

Abstract
The JmjC domain histone H3K36me2/me1 demethylase NDY1/KDM2B is overexpressed in various types of cancer. Here we show that knocking down NDY1 in a set of ten cell lines derived from a broad range of human tumors inhibited their anchorage-dependent and anchorage-independent growth by inducing senescence and/or apoptosis in some and by inhibiting G1 progression in all. We further show that the knockdown of NDY1 in mammary adenocarcinoma cell lines decreased the number, size and replating efficiency of mammospheres and downregulated the stem cell markers ALDH and CD44, while upregulating CD24. These findings combined, suggest that NDY1 is required for the self-renewal of cancer stem cells and are in agreement with additional findings showing that, tumor cells in which NDY1 was knocked down undergo differentiation and a higher number of them is required to induce mammary adenocarcinomas, upon orthotopic injection in animals. Mechanistically, NDY1 functions as a master regulator of a set of microRNAs that target several members of the polycomb complexes PRC1 and PRC2 and its knockdown results in the de-repression of these microRNAs and the downregulation of their polycomb targets. Consistent with these observations, NDY1/KDM2B is expressed at higher levels in basal-like triple negative breast cancers and its overexpression is associated with higher rates of relapse after treatment. In addition, NDY1-regulated microRNAs are downregulated in both normal and cancer mammary stem cells. Finally, in primary human breast cancer, NDY1/KDM2B expression correlates negatively with the expression of the NDY1-regulated microRNAs, and positively with the expression of their PRC targets.
Keywords: PRC1, PRC2, breast cancer, methylation/demethelation coupling
INTRODUCTION
NDY1/KDM2B is a jumonji domain-containing histone lysine demethylase with specificity against histone H3K36me2/me1 and H3K4me3 (1,2). Its involvement in oncogenesis was originally suggested by studies that identified NDY1/KDM2B and its homolog NDY2/KDM2A as targets of provirus integration in retrovirus-induced rodent lymphomas (1). Subsequently, it was shown that NDY1/KDM2B inhibits replicative senescence and promotes the immortalization and proliferation of mouse embryo fibroblasts (MEFs) in culture (2,3). This is due, at least in part, to the repression of the INK4A-INK4B/Arf locus, via the NDY1-dependent coupling of histone H3K36me2/me1 demethylation, with histone H3K27 trimethylation and histone H2AK119 ubiquitination. Mechanistically, it has been shown that the coupling to histone H3K27 trimethylation is mediated by the recruitment of EZH2, the enzymatic subunit of polycomb repressive complex 2 (PRC2) and that the coupling to histone H2AK119 ubiquitination is mediated by the recruitment of polycomb repressive complex 1 (PRC1) (2). Notably, we recently showed that NDY1/KDM2B is induced by FGF-2 via DYRK1A activation and CREB phosphorylation. The induced NDY1 binds the miR-101 gene in concert with EZH2, and this results in the repression of miR-101 and the upregulation of its target EZH2. The upregulation of EZH2 reinforces this pathway and locks the cells in a state characterized by the overexpression of both NDY1 and EZH2. One type of human cancer where EZH2 is overexpressed, often via the activation of the FGF-2-NDY1-miR-101-EZH2 axis is bladder cancer (4). Other recent studies have also shown that NDY1/KDM2B binds CpG islands (CGIs) via its CXXC motif and recruits PRC1, which catalyzes histone H2AK119 ubiquitination (1,2,5–7). Consistent with these observations, NDY1/KDM2B facilitates stem cell reprogramming of differentiated cells in culture (1,8,9) and protects cells from oxidative stress and ROS-induced DNA damage by regulating the expression of antioxidant genes (2,3,10). Finally, it is expressed at high levels in various types of stem cells and its expression declines with differentiation (2,6) The preceding data, combined with observations showing that NDY1 is overexpressed in human T-cell and B-cell acute lymphoblastic leukemia (T-ALL and B-ALL), acute myeloid leukemias (AML) (4,11), seminomas (12) and mammary (13,14) and pancreatic adenocarcinomas (15), strongly suggest that NDY1 functions as an oncogene.
In the present study, we knocked down NDY1 in a set of ten cell lines derived from a broad range of human tumors. In all these cell lines, the NDY1 knockdown consistently induced G1 delay and senescence and/or apoptosis and interfered with anchorage-dependent and anchorage-independent growth. Focusing in a set of two basal-like and two luminal mammary adenocarcinoma cell lines, we showed that the knockdown of NDY1 promotes changes in the expression of cancer stem cell markers, suggesting loss of stemness. Mammosphere assays and orthotopic inoculation of decreasing numbers of the tumor cells in mice confirmed this suggestion by showing that the knockdown of NDY1 results in a decrease in the number of tumor initiating cells. Consistent with this observation, the knockdown of NDY1 promoted the differentiation of basal-like tumor cells to luminal cells and this change in phenotype remained stable upon orthotopic transplantation of the tumor cells into the mouse mammary gland. Studies addressing the molecular mechanisms of these phenomena, revealed that NDY1 functions in concert with EZH2 to repress a set of microRNAs, which target the PRC1 and PRC2 subunits, BMI1, RING1B, SUZ12 and EZH2 itself. Although EZH2 and BMI1 also function in this pathway, they cannot rescue the defect induced by the loss of NDY1, which functions as the master regulator of PRC1 and PRC2 by integrating the activities of these complexes. Consistent with these observations, NDY1/KDM2B is expressed at higher levels in basal-like than in luminal mammary adenocarcinomas and its expression is associated with higher rates of relapse after treatment. More important, NDY1-regulated microRNAs are expressed at low levels in normal and neoplastic mammary stem cells, and their expression correlates negatively with the expression of NDY1/KDM2B, as well as the expression of their polycomb targets.
MATERIALS AND METHODS
Detailed descriptions of the materials and the methods are presented in the supplement.
Cell culture
Cancer cell culture conditions as well as pre-miRs and anti-miRs are listed in supplemental materials and methods. SKOV3, OVCAR3 and OVCAR4 cell lines were kindly provided by Dr Athan Kuliopulos and SUM159 cells by Dr Stephen Ethier. All other cell lines were obtained from ATCC. The authors carried out no additional authentication within the last 6 months. Retroviral and lentiviral packaging and infections were carried out using standard methods. Cell proliferation was measured with the MTT assay. Soft agar and sphere formation assays were performed as previously described (4,16). All studies with cancer cell lines were limited to established cell lines. Patient-derived cell lines were not employed.
Real-time RT- PCR for miRNAs and mRNAs
RNA isolation and real time RT-PCR were carried using standard procedures. The miRNA levels were normalized based on U6 small nuclear RNA (internal controls). The mRNA levels were normalized based on GAPDH or 18S rRNA (internal control).
