

Abstract
Multiple myeloma (MM) is a B lymphocyte malignancy that remains incurable despite extensive research efforts. This is due, in part, to frequent disease recurrences associated with the persistence of myeloma cancer stem cells (mCSCs). Bone marrow mesenchymal stromal cells (BMSCs) play critical roles in supporting mCSCs through genetic or biochemical alterations. Previously, we identified mechanical distinctions between BMSCs isolated from MM patients (mBMSCs) and those present in the BM of healthy individuals (nBMSCs). These properties of mBMSC contributed to their ability to preferentially support mCSCs. To further illustrate mechanisms underlying the differences between mBMSCs and nBMSCs, here we report that (i) mBMSCs express an abnormal, constitutively high level of phosphorylated Myosin II, which leads to stiffer membrane mechanics, (ii) mBMSCs are more sensitive to SDF-1α-induced activation of MYL2 through the G(i./o)-PI3K-RhoA-ROCK-Myosin II signaling pathway, affecting Young’s modulus in BMSCs and (iii) activated Myosin II confers increased cell contractile potential, leading to enhanced collagen matrix remodeling and promoting the cell–cell interaction between mCSCs and mBMSCs. Together, our findings suggest that interfering with SDF-1α signaling may serve as a new therapeutic approach for eliminating mCSCs by disrupting their interaction with mBMSCs.
Keywords: multiple myeloma, SDF-1α, mesenchymal stromal cells, RhoA-ROCK-Myosin II
Multiple myeloma (MM) is a malignant disorder of postgerminal center B-cells.1 MM is generally characterized by the clonal expansion of neoplastic plasma cells in the bone marrow (BM), presence of monoclonal proteins in blood and/or urine and organ dysfunction such as kidney failure, bone density loss and subsequent bone fractures, spinal compressions with severe pain, etc.1,2 Despite recent advances in cancer treatments, MM remains an incurable disease owing to its proclivity for recurrence, which is believed to be caused by minimal residual disease or existence of a myeloma cancer stem cell (mCSC) niche in the BM.1,3
BM constitutes a suitable niche for mCSCs, favoring their self-renewal, differentiation and development of drug resistance through direct and indirect communications with various cell types present in the BM microenvironment.4–7 Among them, BM mesenchymal stromal cells (BMSCs) have been extensively studied in the context of MM disease progression and resistance to chemotherapeutics. Several studies had shown that BMSCs communicate with mCSCs through direct cell–cell interactions and paracrine signaling.4,8–13 In addition, as with other cancer-associated stromal cells in a number of cancer types, genetic and biochemical abnormalities in BMSCs have also been reported.5–7,9,12,14,15 However, distinctions between MM BMSCs (mBMSCs) and normal BMSCs (nBMSCs) are not well defined with respect to biomechanics. To partially address this question, our previous study reported marked differences in cell stiffness observed between mBMSCs and nBMSCs, although the underlying mechanism leading to these differences and how they influenced mBMSCs to promote mCSC pathophysiologic functions remained largely unknown.15
Stromal cell-derived factor-1 (SDF-1α) or C-X-C motif chemokine 12 (CXCL12) is a well-studied member of the chemokine family that specifically activates C-X-C motif chemokine receptor 4 (CXCR4).16 One of the major functions of SDF-1α is to promote chemotaxis of cancer cells.16 Particularly, SDF-1α is known to promote homing of MM cells to the BM,17,18 and to facilitate cell–cell interactions between mCSCs and BMSCs, leading to enhanced mCSC survival and proliferation.19,20 It is, however, still unknown how SDF-1α affects the biophysical properties of BMSCs and regulates their interaction with mCSCs.
Thus, the aims of our study were three-pronged: (i) to investigate if the differences in cell stiffness (defined in terms of cells’ Young’s modulus) are constitutive, (ii) how the cell stiffness contributes to the cancer microenvironment and (iii) how SDF-1α affects the mechanical properties of BMSCs. Herein, we present the first experimental evidence that SDF-1α can increase Young’s modulus in BMSCs by activating the G(i./o)-PI3K-RhoA-ROCK-Myosin II signaling pathway. Moreover, mBMSCs express a constitutively elevated, as compared to nBMSCs, level of activated myosin (MYL2). Finally, our data indicate that the activated myosin influences the contractile potential of the cells, which regulates cell–matrix and cell–cell interactions between mCSCs and mBMSCs.
Material and Methods
Myeloma cell line culture
RPMI 8226 MM cell line was purchased from ATCC, Manassas, VA and U266B-1 cell line was a generous gift from Dr. Jessica Ann Shafer, MD (Texas Children’s Cancer Center, Houston, TX). U266B-1 cells were authenticated at the UT MD Anderson Cancer Center (Houston, TX). The two cell lines were routinely maintained in RPMI-1640 medium (GE Healthcare HyClone™, Logan, UT) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin (Life Technologies, Grand Island, NY). Cells were maintained in a 37°C incubator with a humidified atmosphere containing 5% CO2 (Thermo Fisher Scientific, Waltham, MA).
