

Abstract
The large and complex eukaryotic nucleus is the arbiter of DNA replication, RNA transcription, splicing, and ribosome assembly. With the advent of in vitro nuclear reconstitution extracts derived from Xenopus eggs in the 1980s, it became possible to assemble multiple nuclei in vitro around added DNA or chromatin substrates. Such reconstituted nuclei contain a nuclear lamina, double nuclear membranes, nuclear pores, and are competent for DNA replication and nuclear import. In vitro nuclear reconstitution has allowed the assembly of “wild-type” and “biochemically mutant” nuclei in which the impact of individual components can be assessed. Here, we describe protocols for preparation of the nuclear reconstitution extract, nuclear reconstitution in vitro, assessment of nuclear membrane integrity, and a more specialized assay for nuclear pore assembly into preformed pore-free nuclear intermediates.
INTRODUCTION
In vitro assays allow one to alter or study reactions in more detail. The nuclear reconstitution extract derived from Xenopus eggs in essence serves as a miniature in vitro reactor for analyzing the biochemical reactions and properties encountered in the more organized and complex environment of intact cells (Forbes, Kirschner, & Newport, 1983; Lohka & Masui, 1983, 1984; Newport, 1987; Newport & Spann, 1987; Wilson & Newport, 1988).
Prior to the advent of nuclear reconstitution, an experimental attack on the structure and assembly of the eukaryotic nucleus was almost impossible. The tools available at the time included only nuclear isolation from cells, followed by crude fractionation via sequential enzymatic treatments such as DNAse, RNAse, high salt, and detergent. These approaches were reductive and ones for which the results obtained, in the form of a “nucleoskeleton” or purified proteins, were difficult to assess as being comparable or related to the original in vivo situation. Similarly, the knowledge of nuclear envelope components was greatly limited. Lamins had been discovered, but only one nucleoporin was known (gp210), no inner nuclear membrane proteins had been discovered, the connections between lamins and chromatin were in their infancy, and authentic nuclear transport had never been achieved. Nuclear localization signals had been identified, but the protein(s) that recognized the NLSs or even where such NLS recognition took place were unknown (Dingwall, Sharnick, & Laskey, 1982; Kalderon, Richardson, Markham, & Smith, 1984). It was assumed that there must be a receptor in the nuclear pore complex; even this was later proven not to be the case with the discovery of soluble nuclear import and export receptors (Adam, Lobl, Mitchell, & Gerace, 1989; Finlay, Newmeyer, Price, & Forbes, 1987; Gorlich, Prehn, Laskey, & Hartmann, 1994; Gorlich, Vogel, Mills, Hartmann, & Laskey, 1995; Newmeyer, Finlay, & Forbes, 1986; Newmeyer & Forbes, 1988). At some level, the nucleus of the time was almost a black box with in vitro replication, transcription, and splicing analysis in the test tube far outpacing what we knew of nuclear structure. With few methods for analysis, it seemed this situation would not soon change.
The advent of the ability to reconstitute nuclear structure, both in vivo and in vitro, by two separate groups in the 1980s thus represented a significant and serendipitous advance in the ability to analyze many nuclear structures and reactions in a more natural manner. The ability of extracts to reconstitute nuclei stemmed largely from the fact that organisms with external fertilization and/or embryonic development, such as frogs and flies, construct their yolky eggs to be replete with all the components needed for many rounds of fast cell division and growth. Additionally, these stored cellular components are provided in a disassembled state and in millions of copies, enough to form several thousand cells in the early embryo, almost avoiding the need for early transcription. The last bit of evolutionary serendipity that made Xenopus egg extracts such a powerful tool was the discovery that mere addition of DNA—or chromatin for a faster reaction—was enough to set in motion reconstitution of hundreds of normal appearing nuclei, both within the early egg if the DNA was injected or within the test tube if DNA was added to egg extract. It became clear from these studies that much of the complex structure of the nucleus assembled spontaneously around DNA, one sequential step after another, and did so with complete disregard for eukaryotic DNA sequence (Forbes et al., 1983). Long linear dsDNA of heterogeneous sequence from any source would set nuclear reconstitution in motion (Blow & Laskey, 1986; Dasso & Newport, 1990; Dasso, Smythe, Milarski, Kornbluth, & Newport, 1992; Finlay & Forbes, 1990; Finlay et al., 1987; Forbes et al., 1983; Lohka & Masui, 1983, 1984; Newmeyer et al., 1986; Newmeyer & Wilson, 1991; Newport, 1987).
Newly laid Xenopus laevis eggs are naturally arrested in second meiotic metaphase by a Cytostatic Factor (CSF). This arrested meiotic stage is biochemically and physiologically very related to a mitotic state. Inducing fertilization, or mimicking fertilization by adding Ca++, triggers Xenopus eggs to progress on to an interphase state (Andreuccetti, Denis-Donini, Burrini, & Campanella, 1984; Masui, 2001; Masui & Markert, 1971; Tunquist & Maller, 2003). Extract of such eggs is thus said to be in an interphase state. On the other hand, omission of Ca++ and lysis of the eggs in the presence of the Ca++ chelator EGTA arrests the cell cycle of the eggs in a mitotic state. Techniques for generating extracts that are biochemically considered either interphase or mitotic have been developed and used extensively (reviewed in Murray, 1991). DNA or chromatin can be added to crude interphase extracts to reconstitute nuclei in vitro or, alternately, added to a crude mitotic extract to induce spindle assembly in vitro (Dasso & Newport, 1990; Maresca & Heald, 2006). In addition, extracts prepared even more carefully can reproduce up to three cell-cycle rounds in vitro (Murray, 1991). Further fractionation of interphase extracts, separating the extract into soluble and membrane fractions, can also be performed. When added together, these fractions can be used to more precisely control and analyze the reconstitution of nuclear structure or specific nuclear functions in vitro. A full report of the efforts of all the excellent contributors to this field are far beyond the scope of this chapter, but early contributors and reviews include the following: Desai, Murray, Mitchison, & Walczak, 1999; Finlay et al., 1987; Forbes et al., 1983; Lohka & Maller, 1985; Lohka & Masui, 1983; Newmeyer & Forbes, 1988; Newmeyer & Wilson, 1991; Newport, 1987; Newport & Spann, 1987; Pfaller, Smythe, & Newport, 1991.
Xenopus egg cytosol, when properly prepared, is diluted so minimally that it contains a very high concentration of the soluble factors and proteins needed to assemble nuclei (≥40 mg/mL). This property of the extract confers the experimental advantage of being able to use a small volume for each reconstitution reaction (10 to 50 μL). Another advantage is that a single batch of eggs laid by a Xenopus female frog can produce 2 to 8 mL of extract. Nuclear reconstitution using the Xenopus egg extract additionally enables one to independently study individual steps in the mechanism of assembly occurring during the process of nuclear formation (see, for example, Fichtman, Ramos, Rasala, Harel, & Forbes, 2010; Macaulay & Forbes, 1996). The nuclei assembled in vitro have been found to be comparable in fundamental architecture to nuclei in vivo in a number of aspects (Lau et al., 2009; Newmeyer & Wilson, 1991; Newport, 1987; Newport & Dunphy, 1992; Pfaller et al., 1991). Interestingly, endogenous nuclei can also be isolated from existing cells and added to the egg extract; mammalian nuclei can acquire additional Xenopus nuclear membrane and pore components, becoming hybrid in their nuclear envelope and protein composition (see, for example, Newmeyer & Forbes, 1988; Newmeyer et al., 1986). The mitotic disassembly of isolated nuclei derived from different cell types can also be studied by addition to mitotic egg extracts (Newport & Spann, 1987; Pfaller et al., 1991).
In this chapter, we describe our method of preparation of interphase Xenopus egg extract, sperm chromatin substrate, and applications using the extract. We describe a method for assembling nuclei in vitro. In more specialized protocols, we describe an assay for testing the integrity of the nuclear envelope and a second protocol for analyzing nuclear pore complex assembly into preformed, pore-free nuclear intermediates. The latter has the advantage of allowing one to study nuclear pore assembly separate from nuclear membrane assembly.
In Chapter 9, Eisenhardt et al. describe a slightly different Xenopus egg extract fractionation procedure and complementary protocols that allow the analysis of the function of transmembrane nucleoporins and the targeting of proteins to the inner nuclear membrane. In Chapter 2, Fichtman et al. use an anchored nuclei approach to visualize the NPCs by Field Emission Scanning Electron Microscopy.
8.1 MATERIALS
8.1.1 Equipment
27 G × 0.5-in. gauge needles (cat. no. 305109; BD Biosciences).
1mL syringes (1mL BD Luer-Lok™ disposable syringe; cat. no. 309628; BD Biosciences).
Disposable 2-mL plastic transfer pipettes (cat. no. 357524; BD Biosciences).
Beckman TL100 ultracentrifuge with TLS55 rotor; 11 × 34-mm Ultra-Clear tubes (cat. no. 347356; Beckman).
Tomy high-speed refrigerated centrifuge TX-160 with TMH-21 rotor.
1.5-mL microcentrifuge tubes (1.5 mL MaxyClear Snaplock-lid Microcentrifuge Tube, Polypropylene; cat. no. MCT-150-A; Axygen).