Western blotting
Total cell lysates were generated using a Triton X-100 lysis buffer. Western blots of cell lysates resolved by SDS-PAGE were probed with the antibodies listed in the supplement.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using the Chromatin Immunoprecipitation assay kit (Millipore, cat no. 17-295).
Cell cycle distribution of proliferating cells, apoptosis and differentiation markers
Cell cycle distribution was determined as previously described (10). Apoptosis was measured by Annexin V staining and flow-cytometry (BD Pharmigen, cat no 556419) and/or by the Caspase-Glo® 3/7 Assay (Promega, cat no. G8091). Cells stained for CD24-FITC/EpCAM-PE/CD49f-APC, or ALDH activity (ALDEFLUOR StemCell Technologies, cat no. 01700) (CD24-FITC/CD44-APC were also analyzed by flow-cytometry.
Orthotopic mammary adenocarcinoma xenografts
Using a protocol approved by the Tufts University IACUC committee, MDAMD-231 cells transduced with shControl or shNDY1 were injected into the mammary gland of NOD/SCID mice at concentrations of 106, 105, 104 or 103 cells per gland. Significance of differences in cancer initiating cell numbers between these cells was calculated, using the chi-square test, and Extreme Limiting Dilution Analysis (ELDA) software (17).
Primary human breast cancer. Normal breast and mammary adenocarcinoma stem cells
Data on the expression of NDY1/KDM2B in different types of breast cancer and the rates of relapse after treatment were calculated from the study GSE19783 (18). Significance of differences in the rate of recurrence in patients with NDY1/KDM2B high and low tumors was calculated with the Gehan-Breslow-Wilcoxon test. Finally, the correlations between the expression of NDY1, the NDY1-regulated microRNAs and their polycomb targets were made using RNA isolated from a set of 16 primary human breast cancer specimens (Table S5). The significance of the correlation between the expression of NDY1 and NDY1-regulated microRNAs, as well as between NDY1 and the genes encoding members of the polycomb complexes in these tumors, was calculated using the Pearson statistical test. Due to the small size of the tumor samples, gene expression was examined only at the RNA level. Tumors were obtained from the Tufts Cancer Center tissue bank, following IRB approval (for details see Table S5).
Replicates and statistical analysis
All experiments were performed three times and the data are presented as mean ± SD unless otherwise stated. Two-group comparisons were performed with the Student’s t-test and p-values of 0.05 or less were considered significant (*** p<0.001, ** 0.001<p<0.01, * 0.01<p<0.05).
RESULTS
Knockdown of NDY1/KDM2B in cancer cells inhibits anchorage-dependent and independent growth
To elucidate the role of NDY1/KDM2B in the proliferation and survival of cancer cells, we knocked it down in a broad range of established cancer cell lines. Monitoring these cells revealed that the depletion of NDY1 significantly inhibits both live cell accumulation in culture monolayers and colony formation in soft agar (Fig 1A, 1B and Fig S1A–C), suggesting that NDY1/KDM2B is pro-tumorigenic (19). Four of the cell lines were of mammary epithelial origin and of these two were basal-like (MDAMB-23 and SUM159) and two luminal (T47D and MCF7). Since our focus is on breast cancer, further studies were carried out using these cell lines.
Figure 1.
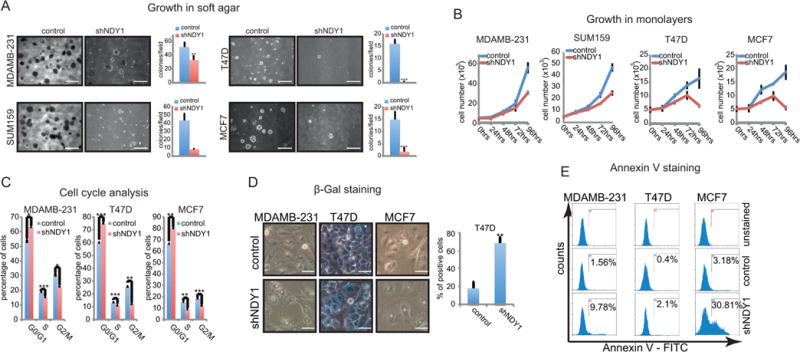
NDY1/KDM2B knockdown inhibits anchorage-dependent and independent growth.
A. Microphotographs of soft agar colonies (bar=500μm) and quantification of the colony numbers expressed as colonies per field of vision.
B. The cells in A were cultured as attached monolayers and the accumulation of live cells over time was monitored with the MTT assay.
C. Cell cycle analyses. Monolayer cultures of the cells in A and B were analyzed for DNA content.
D. shNDY1 promotes senescence in T47D cells, as assessed by β-galactosidase staining at pH 6.0. Microphotographs of the stained cells (bar=30μm) and quantification of the β-galactosidase-positive T47D cells.
E. The knockdown of NDY1/KDM2B in MDAMB-231 and MCF7 cells promotes apoptosis. The cells were stained with Annexin V and analyzed by flow cytometry.
To address the mechanism responsible for the effects of the knockdown on the accumulation of live cells in culture, we first asked whether knocking down NDY1/KDM2B interferes with cell cycle progression. Flow-cytometry of EtBr-stained semi-confluent cell cultures growing under normal tissue culture conditions, revealed that the knockdown of NDY1 induces a partial G1 arrest in all the cell lines (Fig 1C, Fig S1D), and suggested that NDY1 contributes to progression from G1 to S.
The knockdown of NDY1 may interfere with the accumulation of live cells in culture also by promoting senescence or apoptosis. In agreement with our earlier observations in MEFs (1), light microscopy of semi-confluent monolayers, stained for β-galactosidase, revealed that the knockdown elicits a strong senescence-phenotype, which however is limited to T47D cells (68% β-gal-positive) (Fig 1D). Flow-cytometery of Annexin V-stained MDAMB-231-shNDY1, MCF7-shNDY1 and T47D-shNDY1 cells, and their shRNA Controls, revealed that shNDY1 promotes apoptosis, primarily in the first two cell lines (Fig 1E). We conclude that whereas the knockdown of NDY1 inhibits G1 progression in all the tumor cell lines we examined, its ability to induce senescence and apoptosis is selective.
The preceding data addressed the role of NDY1/KDM2B in transformed cells. To determine whether NDY1 is also required for the initiation of transformation, we transduced MCF-10A cells, an immortalized but not transformed mammary epithelial cell line, with shNDY1 or shRNA-control lentiviral constructs and we superinfected them with an H-Ras-V12 retrovirus. Of these cells, only the shControls superinfected with H-Ras-V12 formed colonies in soft agar (Fig S2A and S2B). Cell cycle analysis of sub-confluent monolayer cultures of the same cells showed that the shNDY1 cells accumulate in G1 (Fig S2C). Finally, whereas shRNA control cells transduced with the H-Ras-V12 retrovirus formed mammospheres when cultured in suspension, the shNDY1 cells did not (Fig S2D). These findings combined show that NDY1 is required not only for the maintenance, but also for the initiation of the cell transformation phenotype.
