

Abstract
Chronic rejection of transplanted organs remains the main obstacle in the long-term success of organ transplantation. Thus, there is a persistent quest for development of anti-chronic rejection therapies and identification of novel molecular and cellular targets. One of the potential targets is the pericytes, the mural cells of microvessels, which regulate microvascular permeability, development and maturation by controlling endothelial cell functions and regulating tissue fibrosis and inflammatory response. In this review we discuss the potential of targeting pericytes in development of microvasular dysfunction and the molecular pathways involved in regulation of pericyte activities for anti-chronic rejection intervention.
Keywords: pericytes, chronic rejection, microvasculature
Introduction
The chronic rejection of transplanted organs represents major hurdle for their long-term function and survival. The symptoms of chronic rejection include accelerated graft atherosclerosis (narrowing and eventual occlusion of the arteries), extensive fibrosis, which obliterates the architecture and function of the organ, and damage to the microvasculature. Clinical and rodent model studies indicate that functional microvasculature is required for the effectiveness of immunosuppressive therapies (1, 2) and that microvascular loss or dysfunction leading to local ischemia may be an important factor in development of tissue fibrosis and an underlying cause of chronic rejection (2–6). Recent studies indicate that adenovirus-mediated hypoxia-inducible factor-1 (HIF-1) gene transfer therapy, which through the induction of pro-angiogenic growth factors enhanced microvasculature repair, promotes allograft integrity and alleviates chronic rejection of mouse lung allografts (7). Because, at present, there is no available clinical therapy for chronic rejection, the scientific, medical and pharmaceutical research entities are in relentless pursuit of novel cellular and molecular targets in order to design preventive drugs. In past decades the majority of research in the field has been focused on the most obvious culprits in development of chronic rejection i.e. the immune cells and their regulatory molecules and effectors. However, recently, there are increasing numbers of studies pointing toward greatly underappreciated candidate, the pericytes and the role they play in microvascular dysfunction, fibrosis, inflammation and development of chronic rejection.
1. Pericyte localization, structure and markers
The pericytes (also called the Rouget cells) were discovered in 1871 by German pathologist and bacteriologist C. J. Eberth (8) and subsequently described by Rouget (9) and Zimmerman (10). The pericytes envelop the endothelial wall of the microvessels: capillaries, post-capillary venules, venules and arterioles, and are responsible for microvessel integrity and regulation of blood flow (11,12). The wall of microvessels is built of a single layer of endothelial cells embedded within the basement membrane (which de facto is not a membrane but a thin sheet of collagen, laminin, fibronectin and heparan sulfate fibers), (13, 14), which endothelial cells co-produce and share with the pericytes (Fig. 1; 15). Pericytes have a large, round nucleus, small amount of cytoplasm and long cytoplasmic processes, which embrace endothelial wall of the vessel and may extend to the neighboring vessels. Pericytes are connected to the endothelial cells by three major types of intercellular junctions: 1. the peg-and-socket contacts, which are fingerlike intrusions/protrusions between cells (Fig. 1) in the areas of the low expression regions (LERs) of matrix proteins in the basement membrane; 2. the gap junctions, which form at the peg-socket contacts (Fig. 1) and allow direct chemical communication between the cytoplasm of neighboring cells through the diffusion of ions and various molecules, and 3. the spot–like adherence junctions called the adhesion plaques, which are connected to actin filament bundles and attach cells to each other and to the extracellular matrix (basement membrane) (Fig.1; 14, 16, 17). The density of pericyte coverage (endothelial cell to pericyte ratio) ranges between 1:1 (in brain) and 10:1 (in muscle) and seems to correlate positively with the microvessel permeability barrier requirement within the particular tissue (the tighter the barrier the higher pericyte density). This ratio also depends on the dynamics of endothelium renewal and vertical topography (and corresponding blood pressure) of the microvessels within the body (12, 18).
Figure 1. Microvessel structure and pericytes.
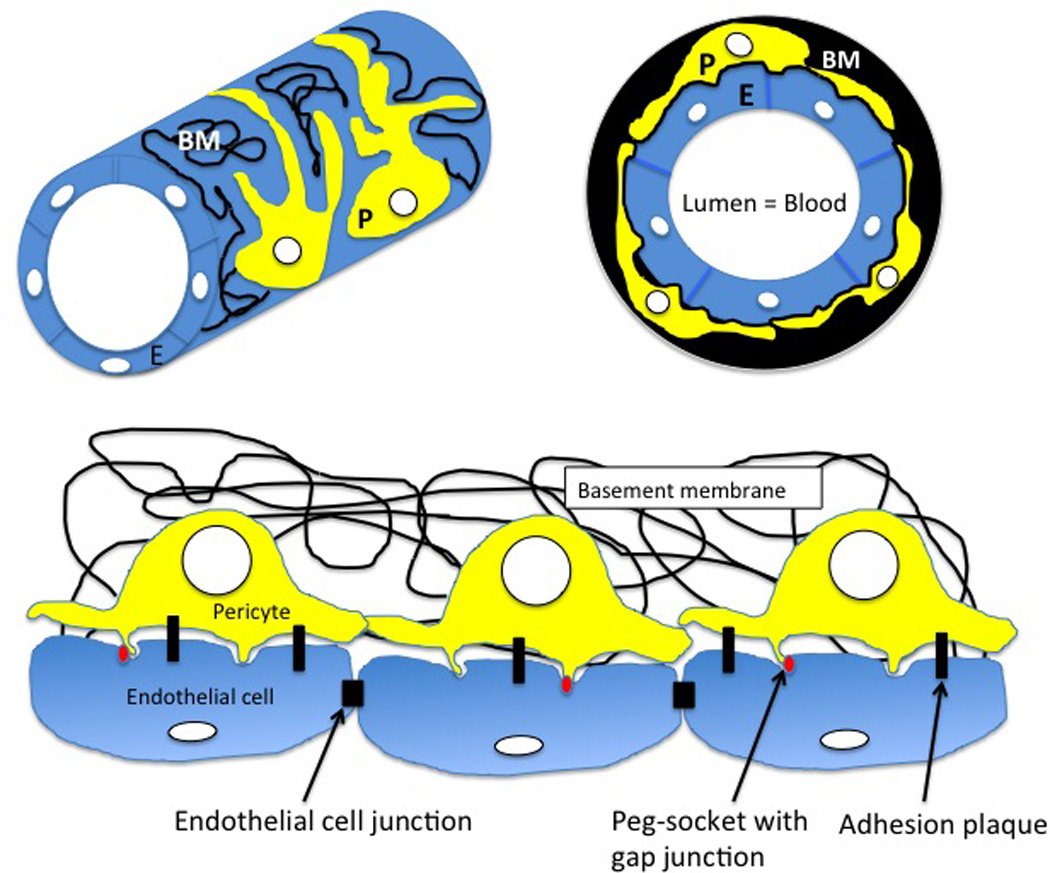
The microvessels: capillaries, post-capillary venules, venules and arterioles are built of a single layer of endothelial cells (E, blue) and covering pericytes (P, yellow), which are both embedded within fibrous, commonly produced, basement membrane (BM, black). Endothelial cells are inter-connected by cell junctions and are connected to the pericytes by adhesion plaques and peg-sockets with the gap junctions (red).
The pericytes develop during embryogenesis from mesenchymal cells present within the differentiating tissues induced by contact with the endothelial tubes of locally forming microvessels. Local differences in cellular environments within tissues and organs may explain the functional variance and tissue specificity between pericytes belonging to different microvessels (16). Recent studies indicate that during vascular development in the perinatal mouse heart the vessel endothelium initiates the pericyte ensheathment through brain-derived neurotrophic factor BDNF/ neurotrophic tyrosine kinase TrkB signaling, which is sensitive, in turn, to the small GTPase RhoA/ROCK kinase inhibitor Y-27632 (Fig. 2), (19).
Figure 2. Pericyte functions regulated by small GTPase RhoA pathway.