2-mL microcentrifuge tubes (2-mL MaxyClear Snaplock-lid Microcentrifuge Tube, Polypropylene; cat. no. MCT-200-C; Axygen).
Pushcap plastic 14mL round bottom centrifuge tubes (cat. no. 14-959-11B; BD Biosciences).
15-mL conical centrifuge tubes (cat. no. 14-959-70C; Corning Life Sciences).
200 and 1000 μl Pipettemen (classic pipettemen brands) and appropriate tips are used throughout.
Large orifice 200 μL pipette tips (cat. no. 1011–8406; USA Scientific). (When not available, 2 mm of the extremity of normal 200-μL pipette tips can be cut off with scissors.)
18 × 18 mm glass coverslips (Premium Cover Glass; cat. no. 12-548A; Fisher Scientific).
Zeiss Axioskop 2 microscope with a 63 × objective, connected to a charge-coupled device camera; images captured on computer.
8.1.2 Reagents and buffers
1. X. laevis frogs
Three to five females for making fresh egg extracts (Section 8.2) and two males to make a stock of demembranated sperm chromatin (Section 8.3).
Note: Key housing conditions for our frog colony are: the frogs are maintained in an 18 °C environment, immersed in tap water supplemented with 100 mM NaCl, and 0.1 mM sodium thiosulfate (cat. no. 72049, Sigma). Large colonies of X. laevis (>20) require substantial amounts of water, which compels the use of tap water. However, frogs are very sensitive to heavy metals, detergents, chlorine, and organic solvents, so care should be taken to avoid these. To purify the tap water and deplete possible contaminants, sodium thiosulfate is used as a chelator for a broad range of heavy metals (mercury, cadmium, etc.) and as a potent dechlorinator. When double distilled water is used (as for fewer frogs), there is no need for adding sodium thiosulfate (Chum, Felt, Garner, & Green, 2013; Wu & Gerhart, 1991). (More information can be found at: http://www.rspca.org.uk/ImageLocator/LocateAsset?asset=document&assetId=1232712646624&mode prd.)
2. Pregnant Mare Serum Gonadotropin
(PMSG, 12.5 × stock; cat. no. 367222; Calbiochem): 5000 U/2 mL in sterile filtered water.
3. 1 × stock Human Chorionic Gonadotropin
(HCG, cat. no. CG-10; Sigma): 10,000 U/10 mL in sterile filtered water.
4. Xenopus buffer (XB)
100 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 50 mM sucrose, pH 7.7, in distilled water.
5. Sucrose cushion
1 × XB with a final concentration of 500 mM sucrose.
6. Cysteine buffer
2% cysteine in distilled water, pH 7.8.
7. Cytochalasin B stock
10 mg/mL in dimethylsulfoxide (cat. no. 1955119; MP Biochemicals).
8. Cycloheximide stock
10 mg/mL in distilled water (cell-culture grade, white powder, cat no. 0219452705; MP Biochemicals).
9. Protease inhibitors
Aprotinin and Leupeptin, combined in a 1000 × stock solution of 10 mg/mL each in distilled water (cat no. A6106 and L2884, respectively; Sigma).
10. 10 × Buffer-X for sperm
100 mM HEPES, pH 7.4, 0.8 M KCl, 150 mM NaCl, 50 mM MgCl2, 10 mM ethylenediaminetetraacetic acid in distilled water.
11. Bovine Serum Albumin
(BSA) (cat. no. A7030; Sigma).
12. Triton X-100 (TX-100)
(cat. no. T8787; Sigma).
13. DTT (dl-Dithiothreitol)
1 M stock solution in distilled water (keep stock aliquoted in 1 mL fractions and frozen at −20 °C) (cat. no. 43815; Sigma).
14. 20 × Energy Mix stock in 1 × XB buffer containing
200 mM phosphocreatine (cat. no. P7936; Sigma; in 10 mM KH2PO4, pH 7.0),
1.6 mg/mL creatine phosphokinase (cat. no. C3755; Sigma; in 10 mM HEPES, pH 7.5, 50% glycerol, 50 mM NaCl, and distilled water),
20 mM ATP (Adenosine 5′-triphosphate disodium salt hydrate—cat. no. A2382; Sigma; in distilled water, pH 7.0),
20 mM MgCl2 (cat. no. M8266; Sigma; in distilled water), and
100 μM EGTA (Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid—cat. no. E3889; Sigma; in distilled water, pH 7.5 with NaOH).
15. Anti-FG Nup antibody mAb414
(cat. no MMS-120P; Covance), directly labeled with an AnaTag™ HiLyte Fluor™ 555 Protein Labeling Kit (cat. no. 72045, Anaspec, CA) or purchased already labeled from Covance. Once labeled, the antibody is referred in the text as HF555-mAb 414.
16. Fixation buffer
48% glycerol, 11% formaldehyde, and 10 mM HEPES, pH 7.5, 5 μg/mL Hoechst 33258 DNA dye (cat. no. 861405; Sigma) and freshly added 100 μg/mL DHCC (final concentration) (3,3-dihexyloxacarbocyanine iodide—cat. no. 318426; Sigma-Aldrich).
8.2 XENOPUS EGG EXTRACTS
Assembly of in vitro nuclei is performed using a high-speed cytosolic extract derived from X. laevis eggs. When demembranated sperm chromatin (from X. laevis males) and membranes (derived from the fractionation of X. laevis eggs; see below) are added to the cytosolic extracts (Newmeyer et al., 1986; Newport, 1987; Pfaller et al., 1991), reconstituted nuclei form and are fully functional in terms of nuclear import and DNA replication.
As an overview of the protocol below, induction of egg laying by X. laevis females is stimulated and precisely controlled by hormone injection (Section 8.2.1). The cytosol is then separated from the membranes by two steps of fractionation and centrifugation (Section 8.2.2). The resulting cytosolic layer is a highly concentrated fraction of soluble proteins (~40 mg/mL) and contains all components necessary for nuclear reconstitution in a cell-free environment. The in vitro assembly of nuclei (Section 8.4) initiates as soon as the cytosol is incubated with the membrane fraction, demembranated sperm chromatin (Section 8.3) and the energy regenerating system. Finally, individual steps in nuclear assembly can be dissected by separate protocols and techniques (Sections 8.5 and 8.6).
8.2.1 Obtaining eggs from frogs: Priming and induction of egg laying
To prime frogs for ovulation, the hormone PMSG is used. This hormone stimulates the growth of oocytes and allows them to reach an adequate size. Only eggs with a certain size will be released during the eventual laying process. Such oocytes are in “Stage VI” of the “Dumont classification” and show a clearly differentiated brown animal hemisphere and a white vegetal hemisphere, separated by an unpigmented equatorial band (Dumont, 1972). The injection of HCG stimulates the production of progesterone, which in turn activates a complex signaling pathway leading the oocytes to enter meiosis (Ferrell, 1999). The cell-cycle progresses until it is stopped in metaphase of the second meiotic division by a CSF; this state is referred to as “CSF arrest” (Masui, 2001; Tunquist & Maller, 2003). This arrest prevents the oocytes from progressing further in the cell cycle as they await laying. Only CSF-arrested eggs will be laid (when laid, they are then called “eggs”). Approximately, 24 h after HCG injection the frogs will lay their eggs naturally.
In the wild, laid eggs resume the cell cycle when fertilized by sperm. In vitro, we induce interphase entry in a population of laid eggs by calcium stimulation, which mimics the burst of calcium that occurs upon fertilization. Calcium is stored in a cortical network of smooth endoplasmic reticulum (ER) vesicles. Simple crushing of the eggs at the beginning of the protocol releases calcium from these specialized storage vesicles and initiates interphase (Andreuccetti et al., 1984; Murray, 1991). A synchronized interphase extract can then be prepared.
PROTOCOL
Each female frog is primed with 0.5 mL PMSG (mix 1 mL from a 12.5 × stock with 11.5 mL sterile distilled water) 2–3 days before inducing the laying process itself, but up to 2 weeks ahead is acceptable (Wu & Gerhart, 1991). Approximately, 2 weeks post-PMSG injection, any unlaid eggs acquire a proapoptotic phenotype (von Ahsen & Newmeyer, 2000).
Frogs are injected in the thigh region (back side), between the skin and the muscle (subcutaneous injection), using a 27 × 0.5 in. gauge needle, then placed in a container of 100 mM NaCl in distilled water at 18–22 °C.
- Approximately, 24 h before egg laying is desired, frogs are injected subcutaneously with 0.5 mL of HCG stock solution (i.e., 500 Units of HCG are injected per frog).Note: When collected, the eggs should be as fresh as possible for extract preparation. Usually, the HCG injection is done in the morning so that the eggs can be collected 24 h later, i.e., the next morning. Then, the remainder of the second day can be used for preparing the extract.
- At the time of injection, place each HCG-injected frog in a separate 10 L container filled with 2 L of distilled water containing 100 mM NaCl at 18 °C (laying solution). Approximately, 24 h later the eggs are collected by pouring off the majority of laying solution and proceeding to Step 5.Note: The 18 °C temperature is ideal for housing the frogs. Inducing the frogs to lay at 18 °C in a laying solution containing 100 mM NaCl is also crucial to preserve good quality in the eggs. After laying, the frogs should rest for 2 to 3 days in a large container of 100 mM NaCl in distilled water. This step is important to allow the frogs to finish laying any remaining eggs (which are discarded) before placing them back in their long-term housing (Wu & Gerhart, 1991).
