PubMed] [Google Scholar]
- 74.Popov VI, Bocharova LS, Bragin AG. Neuroscience. 1992;48:45–51. doi: 10.1016/0306-4522(92)90336-z. [DOI] [PubMed] [Google Scholar]
- 75.Strijkstra AM, Hut RA, de Wilde MC, Stieler J, Van der Zee EA. Neurosci Lett. 2003;344:29–32. doi: 10.1016/s0304-3940(03)00399-9. [DOI] [PubMed] [Google Scholar]
- 76.Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. J Neurosci. 2003;23:6972–81. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arendt T. Int J Dev Neurosci. 2004;22:507–14. doi: 10.1016/j.ijdevneu.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 78.Carro E, Trejo JL, Nunez A, Torres-Aleman I. Mol Neurabiol. 2003;27:153–62. doi: 10.1385/MN:27:2:153. [DOI] [PubMed] [Google Scholar]
- 79.Zhu X, Smith MA, Perry G, Wang Y, Ross AP, Zhao HW, Lamanna JC, Drew KL. J Neurosci Res. 2005;80:862–8. doi: 10.1002/jnr.20526. [DOI] [PubMed] [Google Scholar]
- 80.Freriehs KU, Smith CB, Brenner M, DeGracia DJ, Krause GS, Marrone L, Dever TE, Hallenbeck JM. Proc Natl Acad Sci USA. 1998;95:14511–6. doi: 10.1073/pnas.95.24.14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen Y, Matsushita M, Nairn AC, Damuni Z, Cai D, Frerichs KU, Hallenbeck JM. Biochemistry. 200l;40:11565–70. doi: 10.1021/bi010649w. [DOI] [PubMed] [Google Scholar]
- 82.Knight JE, Narus EN, Martin SL, Jacobson A, Barnes BM, Boyer BB. Mol Cell Biol. 2000;20:6374–9. doi: 10.1128/mcb.20.17.6374-6379.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kruman II, Kolaeva SG, Hjasova EN, Zubrikhina GN, Khachko VN, Petrova AS. Comp Biochem Physiol B. 1986;83:173–7. doi: 10.1016/0305-0491(86)90349-4. [DOI] [PubMed] [Google Scholar]
- 84.Kolaeva SG, Kramarova LI, Ilyasova EN, Ilyasov FE. Int Rev Cytot. 1980;66:147–70. doi: 10.1016/s0074-7696(08)61973-7. [DOI] [PubMed] [Google Scholar]
- 85.Kruman II. Comp Biochem Physiol A. 1992;101:11–8. doi: 10.1016/0300-9629(92)90620-6. [DOI] [PubMed] [Google Scholar]
- 86.Lyman CP. Pharmacol Ther. 1984;25:172–195. doi: 10.1016/0163-7258(84)90005-6. [DOI] [PubMed] [Google Scholar]
- 87.Annorese DA, Swan H, Bamburg JR. Proc Natl Acad Sci USA. 1982;79:6375–9. doi: 10.1073/pnas.79.20.6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Busser J, Geldmacher DS, Herrup K. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Padilla PA, Nystul TG, Zager RA, Johnson AC, Roth MB. Mol Biol Cell. 2002;13:1473–83. doi: 10.1091/mbc.01-12-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hochachka PW. Science. 1986;231:234–41. doi: 10.1126/science.2417316. [DOI] [PubMed] [Google Scholar]
- 91.Pek M, Lutz PL. J Exp Biol. 1997;200:1913–7. doi: 10.1242/jeb.200.13.1913. [DOI] [PubMed] [Google Scholar]
- 92.Perez-Pinzon MA, Rosenthal M, Sick TJ, Lutz PL, Pablo J, Mash D. Am J Physiol. 1992;262:R712–5. doi: 10.1152/ajpregu.1992.262.4.R712. [DOI] [PubMed] [Google Scholar]
- 93.Sick TJ, Rosenthal M, LaManna JC, Lutz PL. Am J Physiol. 1982;243:R281–8. doi: 10.1152/ajpregu.1982.243.3.R281. [DOI] [PubMed] [Google Scholar]
- 94.Gentile NT, Spatz M, Brenner M, McCarron RM, Hallenbeck JM. Neuroichem Res. 1996;21:947–54. doi: 10.1007/BF02532345. [DOI] [PubMed] [Google Scholar]
- 95.Wang SQ, Lakatta EG, Cheng H, Zhou ZQ. J Exp Biol. 2002;205:2957–62. doi: 10.1242/jeb.205.19.2957. [DOI] [PubMed] [Google Scholar]