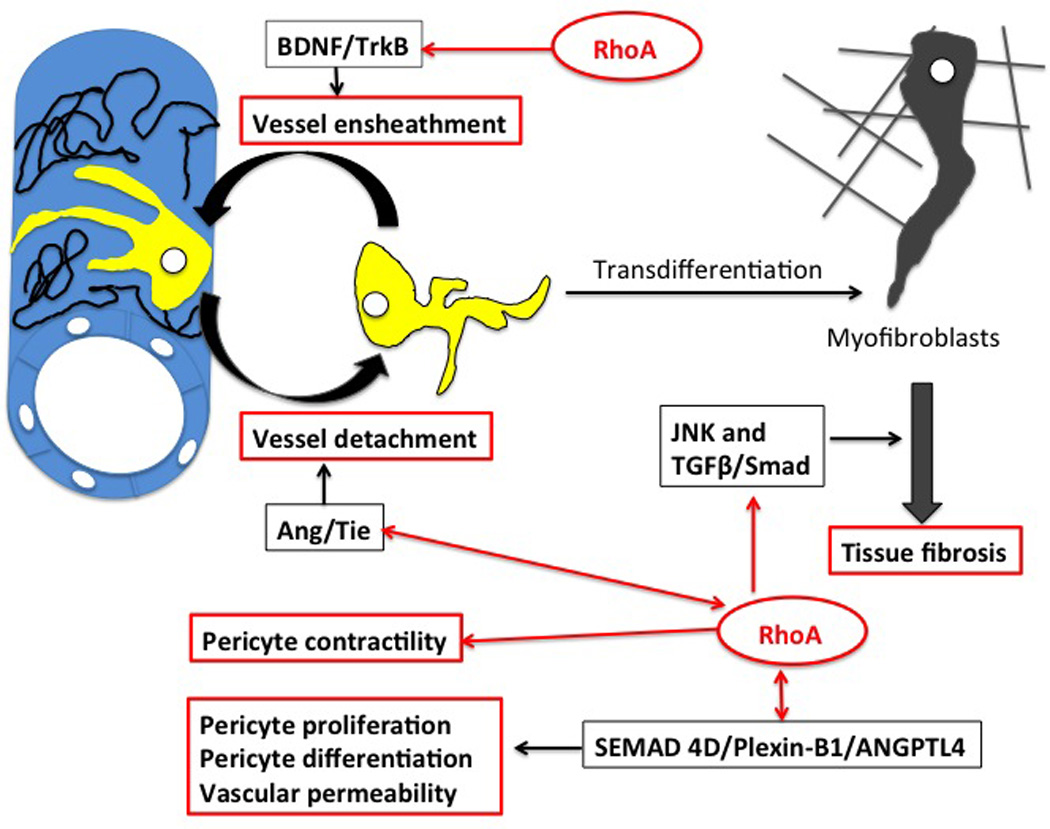
Microvessel ensheathment is initiated by BDNF/TrkB signaling, which is regulated by RhoA pathway and is sensitive to RhoA kinase (ROCK) inhibition. During inflammation and tissue injury pericytes are able to detach from the microvessel wall and transdifferentiate into fibrillar extracellular matrix (ECM)-producing myofibroblasts. Deposition of ECM leads to fibrosis and organ failure. Fibrosis is regulated by JNK and TGFβ/Smad pathways, which cross-talk with RhoA pathway. Inhibition of JNK and TGFβ/Smad through the inhibition of RhoA kinase ROCK abrogates collagen production and fibrosis. Detachment of pericytes from the microvessel wall is regulated by the Ang/Tie pathway, which cross-talks with RhoA signaling. Because RhoA is a master regulator of actin-related cell functions it also regulates pericyte contractility. By crosstalking to Ang/ Tie-2 and SEMAD4D/Plexin-B1/ANGPTL4 pathways, RhoA signaling influences pericyte differentiation and pericyte coverage, which in turn regulate blood flow and overall vessel permeability.
Pericytes are not only involved in de novo formation of microvessels (microvasculogenesis) within differentiating tissues but also in microvessel angiogenesis (sprouting from the preexisting vessels) within fully differentiated tissues and organs. Microvessel angiogenesis starts with the formation of primitive capillary tube (angiogenic sprout) derived from proliferating and migrating endothelial cells, which produce immature basement membrane. Subsequently, endothelial cells recruit the pericytes via fibroblast growth factor (FGF-2), platelet-derived growth factor (PDGF), heparin binding epidermal growth factor (HB-EGF) and Interleukin-6 signaling (20–22). Upon contact with the epithelium, the pericytes suppress endothelial cell proliferation and migration, stabilize the vessel and co-participate in maturation of the basement membrane of the microvessel wall. In turn, the contact with endothelial cells triggers synthesis of contractile proteins in the pericytes (14, 23, 24).
One of the major challenges in pericyte research is the lack of an unequivocal pericyte - specific marker. So far, all molecules, which have been found to be expressed by pericytes are also present in various other cell types. The most common markers used to identify pericytes are described below. Alpha-actin-2 (ACTA2) also called the smooth muscle or aortic smooth muscle actin (α-SMA, SMactin, alpha-SM-actin, ASMA) is one of six different actin isoforms involved in cell structure, contractility and motility. The expression level of α-SMA in pericytes is regulated in vitro and in vivo by various growth factors (25, 26). α-SMA is expressed not only in pericytes (27–34) but also in smooth muscle cells, myofibroblasts, monocytes, macrophages and cardiac microvascular endothelial cells (35–37).
Desmin, a type III intermediate filament required for mechanical elasticity of contractile cells (38) expressed in pericytes (33), is also present in skeletal and smooth muscle cells (39). Chondroitin sulfate proteoglycan (nerve/glia antigen-2/ NG2) also called chondroitin sulfate proteoglycan 4, melanoma chondroitin sulfate proteoglycan and melanoma-associated chondroitin sulfate proteoglycan has multiple signaling and regulatory functions (40) including recruitment of small GTPases (41). In addition to presence in pericytes (42, 43) NG2 is also expressed in neural progenitor cells, human melanoma cells and stem cells (44–46). Murfee et al. (47) analyzed NG2 and α-SMA co-expression in pericytes along the microvessel walls within rat mesenteric tissue, subcutaneous tissue, spinotrapezius muscle, and gracilis muscle. They found that NG2/ α-SMA co-expression was restricted to perivascular cells along arterioles and capillaries but it was absent in the pericytes of venules. This led to the identification of two distinct pericyte subsets: NG2+α-SMA+ pericytes found along arterioles and capillaries and NG2−α-SMA+ pericytes located along postcapillary venules (47). Further studies using confocal intravital microscopy in inflamed tissues showed that these two subsets of pericytes have two distinct functions in interstitial leukocyte migration: NG2−α-SMA+ pericytes facilitate transmigration of neutrophils from post capillary venules (26, 48, 49) and NG2+α-SMA+ pericytes control migration of leukocytes that had exited from the venules (26 49). Aminopeptidase N (ANPEP) also known as APN, CD13 and leukocyte surface antigen gp150, is a cell-membrane metalloprotease involved in metabolism of regulatory peptides and regulation of angiogenesis (50). ANPEP is present in pericytes, epithelial cells, macrophages, granulocytes, synaptic membranes of neural system and stem cells (51). Platelet-derived growth factor receptor beta (PDGFβ), a tyrosine kinase receptor that regulates cell proliferation, growth and differentiation plays a role in the recruitment of pericytes during angiogenesis (52, 53). In addition to its presence in pericytes, it is also expressed by smooth muscle cells, mesenchymal cells and neuronal progenitors. Additional pericyte markers are listed in reference 12 and 54 (although some of these markers still need verification).
In summary, positive identification of pericytes, their subtypes and diverse functions may rely on the multiple criteria, such as morphology, localization and co-expression of several different pericyte markers.
2. Pericyte Stemness/Pluripotency/Transdifferentiation
Research on pericyte functions in various tissues, organs and model systems suggests that they are highly versatile, functionally pluripotent cells with a great phenotypic plasticity. Pericytes express pluripotency and stem cell markers (55–57). Depending on their tissue/ organ-specific location and the specificity of signaling they receive from their surroundings, they exhibit diverse activities, ranging from structural to progenitor-cell like phenotypes and functions. Although pericytes cultured in vitro can differentiate into neural cells, smooth and skeletal muscle cells, adipocytes, chondrocytes and osteoblasts (56, 58–60), the lack of unequivocal pericyte-specific markers hinders the ability to follow their pluripotency, differentiation and ultimate fate in vivo. One of the more successful approaches utilized a transgenic approach combined with kinetic modeling microscopy in murine system enabling the ability to track the fate of fluorescent-labeled pericytes in vivo. These studies (61, 62) demonstrated that pericytes are able to differentiate into collagen-producing myofibroblasts and participate in the development of fibrosis (see below).
3. Role of Pericytes in Fibrosis
Pathogenic fibrosis, which occurs during chronic tissue injury is characterized by profound remodeling and excessive production and deposition of fibrillar extracellular matrix (ECM) containing collagen types I, III and IV, fibronectin, laminins, heparan sulfate proteoglycans and glycosaminoglycans (63, 64). Fibrosis results in disruption of tissue architecture and proper microrperfusion leading to organ failure. It is well established that myofibroblasts are the major ECM-producing cells during inflammation and organ injury. Myofibroblasts develop from myofibroblast progenitors (derived from either resident mesenchymal cells, fibroblasts, fibrocytes, bone marrow-derived cells, epithelial cells or endothelial cells) after they had become activated by pro-fibrotic cytokines and growth factors secreted by lymphocytes upon injury of the endothelium (12, 63, 65, 66). However, recent studies indicate that the ECM-producing myofibroblasts can originate almost exclusively from microvascullar pericytes (61, 67). Lin et al. (61) analyzed the origin of collagen type I, α1 (coll11α1), chondroitin sulfate proteoglycan (NG2 marker) and αSMA-expressing cells in the kidney of transgenic (coll11α1-GFP) mice. Using kinetic modeling microscopy, they showed that kidney injury induces the pericytes to detach from the microvessels and differentiate into the collagen producing-myofibroblasts, the major source of fibrotic ECM. Similarly, detachment of pericytes (labeled with R26R-yellow fluorescent protein) and their contribution to fibrosis was shown in spinal cord injury mouse model (62). Studies of fibrogenesis in kidney, liver and systemic sclerosis also point to the pericytes as the precursors of myofibroblasts (68 69, 70, 71). It is known that fibrosis depends on activation of c-Jun NH2-terminal kinase (JNK) (72, 73) and the TGFβ/Smad pathway (74, 75). Studies show that there is also crosstalk between the RhoA/ ROCK and JNK and TGFβ/Smad pathways (76, 77) and that inhibition of RhoA kinase (ROCK) by fasudil hydrochloride alleviates myocardial fibrosis and production of type-I and type-III collagen in diabetic rats via inhibition of JNK and TGFβ/Smad (Fig. 2; 77). Although further studies are needed to show how this relates to the collagen-deposition function of pericytes, it is plausible that fibrosis-related pericyte functions may be a potential target for novel anti-chronic rejection therapies (78).