- The eggs of each frog (a batch) are kept separate. Each batch is placed in a 250-mL beaker. The eggs must remain immersed in a small volume of laying solution at all times to lower the risk of lysis or any kind of damage during handling and transportation (4 cm of laying solution above the top of the egg layer is sufficient).Note: Eggs within the same batch are likely to be very similar. However, different batches of eggs may differ in quality. It is therefore not good to mix eggs from different frogs as this may increase variability in extract quality and reduce success.
8.2.2 Preparation and fractionation of Xenopus egg extracts
To separate the extract into soluble and membranes fractions, the following protocol is used. The goal is, by a series of centrifugations, to separate and purify: (1) a lighter fraction, which is the clear cytosol that contains soluble proteins and (2) a heavier and cloudy fraction that contains the intracellular membranes (ER and Golgi membranes). Note that the plasma membrane and yolk and pigment granules are removed in an initial low-speed centrifugation step, while mitochondria and glycogen polymers are removed in the second higher-speed centrifugation step. This type of inter-phase extract is sometimes referred to in publications as “US” or “Ultra S” (for “ultracentrifuged S-phase extract”).
PROTOCOL
Steps 4–17 should be carried out on ice unless stated otherwise.
- Solutions are made fresh the same morning as the extract:
- 2% cysteine, pH 7.7–7.8
- The wash and lysis buffer used is 1 × XB buffer.
- The first step consists of removing the gelatinous layer around the eggs by treating with a 2% cysteine solution.
- The excess water is removed from the collected eggs in the 250-mL beaker, making sure that the eggs remain submerged in water (1 cm). At this point, the jelly-coated eggs do not physically touch each other and occupy a large space in the beaker.
- Pour ~100 mL of 2% cysteine, pH 7.7–7.8, into the beaker.
- Very gently swirl the eggs in the beaker with 2–3 swirls.
- Once the eggs have settled, gently remove the excess solution and repeat Steps 2a–c twice more until the jelly is completely removed. The eggs become very fragile without their gelatinous layer, thus pouring, swirling, and removing the solutions must be performed with care. Once dejellied, the eggs are physically in contact with one another and occupy a smaller space in the beaker.Note: This step must be done in relatively short time, i.e., DO NOT let the eggs remain in contact with the 2% cysteine solution >5–6 min total as this will damage the eggs, which will become white, swell, and lyse. Remove every white or nonconventionally shaped egg from the beaker (some are laid this way) using a 2-mL plastic transfer pipette. Dying or damaged eggs release proteolytic enzymes that may trigger a cascade of reactions leading to the death of the entire egg population. Thus, it is necessary to remove the dying eggs during the 5–6 min period.
- Proceed immediately to the following step once the eggs have settled: wash 3 to 4 times with 1 × XB. For this:
- Pour off as much as possible of the 2% cysteine buffer. Immediately, add 50–100 mL of 1 × XB to the beaker and swirl very gently.
- Once the eggs have settled, remove the excess solution and repeat this wash twice more.Note: The cysteine solution has a very specific smell of sulfur. After the last wash, this smell should not be perceptible. If cysteine remains, it may cause problems in future experiments that use the egg extract.
Prepare 2-mL plastic microcentrifuge tubes with snaplock lids that have been prechilled on ice for a few minutes and then filled with 0.5 mL of 1 × XB buffer. The 1 × XB will act as a “cushion,” ensuring that the fragile eggs do not fall abruptly to the bottom of the tube.
Using a 2-mL disposable plastic transfer pipette (with the tip cut off 2 cm with scissors to accommodate the 1 mm eggs), carefully transfer the eggs from the beaker into the 2-mL tubes. (The tube should be filled to no more than 90% of its capacity with eggs plus solution to avoid crushing the eggs with the lid.)
Once the eggs have settled in the tube, this time remove as much as possible of the 1 × XB solution above the eggs. Leaving too much solution will dilute the extract (Fig. 8.1A).
- Add 100 μL of 10 mM cycloheximide to the top of the each tube. The next centrifugation step will mix the cycloheximide into the future extract.Note: Cycloheximide will prevent any further protein synthesis (i.e., cyclin synthesis) and keep the future extract in an interphase state.
Spin the 2 mL microcentrifuge tubes containing the eggs in a TMH-21 horizontal bucket rotor for 15 min, 17,360 rcf (15,000 rpm) at 4 °C using a table top refrigerated centrifuge (Tomy TX-160). Egg lysis during centrifugation will release stored calcium, which sends the extract into interphase.
FIGURE 8.1. Fractionation of Xenopus Eggs.
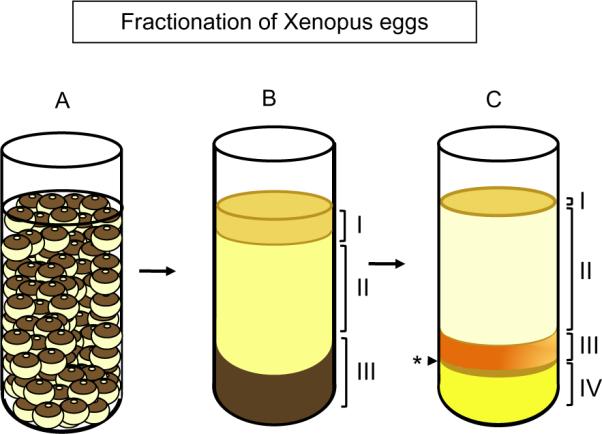
(A) Xenopus eggs prior to the first centrifugation, which will crush and fractionate the eggs. (B) The egg extract fractionated after the first step of centrifugation. Three distinct layers are formed: the yellow lipid layer at the top (I), the intermediate golden crude extract layer (cytosol and membranes) (II), and the black pigment and yolk granule pellet (III). (C) The crude extract after high-speed centrifugation fractionates into four major layers. From the top to the bottom of the tube (I) a thin yellow layer containing remaining lipids, (II) a clear layer (soluble fraction), (III) a cloudy yellow membrane layer, and (IV) a gelatinous pellet with dark and light layers comprised mitochondria, glycogen, and residual pigment and yolk granules. Asterisk indicates the light brown layer containing mitochondria.
THE EGGS WILL FRACTIONATE INTO THREE DISTINCT LAYERS (SEE FIG. 8.1B)
A top opaque bright yellow lipid layer.
A middle cloudy golden layer consisting of crude cytosol (i.e., the soluble part plus membranes).
A bottom dark layer formed largely of black pigment granules and yolk granules.
-
9
Using a 200-μL pipette tip (only the tip) as a tool, scoop out and discard as much as possible of the top lipid layer.
-
10Next, recover the crude cytosol layer gently using a 1000-μL pipette tip and pipette and transfer it into a 2-mL ultracentrifuge tube (Beckman Ultra-Clear tubes—11 × 34 mm).Note: Even if some remaining lipids and pigment granules are collected with the crude cytosol, these will be separated away during the next centrifugation step. An average of 1 mL of crude extract is recovered per ~2 mL of lysed eggs.
-
11Add Cytochalasin B to the crude extract to a final concentration of 50 μg/mL. Mix by pipetting the extract gently up and down a few times, using a 1000 μL tip and pipette.Note: Cytochalasin B treatment decreases the viscosity of the crude cytosol by depolymerizing the cytosolic actin network that would otherwise form in this fraction.
-
12To separate the cytosol from the membranes:
- Combine the crude cytosols (from the same original batch of eggs) into new Ultra-Clear tubes (11 × 34 mm). Fill each Ultra-Clear tube with 2 mL of crude extract (if possible). Repeat this step for the cytosol from each batch of eggs.Note: The tube may collapse during centrifugation if the final volume is less than 1.5 mL/tube.
- Spin down the crude cytosolic extract using a TLS55 Beckman rotor with a TL-100 Beckman ultracentrifuge at 260,000 rcf (55,000 rpm) for 90 min at 4 °C.
THE CYTOSOL WILL FRACTIONATE INTO FOUR DISTINCT LAYERS (FIG. 8.1C)
A thin topmost layer of residual lipids.
An upper clear yellow cytosolic layer.
An underlying cloudy yellow layer comprised membranes.
A gelatinous bottom pellet with a hard consistency, composed of alternating golden and dark layers. These contain mitochondria (a dark layer just underneath the cloudy membrane layer), glycogen (a clear golden layer), and any remaining pigment granules (a bottom dark layer).
-
13Recovery of the clear cytosol and membranes:
- First, remove and discard the top thin lipid layer very gently by aspiration with a glass Pasteur pipette and vacuum.
- The clear cytosol layer should then be recovered with a 200 μL tip and pipette. A gentle swirl while pipetting aids in preventing aspiration of the cloudy membrane fraction. Avoid collecting too much of the cloudy membrane layer while recovering the cytosol. The clear cytosol should be transferred into a new Ultra-Clear tube (11 × 34 mm) and stored on ice until Step 14.