NDY1/KDM2B is required for the maintenance of the cancer stem cell population
Tumor cell lines contain populations of cells that possess tumor-initiating properties. These cells tend to form spheres when grown in suspension in defined serum-free media and they are known as tumor initiating, or cancer stem cells (for Review see (20)). Tumor initiating cells in mammary carcinoma cell lines form mammospheres (16,21). Suspension cultures of MDAMB-231, SUM159, MCF7 and T47D cells, transduced with an shNDY1 lentiviral construct gave rise to fewer and smaller mammospheres than similarly cultured shRNA-control cells (Fig 2A–C). This suggests that NDY1 is required for the maintenance of tumor initiating cells, as well as for their proliferation in the mammosphere environment. Re-culturing cells harvested from mammospheres of two of the cell lines (MDAMB-231 and MCF7), revealed that the difference of shNDY1 and shRNA-control cells in mammosphere-forming potential increases with passage (Fig S3A), suggesting that NDY1 is also required for the long-term maintenance of the tumor initiating phenotype.
Figure 2.
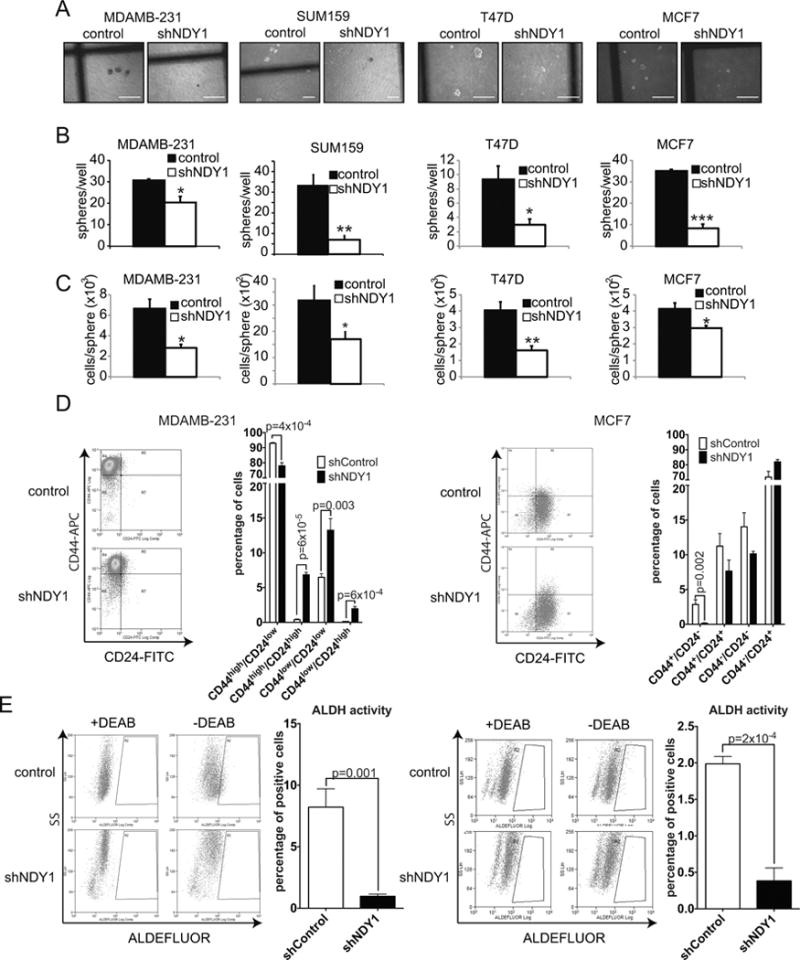
The knockdown of NDY1/KDM2B promotes cancer stem cell depletion.
A. NDY1/KDM2B knockdown inhibits mammosphere formation. Representative microphotographs (bar=300μm).
B. Quantification of the number of mammospheres in the cultures shown in A. Data expressed as spheres per 35mm plate.
C. Cells and spheres were counted in the cultures in A/B. Results are presented as mean cell number per sphere ± SD.
D. Representative plots of two-color flow-cytometric analyses of CD24-FITC/CD44-APC stained MDAMB-231 (Left panel) and MCF7 cells (Right panel). The bar graphs show quantitatively the cumulative data from three independent experiments.
E. ALDH activity of the cells in D. Representative dot plots for MDAMB-231 (Left panel) and MCF7 cells (Right panel). The bar graphs show quantitatively the cumulative data on the percentage of ALDHhigh cells.
The preceding results prompted us to analyze the same cells for the breast cancer stem cell markers CD24/CD44 and ALDH (22,23). Flow-cytometry of anti-CD44 and anti-CD24-stained MDAMB231 and MCF7 cells transduced with shNDY1 or shRNA-control, revealed that shNDY1 causes a decrease in the CD44high/CD24low population and a parallel increase in the double negative and double positive populations (Fig 2D and Fig S3B), indicative of stem cell depletion. Testing the same cells for ALDH activity revealed that shNDY1/KDM2B causes a drop in ALDHhigh cells (Fig 2E). These results combined, support the mammosphere data, suggesting that NDY1/KDM2B is required for the maintenance of the stem cell population.
NDY1/KDM2B is required for the maintenance of the myoepithelial/luminal progenitor cell phenotype, of basal breast cancer cell lines
The stage of differentiation of normal mammary epithelial cells can be determined by the expression of several membrane markers. The expression of the same markers in mammary adenocarcinomas, in combination with the genome wide expression profiles of the tumor cells, have been used to link breast cancer subtypes with the known differentiation stages of normal mammary epithelia. (24–27). Basal, triple negative tumor cells, represented by the MDAMB-231 and SUM159 lines, have a bipotent myoepithelial/luminal progenitor cell phenotype, while luminal tumor cells, represented by the MCF7 and T47D lines, have a differentiated luminal cell phenotype. The results of the mammosphere experiment suggested that NDY1 contributes to the maintenance of mammary tumor cells in an undifferentiated state. To address this hypothesis, MDAMB-231 and SUM159 cells transduced with shNDY1, or shRNA-control lentiviral constructs, were stained with anti-CD49f-APC, anti-EpCAM-PE and anti-CD24-FITC antibodies. Flow-cytometric two-color analysis for CD49f/EpCAM or CD49f/CD24, revealed that the knockdown of NDY1 promotes the downregulation of CD49f in concert with the upregulation of EpCAM and CD24 (Fig 3A). Single color analysis of the same cells confirmed the preceding results (Fig S4A). These results suggest that NDY1/KDM2B is essential for the maintenance of the basal/myoepithelial/luminal progenitor cell phenotype. This suggestion is supported by the results in Fig 3B, which show that whereas the basal markers CD10, NOTCH4 and CK14 (25) are dramatically downregulated in shNDY1-transduced cells, the luminal markers CK18, CK19 and GATA3 (25) are upregulated.
