

Abstract
Recent advances in structural and functional imaging have greatly improved our ability to assess normal functions of the basal ganglia, diagnose parkinsonian syndromes, understand the pathophysiology of parkinsonism and other movement disorders, and detect and monitor disease progression. Radionuclide imaging is the best way to detect and monitor dopamine deficiency, and will probably continue to be the best biomarker for assessment of the effects of disease-modifying therapies. However, advances in magnetic resonance enable the separation of patients with Parkinson’s disease from healthy controls, and show great promise for differentiation between Parkinson’s disease and other akinetic-rigid syndromes. Radionuclide imaging is useful to show the dopaminergic basis for both motor and behavioural complications of Parkinson’s disease and its treatment, and alterations in non-dopaminergic systems. Both PET and MRI can be used to study patterns of functional connectivity in the brain, which is disrupted in Parkinson’s disease and in association with its complications, and in other basal-ganglia disorders such as dystonia, in which an anatomical substrate is not otherwise apparent. Functional imaging is increasingly used to assess underlying pathological processes such as neuroinflammation and abnormal protein deposition. This imaging is another promising approach to assess the effects of treatments designed to slow disease progression.
Introduction
In the past three decades a revolution has occurred in our ability to image brain structure and function, and the insights that such studies can provide into the pathogenesis of diseases and associated complications, and the healthy function and connectivity of the basal ganglia. In this Series paper, we review the application of structural and functional imaging techniques to the investigation of Parkinson’s disease and its complications. We also review the dystonias—disorders in which structural abnormalities are either absent or so subtle that they have evaded definition, and to which the application of these techniques has brought new insights. Neurochemical investigations depend mostly on the use of PET or single photon emission CT (SPECT), in which various substrates or ligands are labelled with positron-emitting or γ-emitting tracers to allow detection and quantitation of radioactivity, from which inferences are drawn about the biological process of interest. Several technical and methodological advances in MRI have occurred in the past few years that have had a profound effect on the use of this approach to study basal ganglia function and neurodegenerative disease.
Imaging for diagnosis of Parkinson’s disease
The principal features of Parkinson’s disease can be attributed to the loss of dopamine produced by neurons projecting from the substantia nigra to the dorsal (motor) striatum. Key differentials include other conditions that result in tremor but are not associated with progressive loss of dopamine—eg, essential tremor—and conditions that result in bradykinesia and rigidity, but in which the pathology is more extensive than that of Parkinson’s disease (atypical parkinsonism, such as progressive supranuclear palsy or multiple system atrophy). The diagnosis of Parkinson’s disease is predominantly clinical; however, imaging might be helpful to confirm the presence of dopamine denervation and to differentiate between Parkinson’s disease and atypical parkinsonism.
Dopamine dysfunction can be assessed with various approaches with either PET or SPECT (figure 1, panel 1). The most widely used approach to assess presynaptic dopamine function is to image with one of several SPECT or PET ligands for the membrane dopamine transporter (DAT). Several options are available for both methods, although the only one to be approved for commercial use is 123I-fluoro-propyl-CIT (FP-CIT; ioflupane, DaTScan [GE Healthcare, Little Chalfont, UK]), which can be used to differentiate between essential tremor and Parkinson’s disease. Although the various ligands differ in kinetics and degree of specificity for the DAT, they all provide similar information. The advantage of SPECT is that it is more readily accessible than is PET, and 123I and 99mTc have longer half-lives. This characteristic enables tracers to be shipped from a distance, rather than depend on an adjacent cyclotron, as is typically necessary for PET. However, the use of DAT ligands has been controversial, because the DAT is subject to compensatory downregulation and, theoretically, to pharmacological regulation. The practical importance of this characteristic is unclear, but it has led to questions of interpretation when DAT binding is used as a biomarker to monitor disease progression or the effects of putative disease-modifying therapies, or both. Although there are subtle differences, the information provided by all presynaptic markers of dopamine function is similar, and, for all of them, Parkinson’s disease is typically characterised by an asymmetric reduction in tracer uptake with a caudal to rostral gradient in which the posterior striatum is maximally affected (figure 1). Although characteristic, this appearance is not specific for Parkinson’s disease, and might be noted in other parkinsonian conditions—in particular multiple system atrophy (MSA). Although not neurochemically specific, the pattern of glucose metabolism assessed with 18F-fluorodeoxyglucose (18F-FDG) PET might reliably differentiate between various parkinsonian syndromes.1
Figure 1. Dopaminergic nerve terminal and various approaches to the assessment of its integrity (A); composite showing PET images from a healthy control individual (B; i–iii) and a patient with mild Parkinson’s disease (B; iv–vi).

FD=6-18F-fluoro-levodopa. DTBZ=11C-dihydrotetrabenazine. MP=11C-d-threo-methylphenidate. VMAT2= vesicular monoamine transporter type 2. DOPA=dihydroxyphenylalanine. DA=dopamine. DOPAC=dihydroxyphenylacetic acid. HVA=homovanillic acid.
Panel 1. Radiotracers to assess dopaminergic function.
6-18F-fluoro-levodopa
Decarboxylated to 6-18F-fluorodopamine
Activity is trapped in synaptic vesicles
Can estimate dopamine turnover with prolonged scan times
Activity can be increased relative to degree of denervation in early disease (possible compensatory upregulation of decarboxylase activity)
Membrane dopamine transporter (DAT)
Measured with a variety of 11C and 18F (PET), 123I, or 99mTc (single photon emission CT) tracers
Selectivity for DAT versus other plasmalemmal monoamine transporters and kinetic properties varies between tracers
DAT activity theoretically subject to pharmacological regulation and compensatory downregulation—practical importance of this characteristic is unclear
Vesicular monoamine transporter type 2
Labelled with 11C-dihydrotetrabenazine (DTBZ) or 18F-fluoropropyl-DTBZ
Expressed by all monoaminergic neurons, therefore not specific for dopamine (however, most binding in striatum is to dopamine nerve terminals)
Theoretically less subject to compensation and regulatory changes than are other tracers, might therefore give a good estimate of nerve terminal density
Dopamine receptors
Most studies are done with ligands for the dopamine D2/3 receptor
Can estimate dopamine release by use of ligands with comparatively weaker affinity for the D2 receptor (binding subject to competition from endogenous dopamine)
Higher affinity ligands used to assess extrastriatal D2 binding
Developments in magnetic resonance techniques (panel 2) have enabled the detection of changes in the substantia nigra and brainstem of patients with Parkinson’s disease, including abnormal shape at ultra-high field, increased iron load, altered diffusion properties, and abnormal anatomical and functional connectivity (figure 2). Ultra-high field 7 Tesla MRI enables better visualisation of basal ganglia contours and shapes because of increased spatial resolution and improved contrast when techniques sensitive to iron deposition are used. At 7 Tesla, changes in the shape and signal intensity of the substantia nigra in T2*-weighted images were reported to accurately differentiate patients with Parkinson’s disease from controls without Parkinson’s disease.2 Results of several studies in Parkinson’s disease suggest increased iron content and neuronal loss in the substantia nigra that are maximum in its lateral segments, and in some studies correlated with the Unified Parkinson’s Disease Rating Scale motor score.3 A combination of measures of iron content with changes in fractional anisotropy in the substantia nigra resulted in better separation of patients with Parkinson’s disease from control individuals compared with each technique separately, with 95% global accuracy.4 Reduced connectivity of the substantia nigra with the striatum and thalamus was also evidenced in patients with Parkinson’s disease using tractography-based methods5 and resting-state functional connectivity (figure 2). Novel MRI techniques might be helpful for differentiation between Parkinson’s disease and atypical parkinsonism (table 1). However, although such approaches have shown sensitivity and specificity in distinguishing Parkinson’s disease from atypical parkinsonism, their use in clinical practice is limited by the scarcity of normative databases in clinical centres, and the significance of the imaging changes is poorly understood. Many automated methods used to establish concentration, density, or volume of brain tissue depend on T1 relaxation time, and could consequently be affected by any tissue property that affects this parameter, even in the absence of volumetric changes. Validation of new quantitative MRI techniques using cellular and molecular approaches in patients and animal models will be crucial to a better understanding of basal ganglia function and disease.