- The cloudy membrane layer (approximately 250–500 μL) should next be collected using a large orifice 200-μL tip and pipette, followed by placement of the cytosol into a 14-mL round bottom push-cap tube and storage on ice until Step 16.Note: When collecting the membrane fraction, avoid the dark-brown viscous layer just underneath as it contains mitochondria. After the in vitro nuclear reconstitution assay, the nuclear envelopes are often detected by staining with DHCC dye. If too many mitochondria are present, the DHCC will stain the mitochondrial membranes and the surface occupied by the coverslip will be covered by a myriad of tiny dots (i.e., the mitochondria). In addition, leaving excess mitochondria in the extract is undesirable as the mitochondria could trigger entry into an apoptotic state (von Ahsen & Newmeyer, 2000).
-
14
To the cytosolic fraction (Step 13c, stored on ice), add Aprotinin and Leupeptin to a final concentration of 10 μg/mL each. To mix, pipet the extract up and down using a 1,000 μL tip and pipette.
-
15
In order to remove residual lipids and membranes from the clear cytosol collected and treated in Steps 13 and 14, the cytosol should be spun a second time, now at 260,000 rcf (55,000 rpm) for 30 min at 4 °C using a TLS55 rotor (in 11 × 34 mm Ultra-Clear tubes) and a TL100 Beckman ultracentrifuge. The ultracentrifuged cytosol should then be aliquoted into 100 μL aliquots, quickly frozen in liquid nitrogen and stored at −80 °C
-
16The membrane layer from Step 13 should next be salt-washed using 1 × XB buffer supplemented with 0.5 M KCl (high-salt XB buffer) to remove much of the proteins bound to the membranes. For washing:
- Transfer the membranes (~250–500 μL) into a 14-mL push-cap tube.
- Fill the tube with 8 mL of high-salt XB buffer.
- Pipet the mixture of membranes and high-salt XB buffer gently up and down using a large orifice 1000-μL tip and pipette.
-
17Purify the salt-washed membranes through a sucrose cushion. To do this:
- Place 2 mL of the mixture of membranes and high-salt XB buffer in a new Ultra-Clear tube (11 × 34 mm).
- Using a 200-μL pipette and tip, go to the bottom of the tube and slowly add 400 μL of a sucrose cushion (500 mM sucrose in 1 × XB).Note: The salt-washed membranes will be on top of the sucrose cushion at this point.
- Spin the mixture at 70,000 rcf (30,000 rpm) for 30 min at 4 °C using a TLS55 rotor and a TL100 Beckman ultracentrifuge.
- After this step of centrifugation, the membranes will be in a loose pellet at the bottom of the tube. Remove most of the liquid over the membranes, leaving a minimal amount of liquid at the bottom of the tube with the pellet of membranes.Note: Leaving too much liquid may dilute the membranes. Leaving a small amount of the liquid, however, is required to enable one to more readily pipet up the membrane pellet (e.g., leave ~20 μL of the top layer over the pellet).
- Flick the tube to mix the liquid and membranes; then use a large orifice 200 μL tip and pipette to recover the membranes. The resuspended membranes should then be aliquoted into 5 μL fractions), quickly frozen in liquid nitrogen and stored at −80 °C. We define this as approximately a 20 × membrane stock and use it accordingly.
8.3 DEMEMBRANATED SPERM CHROMATIN
The source of DNA used for in vitro nuclear reconstitution experiments is sperm chromatin prepared from male Xenopus frog testes. These demembranated sperm contain a packet of chromatin composed of all the sperm chromosomes. This chromatin can form a functional nucleus when added to egg cytosol supplemented with membranes and is a convenient way to store and add chromatin. Thus, sperm chromatin is the commonly used as a source of chromatin for in vitro nuclear reconstitution; however, other sources of DNA or chromatin can also be used (Newport, 1987).
PROTOCOL
Steps 1 to 6 are carried out at room temperature (unless stated otherwise). Steps 7 to 12 are carried out on ice or at 4 °C (unless stated otherwise).
- The Buffer-X solutions used below contain high concentrations of sucrose, which takes considerable time to dissolve. It is recommended that these solutions be prepared the day before the sperm prep.
- Buffer-X + 0.2 M sucrose (prepare 100 mL)
- Buffer-X + 0.2 M sucrose + 0.4% TX-100 + 1 mM DTT + 1μg/mL Aprotinin/Leupeptin (prepare 2 mL)
- Buffer-X + 0.2 M sucrose + 3% BSA + 1 mM DTT 1 + μg/mL Aprotinin/Leupeptin (prepare 10 mL)
- Buffer-X + 0.5 M sucrose + 3% BSA + 1 mM DTT 1 μg/mL Aprotinin/Leupeptin (prepare 5 mL)
- Buffer-X + 2 M sucrose (prepare 5 mL)
- Buffer-X + 2.3 M sucrose (prepare 10 mL)
- Buffer-X + 2.5 M sucrose (prepare 5 mL)
Two male frogs are sufficient to yield a large amount of demembranated sperm chromatin, enough for ~500 reconstitution reactions. For a frog euthanasia procedure and recovery of testes, please refer to Chan and Forbes (2006).
- Cleaning the testes:
- Place the testes obtained in a Petri dish containing Buffer-X + 0.2 M sucrose.
- Remove any remaining blood, blood vessels, and fat from the testes with forceps.
- Move the testes into a new Petri dish containing Buffer-X + 0.2 M sucrose.
- Mince the testes with a small scalpel into very small pieces (1–2 mm). Transfer the minced testes and all the buffer from the Petri dish used in Step c to a 15-mL conical tube.
- Rinse this Petri dish (Step c) with 2 mL of fresh Buffer-X + 0.2 M sucrose. KEEP and transfer the wash to the same 15-mL conical tube.Note: The washes contain some freed sperm released when testes were minced.
- To release additional sperm from the testes:
- Mash the minced testes that have settled to the bottom of the conical tube in Step 3, using a thin glass stirring rod or a thin spatula.
- Vortex the conical tube vigorously for 1 min to maximize the release of sperm.
- Pellet the larger pieces of tissue at 100 rcf (1,000 rpm) for 10 s, at RT, in a low-speed, swinging-bucket clinical tabletop centrifuge.
- Recover and transfer the supernatant into one new 15-mL conical tube (i.e. KEEP and set aside for further use)—Store on ice.
- KEEP the pellet, and add 2–4 mL of Buffer-X + 0.2 M sucrose to this pellet.
- Start over at Step 4a and repeat Step 4 until the supernatant becomes clear (see note below).Note: The supernatants are cloudy when they contain freed sperm. Keep the pellet after every centrifugation. The pellet still contains plenty of trapped sperm. Step 4 must be repeated until the supernatant becomes clear. The turbidity of supernatants indicates the presence of sperm. Depending on the testes quality, Step 4 may be repeated 3–10 times (there is no rule for this; the turbidity is the only reliable indicator).
- Combine all the supernatants collected in Step 4d together (they contain the freed sperm in suspension).Note: Depending on how many times Step 4 has been repeated, more than one 15-mL conical tube may be required for collecting the supernatants.
- To remove remaining large pieces of tissues collected with the sperm in suspension:
- Spin down the combined supernatants in Step 5 at 185 rcf (1350 rpm) for 1 min using a clinical tabletop centrifuge
- The supernatant is recovered and transferred to a new 14-mL round bottom tube. (KEEP)
- This pellet is of no use: DISCARD
- The freed sperm in suspension (recovered in Step 6) will be, now pelleted to remove damaged sperm, contaminant cells and undesired particles. For this:
- Spin down the supernatant at 2,600 rcf (4,000 rpm) for 10 min at 4 °C (using a Sorvall HB6 swinging-bucket rotor).
- KEEP the pellet, which now contains the freed sperm.
- DISCARD the supernatant that contains damaged sperm and other useless particles.
- Resuspend the sperm pellet in 0.8 mL of Buffer-X + 2 M sucrose.Note: Red blood cells are major contaminants of sperm. Step 7 does not remove the red blood cells from the sperm (for this, see next step).
- The red blood cells are separated from the sperm by spinning the sperm solution through a sucrose gradient. For this:
- Take 4 Ultra-Clear tubes (11 × 34 mm) and add to each one:
- 0.25 mL of Buffer-X + 2.5 M sucrose.
- Slowly overlay with 1.7 mL of Buffer-X + 2.3 M sucrose.
- Finally overlay this with 0.2 mL of the resuspended sperm solution.
- Spin, using a TL100 Beckman ultracentrifuge with a TLS-55 rotor at 93,000 rcf (33,000 rpm) for 25 min at 4 °C.
- DISCARD the top half of the gradient by aspiration (it contains the red blood cells).
- KEEP and transfer the lower half of the gradient to a new 14-mL round bottom tube (this lower half contains the sperm).
- To recover any remaining sperm possibly left in the previous set of four Ultra-Clear tubes: add to each tube, 1 mL of Buffer-X + 0.2 M sucrose and rinse each tube walls with this. Keep and transfer the washes into the same 14-mL round bottom tube used in Step 8d.
- Repeat two more times Step e and combine the washes into the same 14-mL round bottom tube used in Step 8d.