Figure 3.
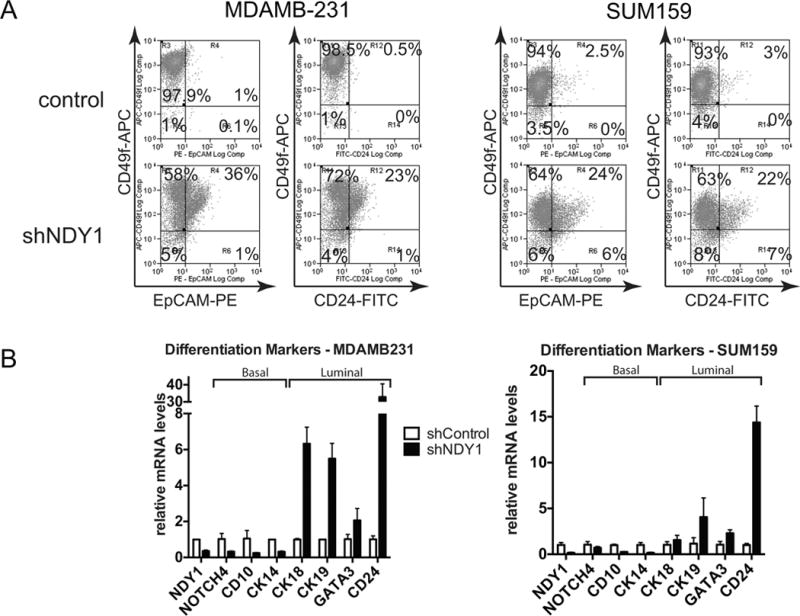
NDY1/KDM2B is required for the maintenance of the myoepithelial/luminal progenitor cell phenotype, of basal breast cancer cell lines.
A. (Left panels) (Representative plots of flow-cytometric analysis of CD49f-EpCAM-stained cells. Right panels) Representative plots of flow-cytometric analysis of CD49f-CD24-stained cells.
B. Changes in the expression of basal and luminal markers, following the knockdown of NDY1/KDM2B in MDAMB231 (Left panel) and SUM159 cells (Right panel). Expression was measured by quantitative real time RT-PCR, with GAPDH as the internal control.
In parallel experiments, two color and single color analyses of MCF7 and T47D cells transduced with shNDY1 or shRNA-control constructs also showed that shNDY1 downregulates CD49f and upregulates CD24 (Fig S4B and S4C). The knockdown of NDY1 in these cells promoted the upregulation of GATA3, CK18 and CK19, as in MDAMB-231 and SUM159 cells. However, it also promoted the upregulation rather than the downregulation of NOTCH4 and CD10 (Fig S4D). This suggests that NDY1 may be required for the maintenance of both the basal and the luminal differentiation state.
The hypothesis that NDY1/KDM2B may be expressed at higher levels in basal-like versus luminal breast cancer, which was suggested by the preceding data, was confirmed by comparing its expression in basal-like vs all other types of human mammary adenocarcinomas in the study GSE19783 (18) (Fig S4E). Comparison of the relapse rates of NDY1/KDM2B high and low tumors, following treatment-induced complete remission in the same study, revealed that the high NDY1 tumors tend to relapse earlier. Given the relatively small number of samples, the marginally significant difference of the relapse rates (p=0.13) is suggestive of a significant relationship.
Restoring the expression of wild type NDY1/KDM2B in shNDY1-transduced tumor cell lines rescues the NDY1/KDM2B phenotype
Infection of shNDY1-transduced T47D cells with a murine NDY1/KDM2B retroviral construct restored NDY1/KDM2B expression (Fig S5A). The biological outcome of this rescue experiment was the restoration of cell proliferation (Fig S5B), cell cycle progression (Fig S5C), growth in soft agar (Fig S5D, S5E), mammosphere formation (Fig S5F, S5G) and CD24 expression (Fig S5H). These findings confirmed that the shNDY1 phenotype is due to the knockdown of NDY1/KDM2B and not to off-target effects of shNDY1.
NDY1/KDM2B represses microRNAs that target polycomb complex subunits and functions as a master regulator of PRC1 and PRC2
The preceding experiments suggested that NDY1/KDM2B regulates cancer stem cell self-renewal and differentiation. Probing western blots of shNDY1 or shRNA-control MDAMB-231, MCF7 and T47D cells with antibodies against a panel of PRC1 and PRC2 subunits, revealed that the knockdown of NDY1 downregulates the PRC2 subunits EZH2 and SUZ12, but not EED, and the PRC1 subunits BMI1 and RING1B (Fig 4A). Given the function of PRC1 and PRC2 (for Review see (28–30)), these data provide potential mechanistic support to the hypothesis that NDY1 regulates cancer stem cell self renewal. In agreement with these results, knocking down NDY1 induced the expression of p21CIP1, a known target of PRC1 (31), and RUNX3, WNT1, HOXA4 and PLCβ4, known targets of PRC2 (32) (Fig 4B). The up-regulation of WNT1 was surprising, because WNT signaling has been linked to stem cell self-renewal (for Review see (33). However, additional experiments showed that despite the upregulation of Wnt1, the knockdown of NDY1 inhibited the Wnt pathway, probably because of changes in other regulators of Wnt signaling (Fig S6).
Figure 4.
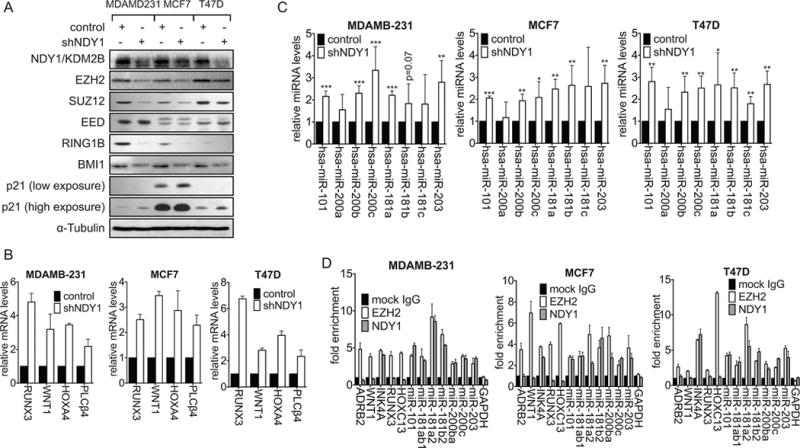
NDY1/KDM2B-regulated miRNAs repress PRC1 subunits BMI1 and RING1B and PRC2 subunits EZH2 and SUZ12.