Panel 2. MRI techniques.
Structural imaging
Provides information about brain morphology—eg, volume, shape, surface, thickness of grey matter
Conventional techniques (T1-weighted, T2-weighted, IR) allow segmentation of the putamen, caudate nucleus, globus pallidus, thalamus
New contrasts improve visualisation of the substantia nigra (magnetisation transfer [MT] and multiparameter mapping) and locus coeruleus (neuromelanin-sensitive imaging)
Magnetic resonance microscopic imaging with ultra-high field MRI (7T and above) provides increased spatial resolution and improved contrasts
Quantitative MRI techniques
-
Relaxometry:
Calculates relaxation times and rates, which are characteristic of tissue composition and depend on its molecular structure
T1 shows interactions between protons and their surroundings (the lattice) and depends mainly on the mobility of water within its microenvironment
T2/T2* are used as estimates of iron content
-
MT imaging:
Relies on the transfer of energy between highly bound protons and mobile protons of free water
Better visualisation of basal ganglia/brainstem nuclei by increase of contrast with surrounding white matter
Considered as a quantitative measure of myelin content (degree of myelination and axonal density)
Diffusion imaging
Diffusion-weighted imaging estimates water diffusion through the application of magnetic field gradient pulses
Diffusion tensor imaging necessitates the application of strong diffusion gradients in at least six directions
-
Metrics:
Mean diffusivity: displacement of molecules and presence of obstacles to diffusion
Fractional anisotropy, or generalised fractional anisotropy: orientation of diffusion in structures such as axons in fibre bundles, anisotropy relates to membranes, myelin, longitudinal filaments, and cytoskeleton
Axial/longitudinal diffusivity: diffusion along the main direction of diffusion attributed to axonal damage
Radial/transverse diffusivity: diffusion perpendicular to the main direction of diffusion thought to indicate myelin damage
Susceptibility-weighted imaging and quantitative susceptibility mapping rely on magnetic susceptibility differences in brain tissues
Uses both magnitude and phase images from a gradient echo MRI sequence
Is used to quantify iron concentration
Functional MRI
Relies on blood oxygenation level-dependent (BOLD) signal
Measures changes in relative amount of oxy/deoxyhaemoglobin associated with neuronal activity
Reflects local field potentials, which indicate perisynaptic events
Brain connectivity
Anatomical connectivity assessed using tractography, allowing reconstruction of fibre bundles in the brain
Metrics: diffusion metrics within the tracts, number of tracks, connection probability between regions
Functional connectivity
Assessed with fMRI to ascertain temporal correlations of low-frequency, spontaneous BOLD signal fluctuations between spatially remote regions
Can be measured at rest (resting state fMRI [rsfMRI]) or during task performance
Metrics: correlation coefficient, integration (quantifies how signals covary between regions belonging to a particular network), small-world network indices
Proton magnetic resonance spectroscopy
Provides information about the concentrations of metabolites in the brain
-
Methods
Single voxel spectroscopy
Chemical shift imaging: metabolite concentrations are obtained from slices
-
Main metabolites studied (significance)
N-acetylaspartate (neuronal number and health)
Choline-containing compounds (demyelination and cell proliferation)
Myo-inositol (osmotic stress or astrogliosis)
Lactate (frequently associated with the presence of pathology)
Neurotransmitters (≥3T): Glx=Glu/Gln, GABA: J-coupling methods
Perfusion imaging
Provides information about brain perfusion
Measured by use of arterial spin labelling techniques
Provides information on regional cerebral blood flow
IR=inversion recovery. MTR=magnetisation transfer ratio. T1=T1 relaxation time. T2=T2 relaxation time, T2*=gradient echo T2 relaxation time. R1=longitudinal relaxation rate. R2=transverse relaxation rate. R2*=T2* relaxation rate. GABA= γ-aminobutyric acid.
Figure 2. Main MRI methods used to study Parkinson’s disease.

In the cortex, changes in cortical thickness (CTh) and grey matter volume or density (VBM) were reported (green boxes). Functional MRI (fMRI) was used to assess changes in brain activation levels during task performance (dark blue box). Changes in structural connectivity were evidenced using diffusion-based tractography and in functional connectivity at rest with resting state fMRI, which assesses correlation (r) between signal fluctuations in distant brain regions (light blue box). In the substantia nigra (red shape), increased iron load was assessed with T2* mapping (T2*) and more recently with quantitative susceptibility mapping (QSM), and microstructural changes with fractional anisotropy, which is decreased (red box). Neuromelanin imaging was used to study the locus coeruleus area (white arrow, yellow box).
rsfMRI=resting state fMRI. NM=neuromelanin imaging. FA=fractional anisotropy. T1=T1-weighted. T2*=gradient echo T2 mapping.
Table 1.
Distinguishing features of Parkinson’s disease versus progressive supranuclear palsy and multiple systems atrophy
| Parkinson’s disease | Progressive supranuclear palsy | Multiple systems atrophy | |
|---|---|---|---|
| Diffusivity | Variable changes reported in striatum and thalamus, most studies report no difference from controls | Increased in midbrain, basal ganglia, thalamus, cortex | Increased in striatum and middle cerebellar peduncles |
| Changes seen on routine clinical imaging sequences | ·· | Midbrain atrophy: “hummingbird” or “penguin” sign; atrophy of superior cerebellar peduncle | Hypointense putamen with hyperintense rim; loss of pontocerebellar fibres (“hot-cross bun” sign) |
| Other special sequences | Magnetisation transfer ratio and fractional anisotropy decreased, iron load increased in substantia nigra | Magnetisation transfer ratio decreased in striatum, globus pallidus, substantia nigra, and white matter | Magnetisation transfer ratio decreased in putamen, globus pallidus, and substantia nigra; iron sensitive sequences (relaxometry, phase-contrast susceptibility) show increased deposition in putamen |
| PET or SPECT: presynaptic dopamine tracers | All decreased, affecting caudal more than rostral striatum | All decreased, but rostral-caudal gradient might be lost | All decreased, rostral-caudal gradient is variable |
| PET or SPECT: striatal dopamine receptors | D2 binding increased in putamen in untreated patients, could normalise on therapy; might see slight reduction in caudate on prolonged dopaminergic therapy | D2 binding reduced | D2 binding reduced |
| PET or SPECT: glucose metabolism or cerebral blood flow | Increased in basal ganglia and cerebellum, decreased in premotor and posterior parietal cortex | Reduced in brainstem and medial frontal cortex | Reduced in putamen and cerebellum |
SPECT=single photon emission CT.