Isolation and expansion of BM mesenchymal stem cells
BMSCs from BM samples of 13 patients (myeloma patients, n =7; monoclonal gammopathy of undetermined significance, MGUS, patient n =1; aged-matched healthy donors, n =5) were isolated and maintained for up to five passages. The age-matched control nBMSCs were obtained from individuals without cytopenia (>55 years old) who received a BM evaluation for lymphoma staging, and were determined to be negative for lymphoma involvement. We specifically selected these controls to be age-matched for the myeloma population to control for the possibility of aging-related functional changes in BMSCs. Use of these samples was approved by the Institutional Review Board of the Houston Methodist Research Institute. BM mononuclear cells from the myeloma or age-matched controls were obtained with Ficoll density gradient medium (1.077 g/ml; Sigma, St. Louis, MO). Cells were plated in 175-cm2 tissue culture flasks in MesenPro RSTM with 2% growth supplement (Invitrogen, Grand Island, NY). After a 72-hr incubation at 37°C in a 5% CO2 humidified atmosphere, nonadhering cells were removed and the adherent cells were cultured in fresh growth medium for up to five passages, or cryopreserved using the growth medium supplemented with 40% FBS and 10% DMSO (Sigma). For further expansion, BMSCs were detached with a mixture of collagenase/hyaluronidase (STEMCELL Technologies, British Columbia, Canada) and trypsin solution diluted to 0.01% (Life Technologies), and plated in 175-cm2 tissue culture flasks or 100-mm dishes coated with rat tail collagen type I (0.2 μg/ml in PBS) and Matrigel (0.02 mg/ml in PBS) (BD Biosciences, Bedford, MA). This condition for tissue culture vessel coating was able to support the proliferation of primary BMSCs, while not allowing for their differentiation. The resultant BMSCs were characterized and strong expression of CD44, CD90, CD73 and CD105, and absence of CD45 and CD138 was confirmed (Supporting Information Fig. 1).
Hoechst staining for side population
A side population (SP) of cancer cells is characterized by their ability to efflux Hoechest 33342 dye, which can be detected by flow cytometry. Isolation of SP cells has been recognized as an approach to isolate cells with stem-cell-like features,21,22 and has been successfully used to identify MM stem cells.13,23 To collect MM SP cell, Hoechst staining was performed as described previously.13 In brief, RPMI 8226 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) supplemented with 10 mM HEPES (Invitrogen), 2% FBS and Hoechst 33342 dye (10 μg/ml final concentration). After incubation at 37°C for 60 min, cells were centrifuged and resuspended in cold Hanks’ balanced salt solution (HBSS) buffer containing 2 μg/ml propidium iodide (PI) used to exclude dead cells. The cell sample was kept on ice cell sorting. Control experiments were performed simultaneously by co-incubating the cells with 50 μM verapamil to block Hoechst efflux. During cell sorting, the Hoechst dye was excited with a UV laser at 350 nm and the light emission was measured with Hoechst blue and red filters. Sorted SP cells were collected and used for further experiments.
Micropipette aspiration/cell stiffness assay
The cell aspiration assay was conducted as described previously with minor modifications.24,25 Briefly, borosilicate capillary pipettes (Kimble Chase, Vineland, NJ) were pulled and forged using a Shutter P-97 puller with the following program parameters: heat 483, pull 120, velocity 100 and time 250. Then, the pipettes were coated with SufaSil (Pierce Bio-technology, Rockford, IL) as suggested by the manufacturer. Pipette manipulation is achieved with a homemade micromanipulator clamped on a microscope (Axiovert 200M inverted microscope on a 40× Ph1 LD A-plan, Zeiss, Thronwood, NY), while the micropipette is connected to a mobile water tank to produce aspirating pressures. The phase-contrast images are taken with a Retiga 2000R (Qimaging, Surrey, BC) and with external triggering via Labview 2009 (National Instrument, Austin, TX) to obtain frame rates of up to ~50 frames per second. Images were subsequently analyzed either manually using the NIH ImageJ draw tool (National Institutes of Health, Bethesda, MA) or with a custom tracking program in Matlab 2009b (The Mathworks, Natick, MA) to identify the edge of the membrane projection and the changes in the membrane deformation in a given time period. The pixel values were converted to μm according to the following ratio: 1 pixel =5.536 μm for a 40× objective lens. Minimum aspiration pressure was sought by gradually lowering the heights of the mobile water tank from the base height (equal to the height of the microscope stage) until the first deformation was seen, and then the length of deformation was recorded for 3 sec. In order to find the Young’s modulus of individual nBMSCs and mBMSCs, we recorded the cell membrane deformation for 100 sec at a constant water tank height. All aspiration assays were performed at a constant pipette-specimen angle. For the aspiration of attached cells, cells were cultured on a 35-mm glass-bottom dish precoated with collagen, and the medium was changed to hypertonic medium (160 mOsm) just before the assay was performed. A Rho-associated coiled coil-forming protein serine/threonine kinase (ROCK) inhibitor, Y-27632 (Y), was used at 10 nM. For the suspension cells, cells were suspended in serum-free DMEM, and 200 μl cell suspension drops were added onto cover slips for single-cell measurement. Fresh drop cell solution was used for measurement of each cell to avoid discrepancy in measurement of membrane dynamics due to temperature fluctuation and evaporation. The final concentration of SDF-1α used for cell stimulation was 100 ng/ml. The previously reported model system26 was used to calculate Young’s modulus.
The displacement of cells into the micropipette as a function of time, L(t) in Eq. (1) is
| (1) |
where Φ is the ratio of the micropipette wall thickness to the pipette radius, a and ΔP are the inner radius and the applied aspiration pressure, respectively. The apparent viscosity, μ, is as below where elastic constants k1 and k2 can be determined by solving Eq. (2). τ is the exponential time constant.
| (2) |
These elastic constants, then, can be related to standard elasticity coefficients by the following equations, Eq. (3): Eo is the instantaneous Young’s modulus and Einf is the equilibrium Young’s modulus.