- 96.Ziegler DR, Oliveira DL, Pires C, Ribeiro L, Leite M, Meadez A, Gonealves D, Tramontina F, Porlela LV, Wofchuk ST, Perry ML, Gonealves CA. Neurosci Res. 2004;50:375–9. doi: 10.1016/j.neures.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 97.Bailey EE, Pfeifer HH, Thiele EA. Epilepsy Behav. 2005;6:4–8. doi: 10.1016/j.yebeh.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 98.Vanltallie TB, Nufert TH. Nutr Rev. 2003;61:327–41. doi: 10.1301/nr.2003.oct.327-341. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki M, Suzuki M, Kitamura Y, Mori S, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A. Jpn J Pharmacol. 2002;89:36–43. doi: 10.1254/jjp.89.36. [DOI] [PubMed] [Google Scholar]
- 100.Suzuki M, Suzuki M, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A. Jpn J Pharmacol. 2001;87:143–50. doi: 10.1254/jjp.87.143. [DOI] [PubMed] [Google Scholar]
- 101.D’Alecy LG, Lundy EF, Khiger MJ, Harker CT, LeMay DR, Shlafer M. Comp Biochem Physiol B. 1990;96:189–93. doi: 10.1016/0305-0491(90)90361-v. [DOI] [PubMed] [Google Scholar]
- 102.Rising CL, D’Alecy LG. Stroke. 1989;20:1219–25. doi: 10.1161/01.str.20.9.1219. [DOI] [PubMed] [Google Scholar]
- 103.Rodriguez de Turco EB, Belayev L, Liu Y, Busto R, Parkins N, Bazan NG, Ginsberg MD. J Neurochem. 2002;83:515–24. doi: 10.1046/j.1471-4159.2002.01121.x. [DOI] [PubMed] [Google Scholar]
- 104.Gum ET, Swanson RA, Alano C, Liu J, Hong S, Weinstein PR, Panter SS. Stroke. 2004;35:590–5. doi: 10.1161/01.STR.0000110790.05859.DA. [DOI] [PubMed] [Google Scholar]
- 105.Belayev L, Marcheselli VL, Khoutorava L, Rodriguez de Tureo EB, Busto R, Ginsberg MD, Bazan NG. Stroke. 2005;36:118–23. doi: 10.1161/01.STR.0000149620.74770.2e. [DOI] [PubMed] [Google Scholar]
- 106.Krilowiez BL. Am J Physiol. 1985;249:R462–70. doi: 10.1152/ajpregu.1985.249.4.R462. [DOI] [PubMed] [Google Scholar]
- 107.Suomalainen H. Experientia. 1967;23:457–8. [Google Scholar]
- 108.Esher RJ, Fleischman AL, Lenz PH. Comp Biochem Physiol A Physiol. 1973;45:933. doi: 10.1016/0300-9629(73)90329-0. [DOI] [PubMed] [Google Scholar]
- 109.Buzadzic B, Spasic M, Saieic ZS, Radojicie R, Halliwell B, Petrovic VM. Free Radic Biol Med. 1990;9:401–6. doi: 10.1016/0891-5849(90)90016-c. [DOI] [PubMed] [Google Scholar]
- 110.Konttinen A, Rajasalmi M, Sarajas HS. Am J Physiol. 1964;207:845–8. doi: 10.1152/ajplegacy.1964.207.4.845. [DOI] [PubMed] [Google Scholar]
- 111.Eddy SF, Storey KB. Biochem Cell Biol. 2003;81:269–74. doi: 10.1139/o03-056. [DOI] [PubMed] [Google Scholar]
- 112.Buck MJ, Squire TL, Andrews MT. Physiol Genomics. 2002;8:5–13. doi: 10.1152/physiolgenomics.00076.2001. [DOI] [PubMed] [Google Scholar]
- 113.Wu P, Peters JM, Harris RA. Biochem Biophys Res Commun. 2001;287:391–6. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]
- 114.Andrews MT. Biochem Soc Trans. 2004;32:1021–4. doi: 10.1042/BST0321021. [DOI] [PubMed] [Google Scholar]
- 115.Traynclis SF, Lipton SA. Nat Med. 2001;7:17–8. doi: 10.1038/83289. [DOI] [PubMed] [Google Scholar]
- 116.Svihla A, Bowman HR, Ritenour R. Science. 1951;114:298–9. doi: 10.1126/science.114.2960.298. [DOI] [PubMed] [Google Scholar]
- 117.Svihla A, Bowman H, Pearson R. Science. 1952;115:272. doi: 10.1126/science.115.2984.272. [DOI] [PubMed] [Google Scholar]
- 118.Lechler E, Penick GD. Am J Physiol. 1963;205:985–8. doi: 10.1152/ajplegacy.1963.205.5.985. [DOI] [PubMed] [Google Scholar]
- 119.Pivorun EB, Sinnamon WB. Cryobiology. 1981;18:515–20. doi: 10.1016/0011-2240(81)90212-1. [DOI] [PubMed] [Google Scholar]
- 120.Deveci D, Stone PC, Egginton S. J Comp Physiol B. 2011;171:135–43. doi: 10.1007/s003600000156. [DOI] [PubMed] [Google Scholar]
- 121.Kondo N, Kondo J. J Biol Chem. 1992;267:473–8. [PubMed] [Google Scholar]
- 122.Srere HK, Wang LC, Martin SL. Proc Natl Acad Sci USA. 1992;89:7119–23. doi: 10.1073/pnas.89.15.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Epperson LE, Martin SL. Physiol Genomics. 2002;10:93–102. doi: 10.1152/physiolgenomics.00004.2002. [DOI] [PubMed] [Google Scholar]