- Now dilute the sperm solution (obtained in 8f ) with Buffer-X + 0.2 M sucrose to reach a volume of 12 mL of solution.
- Next pellet the sperm in suspension (from Step 8g):
- Spin the tube at 4,000 rcf (5,000 rpm) for 10 min at 4 °C using a Sorvall HB6 swinging-bucket rotor.
- KEEP the pellet on ice (i.e., sperm) and DISCARD the supernatant.
- The demembranation process consists in removing the membranes that surround the sperm chromatin. To do this:
- Resuspend the pellet obtained in Step 9b with 1 mL of Buffer-X + 0.2 M sucrose + 0.4% TX-100, 1 × Aprotinin/Leupeptin 1 mM DTT.
- Incubate for 30 min on ice.
- The TX-100 and solubilized membranes are removed by a series of wash/spins:
- In two 1.5-mL microcentrifuge tubes, add 0.5 mL of Buffer-X + 0.5 M sucrose + 3% BSA + 1 × Aprotinin/Leupeptin + 1 mM DTT (this will serve as a cushion).
- Overlay each sucrose cushion with 0.5 mL of the demembranated sperm mixture obtained in Step 10.
- Spin the tubes in a clinical tabletop centrifuge at 360 rcf (1,900 RPM) for 10 min.
- KEEP the pellets (they contains the sperm) and DISCARD the supernatant.
- Resuspend each sperm chromatin pellet with 100 μL of Buffer-X + 0.2 M sucrose + 3% BSA + 1 × Aprotinin/Leupeptin + 1 mM DTT.
- Transfer the sperm chromatin into two new 1.5 mL microcentrifuge tubes and add to each 900 μL of Buffer-X + 0.2 M sucrose + 3% BSA + 1 × Aprotinin/Leupeptin + 1 mM DTT.
- Spin down the tubes in a clinical tabletop centrifuge at 360 rcf (1,900 rpm) for 10 min.
- Repeat Steps d to g twice more.
- Resuspend the TX-100-free pellet of sperm chromatin in 0.5 mL Buffer-X + 0.2 M sucrose + 3% BSA + 1 × Aprotinin/Leupeptin + 1 mM DTT and store on ice.
The sperm chromatin packets (SpC; each being the nuclear contents of one sperm) can be counted using a hemocytometer. After counting, the sperm chromatin should be diluted to a final concentration of 50,000 Units of sperm chromatin/μL (SpC/μL). For dilution, use the same buffer as in Step 11i. This is then aliquoted into 10 μL fractions, quickly frozen in liquid nitrogen and stored at −80 °C. This mix, once stored at −80 °C, is stable for a number of years.
8.4 IN VITRO RECONSTITUTION OF NUCLEI
Reconstitution of nuclei using high-speed Xenopus egg extract, membranes, and sperm chromatin is described in this paragraph. A standard reaction contains ~20 μL of high-speed cytosol, 1 μL of purified membranes, and ~3,000 SpC/μL of final reaction. Some adjustments may be required to determine the best ratio of membranes to cytosol, varying from 0.5 to 2 μL per 20 μL of cytosol, using the considerations in Chan and Forbes (2006) as a guide. To visualize the proceedings of the major points described in the following protocol, an elegant and didactic video can be found online (Cross & Powers, 2008). Addition of an ATP-regenerating system provides the energy necessary to fuel the reconstitution reaction.
PROTOCOL
The protocol described below is sufficient for approximately five reactions of in vitro nuclear reconstitution.
From Step 1 to 5, the reactions must carried out on ice.
- Defrost slowly on ice
- 1 tube of cytosol (100 μL aliquot).
- 1 tube of membranes (5 μL aliquot).
- 1 tube of sperm chromatin (50,000 SpC/μL).
- 1 tube of 20 × ATP-regeneration mix.
Add the cytosol directly to a tube containing the membranes and resuspend the membranes gently using a large orifice 200-μL tip and pipette.
- Separate the mixture of membranes plus cytosol into five 1.5-mL microcentrifuge tubes. To each tube, add 1 μL of 20 × ATP-regeneration mix.Note: At this point in the protocol, any test protein or molecule can be added to study their effect on nuclear membrane or nuclear pore assembly (see Sections 8.5 and 8.6).
LASTLY, to each tube add 1.5 μL from a stock solution of 50,000 SpC/μL to give a final concentration of sperm chromatin at ~3,000 SpC/μL of reaction.
Flick the tube gently five times to mix the sperm chromatin with the membrane/cytosol mixture. Avoid making bubbles while mixing.
Let the reaction incubate for 60 min at room temperature on the bench.
- To determine if a new batch of membranes and cytosol assemble nuclei properly, one can assess the formation of nuclear pore complexes by immunofluorescence. For this, at t=45 min, we add 1 μL of a directly labeled HF555-mAb 414 antibody to an aliquot of the reaction (the antibody dilution is usually 1/50 in the reaction) and mix by gently flicking the tube once or twice.Note: The monoclonal antibody mAb414 recognizes at least four Xenopus FG-nucleoporins (FG-Nups): Nup358, Nup214, Nup153, and Nup62 (Lau et al., 2009). When nuclear pores have been assembled in the nuclear envelope, the antibody will reveal a well-defined punctuate stain on the nuclear rim (mAb414 antibody can be directly labeled using a labeling kit for proteins—See Section 8.1 or bought prelabeled, Covance, CA). A red-labeled antibody is important to use here if the nuclear membranes will be visualized in green using DHCC dye (as below).
Just before the end of the incubation, place a 1 μL drop of fixation buffer containing 100 μg/mL of DHCC on a glass slide (one slide per reaction).
At t=60 min, using a large orifice pipette tip, take a 2 μL aliquot of a reaction and place directly on the drop of fixation buffer. (This is done for each reaction.)
- Overlay the reaction aliquot/fixation buffer mixture with a 18 × 18 mm square coverslip and seal the edges of the coverslip with clear nail polish. Let dry!Note: The fixation buffer/assembly reaction between the glass slide and the coverslip will automatically blend together and spread completely over the surface occupied by the coverslip by capillary action. Maximum care should be taken to gently lay on the coverslip, starting at one edge. Do not press hard on the coverslip and also try to prevent the coverslip from sliding, as this will result in damaged (sheared) nuclei. It is important to keep the tubes and slides in the dark until they can be visualized in Step 11 to avoid bleaching.The samples cannot be stored at 4 °C overnight. The most reliable and clearest results are obtained when visualization is made just after the end of the experiment.
Visualization of the assembled nuclei can be performed using a regular fluorescence microscope, ideally using a 63 × objective.
FINAL NOTE FOR SECTION 8.4
“Good” in vitro reconstituted nuclei have the following characteristics: a round shape, decondensed DNA (showing dark and bright areas upon Hoechst DNA staining), a smooth continuous nuclear envelope when stained by DHCC, and a homogeneous dot-like nuclear rim stain with HF555-mAb414 antibody (staining the FG-Nups). The size of the reconstituted nuclei is usually between 10 and 20 μm (see Control in Figs. 8.2 and 8.3). Smaller or larger nuclei can indicate aberrant assembly.
FIGURE 8.2. Nuclear Membrane Assembly and Integrity.

High-speed interphase Xenopus egg extract is mixed with sperm chromatin and membranes and allowed to incubate at room temperature for 60 min. Here, nuclei formed in the presence and absence of a potential inhibitor of membrane fusion are shown. The recombinant protein added to test its role on nuclear membrane assembly and integrity is GST-transportin (as shown in Lau et al., 2009). The integrity of the resulting nuclear envelope was examined with two assays: exclusion of 70 kDa rhodamine dextran (red) and staining with the membrane dye DHCC (green). The latter reveals the presence or absence of a continuous green nuclear envelope. Chromatin is stained with Hoechst DNA stain (blue). For each condition shown, the nuclear envelope is also shown magnified threefold for better viewing (3 × ; right column). (A) The control consists of addition of a protein that does not affect membrane assembly and integrity (here 20 μM GST is used). A smooth nuclear envelope staining is observed and the representative nucleus shown is impermeable to 70 kDa dextran. (B) A similar intact phenotype is observed when 8 mM BAPTA is added to the reaction. (C) Addition of 20 μM GST-transportin prevents vesicle–vesicle fusion, which results in a permeable nuclear envelope. The phenotype is characterized by a rough nuclear envelope staining around the DNA as well as by a permeable nuclear envelope, as determined by the absence of exclusion of the 70 kDa dextran.
FIGURE 8.3. Nuclear Pore Assembly Assay.

A schematic representation of the assay is depicted at the left. (A) To assemble pore-free nuclear intermediates, sperm chromatin, interphase egg extract, and membranes are incubated in the presence of 8 mM BAPTA for 60 min (Macaulay & Forbes, 1996). The BAPTA intermediate is quite small due to the lack of nuclear pores and resulting lack of nuclear import of vital nuclear proteins. The presence of nuclear pores is detected by staining for FG-nucleoporins (mAb414-TRITC; red), while membranes are detected with DHCC (green), and DNA with Hoechst dye (blue). (B and C) Nuclear pore assembly into these intermediates was tested under different conditions by diluting an aliquot of the pore-free intermediates 1:10 into fresh BAPTA-free cytosol and incubating for a further 60 min. (B) In the control condition, where no inhibitory protein or molecule was added (or when a control protein such as MBP is added), nuclear pore assembly occurred, as seen by the red FG-Nup rim. (C) Addition of 20 μM GST-Xenopus importin beta inhibits nuclear pore assembly, as seen by the lack of FG-Nups at the rim (Harel, Chan, et al., 2003).