| (3) |
Collagen gel contraction assay
BMSCs (1–2 × 105 cells/ml) were embedded in a collagen gel (1.5 mg/ml) as described previously27,28 and casted in 48-well plates for 30 min in a humidified CO2 incubator. For co-culture with MM cells, U266B-1 cells (1 x 104 cells/ml) or SP of RPMI 8226 cells (5 × 103 cells/ml) were mixed in the BMSC collagen gels before solidification. For the floating gel contraction assay, collagen gels were gently detached from the bottom. The plates were scanned after 2 days (for attached gels) or 4 hr (for floating gels). Gel contraction images were analyzed with NIH ImageJ software by measuring areas of each gel in square pixels and by converting the measurements to square centimeters. The area of the inner well was 0.95 cm2 as indicated by the manufacturer (48-well plate; Corning, Tewksbury, MA). The experiments were repeated at least three times independently using triplicates of each experimental group. Pertussis toxin (PT, 50 μg/ml), LY294002 (LY, 20 μM), blebbistatin (bleb, 20 μM), Y-27632 (Y, 10 nM), U0126 (U, 10 μM) or AMD3100 (AMD, 25 μM) were added together with SDF-1α (100 nM) at the start of the floating gel contraction assay. All inhibitors were purchased from TOCRIS Bioscience, Bristol, UK except AMD3100 (Sigma-Aldrich, St. Louis, MO).
mCSC attachment to BMSCs
Adhesion of mCSCs to BMSCs was investigated as described elsewhere with minor modifications.29,30 Briefly, either nBMSCs or mBMSCs (10,000 cells) were plated in 24-well plates and allowed to grow to confluence. RPMI8226 cells expressing the green fluorescent protein (GFP) were stained with Hoechst 33342 dye as described above, 300 SP cells were sorted into each well and incubated in a 5% CO2 incubator at 37°C for 4 hr before the assay. Before physical shaking for 30 min on rotating shakers at 25 rotations per minute, the mCSC-BMSCs were treated with SDF-1α with or without inhibitors (AMD, PT, LY, bleb, Y or U) for 2 hr with or without co-treatment. Following incubation, cells were carefully washed three times with PBS prewarmed to 37°C, and GFP+ SP cells were counted under an inverted fluorescent microscope. Numbers of attached SP cells were normalized to those attached to the control. All experiments were repeated three times independently with six replicates per group.
Western blotting
BMSCs were cultured to 80% confluence in 100-mm tissue culture dishes before harvesting. Before treatment with SDF-1 or SDF-1α/inhibitor combinations, BMSCs were serum-starved for 24 hr. All inhibitors were added to the cells 30 min before SDF-1α stimulation. The cells were lysed and cellular proteins were collected in RIPA buffer (Pierce Biotechnology) supplemented with a protease/phosphatase inhibitor cocktail (Pierce Biotechnology), and their concentration was quantified by a BCA assay (Pierce Biotechnology). Equal amounts of protein (30 μg) were loaded into each well and resolved by SDS-PAGE, followed by Western blotting analyses for p-Erk, p-Akt, p-FAK, p-MYLII, MYLII, Erk, Akt, CXCR4, CXCR7 and β-actin. All antibodies were purchased from Cell Signaling Technology (Boston, MA) unless specified otherwise. Anti-human CXCR4 and CXCR7 antibodies were purchased from Abcam (Cambridge, MA). The dilution factor for all primary antibodies was between 1:1,000 and 1:500, while the secondary antibodies (GE Healthcare, Piscataway, NJ) were diluted to 1:2,000 and 1:3,000. Signal was visualized by chemoluminescence (ECL solution, GE Healthcare).
Detection of active RhoA
BMSC protein extracts (1 mg) were subjected to the Rhotekin Rho-binding domain (RBD) agarose bead pull-down assay (Cell BioLabs, San Diego, CA) according to the manufacturer’s protocol.31 Briefly, cell extracts were reacted with the RBD agarose beads for 1 hr at 4°C followed by serial washing with a provided buffer. The collected agarose beads were then mixed with 2× reducing SDS-PAGE sample buffer and boiled for 5 min. The supernatant was collected and subjected to Western blotting for pRhoA and RhoA. Equal protein loading was confirmed by probing the membranes with an anti-actin antibody.
Statistical analysis
A two-tailed Student’s t-test was used for two-sample comparison. For more than two samples, one-way ANOVA were used. The significance was given by the p-value, which was considered significant when less than 0.05.
Results
mBMSCs exhibit a constitutively high level of tensile stress
To investigate the biomechanical properties of mBMSCs, their cellular membrane dynamics were measured by a micropipette aspiration assay (Fig. 1a, left panel), where nBMSCs were used as a baseline control. The average minimum aspiration pressure to initiate membrane deformation in the attached mBMSCs was 4.3 kPa, while it was 3.1 kPa in the control nBMSCs (p < 0.0001, Fig. 1a, right panel). There was no statistical difference in the length of membrane deformation (Fig. 1a, middle panel). Interestingly, pretreatment of mBMSCs with Y-27632, an inhibitor of ROCK kinase that regulates Myosin II cellular functions, significantly lowered the minimum aspiration pressure in mBMSCs from 4 to 1.5 kPa (p < 0.0001) (Fig. 1b). Subsequently, the Young’s modulus of mBMSCs, as well as control nBMSCs, was kinetically monitored by recording the membrane deformation in a time course. As shown in Figure 1c, Young’s modulus of mBMSCs was on average 400 Pa, and around 200 Pa in nBMSCs (p < 0.01). As SDF-1α is a critical factor regulating cell–cell interactions and can mobilize myeloma cells in vivo, mBMSCs and nBMSCs were treated with SDF-1α and the changes in the Young’s modulus were measured. Treatment with SDF-1α increased the Young’s modulus of mBMSCs from 400 to 530 Pa (p < 0.05), and in nBMSCs from 200 to 450 Pa (p < 0.05). Taken together, these results imply that mBMSCs exhibit a constitutively high level of tensile stress, which can be further enhanced by SDF-1α stimulation.