Imaging to detect preclinical disease and as a marker of disease progression
Imaging is promising for early disease detection and as an independent measure of disease progression, less prone to being affected by factors such as subjectivity, medication, and placebo effects that could confound purely clinical measures. Imaging measures of dopamine function represent the most robust biomarker to assess disease progression, and might provide very helpful secondary outcome measures. Caution is necessary in interpretation of the results, since imaging markers could be altered by compensatory mechanisms or in response to treatment, and it is often difficult to show a tight relationship between change over time in the marker and change in clinical measures of disease severity. Imaging markers indicate activity in dopamine nerve terminals rather than cell counts, and not all of the disability in Parkinson’s disease arises from loss of dopamine. These caveats notwithstanding, functional imaging can give insights into disease progression during clinical and preclinical phases of disease. In a longitudinal study of patients with sporadic Parkinson’s disease, we noted four main outcomes: for all markers of presynaptic dopamine integrity, disease progression is best described by an exponential function that approaches a non-zero asymptote; side-to-side asymmetries, although prominent in early disease, tend to converge over time; by contrast, the rostral-caudal gradient of striatal dopaminergic dysfunction is maintained throughout the illness; and the decay function that describes the rate of decline is similar between striatal subregions.6 Thus, whatever causes the initiation of Parkinson’s disease (resulting in greater involvement of the caudal striatum) is different from factors that contribute to disease progression (which result in similar rates of decline across striatal subregions). Accordingly, strategies to delay disease onset in individuals at risk might not necessarily be the same as those appropriate when disease has become clinically manifest. Expression of the metabolic network characteristic of Parkinson’s disease increases with disease progression, and this might be an alternate or, perhaps more appropriately, a complementary approach to assess disease progression and the effects of potential disease-modifying interventions.1
PET can be used to show subclinical dopamine deficits in clinically unaffected twins of patients with Parkinson’s disease, in patients exposed to nigral toxins, and in patients with a family history of dominantly inherited Parkinson’s disease (see appendix for references; S1–4). Subclinical abnormalities might also be noted in individuals carrying a single mutation of a recessively inherited form of Parkinson’s disease (S5, S6) but the meaning is unclear. Either PET or SPECT can be used to detect abnormalities of dopamine function in other individuals at risk of Parkinson’s disease, such as those with hyposmia (S7) or rapid-eye-movement sleep behaviour disorder (S8).7 Diffusion changes, comprising decreased fractional anisotropy in the midbrain tegmentum and rostral pons, and increased mean diffusivity in the pontine reticular formation and midbrain, have also been reported in idiopathic rapid-eye-movement sleep behaviour disorder (S9). In Parkinson’s disease, MRI with neuromelanin contrast showed a relationship between damage to the locus coeruleus and subcoeruleus complex and abnormal muscle tone during rapid-eye-movement sleep.8 The study of populations at high risk might help improve understanding of the natural history of Parkinson’s disease in its earliest phases, and might ultimately allow introduction of neuroprotective therapies before clinical onset of disease.
Neuropathology and pathogenesis of Parkinson’s disease
Alterations in dopamine release and motor complications
Functional imaging can be used to estimate dopamine release. The binding of ligands with relatively weak affinity for dopamine receptors compete with endogenous dopamine. Thus when the same individual is scanned at two or more points relatively close in time, before and after an intervention, a change in binding is likely to indicate a change in endogenous dopamine—increased synaptic dopamine leads to decreased ligand binding. Dopamine release can be elicited in many ways, including administration of levodopa. Levodopa-induced dopamine release increases with increased severity9 and duration10 of Parkinson’s disease, and is of greater magnitude in patients with dyskinesias.10 A greater magnitude but shorter duration of dopamine release can be shown in patients who go on to develop motor fluctuations at a time when they still have a stable response to drug treatment.11 By contrast with dopamine release after levodopa, release after methamphetamine (which depends on intact DAT) is reduced in proportion to the degree of dopamine denervation (S10). Dopamine release can also be elicited by repetitive transcranial magnetic stimulation in an anatomically discrete fashion. In Parkinson’s disease, the magnitude of striatal dopamine release after repetitive transcranial magnetic stimulation is reduced, but distributed over a greater volume, in keeping with impaired functional segregation of corticostriatal projections.12 With this technique, dopamine release can also be shown in response to expectation, contributing to placebo effects in both Parkinson’s disease13,14 and analgesia,15 as well as to various behavioural stimuli.
Altered connectivity
Researchers in tractography-based studies using diffusion tensor imaging have reported strong analogy between human cortico-striatal connections and anatomical data from tracing studies in non-human primates (S11),16 with partly overlapping, segregated loops in sensorimotor, associative, and limbic networks. Within the striatum, these territories roughly correspond to the posterior putamen (sensorimotor), anterior putamen and caudate nucleus (associative), and ventral striatum (limbic) (figure 3). The pattern of anatomical connection was also paralleled by patterns of functional connectivity in human beings, shown with functional imaging during various behavioural tasks in which cortical regions broadly co-activated with corresponding parts of the striatum.18 Similarly, functional connectivity studies using resting-state fMRI delineated cortico-striatal networks broadly consistent with the predictions of the parallel loop model.19
Figure 3. Three-dimensional representations of the sensorimotor (green), associative (pink/purple) and limbic (yellow) territories of the basal ganglia superimposed on a multiplanar T1-weighted image.
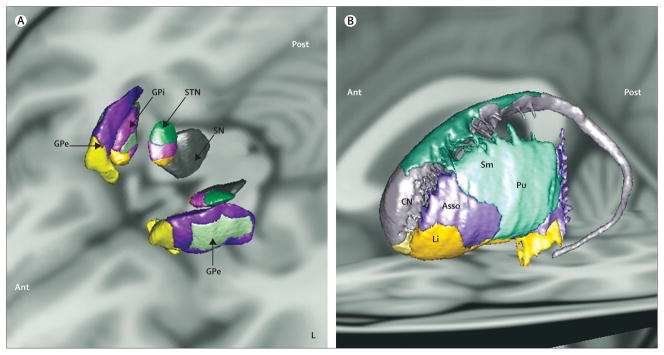
Oblique superior view of the internal and external segments of the globus pallidus, subthalamic nucleus, and substantia nigra (A). Oblique lateral external view of the territories of the striatum (B). The territories are represented using postmortem atlas coregistered to the T1-weighted image of the brain of a control patient as described in Bardinet and colleagues.17 Territories in the substantia nigra are not represented (grey). GPi=internal part of the globus pallidus. STN=subthalamic nucleus. SN=substantia nigra. GPe=external part of the globus pallidus. CN=caudate nucleus. Asso=associative. Sm=sensorimotor. Pu=putamen. Li=limbic. Ant=anterior. L=left. Post=posterior. R=right. Courtesy of Eric Bardinet, Plateforme Stim, Centre de Neuro-Imagerie de Recherche, l’Institut du Cerveau et de la Moelle épinière, Paris, France.
Functional connectivity methods have shown that the interactions of brain networks are abnormal in Parkinson’s disease in the resting state.19–21 Patients with Parkinson’s disease showed decreased coupling in the cortico-striatal sensorimotor network19 and between the striatum and the brainstem,21 and increased coupling (probably compensatory) in the associative network.19 Changes in functional connectivity varied in relation to symptoms22 and were modulated by levodopa.20 In patients with tremor, increased functional connectivity was noted between the internal globus pallidus and putamen and the cerebellothalamic circuit,22 suggestive of increased oscillatory activity in these pathways. Overall, findings with resting-state fMRI suggest that dopamine depletion in Parkinson’s disease leads to a remapping of cerebral connectivity that predominantly affects the sensorimotor circuit and sensorimotor integration and that is differently associated with motor and non-motor symptoms.
Non-dopaminergic changes and non-motor complications in Parkinson’s disease
Whereas the predominant changes in Parkinson’s disease are dopaminergic, several other neurotransmitter systems might be affected, including other monoamines. 6-18F-fluoro-levodopa (FD) is taken up by noradrenergic and serotonergic as well as dopamine neurons. FD uptake in the (noradrenergic) locus coeruleus is increased in early Parkinson’s disease, possibly indicating compensatory upregulation, but decreases to subnormal levels as disease progresses (S12). 11C-RTI-32 binds to the norepinephrine transporter as well as the DAT. RTI-32 binding is decreased in many brain regions of patients with Parkinson’s disease who are depressed, anxious, or both, compared with patients with Parkinson’s disease who are neither depressed nor anxious, including the locus coeruleus and thalamus, where uptake is likely to predominantly show norepinephrine transporter binding.23 Reduced tracer uptake in the thalamus might also partly indicate loss of dopamine input. MRI scans have shown reduced signal intensity in the locus coeruleus compared with controls, which might show loss of neuromelanin-containing neurons.8
Serotonin transporter binding has been measured by 11C-3-amino-4-(2-dimethylaminomethyl-phenylsulfanyl) benzonitrile (11C-DASB). DASB binding is decreased in several regions of the brains of individuals with Parkinson’s disease compared with those of healthy controls, although the deficit does not seem to be related to disease progression.24 In patients with Parkinson’s disease treated with neural transplantation, graft-induced dyskinesias are associated with increased DASB binding;25 on the basis of studies in rodent models, investigators have also suggested that serotonin neurons might be a source of unregulated dopamine release contributing to levodopa-induced dyskinesias.26 Despite expectations to the contrary, depression in Parkinson’s disease is associated with increased DASB binding relative not only to people with Parkinson’s disease who are not depressed,27 but also, in one study, to healthy controls.28 The basis for this association is unclear. Although reduced occupancy due to lower concentrations of endogenous serotonin is a theoretical possibility, DASB binding is not known to be sensitive to changes in serotonin.