4. Pericyte Contractility and blood flow facilitation
Since their initial characterization as “ the contractile cells” or “the microvascular smooth muscle cells” the ability of pericytes to contract and regulate blood flow within capillaries remained controversial for many decades (29, 79, 80). Some studies suggest that pericytes are noncontractile, however through their transformation into smooth muscle cells (34, 81, 82) they progressively, through the intermediate phenotypes, acquire an ultimate contractile phenotype. However, many studies showed that in addition to noncontractile proteins the pericytes also contain several contractile proteins such as smooth muscle- and nonmuscle-specific isoforms of actin, myosin, tropomyosin (31,33, 83–85) and a cyclic guanosine monophosphate (cGMP)-dependent protein kinase that regulates muscle contraction (86). Recent real-time studies of pericytes and dynamics of capillary diameter in mice cortex using intra vital two-photon laser scanning microscopy showed unequivocally that pericytes are contractile and are able to modulate capillary blood flow in the brain (87). The contraction of smooth muscle cells is regulated by the RhoA/Rock signaling pathway, which is a master regulator of actin-related cell functions (88, 89). Recent studies indicate that this is also true for the pericytes and that treatment with the RhoA pathway inhibitor Y-27632 or expression of a dominant-negative or dominant-active Rho severely alters pericyte contractility via interference with their actin filaments (Fig. 2); (23, 90, 91). Interestingly, studies from our laboratory show that the Y-27632 inhibitor abrogates chronic rejection via interference with actin cytoskeleton in rat and mouse cardiac allograft model system (92–95), and our recent (unpublished) data indicate that inhibition of chronic rejection prevents alteration of pericyte shape in the microvessels of transplanted hearts. This may indicate that abrogation of chronic rejection by RhoA pathway inhibitor may depend, at least partially, on the restoration of functions controlled by pericytes such as regulation of blood flow and/or microvasculature permeability.
5. Vessel integrity and pericyte-endothelium cross talk
One of the well-recognized functions of pericytes is their role in securing integrity and regulating permeability of microvessel wall. The loss of pericytes, decrease in pericyte coverage or change of their shape during inflammation distort vessel permeability, lead to vascular hyperdilation and hemorrhage (14, 96). Numerous studies indicate that there is an extensive and multilevel network of crosstalk between the endothelial cells and their pericytes. The major and most comprehensively studied crosstalk circuits consist of transforming growth factor beta (TGFβ), platelet-derived growth factor subunit B (PDGF-B) and the angiopoietin signaling pathways (11, 12 97).
The TGFβ signaling pathway consists of TGFβ superfamily of ligands that include: TGFβ (isoforms β1-3), bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), anti-müllerian hormone (AMH), activin and nodal, and their receptors, which belong to two major types: TGFβ R type I and II. The TGFβ pathway signals either through canonical Smad protein signaling or through non-cannonical non-Smad signaling pathways such as ERK, p38 MAPK, Jun N-terminal kinase (JNK), PI3K-Akt and small GTPases including RhoA/ ROCK and Rac pathways (Fig. 2); (98, 99). Interestingly, RhoA also modulates Smads translocation to the nucleus and acts as a modulator of Smad activation in alpha actin expressing smooth muscle cells, thus regulating TGFβ induced cell differentiation (100). Because pericytes are also contractile and produce alpha actin, this suggests that RhoA may also modulate TGFβ signaling in the pericytes. Although the effects of TGFβ pathway signaling are extremely broad, the principle of canonical Smad signaling is quite simple: the ligands of the TGFβ superfamily interact with a type II receptor, which phosphorylates a type I receptor. These receptors then phosphorylate receptor-regulated R-SMADs, which can now bind the common-mediator Smad (coSMAD). Subsequently, the R-SMAD/coSMAD complexes translocate to the nucleus where they act as transcription factors regulating expression of TGFβ/Smad target genes (101, 102). One of the TGFβ superfamily ligands, the TGFβ protein is highly expressed in various cell types including microvessel endothelial cells and pericytes and it is a secreted protein, which controls a plethora of cell functions including differentiation and proliferation. TGFβ is produced in an inactive pro-form bound to the latent TGF-beta binding protein (LTBP) and latency-associated peptide (LAP), and the release of active TGFβ from this complex is catalyzed by proteinases. Latent TGFβ and TGFβ receptors are expressed both by endothelial cells and pericytes and, at least in bovine cultured cells, a direct contact between these two cell types is necessary for the release of the active form of TGFβ (103). Mouse knockouts of various TGFβ pathway genes including smad4, smad5 and activin receptor-like kinase 1 and 5 genes (alk1 and alk5) result in embryonic lethality and profound defects in vasculature and pericyte development (12, 104–107). Studies in brain endothelium-specific smad4 knockout mice show that the canonical TGFβ pathway is involved in endothelial cell proliferation determining the density of pericyte coverage, via expression of adhesion molecule N-cadherin, which mediates endothelium - pericyte contact (12, 108, 109). In addition, both pericytes and endothelial cells express type I TGFβ activin receptors: receptor-like kinase Alk1 and Alk5, which seem to have antagonistic effects on microvessel maturation and development. Alk-1 signaling promotes cell migration and proliferation and Alk-5 regulates extracellular matrix production and vessel stabilization (12, 110).
The Angiopoietin signaling pathway
Angiopoietins (Ang1, Ang2, Ang3, Ang4) and Tie receptor tyrosine kinases (Tie1 and Tie 2) are involved in all steps of angiogenesis (111,112). Pericytes express Ang1, while endothelial cells and some pericyte subtypes express its main receptor Tie-2 (113, 114). Studies in rodent systems showed that Ang1 and Tie-2 deficient mouse embryos do not form pericytes (115,116). Aditionally, the injection of Ang2, which is an antagonist of Tie-2, into the eyes of normal rats results in a dose-dependent loss of pericytes, while heterozygous Ang2 deficiency prevents diabetes-induced pericyte loss (117). Pfister et al. (118) showed that in a mouse model of diabetic retinopathy, pericyte loss resulted from the overexpression of Ang2 and was completely blocked in Ang-2 deficient mice. However, studies on ang1 conditional knockout, and mice with defective Tie-2 receptors indicate that Ang1/Tie-2 signaling, while critical for regulation of number and diameter of vessels, is dispensable for the pericyte recruitment to newly forming vessels (119, 120). Pericyte detachment from the microvessel wall is not only regulated by Ang /Tie signaling, but probably also by the interactions between focal adhesion kinase (FAK), p21 protein activated kinase (PAK), integrins and the actin–myosin cytoskeleton (65, 118, 121). Ang1/Tie-2 signaling also regulates proliferation and chemotactic migration of endothelial cells during angiogenesis by modulation of RhoA and Rac1-dependent cytoskeletal rearrangements (Fig. 2). Cascone et al. (122) showed that Ang1 modulates the RhoA/Rac1 pathway via phosphoinositide 3-OH kinase/son of sevenless signaling and that chemotactic motility of endothelial cells is drastically reduced upon treatment with phosphoinositide 3-OH kinase inhibitor or in cells carrying dominant-negative mutants of RhoA and Rac1.
PDGF-B pathway signaling
The PDGF family consists of PDGF-A, -B, -C and -D, which bind to the protein tyrosine kinase receptors, PDGF receptor-α and -β. PDGF-B is produced by the endothelium and during angiogenesis is released from endothelial cells binding PDGFRβ on pericytes to recruit them to the vessel sprout. Depending on the organ, PDGF and PDGFRβ deficient mice either partially or completely lack pericytes, while constitutive activation of PDGFRβ promotes pericyte proliferation and inhibits their differentiation (12, 123). On the other hand, over-proliferation of pericytes accompanied by the loss of endothelial cells from the microvessel wall, observed during kidney injury, results from disrupted VEGF/PDGF signaling between the pericytes and endothelium (124). Endothelial cells produce not only PDGF but also the vascular endothelial growth factor (VEGF) receptor 2, while the pericytes produce VEGF in addition to PDGF receptor-β. Lin et al (124) showed that pericyte proliferation accompanied by endothelial cell loss, reduction of capillaries density and development of fibrosis, which occur during mouse kidney injury can be reversed by blocking of VEGF/PDGF signaling using circulating adenovirus-mediated soluble receptors. In addition they showed that during kidney injury pericytes switch the expression of VEGF isomers (from angiogenic VEGF 164 to dysangiogenic VEGF120 and 188). Interestingly, they also showed that this blockade also mitigates recruitment of inflammatory macrophages into injured kidney, which points to an unexpected functional link between microcapillaries, fibrosis and inflammation (65, 124). Using intra vital microscopy in mouse cremaster muscles and an ear skin inflammation model, Wang et al. (125) showed that inflammatory extravasation of the polymorphonuclear neutrophils (PMNs) through the venular wall depends on a direct contact between the PMNs and pericytes. The PMNs induce relaxation of the pericyte cytoskeleton, which results in the formation of spaces between pericytes and enlargement of the low expression regions (LERs) of matrix proteins in the basement membrane. These changes in turn allow PMNs to transverse the vascular wall. The authors also showed that PMNs induced relaxation of the pericyte cytoskeleton is mediated by inhibition of the RhoA/ROCK signaling pathway in pericytes and suggested that pericytes may be a potential target for anti-inflammatory therapies. Recent studies indicate that, at least in tumor-induced angiogenesis, the regulation of proliferation and differentiation of pericytes, as well as overall vascular permeability involves Semaphorin 4D (SEMAD4D) and its receptor Plexin-B1. This regulates the Rho A pathway through the guanine nucleotide exchange factors leukemia-associated Rho GEF (126) and angiopoetin-like protein 4 (ANGPTL4) (127). SEMA4D induces expression of PDGF and ANGPTL4 in endothelial cells in a Plexin-B1/RhoA-dependent manner (Fig. 2). These and other studies, mentioned above, point to the importance of the RhoA pathway in regulation of proliferation and differentiation of pericytes and vascular permeability. Other pathways recently found to be involved in signaling between pericytes and endothelium include: Notch, sonic hedgehog (Shh), stromal-derived factor 1-a (SDF-1a)/CXCR4 and heparin-binding epidermal growth factor (HB-EGF)/EGF receptors (ErbBs) signaling pathways (12).