Figure 1.

mBMSCs have stiffer membrane mechanics than nBMSCs. (a) mBMSCs require a higher minimum aspiration pressure to initiate membrane deformation compared to nBMSCs. (b) Inhibition of ROCK lowered the aspiration pressure for deformation initiation in mBMSCs. (c) Measurements of Young’s modulus of nBMSCs and mBMSCs in the absence or presence of SDF-1α.
mBMSC tensile stress can be increased by the interaction with cancer cells or SDF-1α stimulation
We explored the effects of cell–cell interaction on BMSC biophysical properties using the collagen gel contraction assay in the presence or absence of MM cancer cells. We found that mBMSCs induced 50% gel contraction, while nBMSCs had no effect on the collagen gels (p < 0.0001) (Fig. 2a). Interestingly, addition of U266B-1 MM cells enhanced the mBMSC gel contracting ability by ~40% (p < 0.05), but showed little effect on nBMSCs under similar conditions (Fig. 2b). Because CSCs are known to reside within a specialized BM niche and to be largely resistant to chemotherapeutic agents, we then investigated whether their direct interaction with BMSCs affected the biophysical properties of the stromal cells. To investigate if SP cells (or mCSCs) could induce gel contraction through BMSCs, SP cells were isolated from cultured RPMI 8226 MM cells. Co-culture between mBMSCs and SP cells induced about 50% contraction of the collagen gels, but showed no effect when added to nBMSCs (Fig. 2c). Notably, despite CXCR4 or CXCR7 expression (Supporting Information Fig. 2), MM cells alone or SP cells alone did not induce contraction of the collagen gels (data not shown). These findings demonstrate that there is a specific functional interaction between mBMSCs and MM cancer cells.
Figure 2.

Remodeling of extracellular matrix by mBMSCs. (a) mBMSCs display a higher tensile stress on collagen gels. (b and c) Addition of myeloma cells or myeloma-derived SP cells enhances the contractile potential of mBMSCs. (d) SDF-1α enhances gel contraction by mBMSCs, which is inhibited by AMD, PT, LY, Y or Bleb inhibitors.
We recently reported in silico modeling prediction that the paracrine SDF-1α/CXCR4 loop between mBMSCs and mCSCs may be critical mCSC self-renewal.20 Therefore, we expanded our studies to the floating collagen gel contraction assays, where SDF-1α signaling was regulated by a number of kinase inhibitors. As shown in Figure 2d, untreated (control) mBMSCs resulted in significant gel contraction (~60%), which was further enhanced by 20% following SDF-1α treatment (p < 0.05). Interestingly, the SDF-1α-enhanced gel contraction was inhibited by AMD3100 (AMD, a competitive antagonist of SDF-1α), pertussis toxin (PT, a G-protein-coupled receptor inhibitor, G(i./o)-subunit specific) or LY294002 (LY, a PI3K inhibitor). Conversely, the SDF-1α-induced gel contraction was not inhibited by U0126 (U, MEK inhibitor). Finally, a profound gel contraction inhibition was observed by treating the cells with ROCK inhibitor (Y) or Myosin II inhibitor blebbistatin (Bleb), irrespective of SDF-1α treatment. These results indicate that abnormal tensile stress produced by mBMSCs on collagen gels results from the constitutive activation of the RhoA-ROCK-Myosin II pathway, which can be further enhanced by SDF-1α stimulation that follows the G(i./o)-PI3K to ROCK-Myosin II signaling cascade (see model in Fig. 6).
Figure 6.

A proposed model of biophysical regulation of mBMSCs. Binding of SDF-1α ligand to its cognate receptors CXCR4 or CXCR7 results in the activation of PI3K or MEK. Subsequently, MYL2 is phosphorylated, which leads to changes in cell stiffness, ECM modification and adhesion of mCSCs to mBMSCs. Pathway 1 indicates the CXCR4-Gα(i/o)-PI3K-RhoA-ROCK-MYL2 pathway, while pathway 2 indicates the CXCR4-MEK-RhoA-ROCK-MYL2 pathway. Question marks indicate possible, but unconfirmed, pathways. Arrows indicate activation of downstream molecules. MLCK refers to myosin light chain kinase.
SDF-1α promotes the cell–cell interaction between mCSCs and mBMSCs
Several studies had shown that mCSCs are resistant to chemotherapy and radiotherapy in part because of their direct cell–cell interactions with BMSCs.4,6,15,19,32 Thus, we then evaluated whether SDF-1α promotes cell–cell interactions between mCSCs and BMSCs. As shown in Figure 3a, compared to the nBMSC control, there was an increase of 50% when mCSCs were plated with mBMSCs under the same experimental conditions (p < 0.05). This interaction between mCSCs and mBMSCs was completely inhibited by treatment of cells with AMD, PT, LY, Y or Bleb inhibitors (Fig. 3a). Conversely, treatment of cells with AMD or PT had no significant effects on mCSC/nBMSC interactions, unlike treatment with LY, Y and Bleb inhibitors, suggesting a potential SDF-1α-independent mechanism of cell–cell interaction between mCSCs and nBMSCs.
Figure 3.

Preferential attachment of mCSCs to mBMSCs depends on SDF-1α. (a) mBMSCs promote mCSC adhesion, which is sensitive to inhibition of the SDF-1α signaling pathway. (b) SDF-1α induced mCSC attachment to mBMSCs and can be inhibited by the addition of AMD, PT, LY, Y, U and Bleb inhibitors.