A. Immunoblots of cell lysates harvested from stably-transduced cells were probed with the indicated antibodies.
B. The expression of PRC2 targets in shNDY1 versus shRNA-Control cells was measured in the cells in A by real time RT-PCR with GAPDH as the internal control.
C. The expression of microRNAs targeting the PRC1 subunits BMI1 and RING1B and the PRC2 subunits EZH2 and SUZ12 was measured in the cells in A by real time RT-PCR with U6 as the internal control.
D. ChIP-qPCR analysis of EZH2 and NDY1/KDM2B at the indicated genes and microRNAs in MDAMB-231, MCF7 and T47D cells.
We had previously shown that the upregulation of NDY1 activates an EZH2/miR-101 switch, which ultimately locks the cells in a developmental state characterized by high EZH2 and low miR-101 (4). We therefore hypothesized that shNDY1 may downregulate EZH2 by reversing this pathway. We also hypothesized that it may downregulate SUZ12, BMI1 and RING1B by similar mechanisms. Measuring the expression of a set of microRNAs that target the subunits of PRC1 and PRC2 listed above (34,35) in shNDY1 and shControl-transduced MDAMB-231, MCF7 and T47D cells, confirmed that the knockdown of NDY1 de-represses all the microRNAs in this set (Fig 4C). Chromatin immunoprecipitation experiments revealed that endogenous NDY1/KDM2B binds the genes encoding all these microRNAs, in concert with EZH2. Parallel ChIP experiments showed that whereas EZH2 binds, NDY1 does not bind the promoter region of protein-coding genes that are also repressed by NDY1 via EZH2, such as ADRB2 and WNT1 (Fig 4D).
Superinfection of shNDY1-transduced MDAMB-231 cells with retroviral constructs of EZH2 or BMI1 failed to rescue the defects in monolayer growth, colony formation in soft agar and mammosphere formation, induced by the NDY1/KDM2B knockdown (Fig S7 and S8). In agreement with these results, EZH2 also failed to increase the binding of NDY1/KDM2B and EZH2 to the PRC1 and PRC2-targeting microRNAs that were de-repressed by the NDY1 knockdown (Fig S7E, Left and middle panels) and as a result, it failed to repress these microRNAs (Fig S7E, Right panel) and. These results are consistent with the hypothesis that the binding of EZH2 correlates with histone H3K36me2 and H3K36me1 demethylation by NDY1/KDM2B (2,4) and they support the idea that BMI1 and EZH2 function in concert with NDY1, which is master regulator of PRC1 and PRC2.
The phenotype of the NDY1/KDM2B knockdown in mammary adenocarcinomas depends on the de-repression of NDY1/KDM2B-regulated microRNAs that target subunits of PRC1 and PRC2
To address the hypothesis that the downregulation of EZH2, SUZ12, BMI1 and RING1B in shNDY1-transduced MDAMB-231 cells is due to the de-repression of miR-101, miR-181, miR-200b and miR-203, shControl and shNDY1-transduced MDAMB-231 cells were tranfected with pre-miRs and anti-miRs of these microRNAs respectively. Western blots of cell lysates probed with the indicated antibodies, confirmed that whereas the pre-miRs upregulate EZH2, SUZ12, BMI1 and RING1B, the anti-miRs downregulate them (Fig 5A). We conclude that the de-repression of these miRs in shNDY1 MDAMB-231 cells is responsible for the down-regulation of the PRC1 and PRC2 subunits.
Figure 5.
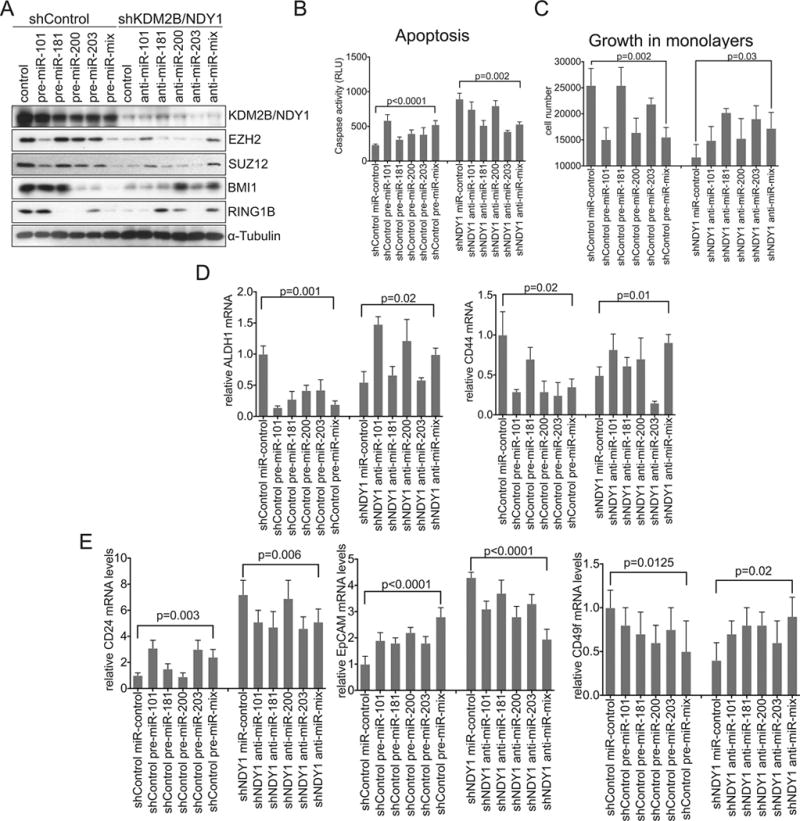
The derepression of NDY1/KDM2B-regulated microRNAs that target PRC1 and PRC2 subunits is at least partially responsible for the phenotype of the NDY1 knockdown.
A. shControl or shDNY1-transduced MDAMB-231 cells were transfected with either pre-miRs or anti-miRs respectively. Immunoblots were probed with the indicated antibodies.
B. Cells in A were analyzed for apoptosis by measuring caspase 3/7 activity.
C. Cells in A/B were seeded at the same density in monolayer cultures and they were counted 5 days later.
D. The cells in A–C were analyzed by real time RT-PCR for the expression of ALDH1 (Left panel) or CD44 (Right panel) with GAPDH as the internal control.
E. Expression of CD24 (left), EpCAM (middle) and CD49f (right) in the cells in A-D was measured by real time RT-PCR with GAPDH as the internal control.