6. Pericytes as master sensors of inflammation and instructors of leukocyte proinflammatory and prosurvival programs
Alteration of microvessel integrity may be one of the most important and underappreciated steps leading to tissue ischemia and the cascade of stimulatory changes driving tissue inflammation and development of chronic rejection. It is known that the leakiness of microvasculature signals T cells, macrophages, monocytes, mast cells and fibroblasts to produce cytokines, chemokines and growth factors, which, in turn, switch on and escalate inflammatory response. One of the most crucial steps in the immune response is the penetration (diapedesis, extravasation) of leukocytes through the capillary walls into the surrounding tissues. Over the years a number of studies have explored mutual endothelium-leukocyte interactions and it is well established that the endothelial wall occupancy and eventual breaching of endothelial wall enhances effector functions of extravasating leukocytes. Recent studies indicate that pericytes play a crucial role in endothelial wall breaching, which is a lengthy (15–40 min) and elaborate process (128). Using intravital confocal microscopy to study neutrophil migration from the venules of the inflamed mouse tissues, Proebst et al. (48) showed that after squeezing between endothelial cells and exiting from the endothelial wall the neutrophils crawl (so called abluminal crawling) along pericyte processes and extravasate the venular wall through the enlarged (by inflammation) gaps between the adjacent pericytes (Fig. 3). Leukocyte crawling depends on the pericyte-expressed intercellular adhesion molecule-1 (ICAM-1) and its leukocyte integrin ligands, Mac-1 and LFA-1. The formation and selection of inter-pericyte exit gaps is coordinated by signaling between the proinflammatory cytokines TNF and IL-1β and their receptors expressed by the pericytes (48). The most recent studies indicate an unexpected and quite refine role of capillary wall pericytes in sensing inflammatory cues and directing leukocyte trafficking into the inflammatory foci and also behaving as per se immune cells (25, 26, 48, 49). In vitro studies of brain-derived pericytes response to artificial inflammation/infection induced by TNF-α, IL-1β, IFN-γ cytokines or LPS stimulation showed that IL-1β induces production of iNOS, RONS (reactive oxygen or nitrogen species) and prostaglandin-endoperoxide synthase 2 (cyclooxygenase-2 or Cox-2). This mechanism is, traditionally, applied by specialized immune cells (such as macrophages) of the innate immune system to kill pathogens at early time points after the infection. On the other hand, the IFN-γ treatment of pericytes induces phagocytosis and expression of MHCII and macrosialin / CD68 / Gp110 protein, which allows macrophage homing and plays a role in cell-cell and cell-pathogen interactions (i.e. induces mechanisms involved in adaptive immune responses involving antigen presentation) by binding to lectins and selectins. The TNF-α treatment of pericytes induces both types of immune responses (synthesis of iNOS and RONS and the phagocytosis rate increase) in the pericytes (25). These findings suggest that pericytes’ ability to behave like the immune cells of the innate and adaptive immune system may contribute to various circuits of immune response. The active role of pericytes in the innate immune response was also shown in vivo in a mouse model of sterile inflammation. Using intravital two-photon microscopy Stark et al. (26) demonstrated that in response to inflammation the arteriolar and capillary NG2+ pericytes produce the chemoattractant MIF (macrophage migration inhibitory factor) and upregulate synthesis of the intercellular adhesion molecule, ICAM-1. These molecules allowed pericytes to attract interstitial neutrophils and macrophages after they had extravasated from postcapillary venules and enhanced their scanning and effector functions at the inflammation sites (Fig. 3). Contact with pericytes “tells” leukocytes to express matrix metalloproteinases, β1 integrins, and formyl peptide receptors (FPR, which bind products of cell and tissue degradation). This allows them to acquire prosurvival signals and directionally migrate toward the inflammation source rich in damage-associated molecular pattern molecules (DAMPs) exposed on cell surfaces following tissue injury (26,49). The authors suggest that the role of NG2+ pericytes is to create the migratory roadways for the movement of extravasated immune cells along arterioles and capillaries and to enhance their ability to scan for and respond to tissue damage and inflammation cues (Fig. 3; 26,49). All these studies indicate that pericytes act as sensors of inflammation, effectors of immunosurveillance and serve various functions of immune cells playing an active role in the immune response and a crucial role in development of chronic rejection.
Figure 3. Pericytes participate in endothelial wall breaching by leukocytes, sensitize them and direct them toward the inflammation foci.
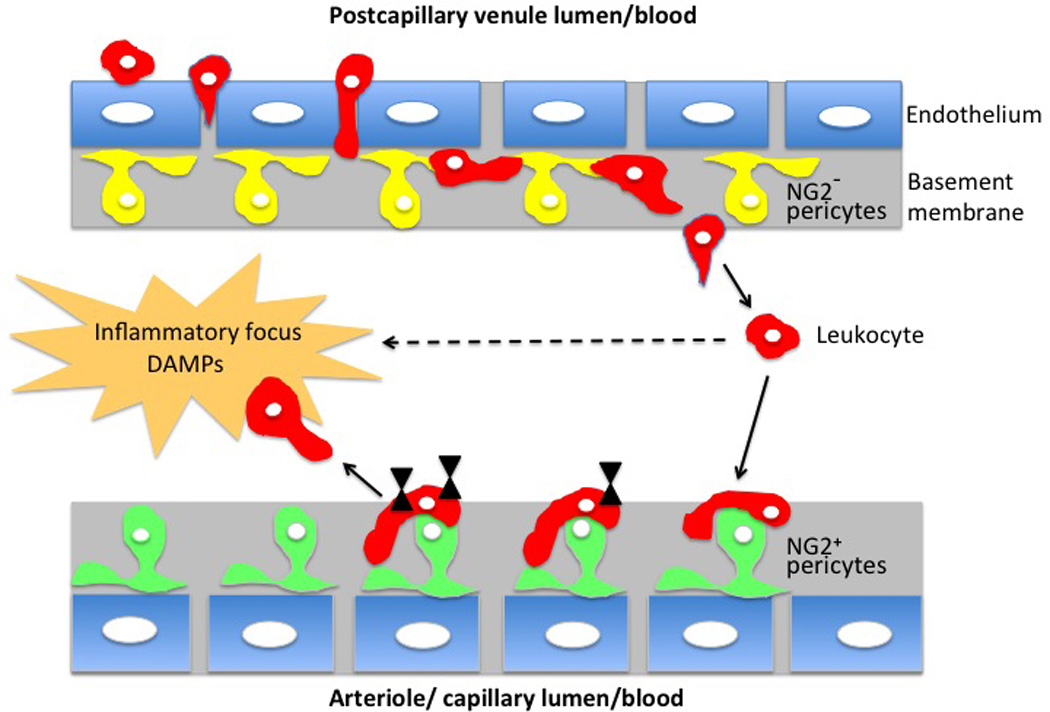
Depending on the type of microvessel there are two subtypes of pericytes: venules are covered by pericytes lacking NG2 marker (yellow), and arterioles and capillaries are covered by NG2 positive pericytes (green). During tissue inflammation the leukocytes (red) enter the venular wall and after squeezing between endothelial cells (blue) migrate (abluminal migration) along the processes of NG2− pericytes until they find large inter-pericyte gaps where they are able to exit (extravasate) the vessel wall. Leukocyte migration is ICAM-1, Mac-1 and LFA-1 dependent and a selection of the large-size exit gaps is TNF/IL-1β dependent. Subsequently, after exiting from the venules some of the leukocytes are attracted toward ICAM-1; MIF positive NG2+ pericytes present on neighboring arterioles and capillaries. Contact with NG2+ pericytes and treking along pericyte processes induce leukocytes to produce matrix metalloproteinses, integrins and FPRs (black triangles), progressively acquiring prosurvival signals, upregulating expression of a promigratory receptors and migrating toward the inflammation sites, which are rich in damage-associated molecular pattern (DAMPs) compounds.
Below we summarize how the pericyte may contribute to chronic rejection and how the available inhibitors of the RhoA pathway may be effective in modulating the chronic rejection-related functions of pericytes (Fig. 4).
Figure 4. RhoA pathway regulation of actin-related pericyte functions involved in development of chronic rejection.