To further investigate the effects of SDF-1α on mCSC/mBMSC cell–cell interactions, cell attachment assays were performed in the presence of SDF-1α with or without co-treatment with the inhibitors. First, SDF-1α treatment enhanced the attachment of mCSCs to mBMSCs by more than twofold (p < 0.001) when compared to the baseline control (Fig. 3b). This enhanced cell binding was completely blocked by pretreatment with AMD, PT, LY, Y and Bleb inhibitors (p < 0.001). Unexpectedly, in contrast to our observations in the gel contraction assays, the SDF-1α-induced cell–cell interactions were also inhibited by MEK inhibitor, U0126. These results imply that the SDF-1α-stimulated cell–cell interactions could be regulated through the activation of CXCR4 G(i./o), with the downstream signaling transduction pathways bifurcating to PI3K and MEK (proposed signaling model, Fig. 6).
SDF-1α activates RhoA through PI3K and MAPK pathways
To dissect signaling events activated by SDF-1α, phosphorylation of myosin light chain II (MYL2, a classical regulatory subunit of myosin II), Akt, Erk and FAK was examined in nBMSCs and mBMSCs. Presence of SDF-1α at as low as 10 ng/ml induced phosphorylation of Erk in both nBMSCs and mBMSCs, while Akt phosphorylation was observed only in mBMSCs when stimulated with 50 ng/ml of SDF-1α (Fig. 4a). Notably, the enhanced phosphorylation of Akt in mBMSCs was not caused by increased levels of either CXCR4 or CXCR7 (Fig. 4a and Supporting Information Fig. 3). Phosphorylation of FAK in nBMSC reached its maximum at 5 min and returned to the basal level at 10 min following SDF-1α stimulation. In contrast, FAK phosphorylation in mBMSCs was higher at 5 min, and its level was maintained for up to 10 min (Fig. 4b). Importantly, treatment with SDF-1α induced a rapid phosphorylation of MYL2 that lasted over 10 min in mBMSCs, but had only a minimal effect in nBMSCs (Fig. 4b). Moreover, assays using the rhotekin Rho-binding domain (RBD) agarose beads31 revealed that phosphorylation of RhoA, an upstream regulator of MYL2, was triggered by SDF-1α treatment in mBMSCs and lasted for over 10 min (Fig. 4c), while it was minimal in nBMSCs. Additionally, phosphorylation of MYL2 induced by SDF-1α was sensitive to LY, U, PT and AMD, suggesting that the activation of RhoA by SDF-1α depends on the G(i./o) subunit, PI3K and MEK. We then investigated further components of the SDF-1α–MYL2 signaling pathway in mBMSCs by comparing Akt and Erk phosphorylation levels subsequent to treatment with inhibitors indicated in Figure 4d. As expected, SDF-1α treatment induced phosphorylation of Akt, Erk and MYL2 over the baseline, while inhibition of PI3K (by LY) or MEK (by U) signaling caused a reduction in MYL2 phosphorylation to a level lower than the baseline. Moreover, treatment of cells with AMD or PT inhibited the SDF-1α-stimulated phosphorylation of Akt and MYL2, but had no effect on the phosphorylation of Erk. Our data further indicate that this sustained Erk phosphorylation was due to the simultaneous stimulation of CXCR7 (Supporting Information Fig. 4). Finally, Bleb directly inhibited the SDF-1α-mediated phosphorylation of MYL2, without affecting phosphorylation of the upstream regulators. Conversely, inhibition of ROCK (by Y) strongly reduced MYL2 phosphorylation, similar to that observed with LY or U. Together, these data indicate that the activation of RhoA-ROCK-MYL2 by SDF-1α mainly depends on the activation of the CXCR4/G(i./o)-PI3K signaling pathway or the CXCR4-MEK pathway.
Figure 4.

SDF-1 signaling pathways in mBMSCs. (a and b) Treatment of mBMSCs with SDF-1α results in higher levels of MYL2, Akt and FAK phosphorylation when compared to nBMSCs. (c) SDF-1α stimulates phosphorylation of RhoA, an upstream regulator of MYL2, in mBMSCs, but not in nBMSCs. (d) Phosphorylation of MYL2 by SDF-1α depends on activation of G(i./o), PI3K, MEK and ROCK.
Constitutive activation of Myosin II in mBMSCs
On the basis of the biochemical and physical differences in nBMSCs and mBMSCs, we compared signaling biomarkers governing their biophysical properties. For these studies, we used BMSCs collected from five nonmyeloma patients, one MGUS patient and seven myeloma patients (Fig. 5). First, we found that the expression of CXCR4, Akt, Erk1/2 and MYL2 varied among the normal and myeloma BMSCs, but there were no significant differences detected between the groups. However, phosphorylation of FAK, Akt and MYL2 was upregulated in mBMSCs when compared to the nBMSC samples. Specifically, phosphorylation of MYL2 (pMYL2) was markedly higher in mBMSCs than in the nonmyeloma-associated BMSCs. Additionally, gene expression analysis of microarray analysis (Supporting Information Fig. 5) highlighted upregulated gene expression involved in the G-protein signaling (negative regulator of GPCR signaling, RGS4, 2.97 fold change), the PI3K/AKT pathway (p ≤6.77E−3) and the RHO family proteins (RHOJ, 3.05 fold change) in mBMSCs. These results confirm that mBMSCs are characterized by a constitutively activated Myosin II, which may foster an abnormally high mBMSC tensile stress observed in the BM of MM patients. We summarized the potential signaling pathways involved in these mBMSC biophysical alterations in Figure 6, as we believe that the SDF-1α signaling in these cells occurs via a simultaneous activation of PI3K and MEK signaling cascades, which lead to activation of MYL2 and regulation of cell stiffness, ECM modification and mCSC adhesion to mBMSCs.
Figure 5.