Cholinergic neurons are also affected in Parkinson’s disease. With use of PET with a substrate for acetyl-cholinesterase activity, widespread changes in forebrain cholinergic innervation can be detected, even in patients with Parkinson’s disease without complications, but these changes are more pronounced in Parkinson’s disease dementia (to a greater degree than in Alzheimer’s disease).29 Cholinergic dysfunction assessed with this technique is also associated with impaired postural and olfactory function, and rapid-eye-movement sleep behaviour disorder.30 PET has also been used to detect widespread reductions in nicotinic acetylcholine receptor binding in Parkinson’s disease, which could relate to depressive (cingulate and occipital cortex, putamen, and midbrain) and cognitive (midbrain, pons, and cerebellum) features.31
Far fewer studies have been done on other neuro-transmitters in Parkinson’s disease than on the monoamines and cholinergic systems. Dyskinesias are associated with reduced opioid32 and with increased adenosine A2A receptor binding.33 Cannabinoid CB1 receptor binding is increased in the striatum and mesolimbic or mesocortical dopamine projections areas, but reduced in the substantia nigra of patients with Parkinson’s disease, but no relation to the presence or absence of dyskinesias exists.34
Neuroinflammation
Microglial cells continuously do repair tasks in the CNS and are the first responders to various types of brain injury. In response to brain insults, these cells are transformed from a resting to an active state, and change their morphology from branched to globular with an increase in cell volume. Thus, activated microglia represent an important biomarker of neuroinflammation. Microglial activation is thought to have an important role in the pathogenesis of Parkinson’s disease (S13). Animal models have shown that microglial activation might precede neuronal degeneration (S14), and results of several post-mortem studies have shown widespread increases of microglial activation in Parkinson’s disease in the substantia nigra, putamen, hippocampus, cingulate, and temporal cortex (S15). These observations strongly suggest a potential role of activated microglia as a biomarker in Parkinson’s disease.
Microglial cells express the so-called translocator protein 18 kDA or TSPO in the outer mitochondrial membrane. Increased TSPO expression has been reported in multiple sclerosis, stroke, and Alzheimer’s disease (S16, S17). PET with 11C-PK11195 has been used as a marker of in-vivo microglial activation in inflammatory (S18), ischaemic (S19), and degenerative (S20) brain lesions. Results in Parkinson’s disease have been controversial. Whereas results of previous PET studies using 11C-PK11195 in Parkinson’s disease showed increased binding in the midbrain35 and different frontal and temporal cortical regions,36 other reports did not identify major differences between binding in Parkinson’s disease and controls.37 Conversely, more consistent increases in 11C-PK11195 binding have been reported in multiple systems atrophy,38 progressive supranuclear palsy,39 corticobasal degeneration,40 and Huntington’s disease.41 Reasons for such variability could include limitations inherent to this particular tracer, such as high non-specific binding, low brain-penetration, high plasma-protein-binding, and difficult preparation.38 These limitations have established the need for better microglial biomarkers which are being tested—eg, 11C-PBR28 (S21), 11C-DPA713 (S22), and 18F-FEPPA (S23). The newer tracers have an improved signal-to-noise ratio compared with 11C PK 11195, but might not be useable in all patients, owing to different affinity patterns, related to a polymorphism in TSPO.42 The biological effects of this polymorphism and related different binding affinity are unknown, but might have important implications for the pathogenesis and assessment of Parkinson’s disease.
Abnormal protein deposition
The assessment of Alzheimer’s disease has been revolutionised by the availability of PET ligands that bind specifically to β amyloid. Use of such agents to diagnose Alzheimer’s disease has been approved by the US Food and Drug Administration, and the technique could offer promise as a marker to establish the effects of putative disease-modifying therapies. The Parkinson’s specialty would similarly benefit from access to a drug that could image aberrant deposits of α synuclein. Finding such a drug has been a more challenging problem than in Alzheimer’s disease, exacerbated by the fact that α-synuclein is deposited in small amounts and is predominantly intracellular, thus, even if a selective drug that crosses the blood–brain barrier were available, the signal would, presumably, be low. Benzoxazole compounds that bind to β-amyloid might also bind to α synuclein and can be labelled with either 11C (S24) or 18F (S25), but are not selective and have not yet gained widespread acceptance for the study of Lewy body disorders. 123I-SIL23 is a phenothiazine that binds to synuclein fibrils in vitro (and in vivo in mouse models),43 but its affinity is relatively low, as is its specificity for synuclein compared with Aβ and tau. Several compounds with improved specificity for abnormally phosphorylated tau have been described.44,45 Varying imaging evidence exists for amyloid deposition associated with cognitive impairment in parkinsonism, noted to a greater extent in dementia with Lewy bodies than in Parkinson’s disease-dementia, also in part reflective of apolipoprotein E status.46
Imaging cognitive and behavioural complications in Parkinson’s disease
The parallel loop model described has provided a functional framework for understanding of the roles of the sensorimotor, associative (cognitive), and limbic (motivation, emotion) circuits of the basal ganglia in cognitive and behavioural complications in Parkinson’s disease. The posterior striatum is involved in the control of habitual movements such as well learned sequences (S26). The rostral associative striatum is involved in goal-directed behaviour (S27) such as learning novel motor sequences, instrumental conditioning, action-contingency, and executive functions (eg, planning and set-shifting [S28]). The ventral striatum is involved in reward-based and reinforcement learning (S29). According to this circuit model, the associative circuit might contribute to some aspects of cognitive deficits and the limbic circuit to behavioural deficits such as impulse control disorders.
Cognitive impairment and dementia are important aspects of Parkinson’s disease. Mild cognitive impairment has been described that often progresses to dementia.47 Parkinson’s disease has a heterogeneous cognitive profile and the topography of structural lesions varies between studies. A common framework for cognitive deficits in Parkinson’s disease was proposed in which fronto-subcortical executive deficits could be related to mild and stable dopaminergic deficits, whereas dementia could be associated with more widespread posterior cortical atrophy and with Lewy body deposition or plaques and cholinergic deficiency.48 Dementia in Parkinson’s disease is also associated with changes in hippocampus49 and frontal-temporal regions.50 Atrophy in these regions accelerates with disease progression, especially in patients who develop cognitive impairment.50 MRI anatomical correlates were also reported for olfactory dysfunction (grey matter atrophy in olfactory regions; S30), depression (grey and white matter in limbic regions; S31), and visual hallucinations (widespread limbic, paralimbic, and neocortical atrophy; S32). Non-motor symptoms are associated with distinct resting activity patterns in Parkinson’s disease. Apathy was predicted by signals in the left supplementary motor cortex, the right orbitofrontal cortex, and middle frontal cortex, whereas depression was predicted by signals in the right subgenual cingulate (S33).
Dopamine agonists used in the treatment of Parkinson’s disease have been implicated in the development of impulse control disorders (ICDs) including hypersexuality, compulsive shopping, compulsive eating and, most commonly, pathological gambling (S34–S36).51 Initial estimates of the prevalence of all ICDs in Parkinson’s disease were reported to range from 5·9–6·6%. The DOMINION study51 included about 3090 patients from 46 sites in North America and reported a 6 month prevalence of all ICDs in Parkinson’s disease of about 13·6%.51 Pathological gambling had a 6 month prevalence of 6·4%, by contrast with the estimated 1·6% prevalence of pathological gambling in the whole North American population. Compulsive shopping was the most prevalent of the ICDs in Parkinson’s disease, found to occur in 7·2% compared with 5·8% estimated in the North American population.