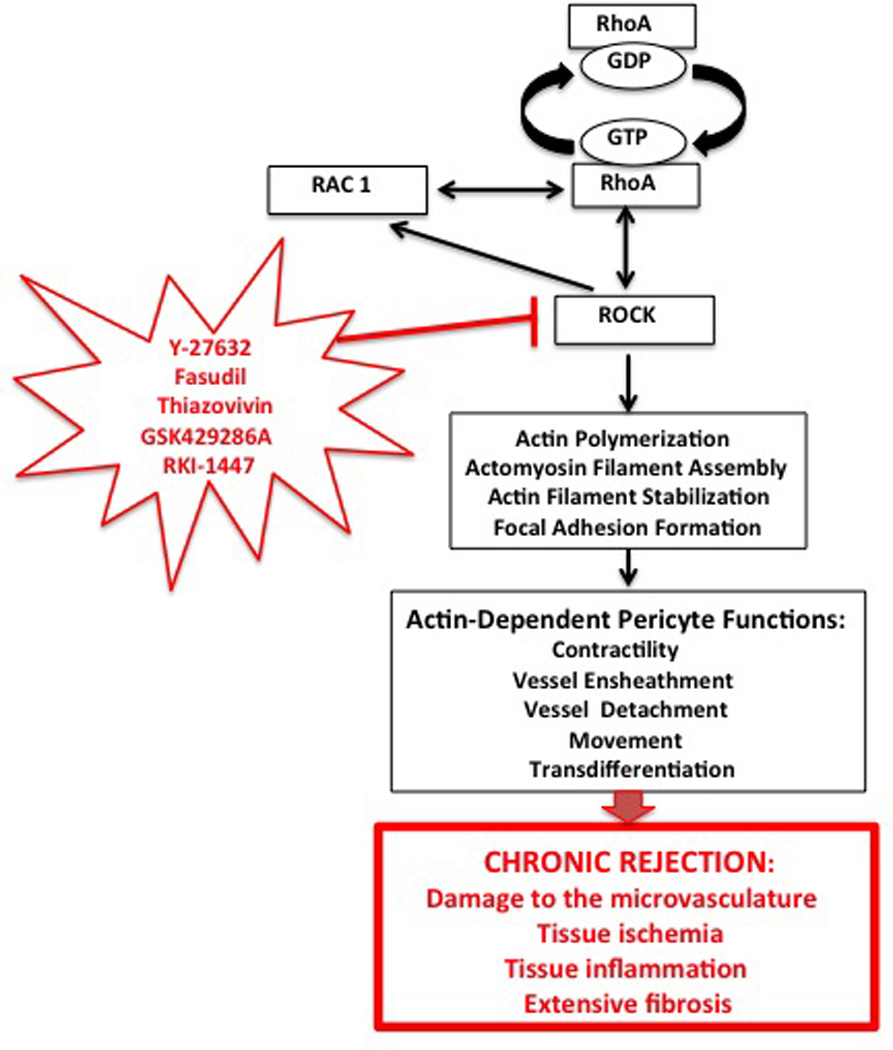
RhoA regulates, through its downstream effector ROCK (containing ROCK 1 and ROCK 2 isoforms) actin filament polymerization and organization, which in turn regulate various functions of pericytes. In addition, RhoA (either directly or through ROCK) cross talks with Rac1 pathway. There are several inhibitors of ROCK (ROCK 1, ROCK 2 or both), which are known, from rodent or human clinical studies, to influence actin-related cellular functions in immune response and transplantation (129, 130). Because many of pericyte functions such as movement, contractility, interaction with vessel endothelium and transdifferentiation into collagen producing myofibroblasts also depend on actin cytoskeleton, these inhibitors, via targeting pericytes, have a potential to be used as the anti-inflammation and anti-chronic rejection therapeutics.
Pericyte functions in inflammation and tissue injury leading to development of chronic rejection, which are known (or predicted) to be sensitive to RhoA pathway inhibitors.
- Vessel integrity and permeability
-
-Pericyte ensheathment of the vessels is initiated by the vessel endothelium through the BDNF/ TrkB signaling, which in turn is sensitive to small GTPase RhoA/ROCK kinase inhibitor Y-27632 (19)
-
-Pericyte coverage and recruitment is regulated by Angiopoietins (Angs) via endothelial cell interaction. The Ang1 modulates RhoA/Rac1 pathway and endothelial cells functions are changed in cells carrying dominant-negative mutant of RhoA (122)
-
-
- Leukocyte extravasation and transmigration
-
-Extravasation of the polymorphonucler neutrophils (PMNs) through the venular wall depends on relaxation of pericyte cytoskeleton and is mediated by the RhoA/ROCK signaling pathway (125)
-
-Transmigration of neutrophils from post capillary venules and migration of leukocytes that had exited from the venules depends on NG2, which has multiple signaling and regulatory functions one of them being a recruitment of small GTPases (such as RhoA), (41).
-
-
-
Pathogenic fibrosis
-
-
Excessive deposition of ECM by myofibroblasts, which originate from microvascular pericytes is regulated by JNK and TGFβ/Smad pathways. There is a crosstalk between RhoA/ ROCK and JNK and TGFβ/Smad pathways and inhibition of RhoA kinase ROCK by Fasudil alleviates myocardial fibrosis (77).
-
-
In summary, although the pericytes and their RhoA pathway regulated functions emerge as the major players in development of chronic rejection it remains to be determined whether they can be used as a critical targets of anti-chronic rejection therapies. Most probably the simultaneous inhibition of several molecular and cellular targets will be required to inhibit chronic rejection of transplanted organs.
Acknowledgments
We are extremely grateful to Dr Rachel K. Miller for critical reading of the manuscript.
Funding Disclosure: there is no funding to disclose
Abbreviations
- ACTA2
Alfa-actin-2
- Alk
Activin receptor-like kinase
- AMH
Anti-müllerian hormone
- Ang
Angiopoietin
- ANPEP
Aminopeptidase N
- Akt
Protein kinase B
- α-SMA
Alfa smooth muscle actin
- BDNF
Brain-derived neurotrophic factor
- CD68
Cluster of differentiation 68
- cGMP
cyclic guanosine monophosphate
- DAMPS
damage-associated molecular pattern molecules
- ECM
Extracellular matrix
- FGF-2
Fibroblast growth factor
- FPR
formyl peptides receptor
- GDFs
Growth and differentiation factors
- Gp110
Envelope glycoprotein
- GSK429286A
inhibitor of ROCK1 and ROCK2
- HB-EGF
Heparin binding epidermal growth factor
- HIF-1
Hypoxia-inducible factor-1
- ICAM-1
Intercellular Adhesion Molecule 1
- IFN-γ
Interferon gamma
- iNOS
Inducible Nitric oxide synthase
- IL-1β
Interlukin 1 beta
- JNK
c-Jun NH2-terminal kinase
- LAP
Latency-associated peptide
- LERs
Low expression regions
- LPS
Lipopolysaccharide
- LTBP
Latent TGF-beta binding protein
- MHCII
Major histocompatibility complex) class II
- MIF
Macrophage migration inhibitory factor
- NG2
Nerve/glia antigen-2
- PDGF
Platelet-derived growth factor
- PDGFβ
Platelet-derived growth factor receptor beta
- PI3K
Phosphatidylinositde-3 Kinase
- PMNs
Polymoprhonuclear neutrophils
- Rac1
Ras-related C3 botulinum toxin substrate 1
- RhoA
Ras homolog gene family, member A
- RKI-1447
inhibitor of ROCK1 and ROCK2
- ROCK
Rho-associated protein kinase (ROCK1 and ROCK2)
- RONS
Receptor tyrosine kinases
- SEMAD4D
Semaphorin 4D
- SMAD
homolog of both the Drosophila protein, mothers against decapentaplegic (MAD and the Caenorhabditis elegans protein SMA (small body size)
- coSMAD
common-mediator Smad
- R-SMAD
receptor-regulated Smad
- TGFβ
Transforming growth factor beta
- Trk
Thyrosine kinase
- VEGF
Vascular endothelial growth factor
- Y-27632
Selective ROCK1 (p160ROCK) inhibitor
Footnotes
Authors’ Contribution:
Malgorzata Kloc - Concept, manuscript writing, figures drawing, mkloc@houstonmethodist.org
Rafik M. Ghobrial - manuscript co-writing, RMGhobrial@houstonmethodist.org
XC Li- manuscript co-writing, XCLi@HoustonMethodist.org
J.Z. Kubiak - manuscript co-writing, jacek.kubiak@univ-rennes1.fr
Disclosures
None of the authors have a conflict of interest to disclose.