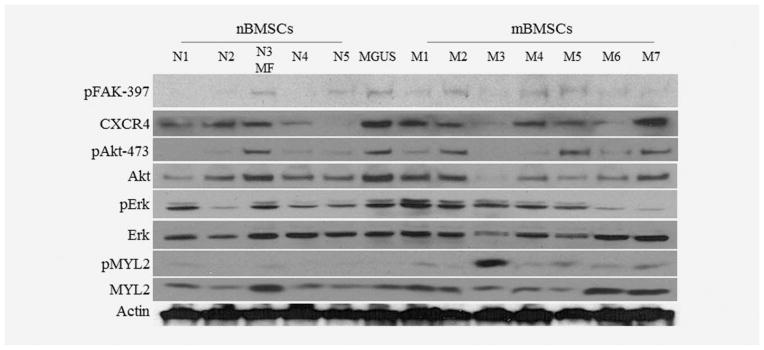
Phosphorylation of FAK, Akt and MYLII is constitutively elevated in mBMSCs. Phosphorylation of FAK, Akt and MYLII is constitutively elevated in mBMSCs (M1 to M7) compared to nBMSCs (N1 to N5). MF and MGUS refer to myeloid fibrosis and monoclonal gammopathy of undetermined significance, respectively.
Discussion
In our study, we demonstrated that stiffer cell membrane mechanics in mBMSCs were associated with higher tensional stress, which was regulated by the RhoA-ROCK-MYL2 signaling pathway (Fig. 6). We also showed for the first time that compared to nBMSCs, mBMSCs exhibit a higher level of phosphorylated MYL2 and FAK, indicating a constitutive tensional stress in mBMSCs. We also demonstrated that mBMSCs were significantly more effective in inducing collagen I gel contraction, while nBMSCs did not induce contraction of gels under similar conditions. Previous reports showed that tissue stiffening is associated with cancer-induced activation of integrins and focal adhesions in cancer-associated stromal cells, resulting in increased tensional stress in the cancer microenvironment.33,34 Moreover, this increase in the tensional stress is associated with enhanced angiogenesis, cancer cell proliferation and metastasis.33–35 Thus, our results suggest that matrix stiffening can occur in BM of MM patients, which, in turn, could promote disease pathogenesis.
In the next set of experiments, we correlated the significance of stiffer mBMSC biomechanics with stronger mCSC to mBMSC cell–cell binding compared to nBMSCs. Our results fit well in the breadth of experimental data showing that MM cells acquire various advantages, such as maintenance of CSCs, acquisition of drug resistance and differentiation, by physically coupling with BMSCs.4,5,12,13,19,36 Treatment with SDF-1α increased the stiffness of both nBMSCs and mBMSCs as did cell–cell interactions and the collagen I gel contraction. SDF-1α was originally identified as a stromal cell-secreted growth factor and as a chemoattractant for MM CSCs homing to the BM.37 Moreover, SDF-1α is known to promote a physical cell–cell contact between BMSCs and mCSCs, leading to a hypothesis that SDF-1α antagonist, AMD3100, could be used as an adjuvant MM therapy to decrease survival of MM stem cells.19
In light of its obvious importance in the progression of MM, it is also important to consider biological sources of SDF-1α within the tumor microenvironment. Others previously reported that mBMSCs produce significantly lower levels of SDF-1α than do nBMSCs as MM disease progresses.12 In contrast, another study clearly illustrated that SDF-1 was produced by the MM cells themselves.38 In our setting, we found that SDF-1α secretion was higher in CSCs than in non-CSCs (data not shown). Others had shown that cancer cells modify their microenvironment through continuous active and passive interactions with their host cells.9,12,34,39–41 Hence, cancer-associated stromal cells, including BMSCs, are known to undergo genetic, biochemical and physical alterations as tumors progress. On the basis of our results, we then propose that mCSC-borne SDF-1α may promote BMSCs to acquire stiffer biomechanics.
CXCR4, a G-protein-coupled receptor (GPCR), consists of three G-protein subunits, Gα, Gβ and Gγ.42,43 Among the three subunits, the Gα subunit is the major constituent of GPCRs, propagating diverse downstream signaling pathways upon its activation. Consequently, the same ligand–receptor interaction can conceivably result in different cellular activities.42 Our data suggest that SDF-1α activates the G(i./o)-RhoA-ROCK-Myosin II signaling pathway in BMSCs, leading to increase in cell stiffness. Also, activation of G(i./o) in the CXCR4 signaling cascade can be propagated through either PI3K alone or through both MEK and PI3K pathways simultaneously following the interaction of mBMSCs with collagen gels or mCSCs, respectively. Interestingly, we noted that compared to nBMSCs, mBMSCs were more sensitive to SDF-1α-induced activation of Akt and FAK that are downstream targets of PI3K. At the same time, we did not observe qualitative differences in the expression of CXCR4 between nBMSCs and mBMSCs. Therefore, data imply that presence of MM cells in the BM causes loss of negative regulators of the SDF-1α-CXCR4 signaling pathway. Supporting this notion, regulators of G-protein signaling (RGS), as well as growth factor independent-1 (GFI1), were identified as negative regulators of CXCR4 in multiple cell types.44–48 Finally, CXCR7 can desensitize the CXCR4 signaling pathway, especially with regard to the Gαi protein-dependent signaling.49
In conclusion, we identified that higher activation of the RhoA-MYL2 pathway confers stiffer cell membranes and higher contractility in mBMSCs, resulting in stronger cell–cell interactions between the BM stromal cells and mCSCs and in remodeling of the BM extracellular matrix. Also, we demonstrated that mBMCSs are more sensitive to SDF-1α-CXCR4 activation, which enhances the biomechanical characteristics of mBMSCs, thus supporting mCSCs. Our data lend support for further detailed studies aimed to characterize how the physical components of BMSCs support mCSCs, and to potentially identify novel therapeutic targets. Finally, our data suggest that caution should be exercised when MM patients received autologous BM transplants following lethal chemotherapy or radiotherapy because this transplanted BM may contain not only the minimal residual disease but also the mCSC-supporting BM stromal cells.