Proposed pathogenic mechanisms for the emergence of ICDs in Parkinson’s disease can be broadly attributed to three potentially interacting factors: premorbid susceptibility; the contribution of Parkinson’s disease itself, whether as a manifestation of a particular disease phenotype or genotype, or as a compensatory mechanism for the underlying disease process; the contribution of dopaminergic agents to the behaviour and their potential interaction with either of the previous factors. Dopaminergic medications could induce impulsive behaviours at several levels, including differential dopamine receptor stimulation preferentially affecting limbic circuits that regulate reward, reinforcing effects of continuous dopamine receptor stimulation conferring excessive salience or impairing its eventual withdrawal, and a more specific priming effect of dopamine agonists inducing excess release of dopamine in ventral striatal circuits that could increase salience and thus trigger craving for the relevant stimulus.52,53 Results of functional imaging studies comparing patients with Parkinson’s disease with and without ICDs have shown specific differences in frontostriatal function during reward tasks that mimic some of the underlying constructs of the pathological behaviour.53,54 From these studies, it seems that tonic stimulation of dopamine receptors via agonists could impair reward processing and inhibitory control mechanisms in ways that promote pathological repetition of behaviours. Although dopamine replacement treatment is titrated to compensate for the deficiencies in the motor basal ganglia network and to alleviate motor symptoms, this treatment could result in a dopamine overdosing effect in corticobasal ganglia pathways mediating cognitive and behavioural functions.55
There is also support for the proposal that ICDs might develop because of an interaction between increased stimulation of dopamine receptors and a pre-existing underlying vulnerability. Particular personality traits are associated with specific ICDs (both in populations with and without Parkinson’s disease) such as impulsivity and novelty-seeking (S37). Susceptibility to chemical and behavioural addictions is associated with decreased D2 receptor availability,56 and researchers have described dopaminergic correlates of impulsive personality trait even in healthy individuals without addiction.57 Using PET with 18F-fallypride, the authors found that amphetamine-induced dopamine release was greater in those with high impulsivity. Both amphetamine-induced striatal dopamine release and impulsivity were negatively correlated with binding in the midbrain, leading the authors to propose that striatal dopamine release in response to reward is governed by midbrain autoreceptor function. Individuals with low midbrain (auto) receptor density have increased dopaminergic responses to reward and are more impulsive.
Steeves and colleagues58 used 11C-raclopride PET to compare D2 receptor availability during a gambling task in patients with Parkinson’s disease with and without pathological gambling. Patients with Parkinson’s disease with pathological gambling had lower D2 binding at baseline, but increased release of dopamine in the ventral striatum during the gambling task (figure 4), as is seen in those with chemical addictions in response to the drug to which they are addicted.56 Results of a subsequent study by O’Sullivan and colleagues54 extended this observation by showing increased release of dopamine in the ventral striatum during reward cues, implying that patients with Parkinson’s disease who develop an ICD could represent a susceptible group of individuals whose ICD has been unmasked by agonists.
Figure 4. Striatal dopamine.

The top figures show the significant areas of striatal dopamine release during gambling compared with control task in patients with Parkinson’s disease (PD) with pathological gambling (PG; A) and without pathological gambling (B) in the ventral striatum (lower figure) and dorsal striatum (upper figure).57 Dopamine receptor binding is higher in the left orbitofrontal and anterior cingular cortex during a reward-based task (C) and higher in anterior cingular cortex of patients with PD with PG compared with patients with PD without PG (D).59
Prefrontal mechanisms also have an important role in prevention of ICDs. This idea is supported by H2O PET studies60 in which patients with Parkinson’s disease with and without pathological gambling were scanned before and after administration of dopamine agonists to measure changes in regional cerebral blood flow (rCBF) during a card selection game with feedback. In patients without pathological gambling, dopamine agonists increased activity in medial prefrontal areas associated with impulse regulation. The gamblers, by contrast, showed a significant agonist-induced reduction in rCBF. Results of a subsequent PET study using the extrastriatal dopamine receptor ligand 11C-FLB-45759 showed significant abnormalities in D2 receptor binding in the orbitofrontal cortex and anterior cingulate cortex in patients with Parkinson’s disease with pathological gambling compared with control patients with Parkinson’s disease, again supporting the key role of prefrontal control in the development of ICDs (figure 4).
Dystonia
Neuroimaging has provided strong evidence that the basal ganglia and related thalamocortical network are major determinants in the pathophysiology of primary dystonia. In secondary dystonia resulting from stroke or other brain damage, lesions were most frequently recorded in the basal ganglia (S38) and thalamus (S39), although were occasionally noted in other regions including parietal cortex, cerebellum (S40), brainstem (S41), and upper spinal cord (S42). In primary sporadic dystonias in which conventional imaging was negative, voxel-based (eg, voxel-based morphometry) and diffusion imaging techniques have shown structural abnormalities in similar regions of brain.61 Using 18F-FDG PET,61 increased glucose metabolism was reported in primary sporadic dystonias,62 and abnormally increased metabolism in a network including the lenticular nucleus, cerebellum, and supplementary motor area in sporadic and genetic DYT1 dystonia.62–64 Functional changes were also recorded during task performance in sensorimotor-related areas.65 Tasks that induced dystonia were frequently associated with increased activation in motor regions and associated areas, whereas tasks that did not induce dystonia, probably showing primary changes in the brain or longstanding consequences of dystonia, were associated with more variable activation changes. Abnormalities in movement imagination and in sensory representation in dystonias have also been reported,66 supporting the view that afferent inputs are inadequately processed at several levels of the CNS, as shown with electrophysiological techniques.67 Signal changes on MRI might be recorded with extensive practice and overtraining—a finding that might be highly relevant to dystonia, in which overuse of the affected body part is a common finding. Potential explanations for such signal changes at the cellular and molecular level include differences in number and morphology of neurons and synapses, and increases in size and number of glial cells. Likewise, white matter diffusion metrics might indicate the number, diameter, branching, trajectories, and myelination of axons, and fibre-packing density.68 Primary cellular changes—such as altered protein folding, abnormal cell trafficking, and altered cytoskeletal and vesicle dynamics that have been described in DYT1 animal models69—could also contribute to differences in water diffusion or relaxation parameters. Results of combined histological and MRI animal studies have suggested that remodelling might better indicate plasticity-related changes than does neurogenesis.70
Several models of basal ganglia dysfunction have been proposed to explain dystonia. These models postulate that dystonia results from a failure of the basal ganglia filtering that enables voluntary movements and suppresses competing ones that could interfere with the selected movement,71 or an imbalance between the direct excitatory and indirect inhibitory output pathways of the basal ganglia.72,73 Although many factors might contribute to altered striatal processing in dystonia, changes in dopamine function might be important.74,75 By contrast with other forms of dystonia in which D2 receptor binding might be decreased, increases are seen in patients with dopa-responsive dystonia and in clinically unaffected mutation carriers,76 in keeping with underlying impairment of dopamine synthesis. F-dopa uptake, which largely indicates the activity of L-aromatic aminoacid decarboxylase, is normal in dopa-responsive dystonia, in accordance with the upstream origin of the deficit in dopamine synthesis.77 Another commonly reported finding in dystonia is the loss of inhibitory processes observed at various levels of the nervous system by use of electrophysiological techniques, such as transcranial magnetic stimulation. Whereas measurements of baseline γ-aminobutyric acid using magnetic resonance spectroscopy were most often normal in patients with writer’s cramp,78 abnormal dynamic transcranial magnetic stimulation-induced changes in γ-aminobutyric acid were noted in these patients.78 Last, abnormal plasticity and learning processes are thought to be important neurophysiological findings in dystonia (S43).67
More recent views have proposed that dystonia involves not only the basal ganglia but also other brain regions and related networks.61 Cortical regions involved include the sensorimotor cortex, the parietal lobe79 (which plays a crucial part in the generation of mental images [S44]), and the premotor cortex during imagination of movement.80 The cerebello-thalamo-cortical network is another potential candidate.61,81 Studies in patients with clinically manifesting and non-manifesting DYT1 and DYT6 dystonia using diffusion-based tractography82 showed reduced connectivity of the cerebellum with the thalamus, suggesting that disruption of cerebellar outflow could be an important factor affecting the occurrence of motor symptoms. A very similar pattern of changes in the cerebellar pathway was reported in DYT1 mutant mice with the torsin1A gene mutation,83 although the relevance of this model to dystonia is debated. Whether these findings are specific to DYT1 and DYT6 dystonia or are also present in sporadic cases is unknown. Several other animal models have also implicated cerebellar dysfunction in dystonia, including the tottering mutant mouse,84 and animals modified by conditional genetics (S45). Conversely, most cerebellar lesions in human beings result in ataxia, and therefore the role of the cerebellum in dystonia is uncertain. Dystonia might result from cerebellar dysfunction and not destruction, or from abnormal interactions of cerebellar and basal ganglia networks. These networks might interact anatomically at the level of the motor cortex or the striatum, as evidenced by histological tract tracing in animals,85 animal models of dystonia,84 and functionally in healthy human volunteers, in whom cortical excitability can be modulated after cerebellar interventions86–88 or after cerebellar degeneration or infarction (S46). The cerebellum might contribute to the deficit in sensorimotor integration recorded in dystonia89 because it processes proprioceptive information, alters somatosensory thresholds in the cortex, and has a key role in both temporal and spatial discrimination (S47).