- 124.Sheikh AM, Chauban V, Tsiouris JA, Mehta PD, Burguess K, Fenko MD, Spivack W, Vaughan M, Malik M. Biochimie. 2003;85:1027–32. doi: 10.1016/s0300-9084(03)00133-0. [DOI] [PubMed] [Google Scholar]
- 125.Lorenzl S, Albers DS, LeWitt PA, Chirichigno JW, Hilgenberg SL, Cudkowiez ME, Beal MF. J Neurol Sci. 2003;207:71–6. doi: 10.1016/s0022-510x(02)00398-2. [DOI] [PubMed] [Google Scholar]
- 126.Birkenmeier G, Muller R, Huse K, Porberg J, Glaser C, Hedrich H, Nicklisch S, Reicnenbach A. Exp Neurol. 2003;184:153–61. doi: 10.1016/s0014-4886(03)00110-9. [DOI] [PubMed] [Google Scholar]
- 127.Saunders AJ, Bertram L, Mullin K, Sampson AJ, Latifzai K, Basu S, Jones J, Kinney D, MacKenzie-Ingano L, Yu S, Albert MS, Moscarillo TJ, Go RC, Bassett SS, Daly MJ, Laird NM, Wang X, Velicelebi G, Wagner SL, Becker DK, Tanzi RE, Blacker D. Hum Mol Genet. 2003;12:2765–76. doi: 10.1093/hmg/ddg310. [DOI] [PubMed] [Google Scholar]
- 128.Nicoleni G, Annesi G, Tomaino C, Spadafora P, Pasqua AA, Annesi F, Serra P, Caracciolo M, Messina D, Zappia M, Quattrone A. Neurosci Lett. 2002;328:65–7. doi: 10.1016/s0304-3940(02)00003-4. [DOI] [PubMed] [Google Scholar]
- 129.Baker AH, Edwards DR, Murphy G. J Cell Sci. 2002;115:3719–27. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 130.Emsley HC, Tyrrell PJ. J Cereb Blood Flow Metab. 2002;22:1399–419. doi: 10.1097/01.WCB.0000037880.62590.28. [DOI] [PubMed] [Google Scholar]
- 131.Macleod MR, O’Collins T, Horky IL, Howells DW, Donnan GA. J Cereb Blood Flow Metab. 2005;25:713–721. doi: 10.1038/sj.jcbfm.9600064. [DOI] [PubMed] [Google Scholar]
- 132.Endres M. J Cereb Blood Flow Metab. 2005;25:1093–1110. doi: 10.1038/sj.jcbfm.9600116. [DOI] [PubMed] [Google Scholar]
- 133.Trubelja N, Vaughan C, Coplan NL. Prev Cardiol. 2005;8:98–101. doi: 10.1111/j.1520-037x.2005.3568.x. [DOI] [PubMed] [Google Scholar]
- 134.Crisby M. Timely Top Med Cardiovasc Dis. 2005;9:E3. [PubMed] [Google Scholar]
- 135.Harkness DR, Roth S, Goldman P. Comp Biochem Physiol A. 1974;48:591–9. doi: 10.1016/0300-9629(74)90742-7. [DOI] [PubMed] [Google Scholar]
- 136.Maniero GD. Dev Comp Immunol. 2002;26:563–74. doi: 10.1016/s0145-305x(02)00006-x. [DOI] [PubMed] [Google Scholar]
- 137.Kandefer-Szerszen M. J Interferon Res. 1988;8:295–302. doi: 10.1089/jir.1988.8.295. [DOI] [PubMed] [Google Scholar]
- 138.Kandefer-Szerszen M, Meezynski S, Kaweeki Z, Kaezor J. J Interferon Res. 1988;8:303–7. doi: 10.1089/jir.1988.8.303. [DOI] [PubMed] [Google Scholar]
- 139.Sidky YA, Daggett LR, Auerbach R. Proc Soc Exp Biol Med. 1969;132:760–3. doi: 10.3181/00379727-132-34305. [DOI] [PubMed] [Google Scholar]
- 140.Sieckmann DG, Cai D, Jaffe H, Hallenbeck J, McCarron RM. In: Life in the Cold Evolution Mechanisms, Adaptation, and Application. Barnes BM, Carey HV, editors. Institute of Arctic Biology; Fairbanks: 2004. pp. 519–525. [Google Scholar]
- 141.Yasuma Y, McCarron RM, Spatz M, Hallenbeck JM. Am J Physiol. 1997;273:R1861–9. doi: 10.1152/ajpregu.1997.273.6.R1861. [DOI] [PubMed] [Google Scholar]
- 142.Peeters C, Hoelen D, Groenendaal F, van Bel F, Bar D. Brain Res. 2003;963:72–80. doi: 10.1016/s0006-8993(02)03843-x. [DOI] [PubMed] [Google Scholar]
- 143.Glantz L, Avromovich A, Trembovler V, Gurvitz V, Kohren R, Eidelman LA, Shuhami E. Exp Neurol. 2005;192:117–24. doi: 10.1016/j.expneurol.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 144.Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Lancet. 2003;362:1028–37. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]