Supplementary Material
What’s new?
Multiple myeloma remains an uncurable disease in part because of the persistence of myeloma cancer stem cells that remain in specific niches in the bone marrow. Here, the authors established a novel function of stromal cell-derived factor (SDF)–1α in altering biomechanics of myeloma-associated bone marrow mesenchymal stromal cells through the activation of myosin II. They further determined that the altered biophysical characteristics play a critical role in regulating the interactions between the stroma and myeloma cancer stem cells. Collectively, the results suggest that matrix stiffening can occur in the bone marrow of multiple myeloma patients which in turn can promote disease pathogenesis.
Acknowledgments
This study was supported in part by NIH grants R01CA151955 and R33CA173382 to Y. Zu and NIH/NLM 5R01LM010185 to Dr. X. Zhou. The authors thank Dr. Philippe Bourin for generously sharing microarray data of BMSCs and Dr. Yekaterina B. Khotskaya for her help with editing of this manuscript.
Abbreviations
- BM
bone marrow
- BMSC
bone marrow mesenchymal stromal cell
- CSC
cancer stem cell
- GFP
green fluorescent protein
- MM
multiple myeloma
- ROCK
Rho-associated coiled coil-forming protein serine/threonine kinase
- SDF-1α
stromal cell-derived factor 1α
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–60. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.Palumbo A, Rajkumar SV, San Miguel JF, et al. International Myeloma Working Group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem-cell transplantation. J Clin Oncol. 2014;32:587–600. doi: 10.1200/JCO.2013.48.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andre T, Meuleman N, Stamatopoulos B, et al. Evidences of early senescence in multiple myeloma bone marrow mesenchymal stromal cells. PLoS One. 2013;8:e59756. doi: 10.1371/journal.pone.0059756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markovina S, Callander NS, O’Connor SL, et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol Cancer. 2010;9:176. doi: 10.1186/1476-4598-9-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pevsner-Fischer M, Levin S, Hammer-Topaz T, et al. Stable changes in mesenchymal stromal cells from multiple myeloma patients revealed through their responses to Toll-like receptor ligands and epidermal growth factor. Stem Cell Rev. 2012;8:343–54. doi: 10.1007/s12015-011-9310-2. [DOI] [PubMed] [Google Scholar]
- 6.Reagan MR, Ghobrial IM. Multiple myeloma mesenchymal stem cells: characterization, origin, and tumor-promoting effects. Clin Cancer Res. 2012;18:342–9. doi: 10.1158/1078-0432.CCR-11-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garderet L, Mazurier C, Chapel A, et al. Mesenchymal stem cell abnormalities in patients with multiple myeloma. Leuk Lymphoma. 2007;48:2032–41. doi: 10.1080/10428190701593644. [DOI] [PubMed] [Google Scholar]
- 8.Zdzisinska B, Bojarska-Junak A, Dmoszynska A, et al. Abnormal cytokine production by bone marrow stromal cells of multiple myeloma patients in response to RPMI8226 myeloma cells. Arch Immunol Ther Exp. 2008;56:207–21. doi: 10.1007/s00005-008-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace SR, Oken MM, Lunetta KL, et al. Abnormalities of bone marrow mesenchymal cells in multiple myeloma patients. Cancer. 2001;91:1219–30. doi: 10.1002/1097-0142(20010401)91:7<1219::aid-cncr1122>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Hideshima T, Chauhan D, Hayashi T, et al. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol Cancer Ther. 2002;1:539–44. [PubMed] [Google Scholar]
- 11.Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–55. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corre J, Mahtouk K, Attal M, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia. 2007;21:1079–88. doi: 10.1038/sj.leu.2404621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Y, Wen J, Mike P, et al. Bone marrow stromal cells from myeloma patients support the growth of myeloma stem cells. Stem Cells Dev. 2010;19:1289–96. doi: 10.1089/scd.2010.0010. [DOI] [PubMed] [Google Scholar]
- 14.Garayoa M, Garcia JL, Santamaria C, et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia. 2009;23:1515–27. doi: 10.1038/leu.2009.65. [DOI] [PubMed] [Google Scholar]
- 15.Feng Y, Ofek G, Choi DS, et al. Unique biomechanical interactions between myeloma cells and bone marrow stroma cells. Prog Biophys Mol Biol. 2010;103:148–56. doi: 10.1016/j.pbiomolbio.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 17.Menu E, Asosingh K, Indraccolo S, et al. The involvement of stromal derived factor 1alpha in homing and progression of multiple myeloma in the 5TMM model. Haematologica. 2006;91:605–12. [PubMed] [Google Scholar]
- 18.Gazitt Y, Akay C. Mobilization of myeloma cells involves SDF-1/CXCR4 signaling and downregulation of VLA-4. Stem Cells. 2004;22:65–73. doi: 10.1634/stemcells.22-1-65. [DOI] [PubMed] [Google Scholar]
- 19.Azab AK, Runnels JM, Pitsillides C, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–51. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su J, Zhang L, Zhang W, et al. Targeting the biophysical properties of the myeloma initiating cell niches: a pharmaceutical synergism analysis using multi-scale agent-based modeling. PLoS One. 2014;9:e85059. doi: 10.1371/journal.pone.0085059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodell MA, McKinney-Freeman S, Camargo FD. Isolation and characterization of side population cells. Methods Mol Biol. 2005;290:343–52. doi: 10.1385/1-59259-838-2:343. [DOI] [PubMed] [Google Scholar]