References
- 1.Ozdemir BH, Demirhan B, Ozdemir FN, Dalgic A, Haberal M. The role of microvascular injury on steroid and OKT3 response in renal allograft rejection. Transplantation. 2004;78:734–740. doi: 10.1097/01.tp.0000130453.79906.62. [DOI] [PubMed] [Google Scholar]
- 2.Babu AN, Murakawa T, Thurman JM, Miller EJ, Henson PM, Zamora MR, et al. Microvascular destruction identifies murine allografts that cannot be rescued from airway fibrosis. J Clin Invest. 2007;117:3774–3785. doi: 10.1172/JCI32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biedermann BC, Sahner S, Gregor M, Tsakiris DA, Jeanneret C, Pober JS, Gratwohl A. Endothelial injury mediated by cytotoxic T lymphocytes and loss of microvessels in chronic graft versus host disease. Lancet. 2002;359:2078–2083. doi: 10.1016/S0140-6736(02)08907-9. [DOI] [PubMed] [Google Scholar]
- 4.Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, et al. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant. 2004;23:527–531. doi: 10.1016/j.healun.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Luckraz H, Goddard M, McNeil K, Atkinson C, Sharples LD, Wallwork J. Is obliterative bronchiolitis in lung transplantation associated with microvascular damage to small airways? Ann Thorac Surg. 2006;82:1212–1218. doi: 10.1016/j.athoracsur.2006.03.070. [DOI] [PubMed] [Google Scholar]
- 6.Khan MA, Nicolls MR. Complement-mediated microvascular injury leads to chronic rejection. Adv Exp Med Biol. 2013;735:233–246. doi: 10.1007/978-1-4614-4118-2_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang X, Khan MA, Tian W, Beilke J, Natarajan R, Kosek J, Yoder MC, Semenza GL, Nicolls MR. Adenovirus-mediated HIF-1α gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J Clin Invest. 2011;121:2336–2349. doi: 10.1172/JCI46192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eberth CJ. Handbuch der Lehre von der Gewegen des Menschen und der Tiere. Vol. 1. Leipzig: 1871. [Google Scholar]
- 9.Rouget C. Memoire sur le developpement, la structures et les proprietes des capillaires sanguins et lymphatiques. Archs Physiol Norm Pathol. 1873;5:603–633. 1873. [Google Scholar]
- 10.Zimmermann KW. Der feinere bau der blutcapillares. Z. Anat. Entwicklungsgesch. 1923;68:3–109. [Google Scholar]
- 11.Armulik A, Abramsson A, Betsholtz C. Endothelial/Pericyte Interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 12.Armulik A, Genove G, Betsholtz C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Developmental Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Kafalides NA, Borel JP. Basement Membranes: Cell and Molecular Biology. San Diego, CA: Elsevier Academic Press; 2005. [Google Scholar]
- 14.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandarino LJ, Sundarraj N, Finlayson J, Hassell HR. Regulation of fibronectin and laminin synthesis by retinal capillary endothelial cells and pericytes in vitro. Exp. Eye Res. 1993;57:609–621. doi: 10.1006/exer.1993.1166. [DOI] [PubMed] [Google Scholar]
- 16.Rucker HK, Wynder HJ, Thomas WE. Cellular mechanisms of CNS pericytes. Brain Res Bull. 2000;51:363–369. doi: 10.1016/s0361-9230(99)00260-9. 2000. [DOI] [PubMed] [Google Scholar]
- 17.Dore-Duffy P, Cleary K. Morphology and Properties of Pericytes. The Blood-Brain and Other Barriers. Methods in Mol Biol. 2011;686:49. doi: 10.1007/978-1-60761-938-3_2. [DOI] [PubMed] [Google Scholar]
- 18.Sims DE. The pericyte—a review. Tissue Cell. 1986;18:153–174. doi: 10.1016/0040-8166(86)90026-1. [DOI] [PubMed] [Google Scholar]
- 19.Anastasia A, Deinhardt K, Wang S, Martin L, Nichol D, Irmady K, Trinh J, Parada L, Rafii S, Hempstead BL, Kermani P. Trkb signaling in pericytes is required for cardiac microvessel stabilization. PLoS One. 2014;9:e87406. doi: 10.1371/journal.pone.0087406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev. 2003;17:1835–1840. doi: 10.1101/gad.266803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricard N, Tu L, Le Hiress M, Huertas A, Phan C, Thuillet R, Sattler C, Fadel E, Seferian A, Montani D, Dorfmüller P, Humbert M, Guignabert C. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation. 2014;129:1586–1597. doi: 10.1161/CIRCULATIONAHA.113.007469. [DOI] [PubMed] [Google Scholar]
- 22.Stratman AN, Schwindt AE, Malotte KM, Davis GE. Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood. 2010;116:4720–4730. doi: 10.1182/blood-2010-05-286872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutcher ME, Herman IM. The pericyte: Cellular regulator of microvascular blood flow. Microvasc Res. 2009;77:235–246. doi: 10.1016/j.mvr.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papetti M, Shujath J, Riley KN, Herman IM. FGF-2 antagonizes the TGF-beta1-mediated induction of pericyte alpha-smooth muscle actin expression: a role for myf-5 and Smad-mediated signaling pathways. Invest Ophthalmol Vis Sci. 2003;44:4994–5005. doi: 10.1167/iovs.03-0291. [DOI] [PubMed] [Google Scholar]
- 25.Pieper C, Marek JJ, Unterberg M, Schwerdtle T, Galla HJ. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014;1550:1–8. doi: 10.1016/j.brainres.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Brühl ML, Gärtner F, Khandoga AG, Legate KR, Pless R, Hepper I, Lauber K, Walzog B, Massberg S. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and 'instruct' them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51. doi: 10.1038/ni.2477. [DOI] [PubMed] [Google Scholar]
- 27.Nehls V, Drenckhahn D. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol. 1991;113:147–154. doi: 10.1083/jcb.113.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes S, Chan-Ling T. Characterization of smooth muscle cell and pericyte differentiation in the rat retina in vivo. Invest Ophthalmol Vis Sci. 2004;45:2795–2806. doi: 10.1167/iovs.03-1312. [DOI] [PubMed] [Google Scholar]
- 29.Walter JJ, Sane DC. The role of smooth muscle cells and pericytes in angiogenesis. In: Mousa Shaker A., editor. Chapter 3 in Angiogenesis Inhibitors and Stimulators: Potential Therapeutic Implications. © 2000 Eurekah.com. [Google Scholar]
- 30.Nakano M, Atobe Y, Goris RC, Yazama F, Ono M, Sawada H, Kadota T, Funakoshi K, Kishida R. Ultrastructure of the capillary pericytes and the expression of smooth muscle alpha-actin and desmin in the snake infrared sensory organs. Anat Rec. 2000;260:299–307. doi: 10.1002/1097-0185(20001101)260:3<299::AID-AR67>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 31.Skalli O, Pelte MF, Peclet MC, Gabbiani G, Gugliotta P, Bussolati G, Ravazzola M, Orci L. Alpha-smooth muscle actin, a differentiation marker of smooth muscle cells, is present in microfilamentous bundles of pericytes. J Histochem Cytochem. 1989;37:315–321. doi: 10.1177/37.3.2918221. [DOI] [PubMed] [Google Scholar]
- 32.Nees S, Weiss DR, Senftl A, Knott M, Förch S, Schnurr M, Weyrich P, Juchem G. Isolation, bulk cultivation, and characterization of coronary microvascular pericytes: the second most frequent myocardial cell type in vitro. Am J Physiol Heart Circ Physiol. 2012;302:H69–H84. doi: 10.1152/ajpheart.00359.2011. [DOI] [PubMed] [Google Scholar]
- 33.Liu G, Meng C, Pan M, Chen M, Deng R, Lin L, Zhao L, Liu X. Isolation, purification, and cultivation of primary retinal microvascular pericytes: a novel model using rats. Microcirculation. 2014 Feb 5; doi: 10.1111/micc.12121. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34.Birbrair A, Zhang T, Wang Z-M, Messi ML, Enikolopov GN, Mintz A, Delbono O. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Research. 2013;10:67–84. doi: 10.1016/j.scr.2012.09.003. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ando H, Kubin T, Schaper W, Schaper J. Cardiac microvascular endothelial cells express α-smooth muscle actin and show low NOS III activity. American Journal of Physiology - Heart and Circulatory Physiology. 1999;276:H1755–H1768. doi: 10.1152/ajpheart.1999.276.5.H1755. [DOI] [PubMed] [Google Scholar]
- 36.Ludin A, Itkin T, Gur-Cohen S, Mildner A, Shezen E, Golan K, Kollet O, Kalinkovich A, Porat Z, D'Uva G, Schajnovitz A, Voronov E, Brenner DA, Apte RN, Jung S, Lapidot T. Monocytes-macrophages that express α-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol. 2012;13:1072–1082. doi: 10.1038/ni.2408. [DOI] [PubMed] [Google Scholar]
- 37.Nagamoto T, Eguchi G, Beebe DC. Alpha-smooth muscle actin expression in cultured lens epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:1122–1129. [PubMed] [Google Scholar]
- 38.Kiss B, Kellermayer MS. Stretching desmin filaments with receding meniscus reveals large axial tensile strength. J Struct Biol. 2014 doi: 10.1016/j.jsb.2014.04.004. S1047-8477(14)00090-2. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Mericskay M, Agbulut O, Butler-Browne G, Carlsson L, Thornell LE, Babinet C, Paulin D. Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J Cell Biol. 1997;139:129–144. doi: 10.1083/jcb.139.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trotter J, Karram K, Nishiyama N. NG2 cells: properties, progeny and origin. Brain Res Rev. 2010;63:72–82. doi: 10.1016/j.brainresrev.2009.12.006. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisenmann KM, McCarthy JB, Simpson MA, Keely PJ, Guan JL, Tachibana K, Lim L, Manser E, Furcht LT, Iida J. Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack-1 and p130cas. Nat Cell Biol. 1999;1:507–513. doi: 10.1038/70302. 1999. [DOI] [PubMed] [Google Scholar]