8.5 ASSAYING ASSEMBLY AND INTEGRITY OF THE NUCLEAR ENVELOPE
The following assay is done to assay nuclear membrane assembly and integrity in the presence or absence of a component to be tested for an effect on membrane assembly. Normally, at the end of mitosis in vivo, membranes are recruited to the chromatin primarily from ER sheets. These then fuse side to side to surround the set of chromosomes with an intact double nuclear membrane (Anderson & Hetzer, 2007; Antonin, Ellenberg, & Dultz, 2008; Kutay & Hetzer, 2008). In vitro, in Xenopus egg extract, nuclei assemble spontaneously: a double nuclear membrane with nuclear pores forms around added chromatin (Forbes et al., 1983; Hetzer et al., 2001; Lohka & Masui, 1984; Newport, 1987). In this in vitro system, purified membrane vesicles, formed during fractionation of the extract, are the source of the membrane/ER-derived vesicles for the future nuclear membranes. When one observes nuclei very early in assembly in vitro or, alternately, if the vesicle–vesicle fusion machinery is blocked artificially, the nuclear membrane shows a discontinuous pattern when stained by fluorescent DHCC dye. Blocking vesicle–vesicle fusion can be achieved, for example, by the addition of GTP-gamma-S, a nonhydrolyzable analogue of GTP. GTP-gamma-S allows vesicles to be recruited to the DNA/chromatin, but prevents their fusion to form a continuous nuclear envelope (Boman, Delannoy, & Wilson, 1992; Hetzer, Bilbao-Cortes, Walther, Gruss, & Mattaj, 2000; Macaulay & Forbes, 1996). The gaps between the nonfused vesicles render the unfused nuclear membranes permeable to large macromolecules that cannot normally diffuse through fully fused nuclear membranes containing nuclear pore complexes. In contrast, the addition of BAPTA (a calcium chelator) to the reaction results in a fully sealed double nuclear membrane without nuclear pores. BAPTA thus creates intermediates that show a continuous and impermeable nuclear envelope, but no mature nuclear pores (Macaulay & Forbes, 1996). These intermediates are later used in Section 8.6.
To determine the integrity of the nuclear envelope, its ability to exclude 70 kDa rhodamine-labeled dextran is assayed. The 70 kDa dextran is of a size that is too large to diffuse through normal nuclear pores and will not pass through completely formed intact nuclear membranes; 70 kDa dextran will thus appear excluded from nuclei that have achieved complete nuclear membrane enclosure. However, this 70 kDa dextran can diffuse into nuclei if there are gaps in the nuclear membranes or, alternately, in cases where nuclear membrane assembly never proceeded beyond the vesicle-binding stage, such as in GTP-gamma-S reactions (Lau et al., 2009).
PROTOCOL
Initiate nuclear assembly reactions, as described in Section 8.4 through Step 3.
- Add a protein or molecule of interest to an assembly reaction, while adding a similar amount of the same buffer or solvent to a control reaction. Mix the reaction by gently flicking the tube five times. Let incubate for 10–15 min at RT.Note: The volume of the compound of interest to be tested should not exceed 1/5 of the final volume of the reaction. Empirically, 1–2 μL volume of the added compound is preferred.
To each tube, add 1 μL of 20 × ATP-regenerating system.
To each tube, add a final concentration of sperm chromatin of ~3,000 SpC/μL of reaction. Mix gently by flicking the tubes 3 to 5 times.
Allow assembly for 60 min at room temperature.
- At t=60 min, add 100 μM of WGA (final concentration) to each tube.Note: Even though fully formed nuclear pores are expected to exclude 70 kDa rhodamine dextran, lectin WGA can be added after nuclear reconstitution to ensure a tight seal of the nuclear pores, as WGA inhibits transport through the nuclear pore (Finlay et al., 1987, 1989). In cases where the reconstituted nuclei will also be assessed for nuclear import or other functions, WGA addition should be omitted.
Mix by gently flicking the tube several times.
Let incubate for 10 min at RT.
Transfer the tubes to ice for 15 min.
- Add to each tube the equivalent of 2.5 μg of 70 kDa rhodamine-labeled dextran. (Do not exceed 1–2 μL in volume.) Mix by gently flicking the tube 5 times and let incubate on ice for 15 min.Note: Exclusion of rhodamine-labeled 70 kDa dextran (70 kDa Rhod-Dex) appears as if the interior of an intact nucleus resembles a black disc surrounded by a homogeneous red background. Adding too much 70 kDa rhodamine-labeled dextran should be avoided as this results in excessive background noise above and below the excluding nuclei obscuring microscopic observation. The ideal 70 kDa rhodamine-labeled dextran concentration for use can be identified using control or BAPTA nuclear reconstitution reactions.
After incubation on ice, the reaction should be diluted 1:1 in XB buffer containing 7.4% formaldehyde to fix the nuclei.
On a glass slide, place 1 μL of the fixation buffer above containing 100 μg/mL of DHCC and 5 μg/mL Hoechst. Place 3 μL of the assembly reaction onto this drop of fixation buffer, using a large orifice 200 μL tip. Then, for visualization, follow Steps 10 and 11 of Section 8.4. (Other methods for determining nuclear membrane integrity can be found, for example, in Fichtman et al, 2010 and in the Anderson paper.)
Typical results obtained are depicted in Fig. 8.2.
8.6 A NUCLEAR PORE COMPLEX ASSEMBLY ASSAY USING PORE-FREE NUCLEAR INTERMEDIATES
The effect of a protein or molecule of interest on nuclear pore assembly can be assayed using nuclear intermediate structures that contain complete nuclear membranes, but lack mature nuclear pores (pore-free nuclear intermediates). For this, an in vitro nuclear assembly reaction is first performed in the presence of the calcium chelator BAPTA (Harel, Chan, et al., 2003; Lau et al., 2009; Macaulay & Forbes, 1996). If these BAPTA intermediates are diluted 1:10 into fresh BAPTA-free cytosol, nuclear pore assembly ensues in the pore-free nuclear intermediates shortly afterward. The newly formed nuclear pore complexes can be detected via staining of their FG-Nups using a directly labeled fluorescent antibody, such as mAb414. If, however, a protein or molecule that blocks NPC assembly is added, the pore-free nuclear intermediates will remain free of nuclear pores and fail to stain with mAb414. The protocol for this assay is described below (see Fig. 8.3).
Note before starting the protocol
For this type of experiment, pore-free BAPTA nuclear intermediates are first assembled in 20 μL of cytosol for 60 min as described below. Then 5 μL of the BAPTA reaction containing pore-free intermediates are diluted into 45 μL of fresh BAPTA-free cytosol with energy mix, plus or minus a putative inhibitor. Therefore, from one 20 μL-BAPTA reaction, 4 nuclear pore assembly reactions can be performed.
PROTOCOL
Initiate the desired number of in vitro nuclear assembly reactions: relating to the note above and by following instructions described in Section 8.4 through Step 3.
At that point, add BAPTA to each tube to a final concentration of 8 μM from the BAPTA stock solution described in Section 8.1. Mix by flicking the tube gently 5 times.
To each tube, add 1 μL of 20 × ATP-regenerating system.
To each tube, add the desired amount of demembranated sperm chromatin needed to achieve 3,000 SpC Units/μL of reaction. Mix by flicking gently.
- Incubate for 60 min at room temperature.Note: At t=40 min, in order to verify that successful formation of pore-free nuclear intermediates has occurred, check a 5 μL aliquot for the lack of FG-Nups using the HF555-mAb414 antibody and DHCC (as described in Steps 7–11 Section 8.4). The DNA in BAPTA nuclear intermediates should appear slightly decondensed and look “worm-shaped” or like a very small oblong/ellipsoidal structure. These BAPTA pore-free nuclear intermediates should also show a smooth and continuous DHCC staining of the nuclear membrane, with no mAb414 staining of FG-Nups.
- At t=60 min, dilute the BAPTA reactions 1:10 into fresh cytosol (BAPTA-free cytosol) as follows:
- A few minutes before t=60 min for the BAPTA reaction above, prepare a new 1.5-mL microcentrifuge tube containing: 45 μL freshly defrosted cytosol (important) and 2.5 μL of 20 × Energy mix for each experimental condition or control planned. Add the desired amount of protein or molecule of interest for testing for an effect on nuclear pore assembly to the appropriate tubes. Mix each tube by gently vortexing for 1 s or flicking a few times.Note: The compound of interest should be added to the fresh cytosol before proceeding to Step 6b, as it is known that NPC assembly commences as soon as the pore-free nuclear intermediate reaction is diluted into fresh control cytosol.
- Take 5 μL from the 60′ BAPTA reaction of Step 5 and add it to each of the prepared tubes from Step 6a. Mix the reactions by flicking the tubes twice, very gently (the BAPTA nuclear intermediates are extremely fragile).