Pre-miR expression in shControl cells enhanced, while anti-miR expression in shNDY1 cells inhibited the activation of caspases 3 and 7 (Fig 5B). Moreover, pre-miRs inhibited, while anti-miRs enhanced cell proliferation in the same cells (Fig 5C). Finally, the pre-miRs reproduced partially the NDY1/KDM2B knockdown phenotype on the expression of the stem cell markers CD44 and ALDH1 in shControl cells, while the anti-miRs partially blocked the NDY1 knockdown phenotype in shND1 cells (Fig 5D, 5E). We conclude that NDY1/KDM2B plays an obligatory role in the regulation of miR-101, miR-181, miR-200b and miR-203 and that PRC1 and PRC2, which are under the direct control of these microRNAs, function as critical effectors of tumor cell survival, proliferation, stem cell maintenance and differentiation.
Loss of NDY1/KDM2B inhibits growth, depletes the stem cell compartment and promotes differentiation of cancer cells in an orthotopic model of breast cancer
To determine the effects of shNDY1 in vivo, shNDY1 and shControl-transduced MDAMB-231 cells were inoculated into the mammary gland of NOD/SCID mice. Monitoring tumor growth for up to 70 days post-injection, revealed that the growth of cells transduced with the shNDY1 construct was significantly delayed (Fig 6A–6C). More important, tumors arising in animals inoculated with shNDY1-transduced cells, express lower levels of the stem cell marker ALDH1 and basal marker CD49f, but higher levels of CD24 and EpCAM (Fig 6D) as measured by qRT-PCR in mRNA isolated from these tumors. The lower levels of ALDH1 in the harvested tumors suggest that the reduced expression of ALDH1 in shNDY1-transduced cells is stable, despite microenviromental signals and the plasticity of the cancer stem cells. However, we should point out that ALDH1 expression in the xenografts was examined only at the RNA and not at the protein level. We conclude that the loss of NDY1/KDM2B inhibits the growth and promotes the differentiation of mammary adenocarcinoma cell lines from the basal-like to the luminal phenotype and interferes with the self-renewal of cancer stem cells, both in culture and in animals.
Figure 6.
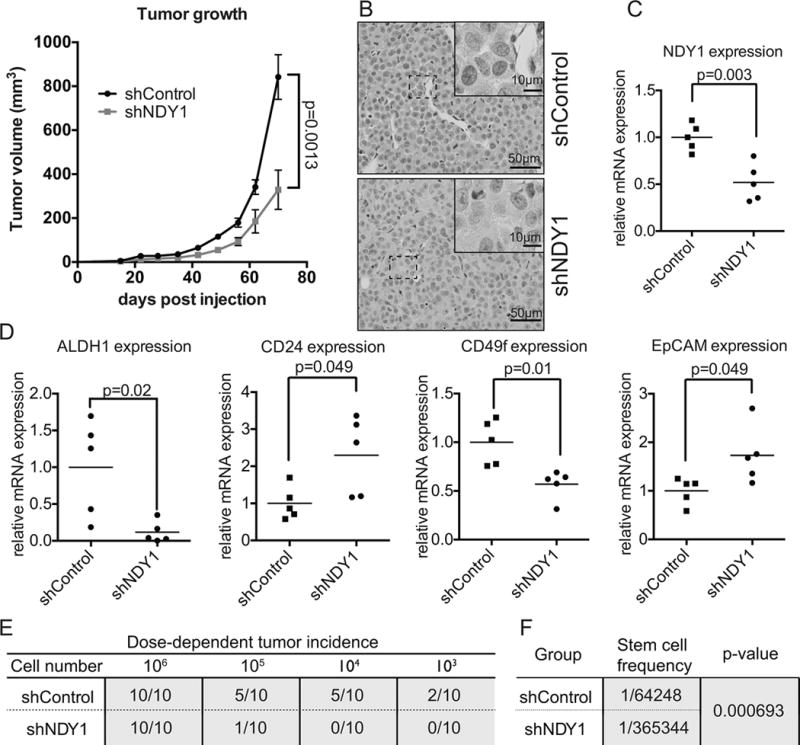
ShNDY1 inhibits growth, promotes differentiation and reduces tumor-initiating cells in the basal-like mammary adenocarcinoma cell line MDAMB-231.
A. NOD/SCID mice injected orthotopically with shControl or shNDY1-transduced MDAMB-231 cells were monitored for tumor growth (mean ± SEM).
B. Representative microphotographs of H&E stained tumor sections. The tumors arising in animals inoculated with either shControl or shNDY1-transduced cells exhibit a solid growth pattern and are composed of sheets and nests of large cells with indistinct cell borders, vesicular nuclei with prominent nucleoli, frequent mitoses, and scant stroma. There are areas of geographic tumor necrosis and frequent apoptotic tumor cells.
C. mRNA levels of NDY1/KDM2B in the tumors were measured by real time RT-PCR.
D. mRNA levels of ALDH1, CD24, CD49f, and EpCAM, were measured in the tumors by real time RT-PCR.
E. Tumor incidence in mice inoculated orthotopically with diminishing numbers of shControl or shNDY1-transduced MDAMB-231 cells.
F. Stem cell frequency in shControl and shNDY1-transduced MDAMB-231 cells. p-value was calculated by ELDA.
To determine whether shNDY1 lowers the number of tumor initiating cells (cancer stem cells), as predicted by the in vitro experiments and by the downregulation of ALDH1 in the shNDY1 tumor xenografts, we repeated the orthotopic inoculation of shControl and shNDY1 cells, using four different dilutions of tumor cells (Fig 6E). Extreme limiting dilution analysis (ELDA) (17) of the results revealed that knocking down NDY1 decreases the cancer stem cell number from 1/64248 in shControl to 1/365344 in shNDY1 cells, a ~5.6 fold decrease (p<0.000693). This confirms that shNDY1 depletes the cancer stem cell compartment of mammary adenocarcinoma cell lines.
The NDY1/EZH2/miRNA/PRC axis is active in primary human breast cancer and breast cancer stem cells
To determine whether the NDY1/EZH2/miRNA/PRC axis we observed in tumor cell lines is active in primary human breast cancer, we examined the expression of the NDY1-regulated miRs and their polycomb targets in a set of 16 frozen primary breast cancer specimens, obtained from the Tufts Cancer Center tissue bank. Measurement of the Pearson correlation between NDY1, microRNAs and PRC subunits, revealed that the expression of NDY1 exhibits a negative correlation with the expression of the NDY1-regulated microRNAs and a positive correlation with the expression of the polycomb targets of these microRNAs (Fig 7). The p value of some of the comparisons was smaller than 0.1, but higher than 0.05. Given the small number of samples and the consistency in the directionality of the data, we believe that even these not formally significant data, are borderline significant. Here we wish to point out that, due to the small size of the tumor samples, the expression of the polycomb targets of the microRNAs regulated by NDY1/KDM2B was examined only at the RNA level. However, given that the same microRNAs regulate their polycomb targets at the RNA level in tumor cell lines in culture, we suggest that the observed microRNA/mRNA correlations are likely to reflect similar miroRNA/protein correlations.