- 42.Ozerdem U, Monosov E, Stallcup WB. NG2 proteoglycan expression by pericytes in pathological microvasculature. Microvasc Res. 2002;63:129–134. doi: 10.1006/mvre.2001.2376. [DOI] [PubMed] [Google Scholar]
- 43.Ozerdem U, Stallcup WB. Pathological angiogenesis is reduced by targeting pericytes via the NG2 proteoglycan. Angiogenesis. 2004;7:269–276. doi: 10.1007/s10456-004-4182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Binamé F. Transduction of Extracellular Cues into Cell Polarity: the Role of the Transmembrane Proteoglycan NG2. Mol Neurobiol. 2014 Jan 5; doi: 10.1007/s12035-013-8610-8. [Epub ahead of print]. PMID: 24390567. [DOI] [PubMed] [Google Scholar]
- 45.Huang W, Zhao N, Bai X, Karram K, Trotter J, Goebbels S, Scheller A, Kirchhoff F. Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia. 2014;62:896–913. doi: 10.1002/glia.22648. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Wang J, Rizvi SM, Jager MJ, Conway RM, Billson FA, Allen BJ, Madigan MC. In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest Ophthalmol Vis Sci. 2005;46:4365–4371. doi: 10.1167/iovs.05-0559. [DOI] [PubMed] [Google Scholar]
- 47.Murfee WL, Skalak TC, Peirce SM. Differential arterial/venous expression of NG2 proteoglycan in perivascular cells along microvessels: identifying a venule-specific phenotype. Microcirculation. 2005;12:151–160. doi: 10.1080/10739680590904955. [DOI] [PubMed] [Google Scholar]
- 48.Proebstl D, et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J. Exp. Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alon R, Nourshargh S. Learning in motion: pericytes instruct migrating innate leukocytes. Nat Immunol. 2013;14:14–15. doi: 10.1038/ni.2489. [DOI] [PubMed] [Google Scholar]
- 50.Rangel R, Sun Y, Guzman-Rojas L, Ozawa MG, Sun J, Giordano RJ, Van Pelt CS, Tinkey PT, Behringer RR, Sidman RL, Arap W, Pasqualini R. Impaired angiogenesis in aminopeptidase N-null mice. Proc. Nat. Acad. Sci. 2007;104:4588–4593. doi: 10.1073/pnas.0611653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerlach JC, Over P, Turner ME, Thompson RL, Foka HG, Chen WC, Péault B, Gridelli B, Schmelzer E. Perivascular mesenchymal progenitors in human fetal and adult liver. Stem Cells Dev. 2012;21:3258–3269. doi: 10.1089/scd.2012.0296. [DOI] [PubMed] [Google Scholar]
- 52.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 53.Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–3055. doi: 10.1242/dev.126.14.3047. 1999. [DOI] [PubMed] [Google Scholar]
- 54.Kelly-Goss MR, Sweat RS, Stapor PC, Peirce SM, Murfee WL. Targeting Pericytes for Angiogenic Therapies. Microcirculation. 2014;21:345–357. doi: 10.1111/micc.12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lugassy C, Wadehra M, Li X, Corselli M, Akhavan D, Binder SW, Péault B, Cochran AJ, Mischel PS, Kleinman HK, Barnhill RL. Pilot study on "pericytic mimicry" and potential embryonic/stem cell properties of angiotropic melanoma cells interacting with the abluminal vascular surface. Cancer Microenviron. 2013;6:19–29. doi: 10.1007/s12307-012-0128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montiel-Eulefi E, Nery AA, Rodrigues LC, Sánchez R, Romero F, Ulrich H. Neural differentiation of rat aorta pericyte cells. Cytometry A. 2012;81:65–71. doi: 10.1002/cyto.a.21152. [DOI] [PubMed] [Google Scholar]
- 57.Tsang WP, Shu Y, Kwok PL, Zhang F, Lee KK, Tang MK, Li G, Chan KM, Chan WY, Wan C. CD146+ human umbilical cord perivascular cells maintain stemness under hypoxia and as a cell source for skeletal regeneration. PLoS One. 2013;8:e76153. doi: 10.1371/journal.pone.0076153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collett G, Wood A, Alexander MY, Varnum BC, Boot-Handford RP, Ohanian V, Ohanian J, Fridell YW, Canfield AE. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ. Res. 2003;92:1123–1129. doi: 10.1161/01.RES.0000074881.56564.46. [DOI] [PubMed] [Google Scholar]
- 59.Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007;9:255–267. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- 60.Farrington-Rock C, Crofts NJ, Doherty MJ, Ashton BA, Griffin-Jones C, Canfield AE. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation. 2004;110:2226–2232. doi: 10.1161/01.CIR.0000144457.55518.E5. [DOI] [PubMed] [Google Scholar]
- 61.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Göritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisén J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–242. doi: 10.1126/science.1203165. 2011. [DOI] [PubMed] [Google Scholar]
- 63.Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis-a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014 Apr 22; doi: 10.1038/nrrheum.2014.53. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 64.Wight TN, Potter-Perigo S. The extracellular matrix: an active or passive player in fibrosis? Am J Physiol Gastrointest Liver Physiol. 2011;301:G950–G955. doi: 10.1152/ajpgi.00132.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hutchison N, Fligny C, Duffield JS. Resident mesenchymal cells and fibrosis. Biochim Biophys Acta. 2012;1832:962–971. doi: 10.1016/j.bbadis.2012.11.015. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Powell DW, Pinchuk IV, Saada JI, Chen X, Mifflin RC. Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol. 2011;73:213–237. doi: 10.1146/annurev.physiol.70.113006.100646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2012;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fabris L, Strazzabosco M. Epithelial-mesenchymal interactions in biliary diseases. Semin. Liver Dis. 2011;31:11–32. doi: 10.1055/s-0031-1272832. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011;20:297–305. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- 70.Mahoney WM, Jr, Fleming JN, Schwartz SM. A unifying hypothesis for scleroderma: identifying a target cell for scleroderma. Curr. Rheumatol. Rep. 2011;13:28–36. doi: 10.1007/s11926-010-0152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei J, Bhattacharyya S, Tourtellotte WG, Varga J. Fibrosis in systemic sclerosis: emerging concepts and implications for targeted therapy. Autoimmun. Rev. 2011;10:267–275. doi: 10.1016/j.autrev.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Omura T, Yoshiyama M, Matsumoto R, Kusuyama T, Enomoto S, Nishiya D, et al. Role of c-Jun NH2-terminal kinase in G-proteincoupled receptor agonist-induced cardiac plasminogen activator inhibitor-1 expression. J Mol Cell Cardiol. 2005;38:583–592. doi: 10.1016/j.yjmcc.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 73.Hao GH, Niu XL, Gao DF, Wei J, Wang NP. Agonists at PPAR-gamma suppress angiotensin II-induced production of plasminogen activator inhibitor-1 and extracellular matrix in rat cardiac fibroblasts. Br J Pharmacol. 2008;153:1409–1419. doi: 10.1038/bjp.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zyadeh FN, Sharma K, Erickson M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose in mediated by autocrine activation of transforming growth factor-β. J Clin Invest. 1994;93:536–542. doi: 10.1172/JCI117004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martin J, Kelly DJ, Mifsud SA, Zhang Y, Cox AJ, See F, et al. Tranilast attenuates cardiac matrix deposition in experimental diabetes: role of transforming growth factor β. Cardiovasc Res. 2005;65:694–701. doi: 10.1016/j.cardiores.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 76.Ohtsu H, Mifune M, Frank GD, Saito S, Inagami T, Kim-Mitsuyama S, et al. Signal-crosstalk between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration of vascular smooth muscle cells stimulated by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:1831–1836. doi: 10.1161/01.ATV.0000175749.41799.9b. [DOI] [PubMed] [Google Scholar]
- 77.Zhou H, Li YJ, Wang M, Zhang LH, Guo BY, Zhao ZS, Meng FL, Deng YG, Wang RY. Involvement of RhoA/ROCK in myocardial fibrosis in a rat model of type 2 diabetes. Acta Pharmacol Sin. 2011;32(8):999–1008. doi: 10.1038/aps.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greenhalgh SN1, Conroy KP, Henderson NC. Healing scars: targeting pericytes to treat fibrosis. QJM. 2014 Apr 10; doi: 10.1093/qjmed/hcu067. [Epub ahead of print]. PMID: 24659747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kelley C, D’Amore P, Hechtman HB, et al. Microvascular pericyte contractility in vitro: Comparison with other cells of the vascular wall. J Cell Biol. 1987;104:483–490. doi: 10.1083/jcb.104.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shepro D, Morel NML. Pericyte physiology. FASEB J. 1993;7:1031–1038. doi: 10.1096/fasebj.7.11.8370472. [DOI] [PubMed] [Google Scholar]
- 81.Rhodin JAG, Fujita H. Capillary growth in the mesentery of normal young rats. Intravital video and electron microscope analyses. J Submicros Cytol Pathol. 1989;21:1–34. [PubMed] [Google Scholar]
- 82.Meyrick B, Fujiwara K, Reid L. Smooth muscle myosin in precursor and mature smooth muscle cells in normal pulmonary arteries and the effect of hypoxia. Exp Lung Res. 1981;2:303–313. doi: 10.3109/01902148109052325. [DOI] [PubMed] [Google Scholar]