Conclusions and future directions
In the past few years, enormous advances in imaging capability have occurred. Advances in magnetic resonance technology have resulted in improved structural imaging, with the capacity to study diffusion properties, anatomical connectivity, and iron deposition, such that magnetic resonance could now provide a valid approach with which to differentiate between Parkinson’s disease and atypical so-called Parkinson-plus disorders. Furthermore, magnetic resonance is rapidly becoming the preferred approach by which to study functional connectivity, either during tasks selected to activate select circuitry, or by study of patterns of connectivity at rest. Although many such studies are in their infancy and their interpretation might still be a matter of some conjecture, huge strides can be anticipated in magnetic resonance in the next decade. Radioisotope imaging with PET or SPECT is the preferred approach to study neurochemical changes and responses, and could increasingly be used to provide insights into disease mechanisms. Both techniques show promise for bio-marker studies, but many more questions must be resolved before they can be considered as surrogate markers for disease severity. Magnetic resonance and radioisotope approaches provide complementary information. Thus, the emerging use of PET-magnetic resonance technology will enable investigators to study changes in connectivity and neurochemical responses concurrently in the same individual. This multimodal approach, perhaps combined with electroencephalography to study cerebral oscillations concurrently, or with the improved temporal discrimination of magnetoencephalography, is likely to provide much greater insights into brain function. Improved understanding of abnormal oscillations could lead to further appreciation for the role of the cerebellum in Parkinson’s disease and its complications,90 as is increasingly the case for dystonia. Although the diagnosis of Parkinson’s disease is predominantly clinical, imaging offers great promise for study of disease pathogenesis and evolution, as well as factors that contribute to complications of disease and its treatment, and brain function in health.
Supplementary Material
Search strategy and selection criteria.
We searched PubMed with use of various combinations of the terms “Parkinson”, “dystonia”, “basal ganglia”, “dopamine”, “addiction”, “gambling”, “MRI”, “PET”, and “SPECT”. When possible, we gave priority to papers published in the past 5 years, and to review articles for older literature when appropriate, and to papers deemed most relevant by the authors. Our last search was done in October, 2013. We apologise to the many authors whose work was not cited due to space restrictions. Some additional references are provided as a supplemental list in the appendix.
Acknowledgments
AJS’s work was supported by the Canadian Institutes of Health Research, the Michael J Fox Foundation, the Pacific Alzheimer Research Foundation, and the Canada Research Chairs programme, none of whom had any direct involvement in the preparation of or decision to submit this Series paper. SL’s work was supported by the “Investissements d’avenir” (grant number ANR-10-IAIHU-06). APS’s work was supported by the Canadian Institutes of Health Research (MOP 110962) and Canada Research Chairs programme.
Footnotes
Contributors
AJS, SL, and APS wrote the Series paper, and did the literature search and interpretation. AJS prepared the final paper.
Declaration of interests
We declare no competing interests.
References
- 1.Niethammer M, Eidelberg D. Metabolic brain networks in translational neurology: concepts and applications. Ann Neurol. 2012;72:635–47. doi: 10.1002/ana.23631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwon DH, Kim JM, Oh SH, et al. Seven-Tesla magnetic resonance images of the substantia nigra in Parkinson disease. Ann Neurol. 2012;71:267–77. doi: 10.1002/ana.22592. [DOI] [PubMed] [Google Scholar]
- 3.Martin WR, Wieler M, Gee M. Midbrain iron content in early Parkinson disease: a potential biomarker of disease status. Neurology. 2008;70:1411–17. doi: 10.1212/01.wnl.0000286384.31050.b5. [DOI] [PubMed] [Google Scholar]
- 4.Péran P, Cherubini A, Assogna F, et al. Magnetic resonance imaging markers of Parkinson’s disease nigrostriatal signature. Brain. 2010;133:3423–33. doi: 10.1093/brain/awq212. [DOI] [PubMed] [Google Scholar]
- 5.Menke RA, Scholz J, Miller KL, et al. MRI characteristics of the substantia nigra in Parkinson’s disease: a combined quantitative T1 and DTI study. Neuroimage. 2009;47:435–41. doi: 10.1016/j.neuroimage.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 6.Nandhagopal R, Kuramoto L, Schulzer M, et al. Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain. 2009;132:2970–79. doi: 10.1093/brain/awp209. [DOI] [PubMed] [Google Scholar]
- 7.Iranzo A, Valldeoriola F, Lomeña F, et al. Serial dopamine transporter imaging of nigrostriatal function in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study. Lancet Neurol. 2011;10:797–805. doi: 10.1016/S1474-4422(11)70152-1. [DOI] [PubMed] [Google Scholar]
- 8.García-Lorenzo D, Longo-Dos Santos C, Ewenczyk C, et al. The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease. Brain. 2013;136:2120–29. doi: 10.1093/brain/awt152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tedroff J, Pedersen M, Aquilonius S-M, Hartvig P, Jacobsson G, Långström B. Levodopa-induced changes in synaptic dopamine in patients with Parkinson’s disease as measured by [11C]raclopride displacement and PET. Neurology. 1996;46:1430–36. doi: 10.1212/wnl.46.5.1430. [DOI] [PubMed] [Google Scholar]
- 10.de la Fuente-Fernández R, Sossi V, Huang Z, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747–54. doi: 10.1093/brain/awh290. [DOI] [PubMed] [Google Scholar]
- 11.de la Fuente-Fernández R, Lu JQ, Sossi V, et al. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann Neurol. 2001;49:298–303. doi: 10.1002/ana.65.abs. [DOI] [PubMed] [Google Scholar]
- 12.Strafella AP, Ko JH, Grant J, Fraraccio M, Monchi O. Corticostriatal functional interactions in Parkinson’s disease: a rTMS/[11C]raclopride PET study. Eur J Neurosci. 2005;22:2946–52. doi: 10.1111/j.1460-9568.2005.04476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de la Fuente-Fernández R, Ruth TJ, Sossi V, Schulzer M, Calne DB, Stoessl AJ. Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science. 2001;293:1164–66. doi: 10.1126/science.1060937. [DOI] [PubMed] [Google Scholar]
- 14.Lidstone SC, Schulzer M, Dinelle K, et al. Effects of expectation on placebo-induced dopamine release in Parkinson disease. Arch Gen Psychiatry. 2010;67:857–65. doi: 10.1001/archgenpsychiatry.2010.88. [DOI] [PubMed] [Google Scholar]
- 15.Scott DJ, Stohler CS, Egnatuk CM, Wang H, Koeppe RA, Zubieta JK. Individual differences in reward responding explain placebo-induced expectations and effects. Neuron. 2007;55:325–36. doi: 10.1016/j.neuron.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 16.Lehéricy S, Ducros M, Van de Moortele PF, et al. Diffusion tensor fiber tracking shows distinct corticostriatal circuits in humans. Ann Neurol. 2004;55:522–29. doi: 10.1002/ana.20030. [DOI] [PubMed] [Google Scholar]
- 17.Bardinet E, Bhattacharjee M, Dormont D, et al. A three-dimensional histological atlas of the human basal ganglia. II. Atlas deformation strategy and evaluation in deep brain stimulation for Parkinson disease. J Neurosurg. 2009;110:208–19. doi: 10.3171/2008.3.17469. [DOI] [PubMed] [Google Scholar]