Figure 7.
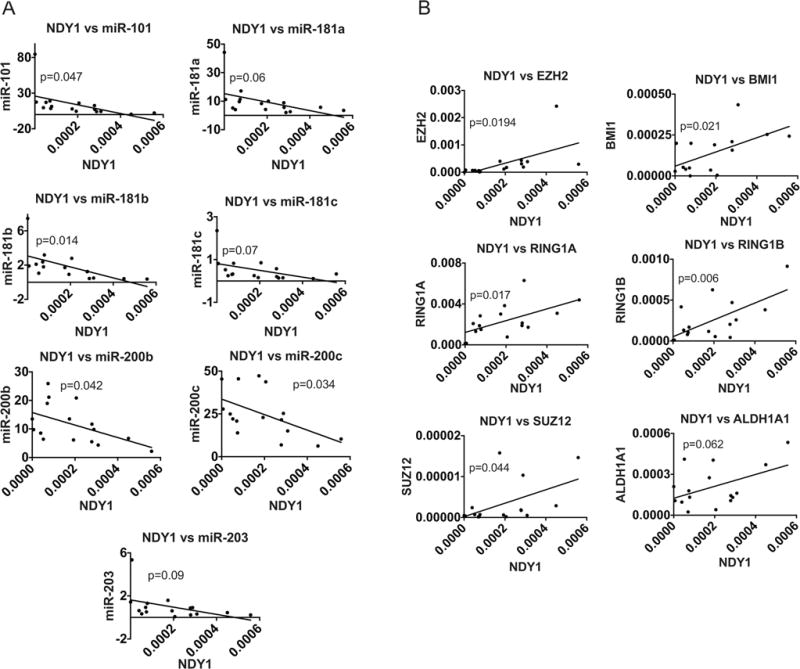
The NDY1/EZH2/miRNA/PRC axis is active in primary human breast cancer. mRNA and miRNA levels in 16 primary human tumors were measured by real time RT-PCR with 18S rRNA (for mRNAs) and U6 (for miRNAs) as internal controls.
A. The expression of NDY1/KDM2B exhibits a negative correlation with the expression of miR-101, miR-281a, miR-181b, miR-181c, miR-200b, miR-200c or miR-203 in a set of primary human mammary adenocarcinomas.
B. The expression of NDY1/KDM2B exhibits a positive correlation with the expression of EZH2, BMI1, RING1A, RING1B, SUZ12 and ALDH1, in the set of mammary adenocarcinomas, shown in A.
DISCUSSION
Earlier studies had shown that NDY1/KDM2B functions as an oncogene in retrovirus-induced rodent T cell lymphomas (1). In addition, they provided evidence that NDY1 functions as an oncogene in human bladder and pancreatic carcinomas (4,15). Experiments in this report focusing on a set of basal-like and luminal mammary adenocarcinoma cell lines, provided support to the hypothesis that NDY1/KDM2B functions as an oncogene, by showing that NDY1 sustains tumor growth, both in culture and in an orthotopic mammary adenocarcinoma xenograft model. In addition, they revealed that the loss of NDY1/KDM2B depletes the cancer stem cell compartment and promotes the differentiation of cancer cells both in culture and in animals. Mechanistically, these activities of NDY1 depend on the concerted action of NDY1 and EZH2, which results in the repression of a set of microRNAs that target multiple subunits of PRC1 and PRC2.
The NDY1/EZH2/microRNA/PRC axis was originally observed and experimentally-validated in established breast cancer cell lines. Even though patient-derived cancer cell lines were not utilized in this study, the relevance of this axis to human breast cancer was confirmed by observations that in primary tumors, the expression of NDY1/KDM2B exhibits a negative correlation with the expression of the NDY1-regulated microRNAs and a positive correlation with the expression of their polycomb targets. Of the microRNA targets of NDY1/EZH2, which were interrogated in this paper, the members of the miR-200 family had been shown previously to be expressed at low levels in normal human and mouse mammary stem cells and in breast cancer stem cells isolated from primary human breast cancer specimens (35,36). These data support the role of NDY1/KDM2B in stem cell self-renewal, in vivo. Further support for the essential role of NDY1 in stem cell biology in vivo was provided by the preliminary analysis of an RNA-Seq experiment in shControl and shNDY1-transduced T47D cells. This analysis, which will be presented in full elsewhere, showed that NDY1 also represses miR-141, a member of the miR-200c cluster, and all the members of the miR-182/96/193 cluster (data not shown), which are also expressed at low levels in the normal and neoplastic mammary stem cells (36).
NDY1/KDM2B regulates PRC1 and PRC2 at multiple levels. Data presented in this report showed that NDY1/KDM2B functions in concert with EZH2 to repress a set of microRNAs that target multiple subunits of both PRC1 and PRC2. Equally important, the overexpression of EZH2 or BMI1, both of which were downregulated in shNDY1-transduced cells, failed to restore the phenotype in NDY1 knockdown cells. This finding is consistent with additional observations showing that EZH2 overexpression is not sufficient to repress at least some of its microRNA targets and that catalytically active NDY1 is required to activate the repressive function of EZH2. We conclude that NDY1 functions as a master regulator of both PRC1 and PRC2.
Basal-like breast cancer, a poorly differentiated neoplasm, is characterized by high risk of recurrence after chemotherapy, and poor prognosis (37). Experiments presented in this paper show that the knockdown of NDY1 inhibits the proliferation and survival of breast cancer cell lines and that it depletes them of cancer stem cells. Consistent with these observations, basal-like breast cancers tend to express higher levels of NDY1 and tumors with high NDY1 tend to recur earlier than tumors with low NDY1, following treatment-induced complete remission (18). Moreover, shNDY1 promotes the differentiation of basal-like tumor cell lines to a luminal cell phenotype. These findings suggest that NDY1/KDM2B promotes the survival and proliferation of tumor cells, as well as the self-renewal of cancer stem cells. Given the relative resistance of cancer stem cells to chemotherapy and their importance in tumor relapse after chemotherapy-induced remission (38), our data highlight the importance of NDY1/KDM2B as a chemotherapeutic target. The fact that luminal type breast cancers have a better prognosis than the basal-like ones is another reason why inhibition of NDY1/KDM2B may alter the response of basal-like tumors to chemotherapy.
Data presented in this report suggest that NDY1/KDM2B may prevent the differentiation of basal-like mammary adenocarcinomas to a differentiated luminal cell phenotype. A partial block at a similar stage of differentiation has been described in the mammary epithelia of BRCA1 mutation carriers (BRCA1+/mut), who ultimately develop basal mammary adenocarcinomas (39). The apparent phenotypic similarity between BRCA1 mutations and NDY1/KDM2B raises the question whether the two function in the same pathway. This question will be addressed in future studies.