- 23.Matsui W, Wang Q, Barber JP, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008;68:190–7. doi: 10.1158/0008-5472.CAN-07-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark DJ, Killian TC, Raphael RM. A microfabricated magnetic force transducer-microaspiration system for studying membrane mechanics. Phys Biol. 2011;8:056008. doi: 10.1088/1478-3975/8/5/056008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lichtenberger LM, Zhou Y, Dial EJ, et al. NSAID injury to the gastrointestinal tract: evidence that NSAIDs interact with phospholipids to weaken the hydrophobic surface barrier and induce the formation of unstable pores in membranes. J Pharm Pharmacol. 2006;58:1421–8. doi: 10.1211/jpp.58.10.0001. [DOI] [PubMed] [Google Scholar]
- 26.Tan SC, Pan WX, Ma G, et al. Viscoelastic behaviour of human mesenchymal stem cells. BMC Cell Biol. 2008;9:40. doi: 10.1186/1471-2121-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia H, Nho RS, Kahm J, et al. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279:33024–34. doi: 10.1074/jbc.M313265200. [DOI] [PubMed] [Google Scholar]
- 28.Park DW, Choi DS, Ryu HS, et al. A well-defined in vitro three-dimensional culture of human endometrium and its applicability to endometrial cancer invasion. Cancer Lett. 2003;195:185–92. doi: 10.1016/s0304-3835(03)00131-9. [DOI] [PubMed] [Google Scholar]
- 29.Bevilacqua MP, Pober JS, Wheeler ME, et al. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest. 1985;76:2003–11. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maretzky T, Reiss K, Ludwig A, et al. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci USA. 2005;102:9182–7. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–72. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- 32.Kirshner J, Thulien KJ, Martin LD, et al. A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood. 2008;112:2935–45. doi: 10.1182/blood-2008-02-142430. [DOI] [PubMed] [Google Scholar]
- 33.Paszek MJ, Zahir N, Johnson KR, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Schrader J, Gordon-Walker TT, Aucott RL, et al. Matrix stiffness modulates proliferation, chemo-therapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanjaya-Putra D, Yee J, Ceci D, et al. Vascular endothelial growth factor and substrate mechanics regulate in vitro tubulogenesis of endothelial progenitor cells. J Cell Mol Med. 2010;14:2436–47. doi: 10.1111/j.1582-4934.2009.00981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanz-Rodriguez F, Hidalgo A, Teixido J. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-mediated multiple myeloma cell adhesion to CS-1/fibronectin and VCAM-1. Blood. 2001;97:346–51. doi: 10.1182/blood.v97.2.346. [DOI] [PubMed] [Google Scholar]
- 37.Alsayed Y, Ngo H, Runnels J, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–17. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin SK, Diamond P, Williams SA, et al. Hypoxia-inducible factor-2 is a novel regulator of aberrant CXCL12 expression in multiple myeloma plasma cells. Haematologica. 2010;95:776–84. doi: 10.3324/haematol.2009.015628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zong Y, Huang J, Sankarasharma D, et al. Stromal epigenetic dysregulation is sufficient to initiate mouse prostate cancer via paracrine Wnt signaling. Proc Natl Acad Sci USA. 2012;109:E3395–E3404. doi: 10.1073/pnas.1217982109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hawsawi NM, Ghebeh H, Hendrayani SF, et al. Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res. 2008;68:2717–25. doi: 10.1158/0008-5472.CAN-08-0192. [DOI] [PubMed] [Google Scholar]
- 41.Tuhkanen H, Anttila M, Kosma VM, et al. Frequent gene dosage alterations in stromal cells of epithelial ovarian carcinomas. Int J Cancer. 2006;119:1345–53. doi: 10.1002/ijc.21785. [DOI] [PubMed] [Google Scholar]
- 42.McCudden CR, Hains MD, Kimple RJ, et al. G-protein signaling: back to the future. Cell Mol Life Sci. 2005;62:551–77. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kucia M, Reca R, Miekus K, et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23:879–94. doi: 10.1634/stemcells.2004-0342. [DOI] [PubMed] [Google Scholar]
- 44.De La Luz Sierra M, Gasperini P, McCormick PJ, et al. Transcription factor Gfi-1 induced by G-CSF is a negative regulator of CXCR4 in myeloid cells. Blood. 2007;110:2276–85. doi: 10.1182/blood-2007-03-081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Airoldi I, Raffaghello L, Piovan E, et al. CXCL12 does not attract CXCR4+ human metastatic neuroblastoma cells: clinical implications. Clin Cancer Res. 2006;12:77–82. doi: 10.1158/1078-0432.CCR-05-1376. [DOI] [PubMed] [Google Scholar]
- 46.Berthebaud M, Riviere C, Jarrier P, et al. RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood. 2005;106:2962–8. doi: 10.1182/blood-2005-02-0526. [DOI] [PubMed] [Google Scholar]
- 47.Estes JD, Thacker TC, Hampton DL, et al. Follicular dendritic cell regulation of CXCR4-mediated germinal center CD4 T cell migration. J Immunol. 2004;173:6169–78. doi: 10.4049/jimmunol.173.10.6169. [DOI] [PubMed] [Google Scholar]
- 48.Lippert E, Yowe DL, Gonzalo JA, et al. Role of regulator of G protein signaling 16 in inflammation-induced T lymphocyte migration and activation. J Immunol. 2003;171:1542–55. doi: 10.4049/jimmunol.171.3.1542. [DOI] [PubMed] [Google Scholar]
- 49.Levoye A, Balabanian K, Baleux F, et al. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–93. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.