- 18.Postuma RB, Dagher A. Basal ganglia functional connectivity based on a meta-analysis of 126 positron emission tomography and functional magnetic resonance imaging publications. Cereb Cortex. 2006;16:1508–21. doi: 10.1093/cercor/bhj088. [DOI] [PubMed] [Google Scholar]
- 19.Helmich RC, Derikx LC, Bakker M, Scheeringa R, Bloem BR, Toni I. Spatial remapping of cortico-striatal connectivity in Parkinson’s disease. Cereb Cortex. 2010;20:1175–86. doi: 10.1093/cercor/bhp178. [DOI] [PubMed] [Google Scholar]
- 20.Esposito F, Tessitore A, Giordano A, et al. Rhythm-specific modulation of the sensorimotor network in drug-naive patients with Parkinson’s disease by levodopa. Brain. 2013;136:710–25. doi: 10.1093/brain/awt007. [DOI] [PubMed] [Google Scholar]
- 21.Hacker CD, Perlmutter JS, Criswell SR, Ances BM, Snyder AZ. Resting state functional connectivity of the striatum in Parkinson’s disease. Brain. 2012;135:3699–711. doi: 10.1093/brain/aws281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmich RC, Janssen MJ, Oyen WJ, Bloem BR, Toni I. Pallidal dysfunction drives a cerebellothalamic circuit into Parkinson tremor. Ann Neurol. 2011;69:269–81. doi: 10.1002/ana.22361. [DOI] [PubMed] [Google Scholar]
- 23.Remy P, Doder M, Lees A, Turjanski N, Brooks D. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain. 2005;128:1314–22. doi: 10.1093/brain/awh445. [DOI] [PubMed] [Google Scholar]
- 24.Politis M, Wu K, Loane C, et al. Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiol Dis. 2010;40:216–21. doi: 10.1016/j.nbd.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 25.Politis M, Wu K, Loane C, et al. Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci Transl Med. 2010;2:38ra46. doi: 10.1126/scitranslmed.3000976. [DOI] [PubMed] [Google Scholar]
- 26.Carta M, Carlsson T, Kirik D, Björklund A. Dopamine released from 3-HT terminals is the cause of L-dopa-induced dyskinesia in parkinsonian rats. Brain. 2007;130:1819–33. doi: 10.1093/brain/awm082. [DOI] [PubMed] [Google Scholar]
- 27.Politis M, Wu K, Loane C, et al. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology. 2010;75:1920–27. doi: 10.1212/WNL.0b013e3181feb2ab. [DOI] [PubMed] [Google Scholar]
- 28.Boileau I, Warsh JJ, Guttman M, et al. Elevated serotonin transporter binding in depressed patients with Parkinson’s disease: a preliminary PET study with 11C-DASB. Mov Disord. 2008;23:1776–80. doi: 10.1002/mds.22212. [DOI] [PubMed] [Google Scholar]
- 29.Bohnen NI, Kaufer DI, Ivanco LS, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. 2003;60:1745–48. doi: 10.1001/archneur.60.12.1745. [DOI] [PubMed] [Google Scholar]
- 30.Müller ML, Bohnen NI. Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep. 2013;13:377. doi: 10.1007/s11910-013-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer PM, Strecker K, Kendziorra K, et al. Reduced alpha4beta2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Arch Gen Psychiatry. 2009;66:866–77. doi: 10.1001/archgenpsychiatry.2009.106. [DOI] [PubMed] [Google Scholar]
- 32.Piccini P, Weeks RA, Brooks DJ. Alterations in opioid receptor binding in Parkinson’s disease patients with levodopa-induced dyskinesias. Ann Neurol. 1997;42:720–26. doi: 10.1002/ana.410420508. [DOI] [PubMed] [Google Scholar]
- 33.Ramlackhansingh AF, Bose SK, Ahmed I, Turkheimer FE, Pavese N, Brooks DJ. Adenosine 2A receptor availability in dyskinetic and nondyskinetic patients with Parkinson disease. Neurology. 2011;76:1811–16. doi: 10.1212/WNL.0b013e31821ccce4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van LK, Casteels C, Lunskens S, et al. Regional changes in type 1 cannabinoid receptor availability in Parkinson’s disease in vivo. Neurobiol Aging. 2012;33:620. doi: 10.1016/j.neurobiolaging.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 35.Ouchi Y, Yoshikawa E, Sekine Y, et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–75. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 36.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with 11C(R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–12. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL. 11C-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord. 2010;16:57–59. doi: 10.1016/j.parkreldis.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Gerhard A, Banati RB, Goerres GB, et al. 11C(R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. 2003;61:686–89. doi: 10.1212/01.wnl.0000078192.95645.e6. [DOI] [PubMed] [Google Scholar]
- 39.Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with 11C(R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
- 40.Gerhard A, Watts J, Trender-Gerhard I, et al. In vivo imaging of microglial activation with 11C(R)-PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19:1221–26. doi: 10.1002/mds.20162. [DOI] [PubMed] [Google Scholar]
- 41.Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–43. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 42.Owen DR, Yeo AJ, Gunn RN, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5. doi: 10.1038/jcbfm.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bagchi DP, Yu L, Perlmutter JS, et al. Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One. 2013;8:e55031. doi: 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–68. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 45.Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donaghy P, Thomas AJ, O’Brien JT. Amyloid PET imaging in Lewy body disorders. Am J Geriatr Psychiatry. 2013 doi: 10.1016/j.jagp.2013.03.001. published online July 3. http://dx.doi.org/10.1016/j.jagp.2013.03.001. [DOI] [PubMed]
- 47.Litvan I, Aarsland D, Adler CH, et al. MDS Task Force on mild cognitive impairment in Parkinson’s disease: critical review of PD-MCI. Mov Disord. 2011;26:1814–24. doi: 10.1002/mds.23823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ibarretxe-Bilbao N, Junque C, Marti MJ, Tolosa E. Brain structural MRI correlates of cognitive dysfunctions in Parkinson’s disease. J Neurol Sci. 2011;310:70–74. doi: 10.1016/j.jns.2011.07.054. [DOI] [PubMed] [Google Scholar]
- 49.Weintraub D, Dietz N, Duda JE, et al. Alzheimer’s disease pattern of brain atrophy predicts cognitive decline in Parkinson’s disease. Brain. 2012;135:170–80. doi: 10.1093/brain/awr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ibarretxe-Bilbao N, Junque C, Segura B, et al. Progression of cortical thinning in early Parkinson’s disease. Mov Disord. 2012;27:1746–53. doi: 10.1002/mds.25240. [DOI] [PubMed] [Google Scholar]
- 51.Weintraub D, Koester J, Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol. 2010;67:589–95. doi: 10.1001/archneurol.2010.65. [DOI] [PubMed] [Google Scholar]
- 52.Ray N, Strafella AP. Dopamine, reward, and frontostriatal circuitry in impulse control disorders in Parkinson’s disease: insights from functional imaging. Clin EEG Neurosci. 2010;41:87–93. doi: 10.1177/155005941004100208. [DOI] [PubMed] [Google Scholar]
- 53.van Eimeren T, Ballanger B, Pellecchia G, Miyasaki JM, Lang AE, Strafella AP. Dopamine agonists diminish value sensitivity of the orbitofrontal cortex: a trigger for pathological gambling in Parkinson’s disease? Neuropsychopharmacology. 2009;34:2758–66. doi: 10.1038/sj.npp.npp2009124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Sullivan SS, Wu K, Politis M, et al. Cue-induced striatal dopamine release in Parkinson’s disease-associated impulsive-compulsive behaviours. Brain. 2011;134:969–78. doi: 10.1093/brain/awr003. [DOI] [PubMed] [Google Scholar]