In summary, data presented in this report, identify a novel microRNA-dependent mechanism by which NDY1/KDM2B regulates PRC1 and PRC2 and they show that through this mechanism NDY1/KDM2B regulates the self-renewal and differentiation of tumor initiating cells in human breast cancer cell lines. These data confirm the importance of NDY1/KDM2B as a therapeutic target for breast and perhaps other human cancers.
Supplementary Material
Acknowledgments
We thank Drs Mark Ewen and Gail Sonenshein, for critical review of the manuscript and all the members of the P.N.T. laboratory for supporting this work with their intellectual and technical expertise. We also thank Dr Athan Kuliopoulos and Dr Stephen Ethier for cell lines and the Tufts Cancer Center tissue bank for breast cancer specimens. Finally, we would like to thank Dr Alexandra Touroutoglou for help with the statistical analyses. PNT wishes to dedicate this article to the memory of Dr Patricia Lorenzo, a former member of the NIH Molecular Oncogenesis Study Section and the University of Hawaii Cancer Center. Patricia was an outstanding scientist and a committed member of the Study Section. She will be remembered for her Science, her courage in the face of terminal illness and the overall strength of her character.
Financial support: PNT: NIH R01 CA109747, CK: Breast Cancer Research Foundation, NIH/NICHD HD073035, NIH/NCI CA25554 and CA170851
Footnotes
The authors declare no conflicts of interest
References
- 1.Pfau R, Tzatsos A, Kampranis SC, Serebrennikova OB, Bear SE, Tsichlis PN. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc Natl Acad Sci USA. 2008;105:1907–12. doi: 10.1073/pnas.0711865105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc Natl Acad Sci USA. 2009;106:2641–6. doi: 10.1073/pnas.0813139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He J, Kallin EM, Tsukada Y, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b) Nat Struct Mol Biol. 2008;15:1169–75. doi: 10.1038/nsmb.1499. 2008 ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 Regulates Cell Proliferation, Migration, and Angiogenesis through an NDY1/KDM2B-miR-101-EZH2 Pathway. Mol Cell. 2011;43:285–98. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu X, Johansen JV, Helin K. Fbxl10/Kdm2b Recruits Polycomb Repressive Complex 1 to CpG Islands and Regulates H2A Ubiquitylation. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 6.He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat Cell Biol. 2013 doi: 10.1038/ncb2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farcas AM, Blackledge NP, Sudbery I, Long HK, McGouran JF, Rose NR, et al. KDM2B links the Polycomb Repressive Complex 1 (PRC1) to recognition of CpG islands. eLife. 2012;1:e00205–5. doi: 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang G, He J, Zhang Y. Kdm2b promotes induced pluripotent stem cell generation by facilitating gene activation early in reprogramming. Nat Cell Biol. 2012;14:457–66. doi: 10.1038/ncb2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi X, et al. The histone demethylases jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell. 2011;9:575–87. doi: 10.1016/j.stem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Polytarchou C, Pfau R, Hatziapostolou M, Tsichlis PN. The JmjC domain histone demethylase Ndy1 regulates redox homeostasis and protects cells from oxidative stress. Mol Cell Biol. 2008;28:7451–64. doi: 10.1128/MCB.00688-08. 2008 ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersson A, Ritz C, Lindgren D, Eden P, Lassen C, Heldrup J, et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–203. doi: 10.1038/sj.leu.2404688. 2007 ed. [DOI] [PubMed] [Google Scholar]
- 12.Korkola JE, Houldsworth J, Chadalavada RSV, Olshen AB, Dobrzynski D, Reuter VE, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–7. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- 13.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Cancer Genome Atlas – Data Portal [Internet] tcga-data.nci.nih.gov. [cited 2013 Mar 27]. Available from: https://tcga-data.nci.nih.gov/tcga/
- 15.Tzatsos A, Paskaleva P, Ferrari F, Deshpande V, Stoykova S, Contino G, et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J Clin Invest. 2013 doi: 10.1172/JCI64535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Enerly E, Steinfeld I, Kleivi K, Leivonen S-K, Aure MR, Russnes HG, et al. miRNA-mRNA Integrated Analysis Reveals Roles for miRNAs in Primary Breast Tumors. PLoS ONE Public Library of Science. 2011;6:e16915. doi: 10.1371/journal.pone.0016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596–602. doi: 10.1038/304596a0. 1983rd ed. [DOI] [PubMed] [Google Scholar]
- 20.Ablett MP, Singh JK, Clarke RB. Stem cells in breast tumours: are they ready for the clinic? Eur J Cancer. 2012;48:2104–16. doi: 10.1016/j.ejca.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and In vitro Propagation of Tumorigenic Breast Cancer Cells with Stem/Progenitor Cell Properties. cancerresaacrjournalsorg. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 22.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–13. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sotiriou C. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA National Academy of Sciences. 2003;100:10393–8. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1017626108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prat A, Perou CM. Mammary development meets cancer genomics. Nat Med. 2009;15:842–4. doi: 10.1038/nm0809-842. [DOI] [PubMed] [Google Scholar]
- 27.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–13. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 28.Luis NM, Morey L, Di Croce L, Benitah SA. Polycomb in stem cells: PRC1 branches out. Cell Stem Cell. 2012;11:16–21. doi: 10.1016/j.stem.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christophersen NS, Helin K. Epigenetic control of embryonic stem cell fate. J Exp Med. 2010;207:2287–95. doi: 10.1084/jem.20101438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell. 2007;1:87–99. doi: 10.1016/j.stem.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–9. doi: 10.1126/science.1165395. 2008 ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coombs GS, Covey TM, Virshup DM. Wnt signaling in development, disease and translational medicine. Curr Drug Targets. 2008;9:513–31. doi: 10.2174/138945008784911796. [DOI] [PubMed] [Google Scholar]
- 34.Cao Q, Mani R-S, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, et al. Coordinated Regulation of Polycomb Group Complexes through microRNAs in Cancer. Cancer Cell. 2011;20:187–99. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761–72. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell Elsevier. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertucci F, Finetti P, Birnbaum D. Basal breast cancer: a complex and deadly molecular subtype. Curr Mol Med. 2012;12:96–110. doi: 10.2174/156652412798376134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, et al. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J. 2013;27:13–24. doi: 10.1096/fj.12-218222. [DOI] [PubMed] [Google Scholar]
- 39.Buckley NE, Mullan PB. BRCA1–conductor of the breast stem cell orchestra: the role of BRCA1 in mammary gland development and identification of cell of origin of BRCA1 mutant breast cancer. Stem Cell Rev. 2012;8:982–93. doi: 10.1007/s12015-012-9354-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.