Let the reactions proceed for an additional 60 min at room temperature.
To visualize the effects of the tested proteins or molecules on the assembly of nuclear pores follow the steps as described in Section 8.4, Steps 7–11.
Representative results using this technique are shown in Fig. 8.3.
ADDITIONAL NOTES FOR SECTION 8.6
If the protein/molecule added to the reaction does not block NPC assembly, the pore-free BAPTA intermediates, once diluted 1:10 into fresh cytosol, should convert into regularly shaped large nuclei, as are described in the last note of Section 8.4.
The concentration of nuclei present in the final reaction, after dilution of the BAPTA reaction 1:10 into fresh cytosol, will be 1/10th the normal 3,000 SpC/μL. However, do not compensate for this by adding excess SpC/μL in the initial reaction because this will overload the reaction's capacity for correct nuclear assembly. One must compensate by observing more microscopic fields per assay.
CONCLUSION
In the 1980s, upon observing nuclear reconstitution in vivo and in vitro in the Xenopus egg or its extract, scientists realized that the cell was capable of performing a true molecular tour de force. Now it is the modus operandi for examining nuclear assembly in a cell-free environment. Greater mastery of this technique has opened up a wide field for developing new approaches derived from the in vitro reconstitution assay itself. Two assays we describe in this chapter, based on the first protocol, allow one to study independently nuclear membrane and nuclear pore assembly events. To determine the role of a factor in such assembly events, a protein or a molecule of interest can be added for testing on the in vitro reaction. One is not limited, however, to the sole addition of factors. Depletion techniques are another available option (Finlay & Forbes, 1990). Immunodepletion is one of the most used and has been used, to cite just a few examples, to characterize the absolute requirement of the Nup 107–160 complex, the pore-targeting protein ELYS, the GLFG nucleoporin Nup 98, and the nuclear pore scaffold protein Nup188 for nuclear pore assembly (Franz et al., 2007; Harel, Orjalo, et al., 2003; Powers, Macaulay, Masiarz, & Forbes, 1995; Theerthagiri, Eisenhardt, Schwarz, & Antonin, 2010; Walther et al., 2003). Nuclear reconstitution following specific protein immunodepletion has similarly been used to analyze the requirement for different transmembrane nucleoporins (see Chapter 9). In vitro nuclear reconstitution assays have also been widely used to study nuclear transport (Chan & Forbes, 2006; Lachish-Zalait et al., 2009; Savulescu et al., 2011), nuclear membrane and NPC assembly (Anderson & Hetzer, 2007; Fichtman et al., 2010), DNA replication (Gillespie, Gambus, & Blow, 2012; Gillespie, Li, & Blow, 2001; Walter & Newport, 1997), apoptosis (von Ahsen & Newmeyer, 2000), nuclear breakdown and reassembly mechanisms at mitosis (Galy et al., 2008; Newport & Spann, 1987), pol III transcription from nuclei composed of recombinant tRNA genomes (Ullman & Forbes, 1995), and many other areas. High-resolution scanning electron microscopy has also had a strong impact on the study of in vitro nuclear assembly, as described in Chapter 2. All together, these discoveries have shown that the Xenopus egg extract system used for in vitro nuclear reconstitution is a powerful tool for studying the nucleus and its mechanisms of regulation.
References
- Adam SA, Lobl TJ, Mitchell MA, Gerace L. Identification of specific binding proteins for a nuclear location sequence. Nature. 1989;337(6204):276–279. doi: 10.1038/337276a0. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Hetzer MW. Nuclear envelope formation by chromatin-mediated reorganization of the endoplasmic reticulum. Nature Cell Biology. 2007;9(10):1160–1166. doi: 10.1038/ncb1636. [DOI] [PubMed] [Google Scholar]
- Andreuccetti P, Denis-Donini S, Burrini AG, Campanella C. Calcium ultra-structural localization in Xenopus laevis eggs following activation by pricking or by calcium ionophore A 23187. Journal of Experimental Zoology. 1984;229(2):295–308. doi: 10.1002/jez.1402290215. [DOI] [PubMed] [Google Scholar]
- Antonin W, Ellenberg J, Dultz E. Nuclear pore complex assembly through the cell cycle: Regulation and membrane organization. FEBS Letters. 2008;582(14):2004–2016. doi: 10.1016/j.febslet.2008.02.067. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Laskey RA. Initiation of DNA replication in nuclei and purified DNA by a cell-free extract of Xenopus eggs. Cell. 1986;47(4):577–587. doi: 10.1016/0092-8674(86)90622-7. [DOI] [PubMed] [Google Scholar]
- Boman AL, Delannoy MR, Wilson KL. GTP hydrolysis is required for vesicle fusion during nuclear envelope assembly in vitro. The Journal of Cell Biology. 1992;116(2):281–294. doi: 10.1083/jcb.116.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Forbes DI. In vitro study of nuclear assembly and nuclear import using Xenopus egg extracts. Methods in Molecular Biology. 2006;322:289–300. doi: 10.1007/978-1-59745-000-3_20. [DOI] [PubMed] [Google Scholar]
- Chum H, Felt S, Garner J, Green S. Biology, behavior, and environmental enrichment for the captive African clawed frog (Xenopus spp). Applied Animal Behaviour Science. 2013;143(2):150–156. [Google Scholar]
- Cross M, Powers M. In vitro nuclear assembly using fractionated Xenopus egg extracts. Journal of Visualized Experiments. 2008;(18) doi: 10.3791/908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasso M, Newport JW. Completion of DNA replication is monitored by a feedback system that controls the initiation of mitosis in vitro: Studies in Xenopus. Cell. 1990;61(5):811–823. doi: 10.1016/0092-8674(90)90191-g. [DOI] [PubMed] [Google Scholar]
- Dasso M, Smythe C, Milarski K, Kornbluth S, Newport JW. DNA replication and progression through the cell cycle. Ciba Foundation Symposium. 1992;170:161–180. doi: 10.1002/9780470514320.ch11. discussion 180–166. [DOI] [PubMed] [Google Scholar]
- Desai A, Murray A, Mitchison TJ, Walczak CE. The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods in Cell Biology. 1999;61:385–412. doi: 10.1016/s0091-679x(08)61991-3. [DOI] [PubMed] [Google Scholar]
- Dingwall C, Sharnick SV, Laskey RA. A polypeptide domain that specifies migration of nucleoplasmin into the nucleus. Cell. 1982;30(2):449–458. doi: 10.1016/0092-8674(82)90242-2. [DOI] [PubMed] [Google Scholar]
- Dumont JN. Oogenesis in Xenopus laevis (Daudin). I. Stages of oocyte development in laboratory maintained animals. Journal of Morphology. 1972;136(2):153–179. doi: 10.1002/jmor.1051360203. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr. Xenopus oocyte maturation: New lessons from a good egg. Bioessays. 1999;21(10):833–842. doi: 10.1002/(SICI)1521-1878(199910)21:10<833::AID-BIES5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Fichtman B, Ramos C, Rasala B, Harel A, Forbes DJ. Inner/outer nuclear membrane fusion in nuclear pore assembly: Biochemical demonstration and molecular analysis. Molecular Biology of the Cell. 2010;21(23):4197–4211. doi: 10.1091/mbc.E10-04-0309. http://dx.doi.org/10.1091/mbc.E10-04-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay DR, Forbes DJ. Reconstitution of biochemically altered nuclear pores: Transport can be eliminated and restored. Cell. 1990;60(1):17–29. doi: 10.1016/0092-8674(90)90712-n. [DOI] [PubMed] [Google Scholar]
- Finlay DR, Newmeyer DD, Hartl PM, Horecka J, Forbes DJ. Nuclear transport in vitro. Journal of Cell Science Supplement. 1989;11:225–242. doi: 10.1242/jcs.1989.supplement_11.17. [DOI] [PubMed] [Google Scholar]
- Finlay DR, Newmeyer DD, Price TM, Forbes DJ. Inhibition of in vitro nuclear transport by a lectin that binds to nuclear pores. The Journal of Cell Biology. 1987;104(2):189–200. doi: 10.1083/jcb.104.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes DJ, Kirschner MW, Newport JW. Spontaneous formation of nucleus-like structures around bacteriophage DNA microinjected into Xenopus eggs. Cell. 1983;34(1):13–23. doi: 10.1016/0092-8674(83)90132-0. [DOI] [PubMed] [Google Scholar]
- Franz C, Walczak R, Yavuz S, Santarella R, Gentzel M, Askjaer P, et al. MEL-28/ELYS is required for the recruitment of nucleoporins to chromatin and postmitotic nuclear pore complex assembly. EMBO Reports. 2007;8(2):165–172. doi: 10.1038/sj.embor.7400889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy V, Antonin W, Jaedicke A, Sachse M, Santarella R, Haselmann U, et al. A role for gp210 in mitotic nuclear-envelope breakdown. Journal of Cell Science. 2008;121(Pt. 3):317–328. doi: 10.1242/jcs.022525. http://dx.doi.org/10.1242/jcs.022525. [DOI] [PubMed] [Google Scholar]