- 55.Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev. 2006;30:1–23. doi: 10.1016/j.neubiorev.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 56.Wang GJ, Volkow ND, Thanos PK, Fowler JS. Similarity between obesity and drug addiction as assessed by neurofunctional imaging: a concept review. J Addict Dis. 2004;23:39–53. doi: 10.1300/J069v23n03_04. [DOI] [PubMed] [Google Scholar]
- 57.Buckholtz JW, Treadway MT, Cowan RL, et al. Dopaminergic network differences in human impulsivity. Science. 2010;329:532. doi: 10.1126/science.1185778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steeves TD, Miyasaki J, Zurowski M, et al. Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C] raclopride PET study. Brain. 2009;132:1376–85. doi: 10.1093/brain/awp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ray NJ, Miyasaki JM, Zurowski M, et al. Extrastriatal dopaminergic abnormalities of DA homeostasis in Parkinson’s patients with medication-induced pathological gambling: a 11C FLB-457 and PET study. Neurobiol Dis. 2012;48:519–25. doi: 10.1016/j.nbd.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Eimeren T, Pellecchia G, Cilia R, et al. Drug-induced deactivation of inhibitory networks predicts pathological gambling in PD. Neurology. 2010;75:1711–16. doi: 10.1212/WNL.0b013e3181fc27fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neychev VK, Gross RE, Lehéricy S, Hess EJ, Jinnah HA. The functional neuroanatomy of dystonia. Neurobiol Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eidelberg D, Moeller JR, Ishikawa T, et al. The metabolic topography of idiopathic torsion dystonia. Brain. 1995;118:1473–84. doi: 10.1093/brain/118.6.1473. [DOI] [PubMed] [Google Scholar]
- 63.Eidelberg D, Moeller JR, Antonini A, et al. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44:303–12. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- 64.Trost M, Carbon M, Edwards C, et al. Primary dystonia: is abnormal functional brain architecture linked to genotype? Ann Neurol. 2002;52:853–56. doi: 10.1002/ana.10418. [DOI] [PubMed] [Google Scholar]
- 65.Zoons E, Booij J, Nederveen AJ, Dijk JM, Tijssen MA. Structural, functional and molecular imaging of the brain in primary focal dystonia—a review. Neuroimage. 2011;56:1011–20. doi: 10.1016/j.neuroimage.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 66.Nelson AJ, Blake DT, Chen R. Digit-specific aberrations in the primary somatosensory cortex in Writer’s cramp. Ann Neurol. 2009;66:146–54. doi: 10.1002/ana.21626. [DOI] [PubMed] [Google Scholar]
- 67.Quartarone A, Hallett M. Emerging concepts in the physiological basis of dystonia. Mov Disord. 2013;28:958–67. doi: 10.1002/mds.25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zatorre RJ, Fields RD, Johansen-Berg H. Plasticity in gray and white: neuroimaging changes in brain structure during learning. Nat Neurosci. 2012;15:528–36. doi: 10.1038/nn.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bragg DC, Armata IA, Nery FC, Breakefield XO, Sharma N. Molecular pathways in dystonia. Neurobiol Dis. 2011;42:136–47. doi: 10.1016/j.nbd.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lerch JP, Yiu AP, Martinez-Canabal A, et al. Maze training in mice induces MRI-detectable brain shape changes specific to the type of learning. Neuroimage. 2011;54:2086–95. doi: 10.1016/j.neuroimage.2010.09.086. [DOI] [PubMed] [Google Scholar]
- 71.Mink JW. The basal ganglia and involuntary movements: impaired inhibition of competing motor patterns. Arch Neurol. 2003;60:1365–68. doi: 10.1001/archneur.60.10.1365. [DOI] [PubMed] [Google Scholar]
- 72.DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64:20–24. doi: 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- 73.Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends Neurosci. 2012;35:557–64. doi: 10.1016/j.tins.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perlmutter JS, Stambuk MK, Markham J, et al. Decreased [18F] spiperone binding in putamen in idiopathic focal dystonia. J Neurosci. 1997;17:843–50. doi: 10.1523/JNEUROSCI.17-02-00843.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carbon M, Niethammer M, Peng S, et al. Abnormal striatal and thalamic dopamine neurotransmission: genotype-related features of dystonia. Neurology. 2009;72:2097–103. doi: 10.1212/WNL.0b013e3181aa538f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kishore A, Nygaard TG, de la Fuente-Fernandez R, et al. Striatal D2 receptors in symptomatic and asymptomatic carriers of dopa-responsive dystonia measured with [11C]-raclopride and positron-emission tomography. Neurology. 1998;50:1028–32. doi: 10.1212/wnl.50.4.1028. [DOI] [PubMed] [Google Scholar]
- 77.Snow BJ, Nygaard TG, Takahashi H, Calne DB. Positron emission tomographic studies of dopa-responsive dystonia and early-onset idiopathic parkinsonism. Ann Neurol. 1993;34:733–38. doi: 10.1002/ana.410340518. [DOI] [PubMed] [Google Scholar]
- 78.Marjańska M, Lehéricy S, Valabrègue R, et al. Brain dynamic neurochemical changes in dystonic patients: a magnetic resonance spectroscopy study. Mov Disord. 2013;28:201–09. doi: 10.1002/mds.25279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fiorio M, Weise D, Önal-Hartmann C, Zeller D, Tinazzi M, Classen J. Impairment of the rubber hand illusion in focal hand dystonia. Brain. 2011;134:1428–37. doi: 10.1093/brain/awr026. [DOI] [PubMed] [Google Scholar]
- 80.Castrop F, Dresel C, Hennenlotter A, Zimmer C, Haslinger B. Basal ganglia-premotor dysfunction during movement imagination in writer’s cramp. Mov Disord. 2012;27:1432–39. doi: 10.1002/mds.24944. [DOI] [PubMed] [Google Scholar]
- 81.Niethammer M, Carbon M, Argyelan M, Eidelberg D. Hereditary dystonia as a neurodevelopmental circuit disorder: evidence from neuroimaging. Neurobiol Dis. 2011;42:202–09. doi: 10.1016/j.nbd.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Argyelan M, Carbon M, Niethammer M, et al. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 2009;29:9740–47. doi: 10.1523/JNEUROSCI.2300-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Uluğ AM, Vo A, Argyelan M, et al. Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc Natl Acad Sci USA. 2011;108:6638–43. doi: 10.1073/pnas.1016445108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain. 2008;131:2499–509. doi: 10.1093/brain/awn168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci USA. 2010;107:8452–56. doi: 10.1073/pnas.1000496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brighina F, Romano M, Giglia G, et al. Effects of cerebellar TMS on motor cortex of patients with focal dystonia: a preliminary report. Exp Brain Res. 2009;192:651–56. doi: 10.1007/s00221-008-1572-9. [DOI] [PubMed] [Google Scholar]
- 87.Hamada M, Strigaro G, Murase N, et al. Cerebellar modulation of human associative plasticity. J Physiol. 2012;590:2365–74. doi: 10.1113/jphysiol.2012.230540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Popa T, Velayudhan B, Hubsch C, et al. Cerebellar processing of sensory inputs primes motor cortex plasticity. Cereb Cortex. 2013;23:305–14. doi: 10.1093/cercor/bhs016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tinazzi M, Fiorio M, Fiaschi A, Rothwell JC, Bhatia KP. Sensory functions in dystonia: insights from behavioral studies. Mov Disord. 2009;24:1427–36. doi: 10.1002/mds.22490. [DOI] [PubMed] [Google Scholar]
- 90.Wu T, Hallett M. The cerebellum in Parkinson’s disease. Brain. 2013;136:696–709. doi: 10.1093/brain/aws360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.