- 83.Shi X, Han W, Yamamoto H, Tang W, Lin X, Xiu R, Trune DR, Nuttall AL. The cochlear pericytes. Microcirculation. 2008;15:515–529. doi: 10.1080/10739680802047445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Joyce NC, Haire MF, Palade GE. Contractile proteins in pericytes. II. Immunocytochemical evidence for the presence of two isomyosins in graded concentrations. J Cell Biol. 1985a;100:1387–1395. doi: 10.1083/jcb.100.5.1387. 1985a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Joyce NC, Haire MF, Palade GE. Contractile proteins in pericytes. I. Immunoperoxidase localization of tropomyosin. J Cell Biol. 1985b;100:1379–1386. doi: 10.1083/jcb.100.5.1379. 1985b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams M, Kerkar S, Tyburski JG, Steffes CP, Carlin AM, Wilson RF. The roles of cyclic adenosine monophosphate- and cyclic guanosine monophosphate-dependent protein kinase pathways in hydrogen peroxide-induced contractility of microvascular lung pericytes. J Trauma. 2003;55:677–682. doi: 10.1097/01.TA.0000086180.11523.8D. [DOI] [PubMed] [Google Scholar]
- 87.Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci U S A. 2010;107(51):22290–22295. doi: 10.1073/pnas.1011321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai MH, Jiang MJ. Rho-kinase-mediated regulation of receptor-agonist-stimulated smooth muscle contraction. Pflugers Arch. 2006;453:223–232. doi: 10.1007/s00424-006-0133-y. [DOI] [PubMed] [Google Scholar]
- 89.Patel CA, Rattan S. Cellular regulation of basal tone in internal anal sphincter smooth muscle by RhoA/ROCK. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1747–G1756. doi: 10.1152/ajpgi.00438.2006. [DOI] [PubMed] [Google Scholar]
- 90.Kolyada AY, Riley KN, Herman IM. Rho GTPase signaling modulates cell shape and contractile phenotype in an isoactin-specific manner. Am J Physiol Cell Physiol. 2003;285:C1116–C1121. doi: 10.1152/ajpcell.00177.2003. [DOI] [PubMed] [Google Scholar]
- 91.Kutcher ME, Kolyada AY, Surks HK, Herman IM. Pericyte Rho GTPase mediates both pericyte contractile phenotype and capillary endothelial growth state. Am J Pathol. 2007;171:693–701. doi: 10.2353/ajpath.2007.070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ferrati S, Fine D, You J, De Rosa E, Hudson L, Zabre E, Hosali S, Zhang L, Hickman C, Sunder Bansal S, Cordero-Reyes AM, Geninatti T, Sih J, Goodall R, Palapattu G, Kloc M, Ghobrial RM, Ferrari M, Grattoni A. Leveraging nanochannels for universal, zero-order drug delivery in vivo. J Control Release. 2013;172(3):1011–1019. doi: 10.1016/j.jconrel.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 93.Kloc M, Ghobrial RM. Chronic allograft rejection: a significant hurdle to transplant success. Burns & Trauma. 2014;2:3–10. doi: 10.4103/2321-3868.121646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Skelton ST, Tejpal N, Gong Y, Kloc M, Ghobrial RM. Downregulation of RhoA and changes in T cell cytoskeleton correlate with the abrogation of allograft rejection. Transplant Immunol. 2010;23:185–193. doi: 10.1016/j.trim.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang L, Kloc M, Tejpal N, You J, Cordero-Reyes AM, Youker KA, Ghobrial RM. ROCK1 inhibitor abrogates chronic rejection in rat cardiac model system. Open J Organ Transp Surg. 2012;2:46–51. [Google Scholar]
- 96.Fuxe J, Tabruyn S, Colton K, Zaid H, Adams A, Baluk P, Lashnits E, Morisada T, Le T, O'Brien S, Epstein DM, Koh GY, McDonald DM. Pericyte requirement for anti-leak action of angiopoietin-1 and vascular remodeling in sustained inflammation. Am J Pathol. 2011;178:2897–2909. doi: 10.1016/j.ajpath.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. 2009. [DOI] [PubMed] [Google Scholar]
- 98.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buijs JT, Stayrook KR, Guise TA. The role of TGF-β in bone metastasis: novel therapeutic perspectives. Bonekey Rep. 2012;1:96. doi: 10.1038/bonekey.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen S, Crawford M, Day RM, Briones VR, Leader JE, Jose PA, Lechleider RJ. RhoA modulates Smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J Biol Chem. 2006;281:1765–1770. doi: 10.1074/jbc.M507771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 102.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 103.Sato Y, Rifkin DB. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J. Cell Biol. 1989;109:309–315. doi: 10.1083/jcb.109.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 2000;26:328–331. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 105.Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P, Xu X, ten Dijke P, Mummery CL, Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO. J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lan Y, Liu B, Yao H, Li F, Weng T, Yang G, Li W, Cheng X, Mao N, Yang X. Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol. Cell. Biol. 2007;27:7683–7692. doi: 10.1128/MCB.00577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A. Smad5 knockout mice die at mid-gestation due to multiple embryonic and extraembryonic defects. Development. 1999;126:1631–1642. doi: 10.1242/dev.126.8.1631. [DOI] [PubMed] [Google Scholar]
- 108.Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23. doi: 10.1007/s00441-003-0745-x. [DOI] [PubMed] [Google Scholar]
- 109.Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, Han H, Meng A, Wang Y, Yang X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell. 2011;20:291–302. doi: 10.1016/j.devcel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 110.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. 2003. [DOI] [PubMed] [Google Scholar]
- 111.Loughna S, Sato TN. Angiopoietin and Tie signaling pathways in vascular development. Matrix Biol. 2001;20:319–325. doi: 10.1016/s0945-053x(01)00149-4. [DOI] [PubMed] [Google Scholar]
- 112.Eklund L, Saharinen P. Angiopoietin signaling in the vasculature. Exp Cell Res. 2013;319:1271–1280. doi: 10.1016/j.yexcr.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 113.Sundberg C, Kowanetz M, Brown LF, Detmar M, Dvorak HF. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab. Invest. 2002;82:387–401. doi: 10.1038/labinvest.3780433. [DOI] [PubMed] [Google Scholar]
- 114.Cai J, Kehoe O, Smith GM, Hykin P, Boulton ME. The angiopoietin/Tie-2 system regulates pericyte survival and recruitment in diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2008;49:2163–2171. doi: 10.1167/iovs.07-1206. [DOI] [PubMed] [Google Scholar]
- 115.Patan S. TIE1 and TIE2 receptor tyrosine kinases inversely regulate embryonic angiogenesis by the mechanism of intussusceptive microvascular growth. Microvasc. Res. 1998;56:1–21. doi: 10.1006/mvre.1998.2081. [DOI] [PubMed] [Google Scholar]
- 116.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 117.Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U. Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53:1104–1110. doi: 10.2337/diabetes.53.4.1104. [DOI] [PubMed] [Google Scholar]
- 118.Pfister F, Feng Y, vom Hagen F, Hoffmann S, Molema G, Hillebrands JL, Shani M, Deutsch U, Hammes HP. Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes. 2008;57:2495–2502. doi: 10.2337/db08-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tachibana K, Jones N, Dumont DJ, Puri MC, Bernstein A. Selective role of a distinct tyrosine residue on Tie2 in heart development and early hematopoiesis. Mol. Cell. Biol. 2005;25:4693–4702. doi: 10.1128/MCB.25.11.4693-4702.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, Quaggin SE. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Invest. 2011;121:2278–2289. doi: 10.1172/JCI46322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–5101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cascone I, Audero E, Giraudo E, Napione L, Maniero F, Philips MR, Collard JG, Serini G, Bussolino F. Tie-2-dependent activation of RhoA and Rac1 participates in endothelial cell motility triggered by angiopoietin-1. Blood. 2003;102:2482–2490. doi: 10.1182/blood-2003-03-0670. [DOI] [PubMed] [Google Scholar]
- 123.Olson LE, Soriano P. PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev. Cell. 2011;20:815–826. doi: 10.1016/j.devcel.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, Chiang WC, Kuhnert F, Kuo CJ, Chen YM, Wu KD, Tsai TJ, Duffield JS. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am. J. Pathol. 2011;178:911–923. doi: 10.1016/j.ajpath.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang S, Cao C, Chen Z, Bankaitis V, Tzima E, et al. Pericytes Regulate Vascular Basement Membrane Remodeling and Govern Neutrophil Extravasation during Inflammation. PLoS ONE. 2012;7:e45499. doi: 10.1371/journal.pone.0045499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Perrot V, Vazquez-Prado J, Gutkind JS. Plexin B regulates Rho through the guanine nucleotide exchange factors leukemia-associated Rho GEF (LARG) and PDZ-RhoGEF. J Biol Chem. 2002;277:43115–43120. doi: 10.1074/jbc.M206005200. [DOI] [PubMed] [Google Scholar]
- 127.Zhou H, Yang YH, Basile JR. The Semaphorin 4D-Plexin-B1-RhoA signaling axis recruits pericytes and regulates vascular permeability through endothelial production of PDGF-B and ANGPTL4. Angiogenesis. 2014;17:261–274. doi: 10.1007/s10456-013-9395-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 128.Yadav R, Larbi KY, Young RE, Nourshargh S. Migration of leukocytes through the vessel wall and beyond. Thromb. Haemost. 2003;90:598–606. doi: 10.1160/TH03-04-0220. [DOI] [PubMed] [Google Scholar]
- 129.Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 2008;20:242–248. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. http://www.selleckchem.com/ROCK.html. [Google Scholar]