- Gillespie PJ, Gambus A, Blow JJ. Preparation and use of Xenopus egg extracts to study DNA replication and chromatin associated proteins. Methods. 2012;57(2):203–213. doi: 10.1016/j.ymeth.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PJ, Li A, Blow JJ. Reconstitution of licensed replication origins on Xenopus sperm nuclei using purified proteins. BMC Biochemistry. 2001;2:15. doi: 10.1186/1471-2091-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Prehn S, Laskey RA, Hartmann E. Isolation of a protein that is essential for the first step of nuclear protein import. Cell. 1994;79(5):767–778. doi: 10.1016/0092-8674(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Gorlich D, Vogel F, Mills AD, Hartmann E, Laskey RA. Distinct functions for the two importin subunits in nuclear protein import. Nature. 1995;377(6546):246–248. doi: 10.1038/377246a0. [DOI] [PubMed] [Google Scholar]
- Harel A, Chan RC, Lachish-Zalait A, Zimmerman E, Elbaum M, Forbes DJ. Importin beta negatively regulates nuclear membrane fusion and nuclear pore complex assembly. Molecular Biology of the Cell. 2003;14(11):4387–4396. doi: 10.1091/mbc.E03-05-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel A, Orjalo AV, Vincent T, Lachish-Zalait A, Vasu S, Shah S, et al. Removal of a single pore subcomplex results in vertebrate nuclei devoid of nuclear pores. Molecular Cell. 2003;11(4):853–864. doi: 10.1016/s1097-2765(03)00116-3. [DOI] [PubMed] [Google Scholar]
- Hetzer M, Bilbao-Cortes D, Walther TC, Gruss OJ, Mattaj IW. GTP hydro-lysis by Ran is required for nuclear envelope assembly. Molecular Cell. 2000;5(6):1013–1024. doi: 10.1016/s1097-2765(00)80266-x. [DOI] [PubMed] [Google Scholar]
- Hetzer M, Meyer HH, Walther TC, Bilbao-Cortes D, Warren G, Mattaj IW. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nature Cell Biology. 2001;3(12):1086–1091. doi: 10.1038/ncb1201-1086. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Richardson WD, Markham AF, Smith AE. Sequence requirements for nuclear location of simian virus 40 large-T antigen. Nature. 1984;311(5981):33–38. doi: 10.1038/311033a0. [DOI] [PubMed] [Google Scholar]
- Kutay U, Hetzer MW. Reorganization of the nuclear envelope during open mitosis. Current Opinion in Cell Biology. 2008;20(6):669–677. doi: 10.1016/j.ceb.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachish-Zalait A, Lau CK, Fichtman B, Zimmerman E, Harel A, Gaylord MR, et al. Transportin mediates nuclear entry of DNA in vertebrate systems. Traffic. 2009;10(10):1414–1428. doi: 10.1111/j.1600-0854.2009.00968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CK, Delmar VA, Chan RC, Phung Q, Bernis C, Fichtman B, et al. Transportin regulates major mitotic assembly events: From spindle to nuclear pore assembly. Molecular Biology of the Cell. 2009;20(18):4043–4058. doi: 10.1091/mbc.E09-02-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohka MJ, Maller JL. Induction of nuclear envelope breakdown, chromosome condensation, and spindle formation in cell-free extracts. The Journal of Cell Biology. 1985;101(2):518–523. doi: 10.1083/jcb.101.2.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohka MJ, Masui Y. Formation in vitro of sperm pronuclei and mitotic chromosomes induced by amphibian ooplasmic components. Science. 1983;220(4598):719–721. doi: 10.1126/science.6601299. [DOI] [PubMed] [Google Scholar]
- Lohka MJ, Masui Y. Roles of cytosol and cytoplasmic particles in nuclear envelope assembly and sperm pronuclear formation in cell-free preparations from amphibian eggs. The Journal of Cell Biology. 1984;98(4):1222–1230. doi: 10.1083/jcb.98.4.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay C, Forbes DJ. Assembly of the nuclear pore: Biochemically distinct steps revealed with NEM, GTP gamma S, and BAPTA. The Journal of Cell Biology. 1996;132(1–2):5–20. doi: 10.1083/jcb.132.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca TJ, Heald R. Methods for studying spindle assembly and chromosome condensation in Xenopus egg extracts. Methods in Molecular Biology. 2006;322:459–474. doi: 10.1007/978-1-59745-000-3_33. [DOI] [PubMed] [Google Scholar]
- Masui Y. From oocyte maturation to the in vitro cell cycle: The history of discoveries of maturation-promoting factor (MPF) and cytostatic factor (CSF). Differentiation. 2001;69(1):1–17. doi: 10.1046/j.1432-0436.2001.690101.x. [DOI] [PubMed] [Google Scholar]
- Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. Journal of Experimental Zoology. 1971;177(2):129–145. doi: 10.1002/jez.1401770202. [DOI] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Methods in Cell Biology. 1991;36:581–605. [PubMed] [Google Scholar]
- Newmeyer DD, Finlay DR, Forbes DJ. In vitro transport of a fluorescent nuclear protein and exclusion of non-nuclear proteins. The Journal of Cell Biology. 1986;103(6 Pt. 1):2091–2102. doi: 10.1083/jcb.103.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer DD, Forbes DJ. Nuclear import can be separated into distinct steps in vitro: Nuclear pore binding and translocation. Cell. 1988;52(5):641–653. doi: 10.1016/0092-8674(88)90402-3. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Wilson KL. Egg extracts for nuclear import and nuclear assembly reactions. Methods in Cell Biology. 1991;36:607–634. doi: 10.1016/s0091-679x(08)60299-x. [DOI] [PubMed] [Google Scholar]
- Newport J. Nuclear reconstitution in vitro: Stages of assembly around protein-free DNA. Cell. 1987;48(2):205–217. doi: 10.1016/0092-8674(87)90424-7. [DOI] [PubMed] [Google Scholar]
- Newport J, Dunphy W. Characterization of the membrane binding and fusion events during nuclear envelope assembly using purified components. The Journal of Cell Biology. 1992;116(2):295–306. doi: 10.1083/jcb.116.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J, Spann T. Disassembly of the nucleus in mitotic extracts: Membrane vesicularization, lamin disassembly, and chromosome condensation are independent processes. Cell. 1987;48(2):219–230. doi: 10.1016/0092-8674(87)90425-9. [DOI] [PubMed] [Google Scholar]
- Pfaller R, Smythe C, Newport JW. Assembly/disassembly of the nuclear envelope membrane: Cell cycle-dependent binding of nuclear membrane vesicles to chromatin in vitro. Cell. 1991;65(2):209–217. doi: 10.1016/0092-8674(91)90155-r. [DOI] [PubMed] [Google Scholar]
- Powers MA, Macaulay C, Masiarz FR, Forbes DJ. Reconstituted nuclei depleted of a vertebrate GLFG nuclear pore protein, p97, import but are defective in nuclear growth and replication. The Journal of Cell Biology. 1995;128(5):721–736. doi: 10.1083/jcb.128.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savulescu AF, Shorer H, Kleifeld O, Cohen I, Gruber R, Glickman MH, et al. Nuclear import of an intact preassembled proteasome particle. Molecular Biology of the Cell. 2011;22(6):880–891. doi: 10.1091/mbc.E10-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theerthagiri G, Eisenhardt N, Schwarz H, Antonin W. The nucleoporin Nup188 controls passage of membrane proteins across the nuclear pore complex. The Journal of Cell Biology. 2010;189(7):1129–1142. doi: 10.1083/jcb.200912045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Maller JL. Under arrest: Cytostatic factor (CSF)-mediated meta-phase arrest in vertebrate eggs. Genes & Development. 2003;17(6):683–710. doi: 10.1101/gad.1071303. [DOI] [PubMed] [Google Scholar]
- Ullman KS, Forbes DJ. RNA polymerase III transcription in synthetic nuclei assembled in vitro from defined DNA templates. Molecular and Cellular Biology. 1995;15(9):4873–4883. doi: 10.1128/mcb.15.9.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ahsen O, Newmeyer DD. [16]—Cell-free apoptosis in Xenopus laevis egg extracts. In: John CR, editor. Methods enzymology. Vol. 322. Academic Press; New York (USA): 2000. pp. 183–198. [DOI] [PubMed] [Google Scholar]
- Walter J, Newport JW. Regulation of replicon size in Xenopus egg extracts. Science. 1997;275(5302):993–995. doi: 10.1126/science.275.5302.993. [DOI] [PubMed] [Google Scholar]
- Walther TC, Alves A, Pickersgill H, Loiodice I, Hetzer M, Galy V, et al. The conserved Nup107-160 complex is critical for nuclear pore complex assembly. Cell. 2003;113(2):195–206. doi: 10.1016/s0092-8674(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Wilson KL, Newport J. A trypsin-sensitive receptor on membrane vesicles is required for nuclear envelope formation in vitro. The Journal of Cell Biology. 1988;107(1):57–68. doi: 10.1083/jcb.107.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Gerhart J. Raising Xenopus in the laboratory. Methods in Cell Biology. 1991;36:3–18. doi: 10.1016/s0091-679x(08)60269-1. [DOI] [PubMed] [Google Scholar]